

Abstract
The RNA exosome is the major 3′-5′ RNA degradation machine of eukaryotic cells and participates in processing, surveillance and turnover of both nuclear and cytoplasmic RNA. In both yeast and human, all nuclear functions of the exosome require the RNA helicase MTR4. We show that the Arabidopsis core exosome can associate with two related RNA helicases, AtMTR4 and HEN2. Reciprocal co-immunoprecipitation shows that each of the RNA helicases co-purifies with the exosome core complex and with distinct sets of specific proteins. While AtMTR4 is a predominantly nucleolar protein, HEN2 is located in the nucleoplasm and appears to be excluded from nucleoli. We have previously shown that the major role of AtMTR4 is the degradation of rRNA precursors and rRNA maturation by-products. Here, we demonstrate that HEN2 is involved in the degradation of a large number of polyadenylated nuclear exosome substrates such as snoRNA and miRNA precursors, incompletely spliced mRNAs, and spurious transcripts produced from pseudogenes and intergenic regions. Only a weak accumulation of these exosome substrate targets is observed in mtr4 mutants, suggesting that MTR4 can contribute, but plays rather a minor role for the degradation of non-ribosomal RNAs and cryptic transcripts in Arabidopsis. Consistently, transgene post-transcriptional gene silencing (PTGS) is marginally affected in mtr4 mutants, but increased in hen2 mutants, suggesting that it is mostly the nucleoplasmic exosome that degrades aberrant transgene RNAs to limit their entry in the PTGS pathway. Interestingly, HEN2 is conserved throughout green algae, mosses and land plants but absent from metazoans and other eukaryotic lineages. Our data indicate that, in contrast to human and yeast, plants have two functionally specialized RNA helicases that assist the exosome in the degradation of specific nucleolar and nucleoplasmic RNA populations, respectively.
Author Summary
Cells rely on a number of RNA degradation pathways to ensure correct and timely processing and turnover of both coding and non-coding RNAs. Another important function of RNA degradation is the rapid elimination of misprocessed RNA species, maturation by-products, and nonfunctional RNAs that are frequently produced by pervasive transcription. The main 3′-5′ RNA degradation machine in eukaryotic cells is the exosome, which is activated by cofactors such as RNA helicases. In yeast and human, processing, turnover and surveillance of all nuclear exosome targets depend on a single RNA helicase, MTR4. We show here that the Arabidopsis exosome complex can associate with two related RNA helicases, MTR4 and HEN2. MTR4 and HEN2 reside in nucleolar and nucleoplasmic compartments, respectively, and target different subsets of nuclear RNA substrates for degradation by the exosome. The presence of both MTR4 and HEN2 homologues in green algae, mosses and land plants suggest that the functional duality of exosome-associated RNA helicases is evolutionarily conserved in the entire green lineage. The emerging picture is that, despite a high degree of sequence conservation, intracellular distribution, activities and functions of exosome cofactors vary considerably among different eukaryotes.
Introduction
Efficient processing and degradation of RNA is a key process for the post-transcriptional control of gene expression. The main 3′-5′ RNA degradation machine of eukaryotic cells is the exosome, a multi-subunit complex found in both cytoplasm and nuclear compartments [1], [2]. The exosome participates in a plethora of processing and degradation reactions, including the processing of ribosomal RNAs, snoRNAs and snRNAs, the turnover and quality control of mRNAs and the efficient elimination of RNA maturation by-products and diverse RNA species generated from non-genic regions [3]–[5]. In vivo, exosome activity requires the interaction of the exosome complex with associated RNA helicases. In yeast, cytoplasmic and nuclear exosomes are activated by the RNA helicases SKI2 and MTR4, respectively [6]–[8]. In both yeast and human MTR4 is an essential protein required for all functions of the nuclear exosome [9], [10]. Interestingly, Arabidopsis thaliana has two MTR4 homologues, designated AtMTR4 and HEN2 [11]–[13]. We have previously shown that AtMTR4 (encoded by At1g59760) is a predominantly nucleolar protein required for the efficient degradation of misprocessed 5.8S rRNA precursors and specific fragments of the 5′ external transcribed spacer (5′ ETS), a by-product released during processing of three rRNAs from their common precursor transcript [13]. The requirement for AtMTR4 in efficient rRNA production is reflected by the phenotype of mtr4 mutants, which show a characteristic combination of developmental growth defects also observed in ribosomal protein mutants and in other Arabidopsis mutants lacking putative ribosome biogenesis factors such as nucleolin [14]–[19].
HEN2 (HUA enhancer 2, At2g06990) was originally identified in a genetic screen for mutations that enhance the flower morphology defects observed in hua1 and hua2 mutants [12]. A follow-up study showed that hen2 single mutants accumulate, as compared to wild type plants, slightly higher levels of a polyadenylated transcript comprising the two first exons and a large portion of the second intron of the AGAMOUS gene product, suggesting that the HEN2 protein could be involved in the degradation of misprocessed AGAMOUS transcripts [20]. These data and the strong homology with the exosome activator MTR4 prompted us to examine possible roles of HEN2 in exosome-mediated RNA degradation in Arabidopsis thaliana. In contrast to AtMTR4, HEN2 is not required for processing or degradation of 5.8S rRNA precursors or the elimination of the 5′ ETS [13]. We show here that HEN2 is a nucleoplasmic protein that is associated with the Arabidopsis exosome core complex and has a specific role in the exosome-mediated degradation of non-coding RNAs, misprocessed mRNAs, introns and transcripts derived from retrotransposons and non-genic regions. Interestingly and as recently reported for human MTR4 [9], [21], HEN2 associates with homologues of the NEXT (for Nuclear Exosome Targeting) complex components and also co-purifies with the cap-binding complex. MTR4, by contrast, is associated with a distinct set of proteins, many of which appear to be involved in ribosome biogenesis. Our results indicate a high degree of spatial and functional specialization of exosome activating RNA helicases in Arabidopsis.
Results
Both ATMTR4 and HEN2 are associated with plant exosome complexes
Several co-factors of the Arabidopsis exosome such as RRP6L1 (RRP6-LIKE 1), RRP6L2, MTR4 and DIS3/RRP44 have been identified based on sequence homology with yeast counterparts and are genetically linked with nuclear exosome functions [13], [22]–[25]. However, none of them has yet been shown to physically interact with the exosome. To better define the composition of plant exosome complexes, we used myc-tagged and GFP-tagged versions of the exosome core subunit RRP41 as baits in co-immunoprecipitation (IP) experiments. RRP41 fusion proteins were expressed under the control of the 1 kb genomic region upstream of the endogenous RRP41 gene. Both myc-tagged RRP41 and GFP-tagged RRP41 constructs were able to complement the otherwise lethal rrp41-X null mutation. A similar full phenotypic complementation was previously reported for TAP-tagged RRP41 [5]. Complementation of the null mutation by both RRP41-myc and RRP41-GFP suggested that both fusion proteins can be integrated in the core exosome complex. To test this hypothesis and identify potential exosome co-factors, tagged-RRP41 and associated proteins were affinity-purified using superparamagnetic particles coated with anti-myc or anti-GFP monoclonal antibodies, respectively. As shown for myc-tagged RRP41 IP, a specific group of proteins were visualized on silver-stained SDS-PAGE gel as compared with mock IP (Fig. 1). The proteins co-purifying with RRP41 were identified by mass spectrometry (nano LC-MS/MS) analysis. A final list of 14 proteins was established by excluding proteins present in mock purifications and by crossing the datasets of three biological repeats (Table 1). All 14 proteins were identified with both Mascot and PEAKS DB algorithms with a false discovery rate <1%. An exhaustive list of the peptides shared by the three RRP41 IPs is shown in Table S1.
Figure 1. Purification of exosome complexes.
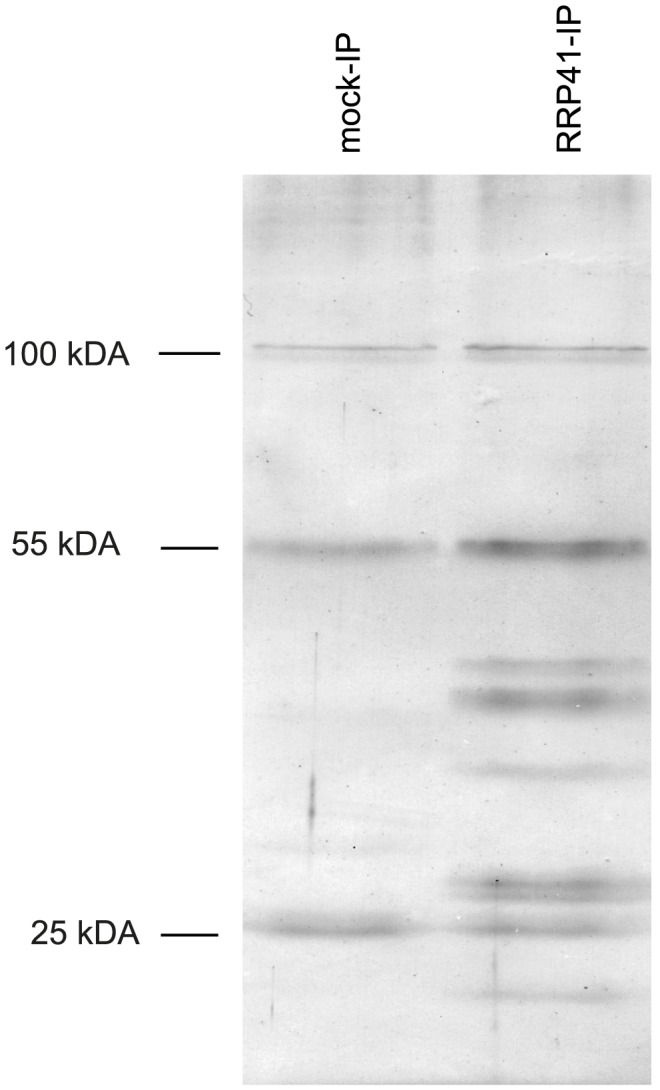
Silver-stained SDS-PAGE of proteins co-immunoprecipitated with RRP41-myc. Similar results were obtained when GFP-tagged RRP41 was used as bait. The three main bands observed in mock IP and also present in RRP41-myc IP correspond to immunoglobulins.
Table 1. Proteins co-purified with tagged RRP41 and identified by LC-MS/MS.
| Experiment 1 | Experiment 2 | Experiment 3 | ||||||
| Accession | Name | Function | Mascotscore | #Spectra | Mascot score | #Spectra | Mascot score | #Spectra |
| AT3G61620 | RRP41 (bait) | EXO9 | 2795.5 | 232 | 1458.4 | 124 | 1104.2 | 84 |
| AT3G07750 | RRP42 | EXO9 | 2682.2 | 230 | 1786.9 | 134 | 981.8 | 59 |
| AT5G38890 | CSL4 | EXO9 | 2338.6 | 221 | 1446 | 150 | 695.8 | 46 |
| AT1G60080 | RRP43 | EXO9 | 2733.2 | 193 | 1699.7 | 118 | 1105.3 | 58 |
| AT3G60500 | RRP45B/CER7 | EXO9 (*) | 3290.3 | 160 | 1528.4 | 138 | 1138.7 | 53 |
| AT2G25355 | RRP40 | EXO9 | 2567.2 | 146 | 1584.9 | 81 | 723.4 | 26 |
| AT4G27490 | MTR3 | EXO9 | 1992.3 | 126 | 1410.7 | 133 | 591.8 | 30 |
| AT1G03360 | RRP4 | EXO9 | 1586.5 | 108 | 988.5 | 73 | 882.3 | 46 |
| AT3G46210 | RRP46 | EXO9 | 1160 | 86 | 655.8 | 68 | 254.1 | 14 |
| AT3G27670 | RST1 | ARM repeat | 996.1 | 36 | 146.2 | 4 | 270.8 | 7 |
| AT3G12990 | RRP45A | EXO9 (*) | 836.8 | 34 | 450.4 | 20 | 283.6 | 12 |
| AT1G59760 | MTR4 | RNA helicase | 188.2 | 6 | 78.2 | 1 | 399 | 6 |
| AT2G06990 | HEN2 | RNA helicase | 222.6 | 5 | 235.2 | 6 | 134.1 | 5 |
| AT2G17510 | RRP44/DIS3 | Ribonuclease | 157.4 | 2 | 166.9 | 4 | 267.3 | 6 |
EXO9, Exosome core complex; ARM repeat, Armadillo-repeat protein; EXO9 (*) alternative isoforms of EXO9 subunit RRP45.
As expected, all nine canonical core subunits of EXO9 (RRP41, RRP42, RRP43, RRP45B, RRP46, MTR3, CSL4, RRP4 and RRP40A) were identified which confirms that we indeed purified intact exosome complexes (Table 1, Table S1). In addition to the nine exosome subunits that were previously characterized [5], five novel proteins were detected, albeit with lower number of spectra reflecting a lower abundance as compared to the canonical EXO9 subunits (Table 1, Table S1).
An isoform of RRP45B, RRP45A, was unambiguously identified by several discriminating peptides (given in bold in Table S1). In Arabidopsis, the RRP45 subunit is encoded by two genes: RRP45A and RRP45B/CER7 [5], [26]. Although single rrp45a and rrp45b mutants are both viable, a double knock out is lethal [26]. The rrp45b/cer7 mutant is characterized by a defect in cuticular wax accumulation, which is not observed in rrp45a mutants, suggesting specialized functions for both isoforms [26]. Only RRP45B/CER7 was previously detected in EXO9 [5], likely because of its higher expression level. Our results show that both isoforms are incorporated in plant exosome core complexes.
A protein of unknown function RESURRECTION 1 (RST1) also consistently co-purified with EXO9 (Table 1, Table S1). Interestingly, RST1 is crucial for cuticular wax accumulation, the very same biological process affected by lack of RRP45B [26], [27]. The potential interaction between RST1 and EXO9 is being assessed and is not the focus of this study.
DIS3/RRP44 was also detected in all three biological replicates indicating that this exoribonuclease can associate with the plant EXO9 core exosome as reported for yeast and animal exosomes [28]–[31]. However, as for the other putative co-factors that we detected by LC-MS/MS, DIS3/RRP44 is not visible on silver-stained gels (Fig. 1) and is detected with only few spectra by LC-MS/MS (Table S1), presumably reflecting a labile association to plant EXO9 under the biochemical conditions used for immunoprecipitation experiments. Since none of the three RRP6L present in Arabidopsis was detected in our analysis, DIS3/RRP44 represents so far the only exoribonuclease whose physical interaction with plant EXO9 can be inferred from mass spectrometry data.
In all three replicates, discriminating peptides indicated that two RNA helicases, AtMTR4 and HEN2, co-purified with EXO9 (Table 1, Table S1). AtMTR4 was previously shown to participate in processing or degradation of ribosomal RNA precursors [13]. HEN2, a close relative of AtMTR4 was also suggested to play a role in RNA degradation [12], [20], but its substrates or its role in exosome-mediated pathways have not been studied yet.
HEN2 is a plant-specific member of the SKI2/MTR4 RNA helicase family
AtMTR4 and HEN2 share 43% identity and 59–60% similarity with yeast MTR4 and with each other, but only 24% identity/39% similarity with SKI2, a cofactor of the cytoplasmic exosome [7], [32], [33]. Structural modeling of HEN2 and AtMTR4 confirmed that both possess an arch domain, a characteristic feature of MTR4/SKI2 RNA helicases [34]–[37]. While the modeled structure of HEN2 matches closely to the structure of yeast MTR4, AtMTR4 has an insertion of nine amino acids in the RNA binding part of the arch domain, the KOW-motif [34], [35]. Interestingly, a similar insertion is present in the KOW motifs of all plant MTR4 proteins investigated (Fig. S1). Other characteristic sequence differences between AtMTR4 and HEN2 concern RecA-domains, the arch domain and the C-terminal helix-loop-helix domain (Fig. S2), respectively, and allow the reliable discrimination of HEN2 and AtMTR4 homologues by sequence alignment algorithms. A search for homologues of AtSKI2, AtMTR4 and HEN2 in all genomes available at www.phytozome.net shows that all three RNA helicases are conserved throughout green algae, mosses and land plants. A phylogenetic analysis of related proteins from animals, fungi and other eukaryotic clades revealed that most organisms possess both a single MTR4 and a single SKI2 protein; however, HEN2 homologues are restricted to the green lineage (Fig. 2). Despite of the short insertion in the KOW motif, plant MTR4 proteins cluster with the MTR4 proteins of other organisms, while HEN2 proteins form a separate clade. Taken together, these data suggest that HEN2 is a plant-specific isoform of the nuclear exosome activator MTR4.
Figure 2. HEN2 proteins are restricted to the green lineage.
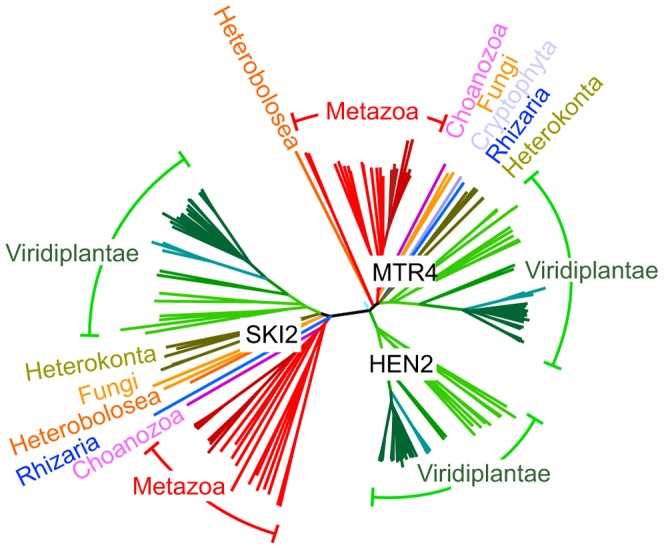
Phylogenetic tree of the MTR4/SKI family of RNA helicases. The basic branch that separates HEN2 homologues from MTR4 and SKI2 homologues is detected with 1000/1000 bootstraps. Protein sequences were retrieved from metazome, phytozome and JGI databases and aligned with ClustalX. Dark red, vertebrates; light red, other eumetazoa; pink, M. brevicollis (Choanozoa); blue, B. natans (Rhizaria); orange, N. gruberi (Heterobolosea); yellow, S. cerevisiae and S. pombe (Fungi); olive, F. cylindrus, P. tricornutum, T. pseudonana; P. cinammomi, P. sojae (Heterokonta); light green, green algae; green, mosses; blue-green, grasses; dark green, dicotyledons. Scale bar = 0.05 amino acid substitutions per site.
AtMTR4 and HEN2 have distinct intranuclear localization patterns
To extend previous localization studies [13], we transiently expressed HEN2 and AtMTR4 GFP fusion proteins in Nicotiana benthamiana leaves, alongside with RFP-labeled XRN2 and Fibrillarin as nucleolar markers, and with SRP34 as a nucleoplasmic marker [38]–[43]. Similar to XRN2-RFP and Fibrillarin-RFP, AtMTR4-GFP was detected in the nucleus, strongly enriched in nucleoli (Fig. S3). HEN2-GFP and SRP34-RFP were detected only in the nucleoplasm (Fig. S4). Next, we determined the intracellular localization of HEN2-GFP in root tips of stable Arabidopsis thaliana transformants. For comparison, we examined roots of plants expressing either AtMTR4-GFP or the exosome core subunits RRP4-GFP and RRP41-GFP. As expected, RRP4-GFP and RRP41-GFP were observed in both cytoplasm and nuclei, and enriched in nucleoli (Fig. 3, Fig. S5). As reported before, AtMTR4-GFP was observed predominantly in nucleoli, and only a faint signal was observed in the nucleoplasm (Fig. 3, Fig. S5) [13]. HEN2-GFP was observed in the nucleoplasm, was enriched in nuclear foci and appeared excluded from nucleoli (Fig. 3, Fig. S5, Fig. S6). These results show that HEN2 is a nucleoplasmic protein, and that AtMTR4 and HEN2 are for the most part located in distinct subnuclear compartments.
Figure 3. MTR4 and HEN2 have distinct localization patterns.
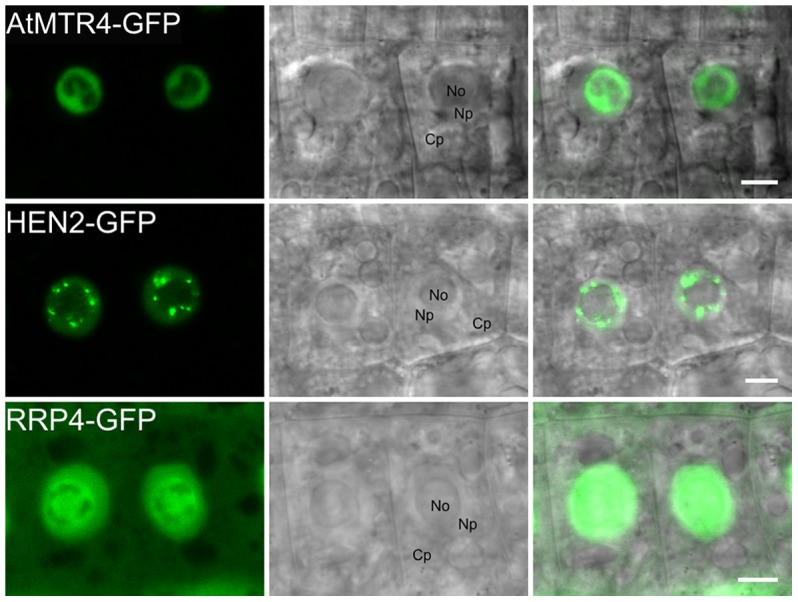
Distribution of GFP-fusion proteins in root cells of stable Arabidopsis transformants. No, Nucleolus; Np, Nucleoplasm; Cp, Cytoplasm. Scale bars 5 µm.
AtMTR4 and HEN2 interact with different sets of proteins
To investigate whether AtMTR4 and HEN2 are associated with specific proteins reflecting their distinct localization and to confirm that both helicases interact with EXO9, we performed IP experiments using plant lines expressing GFP-tagged versions of AtMTR4 or HEN2 in their respective mutant backgrounds. The list of proteins co-purifying with AtMTR4-GFP or HEN2-GFP was established by considering only proteins that were not identified in mock purifications and common to replicate IPs for AtMTR4-GFP and HEN2-GFP experiments, respectively (Tables 2 and 3; Tables S2 and S3).
Table 2. Proteins co-purified with MTR4-GFP and identified by LC-MS/MS.
| Experiment 1 | Experiment 2 | Experiment 3 | ||||||
| Accession | NAME or Annotation | Function | Mascot score | #Spectra | Mascot score | #Spectra | Mascot score | #Spectra |
| AT1G59760 | MTR4 (bait) | 7666.5 | 1332 | 7928.1 | 1352 | 8755.6 | 1999 | |
| AT3G21540 | Transducin family/WD-40 repeat protein | Ribo. bio. | 537.8 | 26 | 606.2 | 29 | 720.0 | 28 |
| AT1G15440 | Periodic tryptophan protein 2 | Ribo. bio. | 718.5 | 27 | 863.7 | 27 | 857.8 | 26 |
| AT3G61620 | RRP41 | EXO9 | 548.2 | 21 | 503.5 | 21 | 592.3 | 26 |
| AT1G63810 | Nrap family, nucleolar protein 6 alpha | Ribo. bio. | 563.3 | 18 | 449.4 | 10 | 907.6 | 26 |
| AT5G11240 | Transducin family/WD-40 repeat protein | 653.5 | 24 | 647.8 | 24 | 730.1 | 24 | |
| AT3G06530 | U3 small nucleolar RNA-associated protein 10 domain | Ribo. bio. | 476.1 | 17 | 597.5 | 19 | 762.8 | 24 |
| AT4G04940 | Transducin family/WD-40 repeat protein | Ribo. bio. | 692.4 | 26 | 576.2 | 22 | 713.4 | 23 |
| AT5G38890 | CSL4 | EXO9 | 360.8 | 14 | 507.5 | 23 | 545.0 | 22 |
| AT3G07750 | RRP42 | EXO9 | 580.3 | 23 | 487.8 | 19 | 576.4 | 22 |
| AT4G07410 | POPCORN | Auxin signaling | 464.9 | 12 | 430.6 | 17 | 618.9 | 21 |
| AT2G18900 | Transducin family/WD-40 repeat protein | 468.6 | 15 | 371.5 | 14 | 639.3 | 21 | |
| AT3G57940 | unknown protein, DUF1726 | Nuc. bind. | 491.5 | 11 | 577.3 | 19 | 737.6 | 19 |
| AT5G16750 | TORMOZ EMBRYO DEFECTIVE | Ribo. bio. | 486.4 | 8 | 611.3 | 13 | 728.9 | 19 |
| AT5G22100 | RNA cyclase family protein | Ribo. bio. | 443.5 | 12 | 441.1 | 16 | 518.3 | 17 |
| AT2G25355 | RRP40 | EXO9 | 429.7 | 15 | 363.8 | 11 | 505.8 | 17 |
| AT4G27490 | MTR3 | EXO9 | 355.5 | 13 | 331.3 | 15 | 342.5 | 15 |
| AT1G10490 | unknown protein, DUF1726 | Nuc. bind. | 454.7 | 11 | 479.9 | 14 | 589.9 | 15 |
| AT1G60080 | RRP43 | EXO9 | 565.1 | 15 | 360.3 | 12 | 558.1 | 15 |
| AT1G03360 | RRP4 | EXO9 | 228.7 | 7 | 192.2 | 5 | 361.8 | 15 |
| AT4G05410 | YAOZHE | Ribo. bio. | 441.3 | 15 | 422.7 | 13 | 372.9 | 13 |
| AT3G60500 | RRP45B/CER7 | EXO9 | 341.9 | 12 | 267.2 | 7 | 474.8 | 12 |
| AT1G63780 | IMP4, putative U3 small nucleolar ribonucleoprotein | Ribo. bio. | 268.3 | 9 | 272.9 | 11 | 307.7 | 11 |
| AT4G28450 | WD-40 repeat protein | Nuc. bind. | 251.6 | 11 | 312.6 | 10 | 285.1 | 11 |
| AT2G47990 | SLOW WALKER1 | Ribo. bio. | 293.0 | 7 | 266.0 | 6 | 358.5 | 11 |
| AT1G06720 | BMS1 domain protein | Ribo. bio. | 269.4 | 5 | 319.4 | 4 | 430.2 | 10 |
| AT4G30990 | ARM repeat family protein | 296.8 | 7 | 334.0 | 6 | 348.0 | 9 | |
| AT3G10530 | Transducin/WD40 repeat protein | Nuc. bind. | 265.2 | 7 | 249.1 | 5 | 298.7 | 9 |
| AT4G28200 | U3 small nucleolar RNA-associated protein 6 family | Ribo. bio. | 338.9 | 7 | 293.3 | 6 | 285.9 | 6 |
| AT3G46210 | RRP46 | EXO9 | 136.5 | 5 | 147.9 | 8 | 154.2 | 5 |
| AT5G30495 | Fcf2 pre-rRNA processing protein | Ribo. bio. | 90.1 | 3 | 113.1 | 6 | 138.8 | 5 |
| AT2G36930 | Zinc finger (C2H2 type) family protein | Nuc. bind. | 75.9 | 1 | 70.8 | 4 | 108.2 | 5 |
| AT5G15750 | Ribosomal protein S4 family protein | Ribo. bio. | 178.6 | 4 | 255.0 | 8 | 237.8 | 4 |
| AT1G69070 | Nop14-like protein | Ribo. bio. | 282.6 | 6 | 112.5 | 3 | 241.9 | 4 |
| AT2G46230 | PIN domain-like family protein | Ribo. bio. | 120.1 | 5 | 42.2 | 1 | 105.0 | 4 |
| AT1G18850 | unknown protein | 141.1 | 3 | 96.4 | 1 | 181.5 | 4 | |
| AT1G30880 | unknown protein | 128.2 | 4 | 139.5 | 3 | 126.4 | 3 | |
| AT4G02400 | U3 ribonucleoprotein (Utp) family protein | Ribo. bio. | 157.7 | 4 | 98.1 | 2 | 119.9 | 3 |
| AT1G31660 | ESSENTIAL NUCLEAR PROTEIN 1 | Ribo. bio. | 163.9 | 6 | 66.4 | 1 | 139.1 | 3 |
| AT1G15420 | contains small-subunit processome Utp12 domain | Ribo. bio. | 80.8 | 2 | 64.0 | 1 | 112.7 | 3 |
| AT4G25340 | FK506 BINDING PROTEIN 53 | Ribo. bio. | 112.2 | 3 | 60.9 | 1 | 64.4 | 2 |
| AT3G57000 | nucleolar essential protein-related, EMG1/NEP1 domain | Ribo. bio. | 130.7 | 3 | 77.4 | 4 | 39.2 | 1 |
| AT2G17250 | NUCLEOLAR COMPLEX ASSOCIATED 4 | Ribo. bio. | 50.6 | 3 | 119.7 | 4 | 43.8 | 1 |
| AT2G03820 | NONSENSE-MEDIATED MRNA DECAY 3 | Ribo. bio. | 35.2 | 1 | 67.7 | 1 | 110.3 | 1 |
Ribo. bio., predicted or known function in ribosome biogenesis; EXO9, subunit of exosome core complex, Nuc.bind., Nucleotide binding.
Table 3. Proteins co-purified with HEN2-GFP at 50 mM and 150 mM ionic strength, and identified by LC-MS/MS.
| 50 mM ionic strength | Experiment 1 | Experiment 2 | ||||
| Accession | Name | Function | Mascot score | #Spectra | Mascot score | #Spectra |
| AT2G06990 | HEN2 (bait) | 2663.5 | 207 | 3365.9 | 209 | |
| AT5G38600 | ZCCHC8 homologue | NEXT | 893.2 | 36 | 953.4 | 40 |
| AT3G61620 | RRP41 | EXO9 | 427.0 | 19 | 530.1 | 17 |
| AT2G13540 | CBP80 | CBC | 507.3 | 12 | 444.9 | 14 |
| AT4G10110 | RBM7 homologue | NEXT | 290.5 | 16 | 417.7 | 11 |
| AT3G07750 | RRP42 | EXO9 | 338.8 | 11 | 528.1 | 10 |
| AT5G38890 | CSL4 | EXO9 | 327.2 | 10 | 341.9 | 9 |
| AT3G60500 | RRP45B/CER7 | EXO9 | 319.9 | 10 | 369.7 | 8 |
| AT4G27490 | MTR3 | EXO9 | 224.6 | 8 | 337.1 | 5 |
| AT2G25355 | RRP40 | EXO9 | 179.3 | 7 | 337.6 | 5 |
| AT1G03360 | RRP4 | EXO9 | 138.1 | 5 | 199.8 | 5 |
| AT1G60080 | RRP43 | EXO9 | 263.7 | 4 | 257.1 | 5 |
| AT3G12990 | RRP45A | EXO9 | 126.4 | 4 | 166.0 | 3 |
| AT5G44200 | CBP20 | CBC | 79.1 | 2 | 49.1 | 2 |
| AT3G46210 | RRP46 | EXO9 | 58.3 | 3 | 100.4 | 1 |
| AT1G67210 | ZCCHC8 homologue | NEXT | 68.2 | 1 | 88.1 | 1 |
| AT1G02140 | MAGO | EJC | 62.3 | 1 | 100.2 | 1 |
EXO9, Exosome core complex; NEXT, putative Nuclear EXosome Targeting Complex; CBC, Cap-Binding Complex; EJC, Exon-Junction Complex.
All canonical nine subunits of EXO9 were identified in both AtMTR4-GFP and HEN2-GFP datasets, which confirmed that both RNA helicases interact with the Arabidopsis exosome complex. Remarkably, EXO9 subunits were the sole common proteins among the 43 and 16 significant proteins present in AtMTR4-GFP and HEN2-GFP IPs, respectively (Tables 2 and 3). A Gene Ontology (GO) analysis for the 34 proteins that were specifically co-purified with AtMTR4-GFP exposed that the most significant biological process GO term was ribosome biogenesis (Benjamini-Hochberg corrected p-value, 1.3E-12), which tagged 9 out of 34 proteins. Further data mining revealed that additional 12 proteins have a proven or predicted role in ribosome biogenesis (Table 2). Nine out of the 13 remaining proteins corresponded to transducin/WD40 repeat proteins and/or proteins involved in nucleic acid metabolism (Table 2, Table S2). These results are in agreement with our previous results [13] and further substantiate the role of AtMTR4 in maturation and/or degradation of ribosomal RNA.
HEN2-GFP co-purified the nine canonical subunits of EXO9, the alternative subunit RRP45A and 6 additional proteins (Table 3, Table S3). One of the six proteins that co-purified with HEN2-GFP was a homologue of the exon junction complex (EJC) component MAGO NASHI. Two proteins were the subunits of the cap binding complex (CBC), CBP80 (AT2G13540) and CBP20 (AT5G44200). Finally we identified three putative RNA binding proteins, two of which encoded by AT5G38600 and AT4G10110 had high spectral counts (Table 3, Table S3). AT5G38600 is a 532 amino acid protein containing the CX2CX4HX4C zinc-knuckle motif (Pfam14392), particularly found in plant proteins [44]. A BLAST analysis against the human proteome identified ZCCHC8 as the best sequence homologue (E-value 4e-26). ZCCHC8 is a zinc-knuckle protein that was recently identified as part of the human Nuclear EXosome Targeting (NEXT) complex [9]. At4G10100 is a small protein of 173 amino acids that shares some similarity is related to the second component of the human NEXT complex, RBM 7, although the similarity is restricted to the N-terminal two-thirds of the 266 amino acids of RBM 7 (26% identity, 27% similarity for the aligned sequence, Fig. S7). To further check whether HEN2, AT5G38600 and AT4G10110 form a NEXT-like complex in Arabidopsis, we slightly increased the stringency of HEN2 immunoprecipitation conditions. By a modest increase of ionic strength from 50 to 150 mM NaCl, the co-purification of EXO9 with HEN2-GFP was lost. However, both AT5G38600 and AT4G10110 were still present in duplicate immunoprecipitations (Table 3, Table S3). Interestingly, a third RNA-binding protein, AT1G67210, was identified in all four HEN2 IPs, albeit with a lower spectral count (Table 3, Table S3). As AT5G38600, AT1G67210 also contains the CX2CX4HX4C zinc-knuckle motif (Pfam14392) and both proteins share 49.6% identities and 57.6% similarities. These results support the existence of a NEXT-like complex in Arabidopsis and raise the interesting possibility that multiple NEXT-like complexes exist in plants.
Taken together, our data show that AtMTR4 and HEN2 are associated with distinct sets of proteins. AtMTR4 co-purifies with the exosome, and with putative ribosome biogenesis factors, which highlights the function of AtMTR4 in pre-rRNA processing and degradation. HEN2 co-purifies with the exosome, the CBC complex and with two types of RNA-binding proteins to form a putative plant NEXT-like complex. These data suggest that the functional link between exosome, CBC and NEXT complexes that was recently established in human cells [21] may be conserved in plants. Furthermore, these results strengthen the evidence for a functional specialization of HEN2 and MTR4.
A selection of known exosome substrates accumulates in hen2 mutants
So far, our data suggested that HEN2 might operate as cofactor of the nucleoplasmic exosome complex. In order to investigate the function of HEN2 for the degradation of exosome substrates, we tested the accumulation of a pseudogene and five non-coding RNAs selected from the list of known polyadenylated plant exosome substrates [5]. Targets comprised the pseudogene At1g79245, the non-coding RNAs MRP and 7SL, the dicistronic precursor of snoRNAs At3g58193 and At3g58196, a non-coding RNA encoded by At2g18440, and intergenic transcripts generated from a repeat region located on chromosome 5 [5]. Additional information and hyperlinks to visualize the selected regions on the SALK transcriptome/exosome website (http://signal.salk.edu/) are provided in Fig. S8. Steady-state levels of the six selected exosome targets were determined by quantitative RT-PCR using oligo-dT primed cDNA samples prepared from seedlings of wild type, mtr4-1, mtr4-2, hen2-2 or hen2-4 mutant plants. We included also samples from RRP41 RNAi lines in which depletion of the exosome core subunit RRP41 is triggered by an inducible RNAi construct [5]. As shown in Fig. 4, all exosome substrates tested in this experiment were over-accumulated in hen2 samples as compared to wild type samples. By contrast, no or only a mild accumulation was observed in mtr4 mutants. These data provided a first indication that HEN2 is involved in the degradation of nuclear exosome targets that are not substrates of AtMTR4.
Figure 4. Loss of HEN2 results in over-accumulation of selected exosome targets.
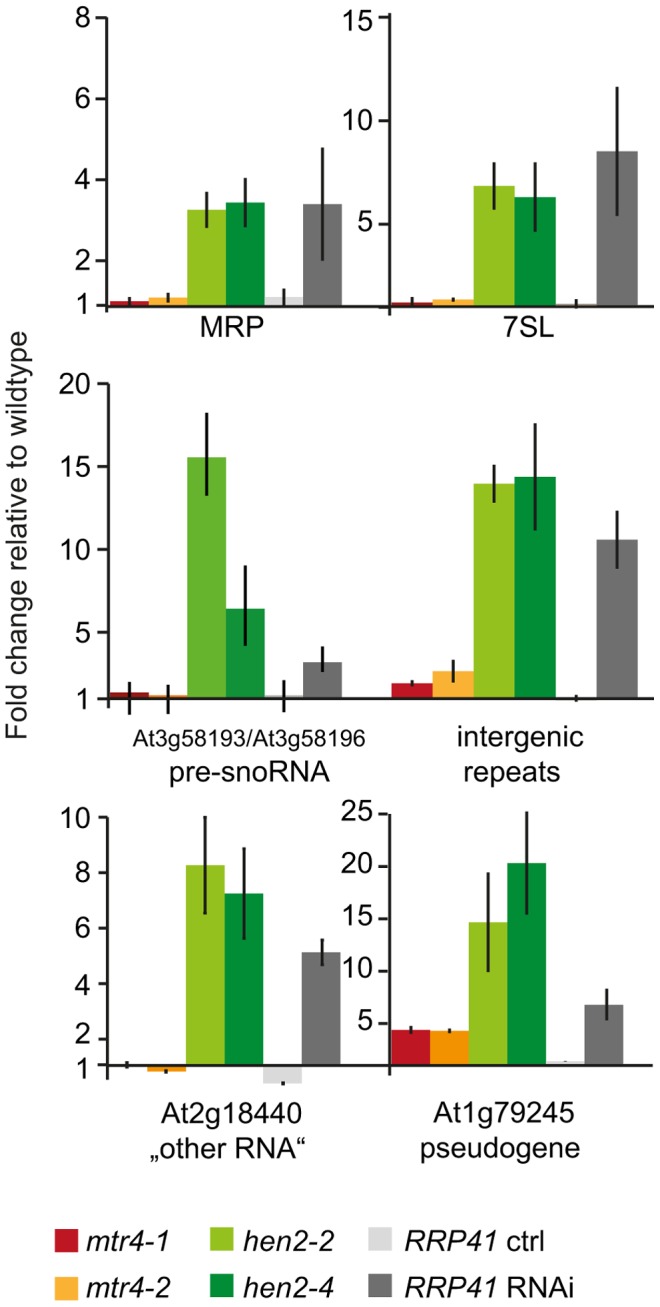
Steady-state levels of exosome targets selected from [5], (see also Fig. S8) in hen2 and mtr4 mutant seedlings were determined by qRT-PCR. Samples from an inducible RRP41 RNAi line grown in absence (RRP41 ctrl) or presence (RRP41 RNAi) of estradiol were included as controls. The histogram shows the fold change relative to wild type. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates.
HEN2 participates in mRNA surveillance
To evaluate the respective contribution of HEN2 and AtMTR4 to the degradation of nuclear exosome substrates in an unbiased manner, we determined the accumulation of polyadenylated transcripts using full-genome (tiling) microarray arrays. For this experiment, cDNA was prepared from two biological replicates of wild type, mtr4-1 and hen2-4 mutants. Each mutant sample was co-hybridized against a wild type sample to NimbleGen A. thaliana 732K whole genome microarrays. The microarray chip contains 1,434,492 strand-specific probes covering both coding and non-coding regions with an average resolution of 175 nt. Probes are 45–85 nt long and designed to achieve a constant Tm of ∼76°C to enhance hybridization consistency across probes. For each biological replicate, expression of each mutant was compared to the expression of the wild type. The statistical analysis, based on a 4-state Hidden Markov Chain, classified probes into four clusters corresponding to over-expressed probes, under-expressed probes, probes with unchanged expression, and noise (not expressed probes), respectively. Interestingly, the analysis did not declare any probe as under-expressed in both biological replicates. This result is in line with the prediction that loss of HEN2 and AtMTR4 impairs RNA degradation, and therefore results predominantly in an increased accumulation of RNA substrates. Indeed, signals for 1860 unique probes were significantly increased in both biological replicates of hen2 samples. 499 probes were identified as overexpressed in both biological replicates of mtr4 samples. A file allowing the visualization of the upregulated probes aligned to the Arabidopsis genome can be found in dataset S1. For the further analysis, we sorted the probes according to their genome coordinates to identify upregulated regions. Only regions with at least two consecutive probes were considered for interpretation. Upregulated regions were then grouped with respect to annotated features taking into account both TAIR10 annotated genes and recently identified genes encoding snoRNAs, miRNAs and lincRNAs [45]–[47]. This procedure identified 387 regions, the majority of which was upregulated exclusively in hen2 samples (Tables S4, S5, S6, S7, S8, S9, S10, S11).
237 of the upregulated regions mapped to protein coding genes. However, for the majority of the cases, (149 regions, 112 of which were only observed in hen2 samples, Table S4), the upregulated transcripts were apparently not mature mRNAs. In fact, most of the upregulated regions corresponded to short portions of protein coding genes (Table S4). The upregulation of short regions located in the 3′ portion of protein coding genes was validated by qRT-PCR analysis for three examples (Fig. 5). As a positive control of exosome-mediated RNA degradation, we used the RRP41 RNAi line. In all three cases, we indeed observed the accumulation of transcripts corresponding to 3′ regions in both the hen2 and the rrp41 samples (Fig. 5). Another group of upregulated regions mapped within the body of the transcripts and beyond mature 3′ ends (Fig. 6, Table S4), indicative of alternative 3′ end processing or readthrough transcription. Furthermore, many upregulated regions contained both exonic and intronic sequences, suggesting the accumulation of incompletely spliced transcripts. To test this possibility, we compared by qRT-PCR the steady-state levels of individual exons, introns, regions comprising unspliced intron-exon junctions and correctly spliced transcripts from selected loci (Fig. 7, Fig. S9). These experiments confirmed the overaccumulation of transcripts comprising unspliced donor or acceptor sites in two independent T-DNA insertion alleles of HEN2 and in RRP41 RNAi plants (Fig. 7A, Fig. S9). For most loci, we detected the upregulation of both unspliced and spliced transcripts (albeit to different levels, please note the scales in Fig. 7 and S9), suggesting that heterogeneous transcripts are produced and targeted for degradation. In order to map the 3′ extremities of the unspliced transcripts, we amplified and sequenced transcripts derived from the At1g79270 locus by 3′ RACE-PCR (Fig. 7B). cDNA synthesis was initiated by oligo dT, and PCR products were amplified with nested forward primers situated in the 3′ region of the first exon, and the adapter sequence of cDNA synthesis primer as a reverse primer. All PCR products amplified from WT, mtr4 or RRP41 control plants corresponded to the fully spliced mature mRNA (Fig. 7B). By contrast, a smaller product of only about 500 bp was amplified from hen2 or RRP41 RNAi plants, and corresponded to a population of transcripts that comprised the unspliced donor site of the first intron (Fig. 7B, Fig. S10), while 3′ extremities were close to the acceptor site. Of the 20 clones that were obtained from the hen2-4 sample 18 were polyadenylated at or close to the intron acceptor site (Fig. S10), indicating that they are indeed marked for degradation by the nuclear exosome. The remaining 2 clones had polyadenylation sites 8 and 52 nt upstream of the acceptor site and likely represent degradation intermediates (Fig. S10). Hence, both qRT-PCR and 3′ RACE results confirmed the results of the microarray analysis and show that polyadenylated transcripts with incorrectly spliced introns accumulate in hen2 plants.
Figure 5. hen2 mutants accumulate short transcripts derived from mRNA genes.
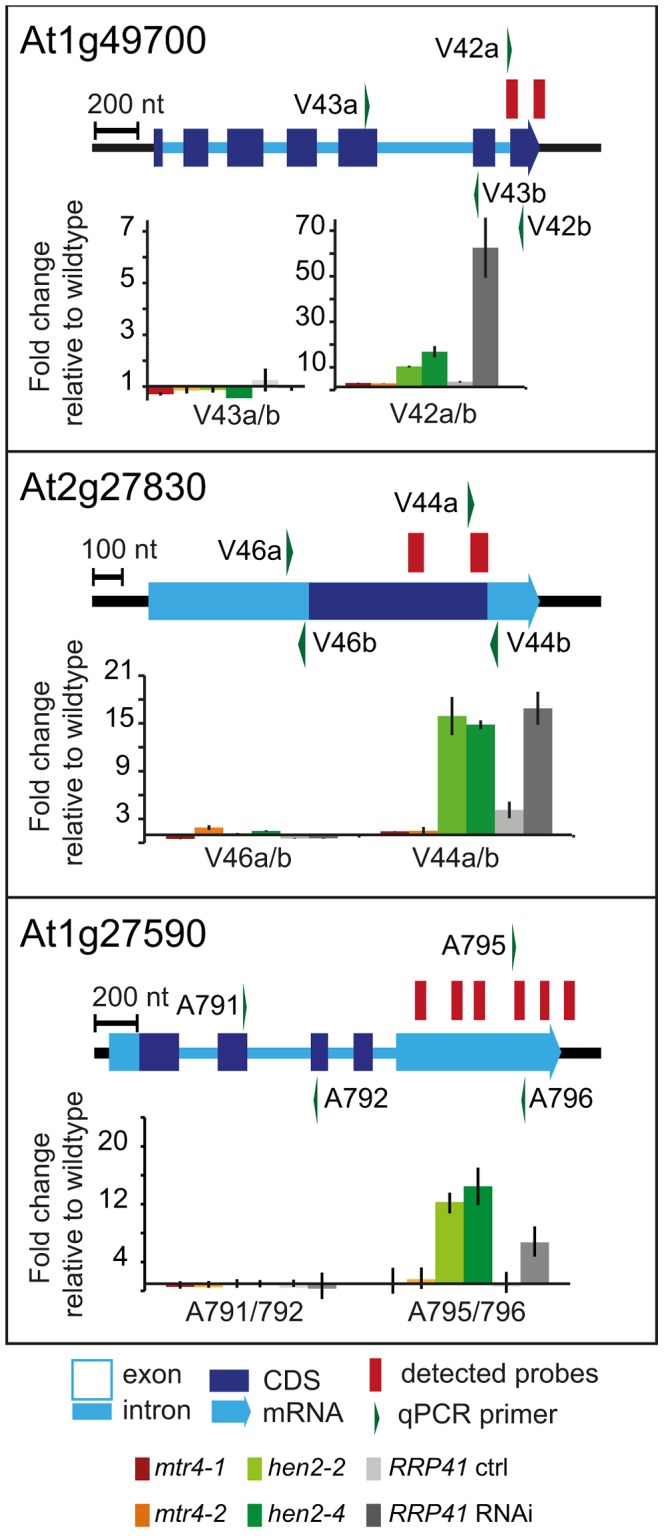
qRT-PCR analysis. A diagram of the genomic locus indicated by the respective AGI number is shown at the top of each panel. Annotated mRNA genes are represented as arrows with dark blue boxes for the CDS, light blue boxes for 3′ and 5′ UTRs, and a light blue line for introns. Red bars above the diagram represent probes detected in the tiling analysis. Green arrows above or below the diagram depict the location of qRT-PCR primers. The corresponding qRT-PCR results for each primer pair are given as fold-change relative to WT in the histograms below each diagram. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates.
Figure 6. Accumulation of 3′ extended mRNAs in hen2 mutants.
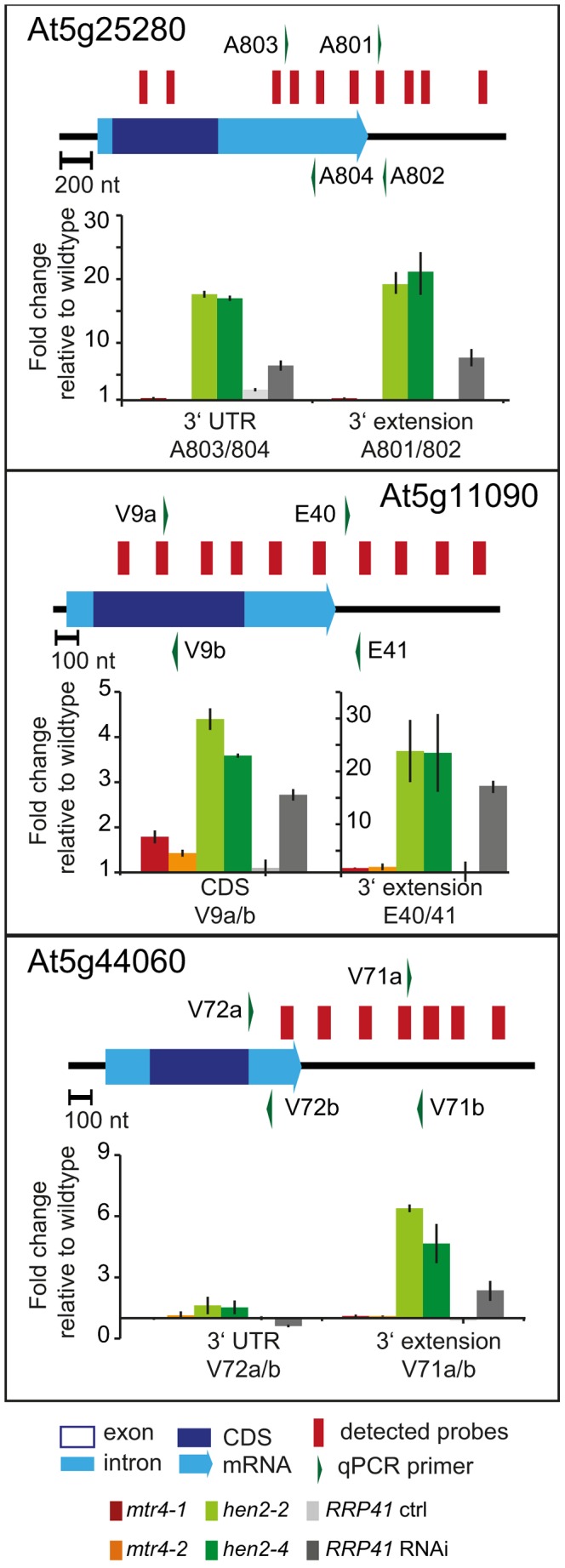
qRT-PCR experiments to test the upregulation of regions that mapped within the body and beyond annotated 3′ ends of mRNAs. Please see legend of Fig. 5 for a detailed explanation of the diagrams.
Figure 7. hen2 mutants accumulate incompletely spliced transcripts.
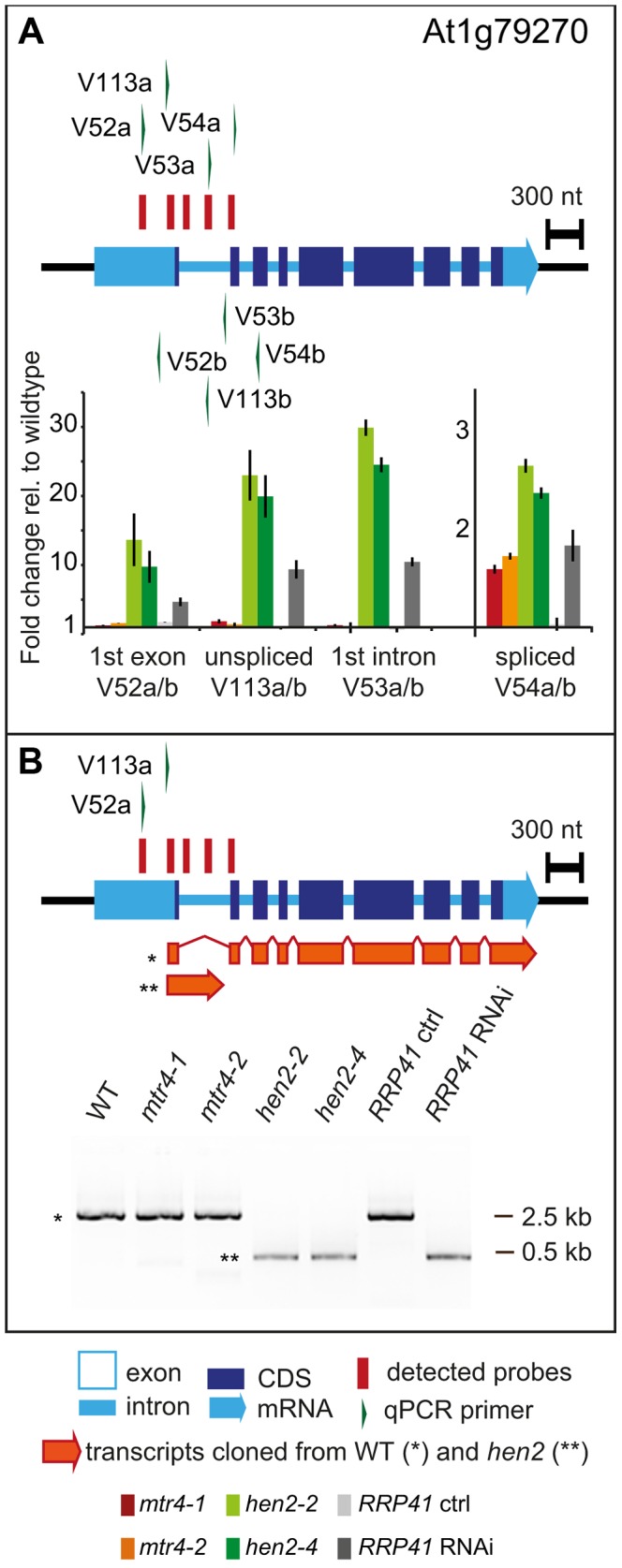
A qRT-PCR. The diagram shows the At1g79170 locus. The At1g79170 mRNA is represented as an arrow with dark blue boxes for the CDS, light blue boxes for 3′ and 5‚ UTRs, and a light blue line for introns. Red bars above the diagram represent probes detected in the microarray analysis. Green arrows above or below the diagram depict the location of qRT-PCR primers. The corresponding qRT-PCR results for each primer pair are given as fold-change relative to WT in the histograms below each diagram. Please note the scales. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates. B 3′ RACE PCR. The diagram shows the At1g79170 locus. Forward primers used for 3′ RACE-PCR on oligo-dT-primed cDNA are shown as green arrows above the diagram. A negative stain of PCR products separated on a 1.5% agarose gel is shown on the bottom. The upper band marked by a star corresponded to the fully spliced mRNA as depicted by the long orange arrow below the diagram. The lower band marked by two stars corresponded to transcripts depicted by the short orange arrow, all of which comprised the unspliced donor site of the first exon/intron junction. The 3′ extremities of these transcripts were located at or upstream of the 3′ acceptor site and were polyadenylated (see Fig. S10).
The tiling data suggested that HEN2 also participates in the elimination of excised introns. In fact, 34 of the 237 protein coding regions that were detected in the microarray analysis corresponded exclusively to intron regions (Table S5), 28 of which were only observed upon loss of HEN2. As for incompletely spliced transcripts, qRT-PCR experiments confirmed the accumulation of intronic regions in two independent alleles of hen2 and in RRP41 RNAi samples (Fig. 8). Finally, 54 regions detected by the tiling analysis likely corresponded to mature mRNAs since they were regions with all probes in exons, and the detected regions spanned at least 50% of the mRNA. 33 mRNAs were detected in hen2, 9 mRNAs in mtr4, and 12 in both hen2 and mtr4 samples (Table S6). The upregulation of the pseudogene At1g79245 (Fig. 4) and several other mRNAs in hen2 and mtr4 samples was validated by qRT-PCR (Fig. S11). However, not all of the tested mRNAs were also found upregulated in RRP41 RNAi samples (Fig. S11). Moreover, none of the positively tested mRNAs was previously identified as an exosome-regulated mRNA [5]. Finally, some mRNAs were only detected in the very same samples that have been used for the microarray, but not in other mutant plants grown in the same culture conditions (Fig. S12). This inconsistence was in sharp contrast to all other types of substrates that were tested in the course of the study, which were reproducibly detected in all replicates, in independent hen2 T-DNA insertion mutants, and in RRP41 RNAi lines. Therefore, we doubt that all of the mRNAs that were detected in our tiling analysis represent true substrates of exosome-mediated decay. Although nuclear degradation can probably contribute to mRNA degradation [3], [5], [48], the upregulation of mRNAs can also be explained by indirect effects, e.g. a differential response of WT and mutant plants to growth conditions. Indeed, data mining revealed that many of the mRNAs detected by the tiling arrays are linked to stress response (Table S12). Hence, the majority of the mRNAs that we detected by the tiling array are probably not bona fide substrates of HEN2 or the nuclear exosome. By contrast, the upregulation of short mRNA-derived transcripts (Fig. 5, Table S4), 3′ extended mRNAs (Fig. 6, Table S4), unspliced transcripts, (Fig. 7, Table S4, Fig. S9, Fig. S10), introns (Fig. 8, Table S5) is consistently detected in all replicates of both mutant alleles of hen2 and in RRP41 RNAi samples, and can be considered as bona fide substrates of the nuclear exosome and the RNA helicase HEN2.
Figure 8. hen2 mutants accumulate regions of excised introns.
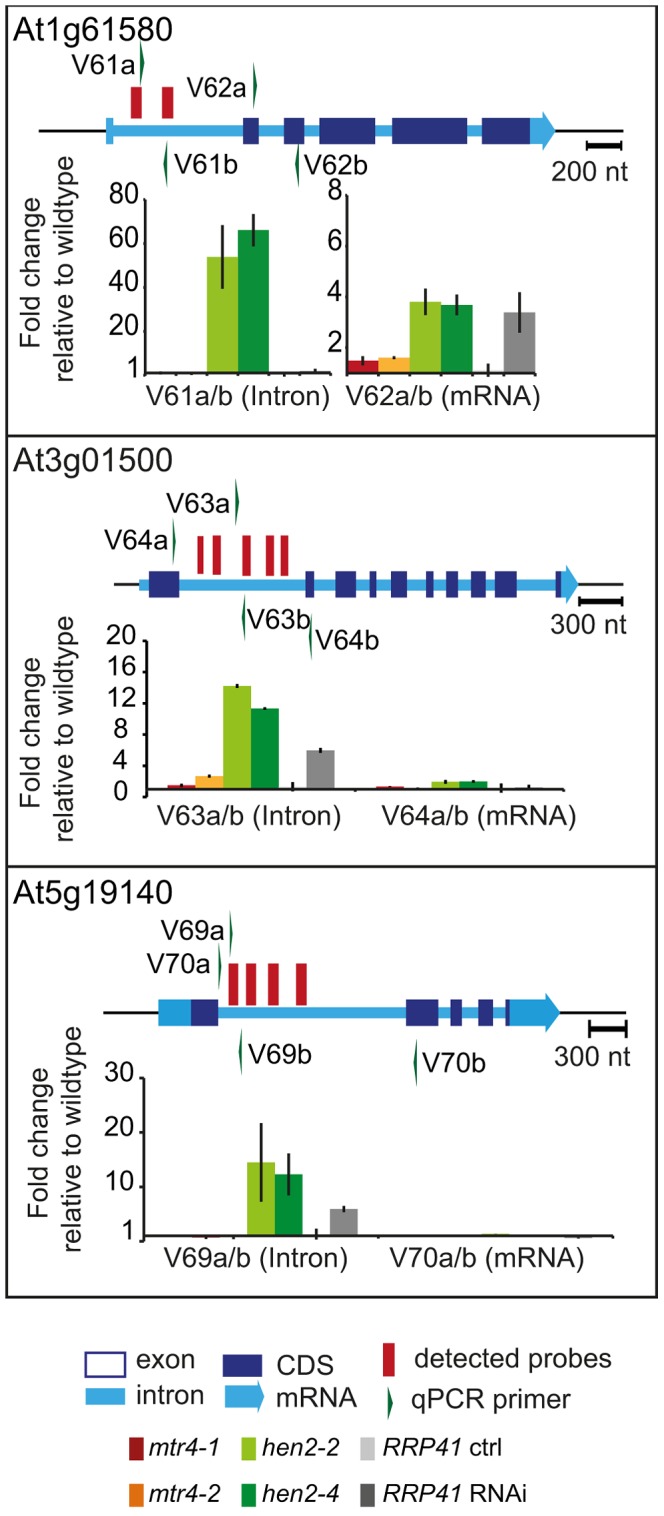
qRT-PCR experiments to validate the upregulation of regions located within introns. Primers pairs V62a, V64a/b and V70ab are exon spanning and produce amplicons only from spliced mRNAs under the conditions used for qRT-PCR. Please refer to the legend of Fig. 5 for a detailed explanation of the diagrams.
Taken together, our data indicate that loss of HEN2 results in the accumulation of short transcripts derived from mRNA regions, 3′ extended transcripts, incompletely spliced mRNA transcripts, and excised introns. The polyadenylated status of the accumulated transcripts indicates that they are tagged for degradation by the exosome, and indeed, all these classes of mRNA-derived transcripts have been described as exosome targets [5]. Hence, the most straightforward explanation for the accumulation of these transcripts in hen2 mutants is that HEN2 is required for the exosome-mediated elimination of different types of probably unfunctional RNAs that are generated from protein coding genes. Only a small number of such transcripts were observed in mtr4 mutants (Table S4, S5) and accumulated at lower levels (Fig. 5–8, Fig. S9), indicating that MTR4, as compared with HEN2, has a rather minor contribution to nuclear mRNA surveillance.
HEN2 is the major RNA helicase for the degradation of non-coding nuclear exosome substrates
Next, we examined the contribution of HEN2 and MTR4 to the degradation of non-coding transcripts. Of the 387 upregulated regions detected in the microarray analysis, 150 regions mapped to non-coding regions. 9 regions mapped to transposable elements (6 in hen2, 3 in mtr4) and were not further investigated (Table S7). 45 regions, all of which were exclusively observed in hen2 mutants, contained one or more snoRNA genes (Table S8), including the regions encoding snoRNAs At3g58193 and At3g58196 (Fig. 4). In fact, almost all of the snoRNA regions detected in our tiling array have also been previously identified as exosome substrates (see Table S8 last column, and [5]). To further investigate the contribution of HEN2 to the degradation of snoRNA precursors, we tested two additional snoRNA regions by qRT-PCR (Fig. 9). The results indicated a strong accumulation of snoRNA precursors in both hen2 mutant lines and in RRP41 RNAi samples. The preferential accumulation of snoRNA precursor transcripts was further confirmed by transferring 3′ RACE-PCR products to membranes followed by hybridisation with radiolabelled probes (Fig. S13). These data strongly indicate that HEN2, but not MTR4, plays an important role for the degradation of snoRNA precursors.
Figure 9. hen2 mutants accumulate snoRNA precursors.
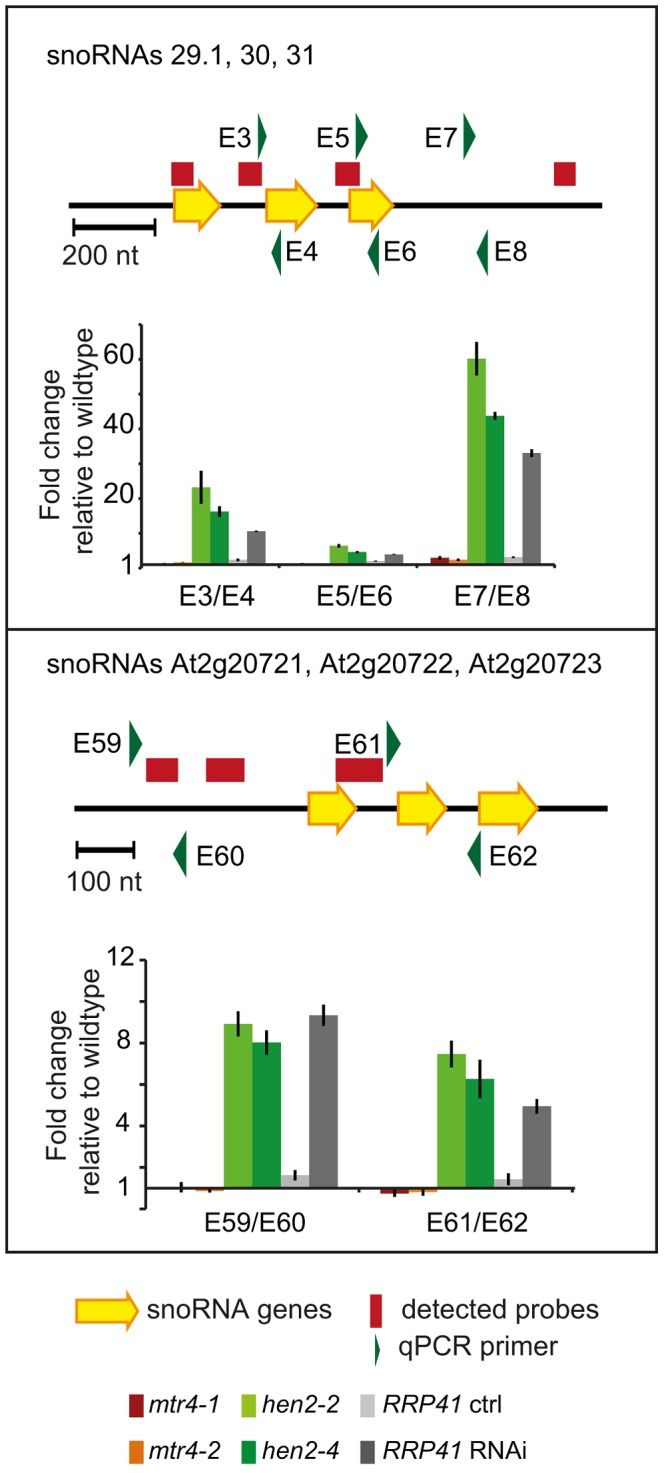
qRT-PCR. A diagram of the genomic locus with the indicated snoRNA genes is shown at the top of each panel. Individual snoRNA genes are represented as yellow arrows. Red bars above the diagram represent probes detected in the tiling analysis. Green arrows above or below the diagram depict the location of qRT-PCR primers. The corresponding qRT-PCR results for each primer pair are given as fold-change relative to WT in the histograms below each diagram. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates.
Other non-coding RNA regions were also unequally distributed between hen2 and mtr4 samples. 22 regions in hen2 samples encoded lincRNAs, putative miRNA precursors or other non-coding RNAs (Table S9), among them a portion of the region encoding GUT15 (for gene with unstable transcript 15)/At2g18440 (Fig. 4). Only 2 of such non-coding RNA regions were observed in mtr4 samples (Table S9), indicating that MTR4 plays a minor role for the degradation of non-ribosomal non-coding RNAs. Similarly, we detected 29 putative antisense transcripts in hen2 samples, while only 2 potential antisense regions were upregulated in mtr4 samples (Table S10). To confirm the polyadenylated status of antisense transcripts, we selected one of the potential antisense regions for 3′ RACE experiments. Cloning and sequencing of the PCR products revealed that 0 of 32 clones obtained from WT samples corresponded to the target sequence (Fig. 10, Fig. S14). By contrast, 28 of 32 clones obtained from hen2 samples and 3 of 32 clones obtained from mtr4 samples corresponded indeed to antisense transcripts derived from the target region (Fig. 10, Fig. S14). Antisense sequences were polyadenylated, a hallmark of exosome-mediated RNA degradation, and between 67 and 208 nt long (Fig. 10, Fig. S14). These data strongly suggest that the antisense transcripts derived from the At5g44306 locus are indeed substrates of polyadenylation-mediated decay facilitated by HEN2 and the RNA exosome. Finally, the microarray analysis detected 43 regions without any annotated genome features, including the intergenic repeat region on chromosome five that was already detected by our initial qRT-PCR experiments (Fig. 4). Similar to the distribution of non-coding RNA regions and potential antisense transcripts, the majority of the non-annotated regions (38 of 42) were exclusively observed in hen2 samples, while only 4 of 42 regions were found in mtr4 samples. These results indicate that the elimination of spurious transcripts generated from antisense or non-annotated regions that are usually described as the “dark matter” of the transcriptome relies mostly on HEN2.
Figure 10. Loss of HEN2 results in accumulation of polyadenylated antisense transcripts.
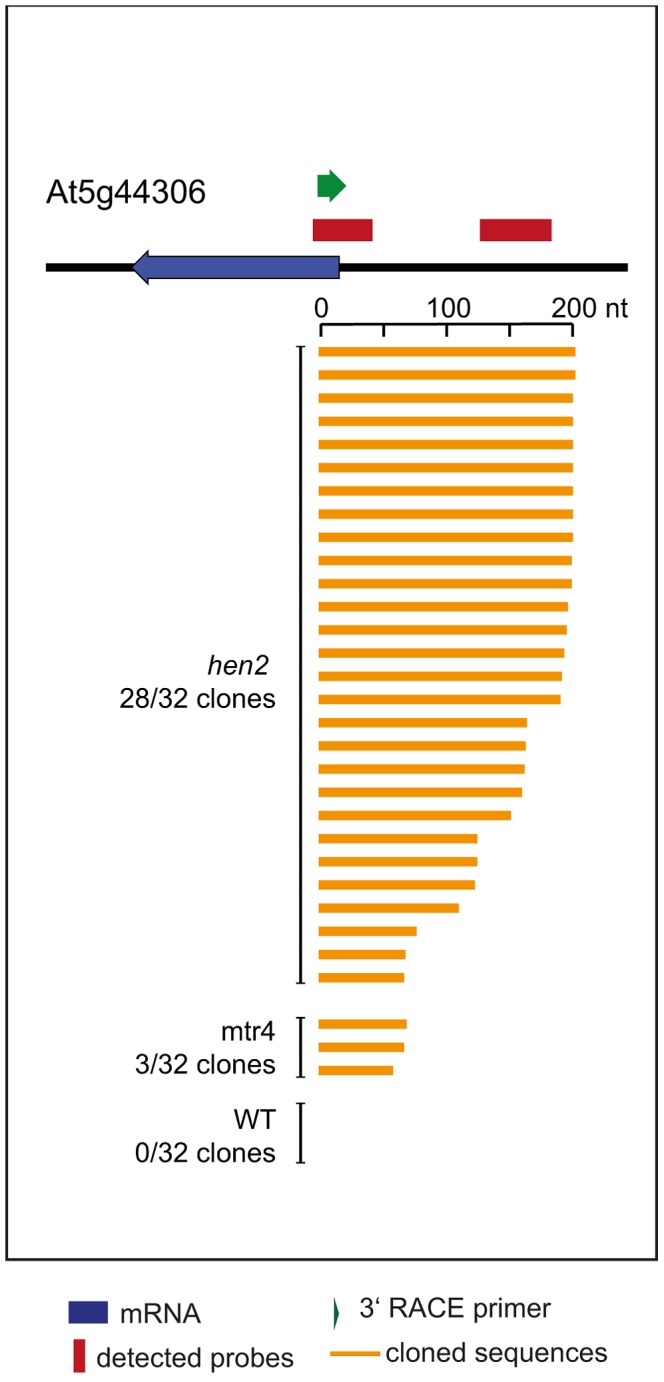
3′ RACE-PCR. A diagram of the At5g44306 locus is shown at the top. A blue arrow represents the At5g44306 mRNA. Red bars above the diagram represent probes detected in the tiling analysis. The location of the primer used for 3′ RACE PCR is indicated by a green arrow above the diagram. Each of the orange horizontal bars below the diagram represents a polyadenylated clone obtained from the indicated sample. 28 of 32 clones obtained from hen2 samples and 3 of 32 clones obtained from mtr4 samples corresponded to antisense transcripts of 67 to 208 nt lenght. 0 of 32 clones obtained from WT samples corresponded to the target sequence.
Taken together, the microarray analysis revealed that a large number of exosome targets, including short or incompletely spliced transcripts derived from mRNA genes, precursors and processing by-products of non-coding RNAs, and spurious transcripts generated from antisense and intergenic regions accumulate specifically in hen2 mutants (Fig. 11). This indicates that HEN2 has a major function in the elimination of many different types of nuclear exosome substrates. A much smaller number of such non-ribosomal exosome substrates accumulated in mtr4 plants (Fig. 11), and average accumulation levels in mtr4 samples were lower than in hen2 samples (Fig. S15). These data indicate that AtMTR4, though it can apparently contribute at least to the degradation of mRNA-derived transcripts, plays a rather minor role in nuclear RNA surveillance.
Figure 11. Degradation of non-ribosomal exosome targets depends largely on HEN2.
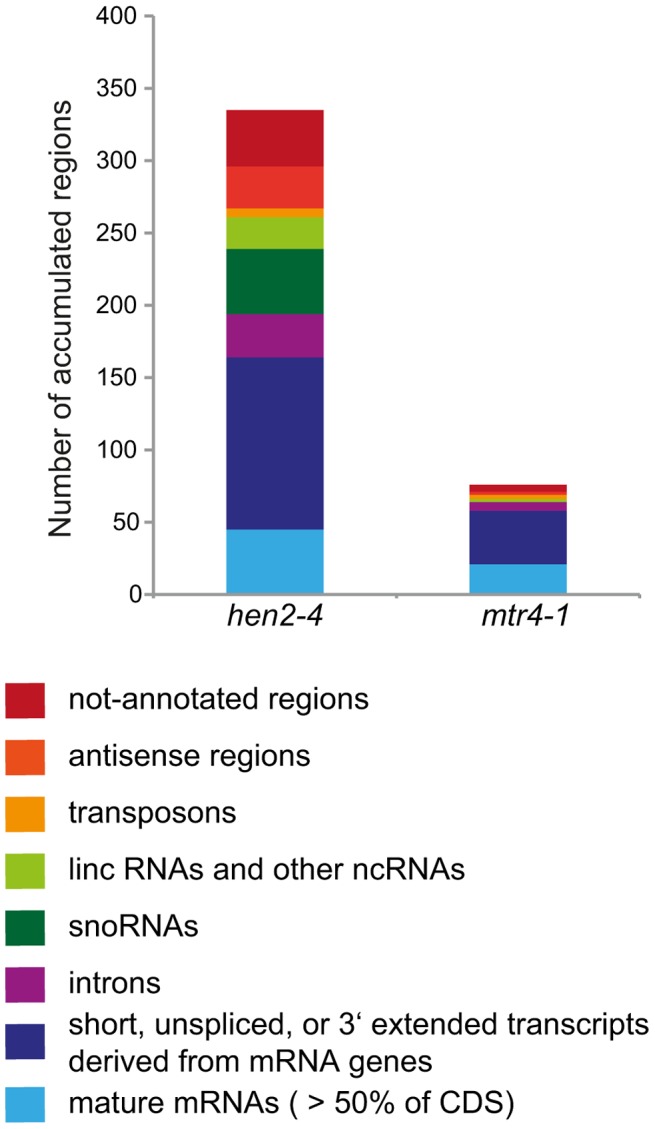
The transcriptomes of WT, hen2-4 and mtr4-1 plants were compared by whole genome microarrays. The histogram shows the total number of regions that were, as compared to wild type, overaccumulated in hen2 or mtr4 samples, respectively. A complete list of the upregulated regions can be found in Tables S4, S5, S6, S7, S8, S9, S10, S11.
HEN2 has an antagonistic effect on post-transcriptional gene silencing mediated by a sense transgene (S-PTGS)
Previous data indicated that mutations in 5′-3′ exoribonucleases in nucleoli (XRN2), nucleoplasm (XRN3) or cytoplasm (XRN4) enhance the efficiency of post-transcriptional gene silencing mediated by sense transgenes (S-PTGS) [49]. Mutations in the exosome core components RRP4 and RRP41, or mutations in the exosome co-factors RRP44/DIS3 and RRP6L1 also enhance S-PTGS, suggesting that both 5′-to-3′ and 3′-to-5′ RNA degradation pathways limit the entry of aberrant transgene RNAs into the S-PTGS pathway [50]. To determine in which nuclear compartment the exosome counteracts transgene PTGS, we analyzed the effect of mtr4 and hen2 mutations on S-PTGS using the Arabidopsis reporter line Hc1 [49]–[52]. Line Hc1 carries a 35S::GUS transgene that triggers S-PTGS at a frequency of 20% at each generation, making this line ideal for identifying mutations that either increase or decrease S-PTGS efficiency. However, only EMS mutants or 35S CaMV promoter-free T-DNA insertion mutants are amenable for such analyses because copies of the 35S CaMV promoter present in the SALK, WISC or GABI T-DNA collections often interfere with the expression of 35S CaMV promoter-driven transgenes, which could report an impact on S-PTGS that is not directly related to the function of the mutated gene [53]. Only the T-DNA mtr4-2 [13] and the EMS hen2-1 mutant [12] fit this requirement. Hc1/mtr4-2 plants triggered S-PTGS at a frequency of 25% (n = 96), which is only slightly higher than S-PTGS frequency in Hc1 controls (Fig. 12A). These data indicate that compromising exosome activity in nucleoli has only a limited effect on transgene S-PTGS. For comparison, loss of the nucleolar exoribonuclease XRN2 was previously shown to trigger S-PTGS at a frequency of 47% [49] (Fig. 12A). In contrast, compromising the nucleoplasmic 5′-to-3′ exonuclease XRN3 had a stronger effect (Fig. 12A) [49]. To determine if loss of the nucleoplasmic protein HEN2 also affects S-PTGS, the hen2-1 mutation, which is in the Landsberg erecta (Ler) ecotype, was crossed to an Hc1/Ler line resulting from ten backcrosses of Hc1 to Ler. Remarkably, no Hc1/Ler plants exhibited S-PTGS (n = 96, Fig. 12B), suggesting that either Ler is less prone to trigger S-PTGS than Col or that Hc1 has lost its capacity to trigger S-PTGS after ten backcrosses to Ler. This later hypothesis was ruled out by crossing Hc1/Ler to xrn4-1 (in Ler). Mutations in the cytoplasmic 5′-to-3′ exonuclease XRN4 are known to enhance S-PTGS in Col [49] (Fig. 12A), so S-PTGS was expected to occur in Hc1/xrn4-1/Ler plants if the Hc1 locus has retained its ability to trigger S-PTGS in Ler. S-PTGS was observed in 100% of Hc1/xrn4-1/Ler plants (n = 96, Fig. 12B), indicating that Ler is less prone to trigger S-PTGS than Col, but that the Hc1/Ler line still is amenable to identify mutations that enhance S-PTGS. Indeed, 23% of Hc1/hen2-1/Ler plants (n = 83) exhibited S-PTGS (Fig. 12B), indicating that compromising exosome activity in the nucleoplasm strongly enhances transgene S-PTGS. The antagonistic effect of HEN2 on S-PTGS was confirmed in Col using an AGO1 transgenic reporter system. In this system, transformation of wild type Col with a T-DNA carrying an ectopic pAGO1::AGO1 construct triggers cosuppression (S-PTGS) of endogenous AGO1 in 50% of the transformants [54] (Fig. 12C). Transformation of hen2-2 (in Col) with the same pAGO1::AGO1 construct triggered AGO1 cosuppression in 72% of the transformants (Fig. 12C). This increase is almost comparable to the effect of xrn4 on AGO1 cosuppression (Fig. 12C), indicating that hen2 strongly affects S-PTGS in Col. Taken together, these data show that HEN2 counteracts S-PTGS in both Col and Ler, likely through its role as a co-factor of the nucleoplasmic exosome.
Figure 12. Effect of mtr4 and hen2 mutations on posttranscriptional silencing.
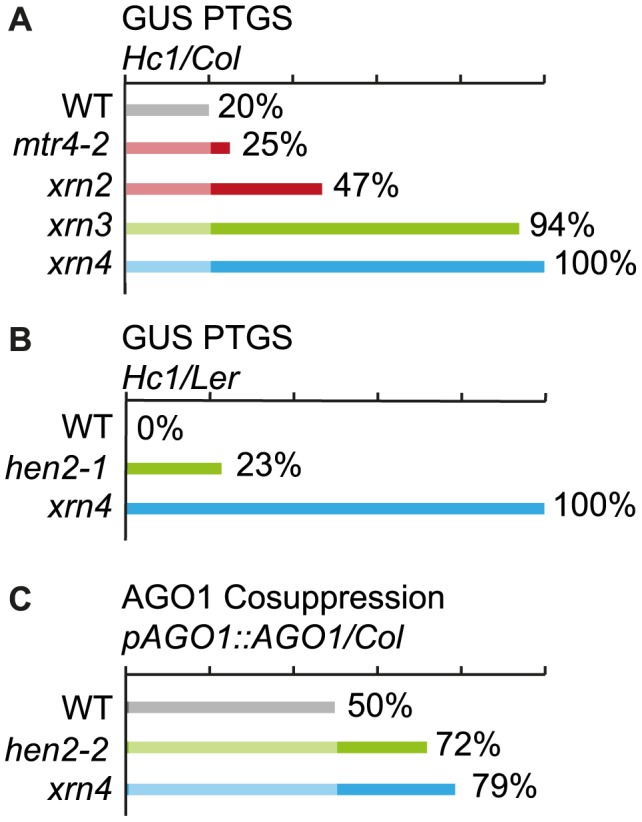
Diagrams show the proportion of plants that undergo systemic silencing of a GUS (panels A and B) or AGO1 (panel C) reporter transgene in the indicated mutants and backgrounds. The color code indicates the intracellular localization of the mutated proteins: red marks the nucleolar proteins; green marks nucleoplasmic proteins, and blue mark cytoplasmic proteins. A. Loss of MTR4 has only a marginal effect on GUS S-PTGS. The effect of the mtr4-2 mutation on silencing of a GUS reporter transgene was tested in the Hc1/Col reporter system as described in [51]. For comparison, we included previously published data from [49] that show the effects of compromised 5′-3′ exoribonucleases XRN2 (nucleolar), XRN3 (nucleoplasmic) or XRN4 (cytoplasmic) in the same reporter system. B. Loss of HEN2 triggers silencing of a GUS PTGS reporter. The effect of hen2-1 (in Ler) on GUS-PTGS was tested in an Hc1-reporter line backcrossed to Ler. Please note that the Hc1/Ler reporter line does not trigger silencing of the GUS reporter in WT. xrn4 in Hc1/Ler was used as a positive control. C. Loss of HEN2 triggers co-suppression of AGO1. To further confirm the role of HEN2 as silencing suppressor, WT, hen2-2 and xrn4 plants (in Col) were transformed with a pAGO1::AGO1 construct that triggers cosuppression of the endogenous AGO1 gene.
Discussion
We show here that two isoforms of MTR4, AtMTR4 and HEN2, assist the Arabidopsis exosome for the degradation of mostly distinct sets of nuclear RNA substrates. Both AtMTR4 and HEN2 co-purify with the Arabidopsis exosome core complex but AtMTR4 and HEN2 occupy primarily distinct intranuclear compartments. The main role of the nucleolar isoform AtMTR4 is to assist in the exosome-mediated degradation of misprocessed rRNA precursors and maturation by-products [13]. Our new data show that the main function of the nucleoplasmic isoform HEN2 is to assist the exosome-mediated processing and/or degradation of snoRNAs and snoRNA precursors, miRNA precursors, lincRNAs, and a large number of spurious transcripts derived from antisense and non-annotated regions. In addition, HEN2 is involved in the degradation of excised introns and incompletely spliced or otherwise mis-transcribed or mis-processed mRNAs. Of the 387 regions detected in this study, 100 have been previously identified as targets of the Arabidopsis core exosome (Tables S4, S5, S6, S7, S8, S9, S10, S11) using a different type of tiling microarray [5]. However, our qRT-PCR data indicate that majority of the HEN2 substrates accumulate also in RRP41 RNAi samples, even though some of them were not detected previously [5], such as At1g79270 (Fig. 6), At3g26510 (Fig. S9) and At1g58602 (Fig. S9). Vice versa, several known exosome substrates were not identified by our tiling analysis but easily detected by qRT-PCR (Fig. S16). These findings indicate that both tiling studies probably underestimate the contribution of HEN2 and the exosome to RNA surveillance. Only a small number of exosome substrates are observed in mtr4 mutants, which indicates that AtMTR4 participates, but plays only a minor role in nuclear RNA surveillance. This is in line with the finding that AtMTR4 and HEN2 have marginal and strong contributions, respectively, to the exosome activity that likely degrades aberrant transgene RNAs in the nucleus to limit their entry in the PTGS pathway [49], [50].
In a previous study, we have shown that hen2 mutants do not accumulate the 5.8S rRNA precursors and the 5′ ETS that are observed upon down-regulation of AtMTR4 [13]. Accordingly, hen2 single mutants do not display any of the developmental defects linked to disturbed ribosome biogenesis or ribosome function that were observed in mtr4 mutants. These data were the first clues that AtMTR4 and HEN2 have rather distinct functions in plants. However, we were not able to obtain double mtr4 hen2 mutants, signifying that simultaneous loss of both AtMTR4 and HEN2 is lethal [13]. The data presented here suggest that AtMTR4 can perform some of the functions of HEN2. For instance, a limited number of mRNA-derived fragments, non-coding RNAs and spurious transcripts accumulated in mtr4 single mutants, indicating that AtMTR4 can contribute, even in presence of HEN2, to the degradation of non-ribosomal exosome targets. In line with this, a small fraction of AtMTR4-GFP can be detected in the nucleoplasm of stable Arabidopsis transformants (Fig. S17, see also [13]). Taking together, these results indicate that a limited overlap between AtMTR4 and HEN2 functions exists. However, our data show that most exosome functions are activated either by AtMTR4 or by HEN2 in the nucleolus and the nucleoplasm, respectively.
It is interesting to note that Arabidopsis has also specific nucleoplasmic and nucleolar isoforms (RRP6L1 and RRP6L2, respectively) of RRP6, a catalytically active exoribonuclease associated with nuclear exosomes in yeast and human [28], [30]. So far, we and others [5] did not detect any of the plant RRP6-like proteins in plant exosome preparations. However, we have previously shown that the downregulation of the nucleolar isoform RRP6L2 leads to a mild accumulation of misprocessed 5.8S rRNA precursors and the 5′ ETS, suggesting that RRP6L2 acts in the same degradation processes as AtMTR4 [13], [55]. By contrast, we did not detect a significant overaccumulation of HEN2 targets upon down-regulation of the nucleoplasmic isoform RRP6L1 (data not shown). A possible explanation is that RRP6L2 and RRP6L1 can substitute for each other in the degradation of HEN2 targets, since the two nuclear RRP6-like proteins appear to have both specific and common roles linked to the degradation of exosome targets [24], [55]. Interestingly, a recent study revealed that RRP6L1, but not RRP6L2 has also a role in transcriptional silencing by retaining PolV transcripts on chromatin, thereby promoting the production of 24 nt siRNAs that direct DNA methylation via the RdDM pathway [56]. Remarkably, this function of RRP6L1 is independent of the core exosome [56]. By contrast, transcriptional silencing at soloLTR loci is mediated by both RRP6L and the core exosome [24]. In addition, RRP6L1 and the exosome core complex have a common role in 21-nt siRNA-dependent posttranscriptional silencing (PTGS), since downregulation of either RRP41 or RRP6L1 alone is sufficient to enhance PTGS in the sensitive Hc1-GUS reporter system that was used in this study [50]. Hence, the role of RRP6L1 in PTGS is likely linked to exosome-mediated RNA degradation, suggesting that HEN2 and RRP6L1 are involved in at least one similar function.
In animals and fungi, a single MTR4 protein is present in nucleoplasm and nucleoli, and essential for both processing/degradation of rRNA precursors and the elimination of all other nuclear exosome substrates [8]–[10], [57]. However, both yeast and human MTR4 proteins are incorporated in more than one exosome activator/adapter complex. Yeast MTR4 is detected in TRAMP4 and TRAMP5 (for TRF4/5 AIR1/2 MTR4 Polyadenylation), each of which comprises a RNA binding protein and a non-canonical poly(A) polymerase [58]–[61]. Although TRAMP4 and 5 have a similar composition and many redundant functions, TRAMP5 seems be more important for the polyadenylation of pre-rRNAs while TRAMP4 might be more important for the degradation of other non-coding RNAs and intergenic transcripts [62]–[64]. The functional specialization between nucleolar and nucleoplasmic exosome activator complexes is clearer in human. In nucleoli, hMTR4 is incorporated in a TRAMP-like complex which polyadenylates rRNA maturation by-products [9]. In the nucleoplasm, hMTR4 is associated with the NEXT (for Nuclear EXosome Targeting) complex, which targets PROMPTS (PROMoter uPstream TranScripts) for degradation by the exosome [9]. Hence, both yeast and animals possess nucleolar and nucleoplasmic exosome activators, which share MTR4 as a central component. By contrast, an exosome activating system with two specialized RNA helicases has evolved early in the green lineage. Interestingly, both the nucleoplasmic fraction of human MTR4 and the Arabidopsis nucleoplasmic-specific RNA helicase HEN2 appear associated with similar RNA binding proteins to form NEXT and NEXT-like complexes, respectively, and with the cap-binding complex [9], [21, this study]. These findings suggest a high degree of functional conservation between the nucleoplasmic fraction of human MTR4 and plant HEN2. By contrast, a TRAMP-like complex comprising a non-canonical poly(A) polymerase remains to be identified in plants. Hence, the emerging picture is that only the core exosome machinery is conserved in all eukaryotes, while exosome-associated activities and activating complexes show intriguing diversity and complexity in fungi, insects, animals and plants.
Methods
Plant material
With the exception of hen2-1 [12] used for the S-PTGS assay, all Arabidopsis thaliana plants were of Columbia ecotype (Col-0). T-DNA insertion lines were retrieved from NASC (http://arabidopsis.info/). mtr4-1, mtr4-2, hen2-2 and hen2-4 lines are described in [13]. RRP41 RNAi lines are described in [5]. The S-PTGS reporter line Hc1 was first described in [51]. Hc1/xrn/col lines are described in [49]. Unless stated otherwise, plants were grown on soil at 20°C with cycles of 16 h light/8 h darkness.
Sequence analysis and phylogeny
Sequences were retrieved from Phytozome (http://www.phytozome.net), Metazome (http://www.metazome.net) and JGI (http://genome.jgi.doe.gov) genome databases, using AtMTR4, HEN2 and AtSKI2 proteins as BLAST queries. Structures of AtMTR4 and HEN2 were modeled with MODELLER Software (http://modbase.compbio.ucsf.edu/ModWeb20-html/modweb.html) using the crystal structures of S. cerevisiae MTR4 (PDB 3L9O and 2XGJ) [34], [35] as templates. Alignments were performed with Chimera (http://www.cgl.ucsf.edu/chimera, for structure-based sequence alignments) and ClustalX (http://www.clustal.org, for phylogenetic analysis). The phylogenetic tree was calculated with the neighbor-joining algorithm built in ClustalX and 1000 bootstraps, and drawn with Figtree (http://tree.bio.ed.ac.uk/software/figtree).
Expression of GFP and myc-tagged fusion proteins
For the expression of RRP4, AtMTR4 and HEN2 GFP fusion proteins under the control of the 35S CaMV promoter, the coding sequences of RRP4, AtMTR4 and HEN2 were amplified from cDNA and cloned into vector pK7FWG2 [65]. For expression of GFP-tagged or myc-tagged RRP41 the genomic sequence of RRP41 including 1 kb upstream of the RRP41 gene was cloned into vectors pGWB604 and pGWB616, respectively [66]. For immunoprecipitations (see below), AtMTR4-GFP was expressed under the control of its own promoter. To do so, a genomic region comprising 1 kb upstream of the MTR4 gene, the first two exons and the first intron were fused to the CDS downstream of the second exon and cloned into pGWB604 [66]. Constructs that allow the expression of RFP-tagged FIB1 [67], XRN2 and SRP43a [68] were kind gifts of Jane Brown and Martin Crespi, respectively. Infiltration of N. benthamiana leaves was performed as described in [50] except that P19 was used as a suppressor of silencing. Arabidopsis plants were transformed by the floral dip method [69]. Root tips of stable transformants were examined 8 days after germination by confocal microscopy.
Co-immunoprecipitation and mass-spectrometry analysis
RRP41-myc, RRP41-GFP, AtMTR4-GFP and HEN2-GFP-fusion proteins were extracted from flowers of stable Arabidopsis transformants and purified using magnetic microparticles coated with monoclonal myc or GFP antibodies (MACS purification system, Miltenyi Biotech) according to the manufacturer's instructions except that SDS was omitted from washing buffers. Co-IP experiments were carried out in triplicates for RRP41 and MTR4 and duplicates for HEN2 with 50 mM and 150 mM NaCl.
For in-gel digestion, samples were separated by SDS-PAGE followed by trypsic digestion and peptide extraction as described in [70]. Otherwise, proteins were eluted directly from magnetic beads in 1× Laemmli buffer, precipitated with 100 mM ammonium acetate in methanol, and resuspended in 50 mM ammonium bicarbonate. After reduction and alkylation steps with 5 mM dithiothreitol and 10 mM iodoacetamide, respectively, proteins were digested overnight with trypsin 1/25 (w/w). Vacuum dried peptides were re-suspended in 15 µl 0.1% FA (solvent A). One third of each sample was injected on a NanoLC-2DPlus system (nanoFlex ChiP module; Eksigent, ABSciex, Concord, Ontario, Canada) coupled to a TripleTOF 5600 mass spectrometer (ABSciex) operating in positive mode. Peptides were loaded on C18 columns (ChIP C-18 precolumn 300 µm ID × 5 mm ChromXP and ChIP C-18 analytical column 75 µm ID × 15 cm ChromXP; Eksigent) and were eluted using a 5%–40% gradient of solvent B (0.1% FA in Acetonitrile) for 60 minutes at a 300 nl/min flow rate. The TripleTOF 5600 was operated in high-sensitivity data-dependant acquisition mode with Analyst software (v1.6, ABSciex) on a 350–1250 m/z range. Up to 20 of the most intense multiply-charged ions (2+ to 5+) were selected for CID fragmentation, with a cycle time of 3.3s (TOP 20 discovery mode).
For protein identification, raw data were converted to Mascot Generic File format (mgf) and searched against a TAIR 10 database supplemented with a decoy database build from reverse sequences. Data were analyzed using Mascot algorithm version 2.2 (Matrix Science, UK) through ProteinScape 3.1 software (Bruker). Search parameters allowed N-acetylation (protein N-terminal), carbamidomethylation (C) and oxidation (M) as variable peptide modifications. Mass tolerances in MS and MS/MS were set to 20ppm and 0.5Da, respectively. 2 trypsin mis-cleavages were allowed. Peptide identifications obtained from Mascot were validated with a FDR <1%. A second algorithm, PEAKS DB (version 5.3, BSI Informatics) was used with the same search parameters to strengthen the identifications. Identified proteins were assessed by the total number of fragmented spectra per protein (spectral count).
Data of three (RRP41, MTR4) or two (HEN2 50 mM NaCl, HEN2 150 mM) replicates were crossed. Protein partners were considered only if present in all co-IP replicates. All proteins observed in the corresponding control replicates, including same-set and sub-set proteins, were discarded. A second filter was set using controls from 15 independent other IP experiments in A. thaliana carried out by other laboratories in the same MS facility. All proteins observed in any of these negative controls were discarded from the final lists of partner proteins.
Go-term analysis of proteins co-purified with MTR4-GFP was performed with DAVID (http://david.abcc.ncifcrf.gov) [71], [72].
qRT-PCR analysis
Plants were grown on MS agar plates supplemented with 0.5% sucrose. Plates for induction of RRP41 RNAi contained 8 µM 17β-estradiol [5]. For each target, at least three biological replicates from WT, mtr4-1, mtr4-2, hen2-2, hen2-4, RRP41 non-induced (RRP41 Ctrl) and RRP41 induced (RRP41 RNAi) were analyzed. Total RNA was isolated from 7 day-old seedlings using TRI-reagent (MRC). cDNA was synthesized from 5 µg of total RNA with SuperScript III reverse transcriptase (Invitrogen) using 37.5 pmol of oligo(dT) per 20 µl reaction according to the manufacturer's instructions. Samples were analyzed as technical triplicates in a LightCycler 480 Real-Time PCR System (Roche). Each qRT-PCR reaction contained 1× LightCycler 480 SYBR Green I Master Mix (Roche), 5 pmol of each primer, 0.5 µl of cDNA in a volume of 10 µl. ACT2, TIP41 and EXP were used as reference mRNAs.
Microarray analysis
Wild type, hen2 and mtr4 plants were grown on MS agar supplemented with 0.5% sucrose. Total RNA was extracted from two biological replicates using Nucleospin RNA plant columns (Machery & Nagel). cDNA synthesis and labeling with Cy3-dUTP or Cy5-dUTP (Perkin-Elmer-NEN Life Science Products) for fluorochrome reversal was performed as described previously [73]. Samples were hybridized to NimbleGen whole genome microrrays (Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), accession no. GPL17057 and GPL11005) as described in [73]. Topological positions and nucleotide sequence of the 1,434,492 strand-specific NimbleGen probes are available in the FLAGdb++ database at http://urgv.evry.inra.fr/FLAGdb++ [45]. Two micron scanning was performed with an InnoScan900 scanner and raw data were extracted using Mapix software (Innopsys).
Statistical analysis
Probes that mapped to repetitive sequences of the Arabidopsis genome were excluded from the analysis. Statistical analyses were performed with the software R. For each experiment, the raw data comprised the logarithm of median feature pixel intensities at wavelengths 635 nm (red) and 532 nm (green), respectively. The dye bias was corrected by a global intensity-dependent normalization for each chromosome and each array using the loess procedure [74] and then averaged over the technical replicates. The outputs of this procedure are two normalized intensity values per probe, one for each of the co-hybridized samples. Normalized raw data were deposited at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/), accession no. GSE48178, and at CATdb (http://urgv.evry.inra.fr/CATdb/), accession no. TIL-Ath-2011_3.
For the statistical analysis, each mutant sample was compared to the co-hybridized wild type sample. Analyses were performed for each biological replicate and independently for each chromosome and each strand (2 mutants×2 biological replicates×2 strands×5 chromosomes = 40 analysis were performed). To determine probes that behave differently between a mutant and a wild type sample, we recast the question as an unsupervised classification problem for each biological replicate, for each chromosome and for each strand. A Hidden Markov Model was developed to model the joint distribution of the two normalized hybridization intensities in order to distinguish four different biologically interpretable clusters of probes: one cluster with similar behavior in both samples (identically expressed), one cluster with higher intensity in the first sample (over-expressed), a symmetric cluster with lower intensity in the first sample (under-expressed) and one cluster with low intensities in both samples (noise) corresponding to the non-transcribed probes. The emission distribution of the noise cluster was modeled by a spherical Gaussian. A Gaussian mixture, components of which were forced to be colinear along the main axis of the ellipse representing each cluster, modeled each of the three other clusters. Data projection on the main axes allowed us to work with unidimensional mixtures and to put a unique Gaussian distribution along the associated perpendicular axis for all components. Model parameters were estimated using an adapted version of the EM algorithm taking the model constraints and the spatial dependency between probes into account. A detailed documentation of the statistical analysis can be found in dataset S2.
Probe classification into the four clusters was based on the conditional probabilities: a probe was assigned in the cluster for which the conditional probability was the highest (MAP rule). A probe was declared over-expressed in the mutant if this assignment was observed in the two biological replicates. 1860 probes were identified as overexpressed in hen2 mutants, and 499 probes were assigned as overexpressed in mtr4 mutants. For the majority of the estimated models, the main axis of the ellipse representing the cluster of probes under-expressed in the mutant was very closed to the main axis of the cluster with identically expressed probes. As a consequence, only a small number of probes were assigned under-expressed. Intersection of the lists with under-expressed probes across the two biological replicates revealed that no probes were underexpressed in both replicates of hen2 or mtr4, respectively.
Bioinformatics analysis
A file allowing the visualization of the upregulated probes aligned to the Arabidopsis genome using seqmonk software (http://www.bioinformatics.babraham.ac.uk/projects/seqmonk/) can be found in dataset S1. Probes with a unique localisation in the genome and for which a significant over-expression was detected by the statistical analysis were sorted by genome coordinates to identify upregulated regions. Only regions with at least two consecutive probes were allowed. Bioinformatic analysis was performed with adapted perl scripts. Sequences of upregulated regions were annotated using TAIR10 genome database, FLAGdb++, and recent studies that identified snoRNA, miRNA genes and linc RNA genes [45]–[47], [75]. Sequence alignments were performed with BLASTn and sim4 tools (for annotation of spliced transcripts) [76], [77]. Only hits with 100% identity were considered. Annotation of upregulated regions was manually curated to remove double assignments (e.g. a region that matches to snoRNA genes located in an intron of a protein coding genes was assigned as snoRNA and removed from the list of introns). For comparison with a previously published list of exosome substrates, we compared the genome coordinates of upregulated regions with coordinates of upregulated regions extracted from [5], taking the difference between different versions of TAIR into account.
Accession numbers (Arabidopsis Genome Initiative, AGI)
ATMTR4: At1g59760; HEN2: At2g06990; RRP41: At3g61620.
Supporting Information
Seqmonk file to visualize the location of significantly upregulated probes in the Arabidopsis genome, and instructions for opening the file using the free seqmonk software.
(ZIP)
Detailed documentation of the statistical analysis of the tiling data.
(PDF)
Plant MTR4 proteins have an insertion in the inner loop of the arch domain. Top: Sequences of MTR4 and HEN2 proteins from selected plant species were aligned to the sequence of S. cerevisiae MTR4 (highlighted in orange). The alignment is shown for 50 aminoacids of the arch domain. Athaliana, Arabidopsis thaliana (thale cress); Thalophila, Thelluniella halophila (Salt cress); Sitalica, Setaria italica (Foxtail millet); Mguttatus, Mimulus guttatus (Monkey flower); Pvulgaris, Phaesolus vulgaris (Common bean); Mtrunculata, Medicago trunculata (Barrel medic); Mesculenta, Manihot esculenta (Cassava); Ptrichocarpa, Populus trichocarpa (Poplar); Csativus, Cucumis sativus (cucumber); Egrandis, Eucalyptus grandis (Eucalyptus); Smoellendorfii, Selaginella moellendorfii (Spikemoss); Ppatens, Physcomitrella patens (Moss); Scerevisiae, Saccharomyces cerevisiae (Bakers yeast). Left: Model of the arch domains of AtMTR4 and HEN2. AtMTR4 and HEN2 structures were modeled using the yeast MTR4 structure as template. Only the arch domain is shown (from the top). Yeast MTR4 in orange, HEN2 in green, AtMTR4 in blue. A dashed red line indicates the insertion of 9 amino acids present in AtMTR4.
(PDF)
Full sequence alignment of MTR4 and HEN2 proteins from selected species. Amino acids are colored by the ClustalX color scheme. Boxes below the alignment illustrate RecA domains (blue), the winged helix domains (yellow), the arch domain (red, the black box depicts the KOW domain) and the C-terminal bundle domain (pink). Characteristic differences between plant MTR4 and HEN2 sequences are marked by stars. Athaliana, Arabidopsis thaliana (thale cress); Thalophila, Thelluniella halophila (Salt cress); Sitalica, Setaria italica (Foxtail millet); Mguttatus, Mimulus guttatus (Monkey flower); Pvulgaris, Phaesolus vulgaris (Common bean); Mtrunculata, Medicago trunculata (Barrel medic); Mesculenta, Manihot esculenta (Cassava); Ptrichocarpa, Populus trichocarpa (Poplar); Csativus, Cucumis sativus (cucumber); Egrandis, Eucalyptus grandis (Eucalyptus); Smoellendorfii, Selaginella moellendorfii (Spikemoss); Ppatens, Physcomitrella patens (Moss).
(PDF)
MTR4-GFP co-localises with nucleolar marker proteins. Transient expression of fluorescent fusion proteins in Nicotiana benthamiana leaves. MTR4-GFP is shown in green, RFP-fusion proteins are shown in red. Fibrillarin-RFP and XRN2-RFP are known nucleolar markers; SRP34a-RFP was used as a nucleoplasmic marker. The phase contrast picture is shown on the right. Scale bars: 15 µm.
(PDF)
HEN2-GFP co-localises with a nucleoplasmic marker protein. Transient expression of fluorescent fusion proteins in Nicotiana benthamiana leaves. HEN2-GFP is shown in green, RFP-fusion proteins are shown in red. Fibrillarin-RFP and XRN2-RFP were used as nucleolar markers; SRP34a-RFP was used as a nucleoplasmic marker. The phase contrast picture is shown on the right. Scale bars: 15 µm.
(PDF)
MTR4 and HEN2 have distinct localization patterns. The distribution of the indicated GFP-fusion proteins in root cells of stable Arabidopsis transformants is shown on the left. The middle column shows DAPI staining. Please note that take-up of DAPI by intact, living plant tissue is slow and can lead to a strong background signal from cell walls. No, Nucleolus; Np, Nucleoplasm; Cp, Cytoplasm. Scale bars: 5 µm.
(PDF)
HEN2-GFP is localized in nucleoplasmic foci. Co-expression of HEN2-GFP and the nucleoplasmic marker protein SRP34a in leaves of stable Arabidopsis transformants. Nucleoplasmic foci were observed in all cell types of all stable transformants. Scale bars: 15 µm.
(PDF)
Sequence alignment of human RBM7 and Arabidopsis At4g10110.
(PDF)
Accumulation of unspliced transcripts in hen2 mutants. qRT-PCR. A Diagram of the genomic locus indicated by the respective AGI number is shown at the top of each panel. Annotated mRNA genes are represented as arrows with dark blue boxes for the CDS, light blue boxes for 3′ and 5‚ UTRs, and a light blue line for introns. Red bars above the diagram represent probes detected in the microarray analysis. Green arrows above or below the diagram depict the location of qRT-PCR primers. The corresponding qRT-PCR results for each primer pair are given as fold-change relative to WT in the histograms below each diagram. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates. A. At3g26510 (with 4 predicted splice variants). qRT-PCR results suggest that hen2 and RRP41 RNAi plants accumulate a population of transcripts some of which still contain the unspliced acceptor site of the first intron (panel V114a/b) and some of which still contain the 2nd intron (panel V112a/b). B. At1g58602. Transcripts comprising the unspliced 2nd exon/intron donor site accumulate in hen2 and RRP41 RNAi plants. C. At3g43160. Both spliced and unspliced transcripts corresponding to the 3′ region of the At3g43160 locus accumulate in hen2 and RRP41 RNAi plants.
(PDF)
Unspliced transcripts from the At1g79270 locus are polyadenylated. Sequences of 3′ RACE PCR products obtained from hen2-4 samples. cDNA synthesis was initiated using oligo-dT as primer. 3′ RACE PCR was performed with V113a (green arrow) as forward primer, and the adapter sequence of the oligo-dT primer as a reverse primer. PCR products obtained from hen2-4 samples were cloned and sequenced. The genomic sequence is given above the line, with intronic sequence in purple. Red arrows mark donor and acceptor splice sites. Non-encoded nucleotides are in green.
(PDF)
Upregulation of mRNAs in hen2 mutants. Accumulation of mRNAs was tested by qRT-PCR using primer pairs located in 5′, central or 3′ regions of the annotated transcripts as indicated below each panel. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates.
(PDF)
mRNAs are not systematically detected in all replicates. Accumulation of mRNAs was tested by qRT-PCR using primer pairs located in 5′ or 3′ regions of the annotated transcripts as indicated below each panel. Panels on the left show the qRT-PCR results for exactly the same samples that have been used for hybridisation to the tiling arrays. Panels on the right show the results obtained in 3 independent replicates grown in the same culture conditions. A possible explanation for the inconsistence between the replicates could be that mRNAs are not bona-fide substrates of exosome-mediated RNA degradation and might rather be upregulated due to indirect effects. Other types of transcripts such as short mRNA-derived regions, introns, unspliced transcripts or several types of non- coding RNAs are consistently observed in all replicates. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates.
(PDF)
Loss of HEN2 or the exosome is associated with increased levels of polyadenylated snoRNA precursors. Oligo-dT primed cDNA was used for 3′ RACE- PCR, with primer E5 and the adapter sequence of the cDNA synthesis primer as forward and reverse primers, respectively. PCR products were separated on 2% agarose gels, transferred to Hybond XL membranes, and hybridized with radiolabeled probes E6 (mid panel) and E8 (lower panel). The diagram below illustrates location of primers and probes with respect to the snoRNA genes in this region (see also Fig. 9).
(PDF)
Polyadenylated transcripts partially antisense to AT5G44306. Sequences were amplified by 3′ RACE from oligo-dT primed cDNA from the indicated samples using a primer (indicated by the purple arrow) situated antisense to the 5′ region of AT5G44306. Non-encoded nucleotides are in green.
(PDF)
mtr4 mutants accumulate exosome substrates to lower levels than hen2 mutants. Boxplot showing averaged intensity values for the overexpressed probes identified in each comparison of the tiling array analysis. The first two rows show the intensity values in WT and hen2 samples for all probes overexpressed in both biological replicates of hen2. The third and fourth rows show the intensity values in WT and mtr4 samples for all probes overexpressed in both biological replicates of mtr4. The averaged intensities in mtr4 samples are significant lower than the averaged intensities in hen2 samples (p-value<1e -3). The fifth and sixth rows show the intensity values in hen2 and mtr4 for the common probes (overexpressed in both mutants). Again, the mean average intensity in mtr4 samples is lower than the mean average intensity in hen2 samples (p-value<1e -3).
(PDF)
The microarray analysis probably underestimates the contribution of HEN2 to nuclear RNA surveillance. A diagram of the genomic locus indicated by the respective AGI number is shown at the top of each panel. Annotated mRNA genes are represented as arrows with dark blue boxes for the CDS, light blue boxes for 3′ and 5′ UTRs, and a light blue line for introns. Red bars above the diagram represent probes detected in the microarray analysis. Green arrows above or below the diagram depict the location of qRT-PCR primers. The corresponding qRT-PCR results for each primer pair are given as fold-change relative to WT in the histograms below each diagram. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates. Upper panel: The upregulation of two stretches in the 5′ region of At1g20100 in RRP41 RNAi lines (indicated by the red double arrows) was detected in a previous tiling microarray study [5]. Our microarray array detected only a portion of this region (indicated by the red bars above the diagram). However, we could confirm the upregulation of the uppermost 600 kb of At1g20100 in both hen2 alleles by qRT-PCR. Middle panel: A portion of the fifth intron of At5g27720 was previously identified as a target of the exosome core complex [5] (indicated by the red double arrows). In our microarray analysis, only one probe in this region was declared statistically significant. Since we considered only regions with at least two consecutive probes, this region was omitted from the data interpretation. Nevertheless, qRT-PCR data show that a portion of the fifth intron is upregulated in hen2 mutants, while levels of pre-mRNA or mature mRNAs are similar to WT. The presence of polyadenylated transcripts corresponding either to the entire intron or to shorter degradation intermediates in hen2 and RRP41 RNAi samples was further confirmed by cloning of 3′ RACE products (not shown). Lower panel: A previous tiling study [5] identified a short stretch derived from the 5′ region of At4g02890 as a target of the exosome core complex (indicated by the red double arrows). This region was not detected in our tiling analysis, but its upregulation was easily detected by qRT-PCR. The discrepancy between the two tiling studies is probably largely explained by the different arrays designs (the NimbleGen 732K array is not designed to detect very small regions) and by different statistical analysis procedures. Moreover, the accumulation of exosome targets may also vary with growth conditions. In fact, several of our validated targets have not been picked up in the previous genome-wide analysis [5], indicating that both tiling studies under-estimate the contribution of exosome-mediated RNA surveillance.
(PDF)
A fraction of AtMTR4-GFP can be detected in the nucleoplasm. Intracellular distribution of AtMTR4-GFP in root cells of a stable Arabidopsis transformant. The nucleoplasmic fraction of AtMTR4-GFP (white arrows) is more visible in individual transformants displaying relative weak transgene expression, which is not representative for the majority of the investigated plant lines.
(PDF)
Mass-spectrometric analysis of RRP41 IP experiments: list of peptides.
(XLSX)
Mass- spectrometric analysis of MTR4 IP experiments: list of peptides.
(XLSX)
Mass- spectrometric analysis of HEN2 IP experiments: list of peptides.
(XLSX)
Analysis of microarray data. Regions corresponding to short stretches of mRNAs, unspliced transcripts, and 3′ or 5′ extended mRNAs.
(XLSX)
Analysis of microarray data. Regions corresponding to introns.
(XLSX)
Analysis of microarray data. Regions corresponding to mRNAs.
(XLSX)
Analysis of microarray data. Regions corresponding to transposable elements.
(XLSX)
Analysis of microarray data. Regions corresponding to snoRNA precursors.
(XLSX)
Analysis of microarray data. Regions corresponding to linc and other RNAs.
(XLSX)
Analysis of microarray data. Potential antisense transcripts.
(XLSX)
Analysis of microarray data. Non-annotated regions.
(XLSX)
Gene descriptions and biological functions for the mRNAs upregulated in hen2 and/or mtr4 samples.
(XLSX)
Primers used in this study.
(XLSX)
Acknowledgments
We deeply appreciate the expertise and help with bioinformatic analysis and valuable advice of Etienne Delannoy, URGV. We thank Jane Brown and Martin Crespi for the RFP reporter constructs.
Funding Statement
This work was funded by Centre National de la Recherche Scientifique (CNRS, France) and a INRA fellowship to CB. This work was realized in the frame of the LABEX (ANR-2010-LABX-36 to DG) and (ANR-2010-LABX-40 to HV) and benefits from funding from the state managed by the French National Research Agency as part of the programme d'Investissements d'avenir. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lykke-Andersen S, Tomecki R, Jensen TH, Dziembowski A (2011) The eukaryotic RNA exosome: same scaffold but variable catalytic subunits. RNA Biol 8: 61–66. [DOI] [PubMed] [Google Scholar]
- 2. Januszyk K, Lima CD (2014) The eukaryotic RNA exosome. Curr Opin Struct Biol 24C: 132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D (2012) Transcriptome-wide analysis of exosome targets. Mol Cell 48: 422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gudipati RK, Xu Z, Lebreton A, Séraphin B, Steinmetz LM, et al. (2012) Extensive degradation of RNA precursors by the exosome in wild-type cells. Mol Cell 48: 409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chekanova JA, Gregory BD, Reverdatto SV, Chen H, Kumar R, et al. (2007) Genome-wide high-resolution mapping of exosome substrates reveals hidden features in the Arabidopsis transcriptome. Cell 131: 1340–1353. [DOI] [PubMed] [Google Scholar]
- 6. Brown JT, Bai X, Johnson AW (2000) The yeast antiviral proteins Ski2p, Ski3p, and Ski8p exist as a complex in vivo. RNA 6: 449–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anderson JS, Parker RP (1998) The 3′ to 5′ degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 3′ to 5′ exonucleases of the exosome complex. EMBO J 17: 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. De la Cruz J, Kressler D, Tollervey D, Linder P (1998) Dob1p (Mtr4p) is a putative ATP-dependent RNA helicase required for the 3′ end formation of 5.8S rRNA in Saccharomyces cerevisiae. EMBO J 17: 1128–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lubas M, Christensen MS, Kristiansen MS, Domanski M, Falkenby LG, et al. (2011) Interaction profiling identifies the human nuclear exosome targeting complex. Mol Cell 43: 624–637. [DOI] [PubMed] [Google Scholar]
- 10. Bernstein J, Patterson DN, Wilson GM, Toth EA (2008) Characterization of the Essential Activities of Saccharomyces cerevisiae Mtr4p, a 3′-5′ Helicase Partner of the Nuclear Exosome. J Biol Chem 283: 4930–4942. [DOI] [PubMed] [Google Scholar]
- 11. Kobayashi K, Otegui MS, Krishnakumar S, Mindrinos M, Zambryski P (2007) INCREASED SIZE EXCLUSION LIMIT 2 encodes a putative DEVH box RNA helicase involved in plasmodesmata function during Arabidopsis embryogenesis. Plant Cell 19: 1885–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Western TL, Cheng Y, Liu J, Chen X (2002) HUA ENHANCER2, a putative DExH-box RNA helicase, maintains homeotic B and C gene expression in Arabidopsis . Development 129: 1569–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lange H, Sement FM, Gagliardi D (2011) MTR4, a putative RNA helicase and exosome co-factor, is required for proper rRNA biogenesis and development in Arabidopsis thaliana. Plant J 68: 51–63. [DOI] [PubMed] [Google Scholar]
- 14. Abbasi N, Kim HB, Park N-I, Kim H-S, Kim Y-K, et al. (2010) APUM23, a nucleolar Puf domain protein, is involved in pre-ribosomal RNA processing and normal growth patterning in Arabidopsis . Plant J 64: 960–976. [DOI] [PubMed] [Google Scholar]
- 15. Petricka JJ, Nelson TM (2007) Arabidopsis nucleolin affects plant development and patterning. Plant Physiol 144: 173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kojima H, Suzuki T, Kato T, Enomoto K, Sato S, et al. (2007) Sugar-inducible expression of the nucleolin-1 gene of Arabidopsis thaliana and its role in ribosome synthesis, growth and development. Plant J 49: 1053–1063. [DOI] [PubMed] [Google Scholar]
- 17. Pontvianne F, Matia I, Douet J, Tourmente S, Medina FJ, et al. (2007) Characterization of AtNUC-L1 Reveals a Central Role of Nucleolin in Nucleolus Organization and Silencing of AtNUC-L2 Gene in Arabidopsis . Mol Biol Cell 18: 369–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Byrne ME (2009) A role for the ribosome in development. Trends Plant Sci 14: 512–519. [DOI] [PubMed] [Google Scholar]
- 19. Rosado A, Sohn EJ, Drakakaki G, Pan S, Swidergal A, et al. (2010) Auxin-mediated ribosomal biogenesis regulates vacuolar trafficking in Arabidopsis . Plant Cell 22: 143–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng Y, Kato N, Wang W, Li J, Chen X (2003) Two RNA binding proteins, HEN4 and HUA1, act in the processing of AGAMOUS pre-mRNA in Arabidopsis thaliana . Dev Cell 4: 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Andersen PR, Domanski M, Kristiansen MS, Storvall H, Ntini E, et al. (2013) The human cap-binding complex is functionally connected to the nuclear RNA exosome. Nat Struct Mol Biol 20: 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lange H, Sement FM, Canaday J, Gagliardi D (2009) Polyadenylation-assisted RNA degradation processes in plants. Trends Plant Sci 14: 497–504. [DOI] [PubMed] [Google Scholar]
- 23. Zhang W, Murphy C, Sieburth LE (2010) Conserved RNaseII domain protein functions in cytoplasmic mRNA decay and suppresses Arabidopsis decapping mutant phenotypes. Proc Natl Acad Sci USA 107: 15981–15985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shin J-H, Wang H-LV, Lee J, Dinwiddie BL, Belostotsky DA, et al. (2013) The role of the Arabidopsis Exosome in siRNA-independent silencing of heterochromatic loci. PLoS Genet 9: e1003411 10.1371/journal.pgen.1003411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kumakura N, Otsuki H, Tsuzuki M, Takeda A, Watanabe Y (2013) Arabidopsis AtRRP44A is the functional homolog of Rrp44/Dis3, an exosome component, is essential for viability and is required for RNA processing and degradation. PLoS ONE 8: e79219 10.1371/journal.pone.0079219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hooker TS, Lam P, Zheng H, Kunst L (2007) A core subunit of the RNA-processing/degrading exosome specifically influences cuticular wax biosynthesis in Arabidopsis . Plant Cell 19: 904–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen X, Goodwin SM, Liu X, Chen X, Bressan RA, et al. (2005) Mutation of the RESURRECTION1 locus of Arabidopsis reveals an association of cuticular wax with embryo development. Plant Physiol 139: 909–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Allmang C, Petfalski E, Podtelejnikov A, Mann M, Tollervey D, et al. (1999) The yeast exosome and human PM-Scl are related complexes of 3′ – 5′ exonucleases. Genes Dev 13: 2148–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Graham AC, Kiss DL, Andrulis ED (2006) Differential distribution of exosome subunits at the nuclear lamina and in cytoplasmic foci. Mol Biol Cell 17: 1399–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tomecki R, Kristiansen MS, Lykke-Andersen S, Chlebowski A, Larsen KM, et al. (2010) The human core exosome interacts with differentially localized processive RNases: hDIS3 and hDIS3L. EMBO J 29: 2342–2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Staals RHJ, Bronkhorst AW, Schilders G, Slomovic S, Schuster G, et al. (2010) Dis3-like 1: a novel exoribonuclease associated with the human exosome. EMBO J 29: 2358–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dorcey E, Rodriguez-Villalon A, Salinas P, Santuari L, Pradervand S, et al. (2012) Context-Dependent Dual Role of SKI8 Homologs in mRNA Synthesis and Turnover. PLoS Genet 8: e1002652 10.1371/journal.pgen.1002652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fabre A, Charroux B, Martinez-Vinson C, Roquelaure B, Odul E, et al. (2012) SKIV2L Mutations Cause Syndromic Diarrhea, or Trichohepatoenteric Syndrome. The American Journal of Human Genetics 90: 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Weir JR, Bonneau F, Hentschel J, Conti E (2010) Structural analysis reveals the characteristic features of Mtr4, a DExH helicase involved in nuclear RNA processing and surveillance. Proc Natl Acad Sci USA 107: 12139–12144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jackson RN, Klauer AA, Hintze BJ, Robinson H, van Hoof A, et al. (2010) The crystal structure of Mtr4 reveals a novel arch domain required for rRNA processing. EMBO J 29: 2205–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Johnson SJ, Jackson RN (2013) Ski2-like RNA helicase structures: Common themes and complex assemblies. RNA Biol 10: 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Halbach F, Rode M, Conti E (2012) The crystal structure of S. cerevisiae Ski2, a DExH helicase associated with the cytoplasmic functions of the exosome. RNA 18: 124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pih KT, Yi MJ, Liang YS, Shin BJ, Cho MJ, et al. (2000) Molecular cloning and targeting of a fibrillarin homolog from Arabidopsis . Plant Physiol 123: 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Barneche F, Steinmetz F, Echeverría M (2000) Fibrillarin genes encode both a conserved nucleolar protein and a novel small nucleolar RNA involved in ribosomal RNA methylation in Arabidopsis thaliana . J Biol Chem 275: 27212–27220. [DOI] [PubMed] [Google Scholar]
- 40. Kastenmayer JP, Green PJ (2000) Novel features of the XRN-family in Arabidopsis: evidence that AtXRN4, one of several orthologs of nuclear Xrn2p/Rat1p, functions in the cytoplasm. Proc Natl Acad Sci USA 97: 13985–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zakrzewska-Placzek M, Souret FF, Sobczyk GJ, Green PJ, Kufel J (2010) Arabidopsis thaliana XRN2 is required for primary cleavage in the pre-ribosomal RNA. Nucleic Acids Res 38: 4487–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tillemans V, Dispa L, Remacle C, Collinge M, Motte P (2005) Functional distribution and dynamics of Arabidopsis SR splicing factors in living plant cells. Plant J 41: 567–582. [DOI] [PubMed] [Google Scholar]
- 43. Lorkovic ZJ, Lopato S, Pexa M, Lehner R, Barta A (2004) Interactions of Arabidopsis RS domain containing cyclophilins with SR proteins and U1 and U11 small nuclear ribonucleoprotein-specific proteins suggest their involvement in pre-mRNA Splicing. J Biol Chem 279: 33890–33898. [DOI] [PubMed] [Google Scholar]
- 44. Marchler-Bauer A, Zheng C, Chitsaz F, Derbyshire MK, Geer LY, et al. (2013) CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res 41: D348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dèrozier S, Samson F, Tamby J-P, Guichard C, Brunaud V, et al. (2011) Exploration of plant genomes in the FLAGdb++ environment. Plant Methods 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sherstnev A, Duc C, Cole C, Zacharaki V, Hornyik C, et al. (2012) Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat Struct Mol Biol 19: 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu J, Jung C, Xu J, Wang H, Deng S, et al. (2012) Genome-wide analysis uncovers regulation of long intergenic noncoding RNAs in Arabidopsis . Plant Cell 24: 4333–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Golisz A, Sikorski PJ, Kruszka K, Kufel J (2013) Arabidopsis thaliana LSM proteins function in mRNA splicing and degradation. Nucleic Acids Res 41 (12) 6232–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gy I, Gasciolli V, Lauressergues D, Morel J-B, Gombert J, et al. (2007) Arabidopsis FIERY1, XRN2, and XRN3 are endogenous RNA silencing suppressors. Plant Cell 19: 3451–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Moreno AB, Martínez de Alba AE, Bardou F, Crespi MD, Vaucheret H, et al. (2013) Cytoplasmic and nuclear quality control and turnover of single-stranded RNA modulate post-transcriptional gene silencing in plants. Nucleic Acids Res 41: 4699–4708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Elmayan T, Balzergue S, Béon F, Bourdon V, Daubremet J, et al. (1998) Arabidopsis mutants impaired in cosuppression. Plant Cell 10: 1747–1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Martínez de Alba AE, Jauvion V, Mallory AC, Bouteiller N, Vaucheret H (2011) The miRNA pathway limits AGO1 availability during siRNA-mediated PTGS defense against exogenous RNA. Nucleic Acids Res 39: 9339–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Daxinger L, Hunter B, Sheikh M, Jauvion V, Gasciolli V, et al. (2008) Unexpected silencing effects from T-DNA tags in Arabidopsis . Trends Plant Sci 13: 4–6. [DOI] [PubMed] [Google Scholar]
- 54. Le Masson I, Jauvion V, Bouteiller N, Rivard M, Elmayan T, et al. (2012) Mutations in the Arabidopsis H3K4me2/3 demethylase JMJ14 suppress posttranscriptional gene silencing by decreasing transgene transcription. Plant Cell 24: 3603–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lange H, Holec S, Cognat V, Pieuchot L, Le Ret M, et al. (2008) Degradation of a polyadenylated rRNA maturation by-product involves one of the three RRP6-like proteins in Arabidopsis thaliana . Mol Cell Biol 28: 3038–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhang H, Tang K, Qian W, Duan C-G, Wang B, et al. (2014) An Rrp6-like Protein Positively Regulates Noncoding RNA Levels and DNA Methylation in Arabidopsis . Mol Cell 54: 418–30 10.1016/j.molcel.2014.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schilders G, Dijk Evan, Pruijn GJM (2007) C1D and hMtr4p associate with the human exosome subunit PM/Scl-100 and are involved in pre-rRNA processing. Nucleic Acids Res 35: 2564–2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Houseley J, Tollervey D (2006) Yeast Trf5p is a nuclear poly(A) polymerase. EMBO Rep 7: 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. LaCava J, Houseley J, Saveanu C, Petfalski E, Thompson E, et al. (2005) RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121: 713–724. [DOI] [PubMed] [Google Scholar]
- 60. Wyers F, Rougemaille M, Badis G, Rousselle J-C, Dufour M-E, et al. (2005) Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737. [DOI] [PubMed] [Google Scholar]
- 61. Vanacova S, Wolf J, Martin G, Blank D, Dettwiler S, et al. (2005) A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol 3: e189 10.1371/journal.pbio.0030189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Holub P, Vanacova S (2012) TRAMP Stimulation of Exosome. Eukaryotic RNases and their Partners in RNA Degradation and Biogenesis, Part A, in: The Enzymes 31: 77–90. [DOI] [PubMed] [Google Scholar]
- 63. Egecioglu DE, Henras AK, Chanfreau GF (2006) Contributions of Trf4p- and Trf5p-dependent polyadenylation to the processing and degradative functions of the yeast nuclear exosome. RNA 12: 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. San Paolo S, Vanacova S, Schenk L, Scherrer T, Blank D, et al. (2009) Distinct roles of non-canonical poly(A) polymerases in RNA metabolism. PLoS Genet 5: e1000555 10.1371/journal.pgen.1000555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Karimi M, Inzé D, Depicker A (2002) GATEWAY vectors for Agrobacterium-mediated plant transformation. Trends Plant Sci 7: 193–195. [DOI] [PubMed] [Google Scholar]
- 66. Nakamura S, Mano S, Tanaka Y, Ohnishi M, Nakamori C, et al. (2010) Gateway binary vectors with the bialaphos resistance gene, bar, as a selection marker for plant transformation. Biosci Biotechnol Biochem 74: 1315–1319. [DOI] [PubMed] [Google Scholar]
- 67. Kim SH, Macfarlane S, Kalinina NO, Rakitina DV, Ryabov EV, et al. (2007) Interaction of a plant virus-encoded protein with the major nucleolar protein fibrillarin is required for systemic virus infection. Proc Natl Acad Sci USA 104: 11115–11120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Nisa-Martínez R, Laporte P, Jiménez-Zurdo JI, Frugier F, Crespi M, et al. (2013) Localization of a bacterial group II intron-encoded protein in eukaryotic nuclear splicing-related cell compartments. PLoS ONE 8: e84056 10.1371/journal.pone.0084056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Clough SJ, Bent AF (1998) Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana . Plant J 16: 735–743. [DOI] [PubMed] [Google Scholar]
- 70. Poirier I, Hammann P, Kuhn L, Bertrand M (2013) Strategies developed by the marine bacterium Pseudomonas fluorescens BA3SM1 to resist metals: A proteome analysis. Aquat Toxicol 128–129: 215–232. [DOI] [PubMed] [Google Scholar]
- 71. Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 72. Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lurin C, Andrés C, Aubourg S, Bellaoui M, Bitton F, et al. (2004) Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 16: 2089–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang YH, Dudoit S, Luu P, Lin DM, Peng V, et al.. (2002) Normalization for cDNA microarray data: a robust composite method addressing single and multiple slide systematic variation. Nucleic Acids Res 30 : e15. PMCID:PMC100354 [DOI] [PMC free article] [PubMed]
- 75. Lamesch P, Berardini TZ, Li D, Swarbreck D, Wilks C, et al. (2012) The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res 40: D1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 77. Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W (1998) A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res 8: 967–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Seqmonk file to visualize the location of significantly upregulated probes in the Arabidopsis genome, and instructions for opening the file using the free seqmonk software.
(ZIP)
Detailed documentation of the statistical analysis of the tiling data.
(PDF)
Plant MTR4 proteins have an insertion in the inner loop of the arch domain. Top: Sequences of MTR4 and HEN2 proteins from selected plant species were aligned to the sequence of S. cerevisiae MTR4 (highlighted in orange). The alignment is shown for 50 aminoacids of the arch domain. Athaliana, Arabidopsis thaliana (thale cress); Thalophila, Thelluniella halophila (Salt cress); Sitalica, Setaria italica (Foxtail millet); Mguttatus, Mimulus guttatus (Monkey flower); Pvulgaris, Phaesolus vulgaris (Common bean); Mtrunculata, Medicago trunculata (Barrel medic); Mesculenta, Manihot esculenta (Cassava); Ptrichocarpa, Populus trichocarpa (Poplar); Csativus, Cucumis sativus (cucumber); Egrandis, Eucalyptus grandis (Eucalyptus); Smoellendorfii, Selaginella moellendorfii (Spikemoss); Ppatens, Physcomitrella patens (Moss); Scerevisiae, Saccharomyces cerevisiae (Bakers yeast). Left: Model of the arch domains of AtMTR4 and HEN2. AtMTR4 and HEN2 structures were modeled using the yeast MTR4 structure as template. Only the arch domain is shown (from the top). Yeast MTR4 in orange, HEN2 in green, AtMTR4 in blue. A dashed red line indicates the insertion of 9 amino acids present in AtMTR4.
(PDF)
Full sequence alignment of MTR4 and HEN2 proteins from selected species. Amino acids are colored by the ClustalX color scheme. Boxes below the alignment illustrate RecA domains (blue), the winged helix domains (yellow), the arch domain (red, the black box depicts the KOW domain) and the C-terminal bundle domain (pink). Characteristic differences between plant MTR4 and HEN2 sequences are marked by stars. Athaliana, Arabidopsis thaliana (thale cress); Thalophila, Thelluniella halophila (Salt cress); Sitalica, Setaria italica (Foxtail millet); Mguttatus, Mimulus guttatus (Monkey flower); Pvulgaris, Phaesolus vulgaris (Common bean); Mtrunculata, Medicago trunculata (Barrel medic); Mesculenta, Manihot esculenta (Cassava); Ptrichocarpa, Populus trichocarpa (Poplar); Csativus, Cucumis sativus (cucumber); Egrandis, Eucalyptus grandis (Eucalyptus); Smoellendorfii, Selaginella moellendorfii (Spikemoss); Ppatens, Physcomitrella patens (Moss).
(PDF)
MTR4-GFP co-localises with nucleolar marker proteins. Transient expression of fluorescent fusion proteins in Nicotiana benthamiana leaves. MTR4-GFP is shown in green, RFP-fusion proteins are shown in red. Fibrillarin-RFP and XRN2-RFP are known nucleolar markers; SRP34a-RFP was used as a nucleoplasmic marker. The phase contrast picture is shown on the right. Scale bars: 15 µm.
(PDF)
HEN2-GFP co-localises with a nucleoplasmic marker protein. Transient expression of fluorescent fusion proteins in Nicotiana benthamiana leaves. HEN2-GFP is shown in green, RFP-fusion proteins are shown in red. Fibrillarin-RFP and XRN2-RFP were used as nucleolar markers; SRP34a-RFP was used as a nucleoplasmic marker. The phase contrast picture is shown on the right. Scale bars: 15 µm.
(PDF)
MTR4 and HEN2 have distinct localization patterns. The distribution of the indicated GFP-fusion proteins in root cells of stable Arabidopsis transformants is shown on the left. The middle column shows DAPI staining. Please note that take-up of DAPI by intact, living plant tissue is slow and can lead to a strong background signal from cell walls. No, Nucleolus; Np, Nucleoplasm; Cp, Cytoplasm. Scale bars: 5 µm.
(PDF)
HEN2-GFP is localized in nucleoplasmic foci. Co-expression of HEN2-GFP and the nucleoplasmic marker protein SRP34a in leaves of stable Arabidopsis transformants. Nucleoplasmic foci were observed in all cell types of all stable transformants. Scale bars: 15 µm.
(PDF)
Sequence alignment of human RBM7 and Arabidopsis At4g10110.
(PDF)
Accumulation of unspliced transcripts in hen2 mutants. qRT-PCR. A Diagram of the genomic locus indicated by the respective AGI number is shown at the top of each panel. Annotated mRNA genes are represented as arrows with dark blue boxes for the CDS, light blue boxes for 3′ and 5‚ UTRs, and a light blue line for introns. Red bars above the diagram represent probes detected in the microarray analysis. Green arrows above or below the diagram depict the location of qRT-PCR primers. The corresponding qRT-PCR results for each primer pair are given as fold-change relative to WT in the histograms below each diagram. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates. A. At3g26510 (with 4 predicted splice variants). qRT-PCR results suggest that hen2 and RRP41 RNAi plants accumulate a population of transcripts some of which still contain the unspliced acceptor site of the first intron (panel V114a/b) and some of which still contain the 2nd intron (panel V112a/b). B. At1g58602. Transcripts comprising the unspliced 2nd exon/intron donor site accumulate in hen2 and RRP41 RNAi plants. C. At3g43160. Both spliced and unspliced transcripts corresponding to the 3′ region of the At3g43160 locus accumulate in hen2 and RRP41 RNAi plants.
(PDF)
Unspliced transcripts from the At1g79270 locus are polyadenylated. Sequences of 3′ RACE PCR products obtained from hen2-4 samples. cDNA synthesis was initiated using oligo-dT as primer. 3′ RACE PCR was performed with V113a (green arrow) as forward primer, and the adapter sequence of the oligo-dT primer as a reverse primer. PCR products obtained from hen2-4 samples were cloned and sequenced. The genomic sequence is given above the line, with intronic sequence in purple. Red arrows mark donor and acceptor splice sites. Non-encoded nucleotides are in green.
(PDF)
Upregulation of mRNAs in hen2 mutants. Accumulation of mRNAs was tested by qRT-PCR using primer pairs located in 5′, central or 3′ regions of the annotated transcripts as indicated below each panel. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates.
(PDF)
mRNAs are not systematically detected in all replicates. Accumulation of mRNAs was tested by qRT-PCR using primer pairs located in 5′ or 3′ regions of the annotated transcripts as indicated below each panel. Panels on the left show the qRT-PCR results for exactly the same samples that have been used for hybridisation to the tiling arrays. Panels on the right show the results obtained in 3 independent replicates grown in the same culture conditions. A possible explanation for the inconsistence between the replicates could be that mRNAs are not bona-fide substrates of exosome-mediated RNA degradation and might rather be upregulated due to indirect effects. Other types of transcripts such as short mRNA-derived regions, introns, unspliced transcripts or several types of non- coding RNAs are consistently observed in all replicates. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates.
(PDF)
Loss of HEN2 or the exosome is associated with increased levels of polyadenylated snoRNA precursors. Oligo-dT primed cDNA was used for 3′ RACE- PCR, with primer E5 and the adapter sequence of the cDNA synthesis primer as forward and reverse primers, respectively. PCR products were separated on 2% agarose gels, transferred to Hybond XL membranes, and hybridized with radiolabeled probes E6 (mid panel) and E8 (lower panel). The diagram below illustrates location of primers and probes with respect to the snoRNA genes in this region (see also Fig. 9).
(PDF)
Polyadenylated transcripts partially antisense to AT5G44306. Sequences were amplified by 3′ RACE from oligo-dT primed cDNA from the indicated samples using a primer (indicated by the purple arrow) situated antisense to the 5′ region of AT5G44306. Non-encoded nucleotides are in green.
(PDF)
mtr4 mutants accumulate exosome substrates to lower levels than hen2 mutants. Boxplot showing averaged intensity values for the overexpressed probes identified in each comparison of the tiling array analysis. The first two rows show the intensity values in WT and hen2 samples for all probes overexpressed in both biological replicates of hen2. The third and fourth rows show the intensity values in WT and mtr4 samples for all probes overexpressed in both biological replicates of mtr4. The averaged intensities in mtr4 samples are significant lower than the averaged intensities in hen2 samples (p-value<1e -3). The fifth and sixth rows show the intensity values in hen2 and mtr4 for the common probes (overexpressed in both mutants). Again, the mean average intensity in mtr4 samples is lower than the mean average intensity in hen2 samples (p-value<1e -3).
(PDF)
The microarray analysis probably underestimates the contribution of HEN2 to nuclear RNA surveillance. A diagram of the genomic locus indicated by the respective AGI number is shown at the top of each panel. Annotated mRNA genes are represented as arrows with dark blue boxes for the CDS, light blue boxes for 3′ and 5′ UTRs, and a light blue line for introns. Red bars above the diagram represent probes detected in the microarray analysis. Green arrows above or below the diagram depict the location of qRT-PCR primers. The corresponding qRT-PCR results for each primer pair are given as fold-change relative to WT in the histograms below each diagram. mtr4-1 in red, mtr4-2 in orange, hen2-2 in light green, hen2-4 in dark green, RRP41 control in light grey, RRP41 RNAi in dark grey. Error bars = SD in three biological replicates. Upper panel: The upregulation of two stretches in the 5′ region of At1g20100 in RRP41 RNAi lines (indicated by the red double arrows) was detected in a previous tiling microarray study [5]. Our microarray array detected only a portion of this region (indicated by the red bars above the diagram). However, we could confirm the upregulation of the uppermost 600 kb of At1g20100 in both hen2 alleles by qRT-PCR. Middle panel: A portion of the fifth intron of At5g27720 was previously identified as a target of the exosome core complex [5] (indicated by the red double arrows). In our microarray analysis, only one probe in this region was declared statistically significant. Since we considered only regions with at least two consecutive probes, this region was omitted from the data interpretation. Nevertheless, qRT-PCR data show that a portion of the fifth intron is upregulated in hen2 mutants, while levels of pre-mRNA or mature mRNAs are similar to WT. The presence of polyadenylated transcripts corresponding either to the entire intron or to shorter degradation intermediates in hen2 and RRP41 RNAi samples was further confirmed by cloning of 3′ RACE products (not shown). Lower panel: A previous tiling study [5] identified a short stretch derived from the 5′ region of At4g02890 as a target of the exosome core complex (indicated by the red double arrows). This region was not detected in our tiling analysis, but its upregulation was easily detected by qRT-PCR. The discrepancy between the two tiling studies is probably largely explained by the different arrays designs (the NimbleGen 732K array is not designed to detect very small regions) and by different statistical analysis procedures. Moreover, the accumulation of exosome targets may also vary with growth conditions. In fact, several of our validated targets have not been picked up in the previous genome-wide analysis [5], indicating that both tiling studies under-estimate the contribution of exosome-mediated RNA surveillance.
(PDF)
A fraction of AtMTR4-GFP can be detected in the nucleoplasm. Intracellular distribution of AtMTR4-GFP in root cells of a stable Arabidopsis transformant. The nucleoplasmic fraction of AtMTR4-GFP (white arrows) is more visible in individual transformants displaying relative weak transgene expression, which is not representative for the majority of the investigated plant lines.
(PDF)
Mass-spectrometric analysis of RRP41 IP experiments: list of peptides.
(XLSX)
Mass- spectrometric analysis of MTR4 IP experiments: list of peptides.
(XLSX)
Mass- spectrometric analysis of HEN2 IP experiments: list of peptides.
(XLSX)
Analysis of microarray data. Regions corresponding to short stretches of mRNAs, unspliced transcripts, and 3′ or 5′ extended mRNAs.
(XLSX)
Analysis of microarray data. Regions corresponding to introns.
(XLSX)
Analysis of microarray data. Regions corresponding to mRNAs.
(XLSX)
Analysis of microarray data. Regions corresponding to transposable elements.
(XLSX)
Analysis of microarray data. Regions corresponding to snoRNA precursors.
(XLSX)
Analysis of microarray data. Regions corresponding to linc and other RNAs.
(XLSX)
Analysis of microarray data. Potential antisense transcripts.
(XLSX)
Analysis of microarray data. Non-annotated regions.
(XLSX)
Gene descriptions and biological functions for the mRNAs upregulated in hen2 and/or mtr4 samples.
(XLSX)
Primers used in this study.
(XLSX)