

Background: Carbonylation of proteins contributes to increased hepatocellular damage during alcoholic liver disease.
Results: In a murine model of alcoholic liver disease, AMPK is covalently modified by reactive aldehydes reducing activity.
Conclusion: Inhibition of AMPK activity by reactive aldehydes contributes to increased steatosis in alcoholic liver disease.
Significance: This is the first report of AMPK carbonylation and inhibition during conditions of increased oxidative stress.
Keywords: Alcohol, AMP-activated Kinase (AMPK), Lipid Peroxidation, Liver, Oxidative Stress
Abstract
The production of reactive aldehydes including 4-hydroxy-2-nonenal (4-HNE) is a key component of the pathogenesis in a spectrum of chronic inflammatory hepatic diseases including alcoholic liver disease (ALD). One consequence of ALD is increased oxidative stress and altered β-oxidation in hepatocytes. A major regulator of β-oxidation is 5′ AMP protein kinase (AMPK). In an in vitro cellular model, we identified AMPK as a direct target of 4-HNE adduction resulting in inhibition of both H2O2 and 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR)-induced downstream signaling. By employing biotin hydrazide capture, it was confirmed that 4-HNE treatment of cells resulted in carbonylation of AMPKα/β, which was not observed in untreated cells. Using a murine model of alcoholic liver disease, treatment with high concentrations of ethanol resulted in an increase in phosphorylated as well as carbonylated AMPKα. Despite increased AMPK phosphorylation, there was no significant change in phosphorylation of acetyl CoA carboxylase. Mass spectrometry identified Michael addition adducts of 4-HNE on Cys130, Cys174, Cys227, and Cys304 on recombinant AMPKα and Cys225 on recombinant AMPKβ. Molecular modeling analysis of identified 4-HNE adducts on AMPKα suggest that inhibition of AMPK occurs by steric hindrance of the active site pocket and by inhibition of hydrogen peroxide induced oxidation. The observed inhibition of AMPK by 4-HNE provides a novel mechanism for altered β-oxidation in ALD, and these data demonstrate for the first time that AMPK is subject to regulation by reactive aldehydes in vivo.
Introduction
Oxidative modification of proteins by reactive aldehydes in the liver has been implicated in an increasing number of disease states including primary biliary cirrhosis, hepatitis C, and chronic alcoholic liver disease (ALD)2 (1–4). A primary marker for measuring increased oxidative stress in cells is the presence of 4-hydroxy-2-nonenal (4-HNE), which is increased in chronic metabolic liver diseases such as non-alcoholic steatohepatitis (NASH) and ALD (5, 6). Reactive aldehydes, such as 4-HNE, arise from peroxidation of lipids within cellular membranes (7). Although overall concentrations of 4-HNE within the cell are likely in the micromolar or submicromolar range, it has been calculated that concentrations within lipid bilayers could approach 100 mm (8, 9). 4-HNE is an electrophile that will react with protein nucleophiles including Cys, Lys, and His residues of proteins such as phosphatase and tensin homolog deleted on chromosome 10 (PTEN), tubulin, and Atk2 (10–13).
Within the liver, 5′ AMP protein kinase (AMPK) is a key mediator in the regulation of cellular energy and β-oxidation. Activation of AMPK is regulated by binding of AMP as well as by phosphorylation at Thr172 by upstream kinases including liver kinase B1 (LKB1) and Ca2+/calmodulin-dependent protein kinase (14, 15). Phosphorylation of AMPK by LKB1 increases the ability of AMPK to phosphorylate and inactivate a major rate-limiting enzyme in fatty acid synthesis, namely acetyl-CoA carboxylase (ACC). ACC catalyzes the conversion of acetyl CoA to malonyl-CoA, which is then utilized in the synthesis of saturated and unsaturated fatty acids (16). At high ratios of AMP/ATP (low energy), AMPK activity is increased, and ACC activity is decreased leading to a decrease in cellular malonyl CoA. If AMP/ATP ratios are low, AMPK activity is decreased, ACC is active, and malonyl CoA is used in de novo fatty acid synthesis (17).
Oxidative stress also regulates AMPK activity. Treatment of cells with H2O2 results in decreased cellular ATP concentrations and subsequent activation of AMPK (18, 19). Thus, phosphorylation and activation of AMPK regulates cellular energy under conditions of increased oxidative stress via β-oxidation in hepatocytes.
Previous reports concerning the effects of ethanol on activation of the AMPK pathway in mice vary depending on the amount of ethanol and the duration of feeding. In some studies, AMPK phosphorylation is increased (20–22), whereas in others AMPK phosphorylation is decreased (23–26). The use of different types of dietary fats as well as different percentages of dietary fat in these studies may be responsible for the discrepancies. In one report, 40% saturated fat plus ethanol resulted in a 2-fold increase in AMPKα phosphorylation (24). Concurrently, 40% PUFA plus ethanol resulted in a slight decrease in AMPKα phosphorylation (24). Using an intragastric overfeeding model, ethanol resulted in an increase in Thr(P)172 AMPK but no corresponding increase in ACC phosphorylation (21). We have previously reported that the addition of ethanol for 6 weeks in conjunction with 30% PUFA suppressed AMPK phosphorylation, whereas ETOH combined with 45% PUFA resulted in increased AMPK phosphorylation but no change in overall phosphorylation of ACC (20). Most recently, in C57BL6/J mice, chronic ETOH decreased AMPK phosphorylation but resulted in an increase in CPT1 mRNA and CPTII protein expression (27). Activity of CPT1, however, did not significantly change, suggesting no change in β-oxidation.
Herein, we describe the effects of increased lipid peroxidation/4-HNE on AMPK signaling in cell culture as well as in the liver of mice chronically fed ethanol for 7.5 weeks. We determine that in HepG2 cells, 4-HNE inhibits activation of AMPK by H2O2 and direct modification of recombinant AMPK by 4-HNE inhibits its activity. This research is further translated into the identification of AMPK as a direct target of lipid peroxidation in the livers of chronic ethanol-fed mice. These results provide a novel mechanism for dysregulation of AMPK signaling under conditions of increased oxidative stress that occur during chronic ethanol administration.
EXPERIMENTAL PROCEDURES
Animal Model and Dietary Information
C57BL/6J male mice (The Jackson Laboratory, Bar Harbor, ME) 6–8 weeks of age in groups of 12 were fed a modified Lieber-DeCarli diet (30% fat-derived calories (Bio-Serv, Frenchtown, NJ) consisting of isocaloric pair-fed control and ethanol-treated animals (28). Ethanol-derived caloric content was ramped from week 1 of 10.8%, with incremental increases weekly to 16.2, 21.5, 26.9, 29.2, 31.8, and 34.7% for the last 1.5 weeks of feeding (3, 4). In the control animals, calories derived from ethanol were replaced isocalorically by carbohydrates in the form of maltodextrin. Fresh control and ETOH diet was provided at 7:00 a.m. daily. Food consumption was monitored daily, and body weights were measured once per week. Upon completion of the study, animals were anesthetized via intraperitoneal injection with sodium pentobarbital and euthanized by exsanguination. Blood was collected from the inferior vena cava, and plasma was separated via centrifugation at 4 °C and assayed for alanine aminotransferase activity (Sekisui Diagnostics). Blood ethanol concentrations were determined by gas chromatography as previously described from samples obtained at 11:45 p.m. (29). Excised livers were weighed, and subcellular fractions were obtained via differential centrifugation as previously described (6). All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Colorado and were performed in accordance with published National Institutes of Health guidelines.
Treatment of HepG2 Cells
HepG2 cells were purchased from Invitrogen and maintained according to the manufacturer's instructions. Cells were maintained at 50–80% confluence in RPMI supplemented with 10% fetal bovine serum, 100 mm Hepes, 100 IU/ml penicillin, and 100 g/ml streptomycin. Cells were plated into 6-well plates at a density of 1 × 106 cells per well. The following day the cells were washed twice in serum-free RPMI and treated with the indicated concentrations of 4-HNE. For hydrogen peroxide treatments, HepG2 cells were treated with 0–50 μm 4-HNE for 60 min, washed once in serum-free media, and stimulated with 1.0 mm hydrogen peroxide for 5 min (Sigma). For 5-aminoimidazole-4-carboxyamide ribonucleoside (AICAR) experiments, cells were pretreated with 50 μm 4-HNE followed by stimulation with 2 mm AICAR for 60 min.
Western Blotting
Cells were lysed for 5 min in 50 mm HEPES, 100 mm NaCl, 1% Triton X-100, 2 mm EDTA, pH 7.7, plus protease inhibitors (Sigma) followed by sonication 3× for 10 s. For each gel, 10 μg of whole cell lysates were loaded per well on 7% SDS-PAGE gels, electroblotted to PVDF, blocked in Tris-buffered saline with 1% Tween (TBST) and 5% nonfat dry milk for 1 h, and incubated overnight in primary antibody. The following primary antibodies were used at 1:1000 dilution in TBST: Thr(P)172AMPK, AMPK, Ser(P)792Raptor, ACC, Ser(P)79ACC, ACSL1 (Cell Signaling Technology, Danvers, MA), Raptor (Millipore, Billerica, MA), CPT1α (Protein Tech, Chicago, IL), and β-actin (1:10,000) (Sigma). The following morning blots were washed 3 times for 5 min in TBST and incubated for 1 h in horseradish peroxidase-conjugated goat polyclonal anti-rabbit secondary antibody or donkey polyclonal anti-mouse secondary antibody (1:5000) (Jackson ImmunoResearch Laboratories, West Grove, PA). Blots were washed 3 times for 5 min in TBST and subsequently developed using chemiluminescence (Pierce Supersignal, Thermofisher Scientific, Rockford, IL). All Western blots were quantified using NIH freeware ImageJ.
Immunohistochemistry
Sections of freshly excised liver tissue were placed in 10% neutral-buffered formalin (Sigma) for 16 h followed by incubation in 70% ethanol overnight. Samples were then processed, embedded in paraffin, and mounted to slides by the University of Colorado C Denver Histology Core. Immunohistochemical characterization of carbonylation and macrophage infiltration was performed using rabbit polyclonal anti-4-HNE, anti-MDA, anti-acrolein (Abcam, Cambridge, MA), and rat anti-mouse polyclonal antibodies directed against F4/80 antigen (AbSerotec/Bio-Rad), respectively (30).
AMPK Activity Assays
The AMPK activity assay was purchased from MBL International (Woburn, MA) and utilized for AMPK activity. Briefly, 0.5 μg of rAMPK was treated in triplicate with increasing molar ratios of 4-HNE (0–25:1) in NH4HCO3 for 30 min at 37 °C. Protein was resuspended in kinase buffer and incubated in a 96-well plate for 30 min at 30 °C. To each well 100 μl of anti-phospho mouse IRS1 Ser789 was added and incubated for an additional 30 min. Wells were washed, 100 μl of HRP-conjugated anti-mouse IgG added and allowed to incubate for 30 min. Wells were washed, and 100 μl of tetramethylbenzidine were added for 30 min followed by the addition of EDTA stop solution. Absorbance was then read at 450 nm.
Measurement of Reduced and Oxidized Glutathione
Glutathione measurements were performed using a modified method from Reed et al. (31). Briefly, fresh liver tissue was homogenized in 0.1 n perchloric acid using a sonicator and centrifuged at 13,000 rpm for 15 min, and the cleared supernatant was further neutralized with sodium bicarbonate and treated with 1:10 iodoacetic acid (14.9 mg/ml) in the dark for 45 min at room temperature. Samples were then derivatized (1:1) with Sanger's reagent (1.5% 1-fluoro-2,4-dinitrobenzen in absolute ethanol) overnight in the dark at room temperature. Derivatized samples were then stored at 4 °C in the dark until analyzed by HPLC. Samples were applied (50-μl injection) to a Supelco LC-NH2 5 μm, 25 cm × 4.1 mm column (Sigma) separated with a gradient mobile phase system. Solvent A (80% methanol) and Solvent B (80% Solvent B, 20% acetic acid/ammonium acetate solution (756 ml of glacial acetic acid, 244 ml of water, and 308 g of ammonium acetate)) were used for the separation of derivatized reduced (GSH) and oxidized (GSSG) glutathione using a Shimadzu LC-10AD dual pump system (flow rate of 1.0 ml/min) coupled to a Shimadzu SPD-M10AV diode array detector (Kyoto, Japan) set to 350 nm. HPLC running conditions were as follows: isocratic period of 50% solvent B for 10 min followed by a linear gradient from 50% solvent B to 95% solvent B for 15 min for a total run time of 35 min. Typical elution times for GSH were 11.55 and 17.05 min for GSSG. Nanomolar concentrations of GSH and GSSG were calculated against a known standard curve of seven points for each molecule.
Biotin Hydrazide Modification of Alkylated AMPK
To evaluate reactive aldehyde adduction, 1 × 107 HepG2 cells were treated with 50 μm 4-HNE for 1 h. Cells were then lysed in 50 mm HEPES, 100 mm NaCl, 2 mm EDTA, 0.5% Triton-100, 2.5 mm biotin hydrazide for 2 h. To remove excess biotin, lysates were dialyzed overnight at 4 °C. Biotinylated proteins were captured using streptavidin-agarose beads (Pierce) on a rotary mixer at 4 °C for 3 h. Beads were washed 5 times in PBS, boiled in SDS loading buffer, and analyzed using SDS-PAGE/Western blotting for AMPKα, -β1, and -β2. For murine studies, whole cell lysates were prepared from liver tissue isolated from mice fed ETOH for 7.5 weeks followed by biotin hydrazide treatment of homogenates containing 0.5 mg of total protein from pair-fed and ETOH-fed mice.
Nile Red Staining of Neutral Lipids
HepaRG cells were plated in 6-well plates to >90% confluence and treated for 1 h with 5% oleic acid conjugated to BSA or with 50 μm 4-HNE followed by a 24-h recovery in serum-containing RPMI 1640 media. Nile Red was used to detect cellular neutral lipid using a modification of previously reported techniques, and Hoechst 33342 was used to measure cellular DNA (32). Cells were collected in 1 ml of phenol red-free, serum-free RPMI 1640, and 50 μl of cell suspension was incubated in sterile 96 well fluorescence plates for 1 h at 37 °C with 200 μl of serum-free, phenol red-free RPMI 1640 containing Nile Red at 20 μg/ml and Hoechst 33342 at 10 μg/ml. Plates were shaken for 10 s before reading, and neutral lipid was assessed by Nile Red fluorescence with excitation 485 nm/emission 565 nm and was normalized against cellular DNA as measured by Hoechst 33342 staining measured with excitation 355 nm/emission 465 nm.
Adduction and Trypsin Digest of Recombinant AMPKα/β/γ by 4-HNE
10 μg of rAMPKα/β/γ was treated with 4-HNE at molar ratios of 0.1, 1:1, 2.5:1, 5:1,10:1, and 20:1 for 30 min at 37 °C. 0.5 μg were removed and loaded onto an SDS-PAGE gel and Western-blotted using anti-4-HNE polyclonal antibodies. To verify AMPK adduction, membranes were then stripped for 15 min using RestoreTM stripping buffer (Pierce), washed twice in TBST, and blocked once again for 1 h in TBST, 5% nonfat dry milk. After blocking, membranes were incubated overnight with mouse anti-AMPK (Pierce) and subsequently processed. The rest of the sample was treated with 10 mm sodium borohydride in 100 mm NaOH for 30 min at 37 °C. Samples were boiled in 5× SDS loading buffer and subjected to SDS-PAGE. Gels were stained for 15 min with Coomassie Blue R250 and destained overnight in 10% acetic acid, 20% MEOH. From the gel, each band was picked and destained in 50 mm NH4HCO3 for an additional 24 h. Gel slices were dehydrated with 100% acetonitrile for 15 min, reduced in 10 mm DTT, 50 mm NH4HCO3 for 45 min at 60 °C, and alkylated in 50 mm iodoacetic acid in 50 mm NH4HCO3 for 45 min in the dark at room temperature. Samples were washed once in 50 mm NH4HCO3 and digested in 0.3 μg of trypsin 50 mm NH4HCO3 overnight. Peptide extraction consisted of sonicating for 20 min in 50% acetonitrile, 50 mm NH4HCO3 followed by sonication in 5% formic acid in acetonitrile for an additional 20 min. Samples were dried down and resuspended in 0.1% trifluoroacetic acid.
A nano-flow HPLC system (Agilent 1200, Palo Alto, CA) coupled with LTQ Orbitrap Velos Hybrid FT mass spectrometer and nanospray ion source (Thermo Fisher, San Jose, CA) was used for LC-MS/MS analysis. An 8-μl volume of samples was run on a ZORBAX 300SB-C18 trap column (5-μm inner diameter × 5 mm, Agilent Technologies, Santa Clara, CA) to remove salts and contaminating analytes, and reverse phase separation of peptides was performed on a C18 column (100-μm inner diameter × 17-cm length) packed in-house using a 4-μm 80 Å pore size matrix (Synergi, Phenomenex, Torrance, CA). Mobile phase A was composed of 99.9% HPLC grade water, 0.1% formic acid, and mobile phase B was composed of 99.9% acetonitrile, 0.1% formic acid. Peptides were eluted from the column using a linear gradient with mobile phase B increasing from 2–90% over 60 min. The positive ion mode was used for data acquisition of MS of ions m/z 300–2000, and MS2 scans were performed for the most intense ions. All data were acquired using Thermo Xcalibur software (Version 2.1.0.1140, Thermo Fisher).
MS/MS Data Analysis
An in-house script was used to convert raw spectral data files into mascot generic file (mgf) format. The mgf files were searched against the human Swissprot database Mascot (Version 2.2, Matrix Science Inc., London, UK). The MS/MS tolerance was set at ±0.6 Da for the searches, and the peptide tolerance was set at ±15 ppm. The search allowed for the missed cleavage of one tryptic site as well as one carbon 13. The carbamidomethylation of cysteine was searched as a fixed modification. Variable modifications included acylation of the protein N terminus, pyroglutamic acid formation of the peptide N terminus, oxidation of methionine, 4-HNE modification of cysteine, histidine, and lysine, and reduced 4-HNE modification of cysteine, histidine, and lysine. Spectral images were obtained from the raw data files using Xcalibur, and Mascot was used to assist peak assignment. Only Mascot scores of >30 were considered significant.
In Silico Molecular Modeling
All simulations were performed using Discovery Studio software (Version 3.1; Accelrys Inc., San Diego, CA). The crystallographic coordinates of the 1.6 Å human-activated AMPK crystal structure (PDB code 2Y94) were obtained from the RCSB Protein Data Bank (33). The 4-HNE modification of the Cys130, Cys174, and Cys227 residues on AMPKα and Cy225 on AMPKβ were typed with the CHARMm force field and subjected to minimization using the conjugate gradient method to a convergence of 0.001 kcal/mol using the Generalized Born implicit solvent model (34, 35).
Statistical Analysis
Western blots were quantified using ImageJ (rsb.info.nih.gov) followed by statistical analysis via a paired Student's t test and Prism 4.0 for Windows (GraphPad Software, San Diego, CA). All data are expressed as the means ± S.E., and p values <0.05 were considered significant.
RESULTS
AMPK Phosphorylation Is Decreased after 4-HNE Treatment in HepG2 Cells
In our previous report we identified 4-HNE-dependent modification and inhibition of PTEN as a contributor to lipid accumulation in HepG2 cells (11). We further demonstrated that Akt2, which is the immediate downstream target of PTEN, was also inhibited by 4-HNE in the same cells. This suggests that lipid accumulation induced by 4-HNE exposure may be due to regulation of an alternative pathway. In the liver Akt2 is a primary regulator of fatty acid synthesis, whereas AMPK is a primary regulator of β-oxidation (17, 36). To evaluate the hypothesis that 4-HNE alters phosphorylation of AMPKα and ACC, HepG2 cells were treated with 50 μm 4-HNE using a time course of 2 h. Cells were lysed, and lysates were examined for increased phosphorylation of AMPK and its immediate downstream target of ACC via Western blotting. From Fig. 1, incubation of HepG2 cells 4-HNE resulted in a time-dependent decrease in Thr(P)172AMPKα corresponding to decreased Ser(P)79ACC.
FIGURE 1.

Time course of 4-HNE-induced decreases in AMPK phosphorylation. Time-dependent inhibition of AMPK phosphorylation. HepG2 cells were treated with and without (+/−) 50 μm 4-HNE from 5–120 min. Cells were lysed and examined via 7% SDS-PAGE, Western blotting using p-AMPKα (Thr172, AMPKα, p-ACC (Ser79), ACC, and actin.
Protein phosphatase 2A (PP2A) regulates AMPK activation by dephosphorylation of Thr(P)172AMPK (37). To determine if the mechanism of 4-HNE inhibition of AMPK phosphorylation is via increased PP2A activity, HepG2 cells were incubated with the PP2A inhibitor okadaic acid with or without 4-HNE. From Fig. 2, A--C, treatment with okadaic acid (OA) resulted in partial but not complete restoration of AMPK and ACC phosphorylation. This indicates that activation of PP2A contributes to 4-HNE-induced decreases in AMPK phosphorylation.
FIGURE 2.

Effects of okadaic acid on 4-HNE-mediated inhibition of AMPK signaling. HepG2 cells were preincubated with okadaic acid (50 nm 60 min) followed by 4-HNE (50 μm for 60 min). A, Western blot examining the effects of okadaic (Ok and OA) acid on 4-HNE-induced AMPK and ACC dephosphorylation. B, quantification of Western blot of pAMPK/(total AMPK/actin). C, quantification of Western blot of pACC/(total ACC/actin). All experiments were performed in triplicate and normalized against total respective proteins expression and actin. Statistical analysis was via one-way analysis of variance with Tukey's multiple comparison test. *, p < 0.05; ***, p < 0.001.
4-HNE Inhibits Hydrogen Peroxide-induced Activation of AMPK in HepG2 Cells
We have previously shown that 4-HNE will inhibit activation of both Akt1 and Akt2 by H2O2 in HepG2 cells (38). Reactive species such as H2O2 are also known activators of AMPK (19, 37, 39). Treatment of rat hepatoma cells with 1 mm H2O2 resulted in a 1.5-fold increase in AMPK phosphorylation (37). To gain a better understanding of the effects of 4-HNE on H2O2-induced AMPK, HepG2 cells were preincubated with increasing concentrations of 4-HNE followed by H2O2 stimulation. As shown in Fig. 3, A–D, in cells treated with 4-HNE/H2O2, a concentration-dependent decrease in AMPKα phosphorylation was observed. This decrease corresponded to a decrease in phosphorylation of the downstream AMPK targets ACC and Raptor (39). Combined, these data demonstrate 4-HNE inhibits the induction of AMPK signaling by H2O2.
FIGURE 3.

Inhibition of H2O2-dependent AMPK signaling by 4-HNE. HepG2 cells were treated in serum-free media with and without (+/−) 50 μm 4-HNE for 60 min, washed in serum-free media, and stimulated with 1 mm H2O2 for 5 min. A, Western blot of phospho/total AMPK, phospho/total ACC, phospho/total Raptor. B, quantification of pAMPK/(total AMPK/actin). C, quantification of Western blot of pACC/(total ACC/actin). D, quantification of Western blot of pRaptor/(total Raptor/actin). Statistical analysis was via one-way analysis of variance with Tukey's multiple comparison test. *, p < 0.05; **, p < 0.01; ***, p < 0.001. Means without a common superscript letter are significantly different.
In addition to H2O2, AMPK can be activated by the adenosine analog AICAR resulting in increased phosphorylation of ACC (23). To determine if 4-HNE broadly inhibits AMPK activity, HepG2 cells were treated with 4-HNE and 2 mm AICAR, and phosphorylation of ACC was evaluated. From Fig. 4, A and B, 4-HNE significantly inhibits AICAR-induced phosphorylation of ACC in HepG2 cells, indicating that reactive aldehydes inhibit AMPK activation.
FIGURE 4.

Effects of 4-HNE on AICAR-induced ACC phosphorylation. HepG2 cells were stimulated with 50 μm 4-HNE (60 min) followed by 2 mm AICAR (60 min). A, Western blot of phospho ACC and total ACC. B, quantification of blots are representative of at least three independent experiments. Statistical analysis was via one-way analysis of variance with Tukey's multiple comparison test. *, p < 0.05; ***, p < 0.001.
4-HNE Treatment Leads to Increased Carbonylation of AMPK in HepG2 Cells
Biotin hydrazide will react with reactive aldehyde groups on proteins forming a covalent biotin hydrazone bond. We have utilized biotin hydrazide modification followed by streptavidin capture to identify carbonylated proteins from cells as well as hepatic tissue (10–12, 38, 40, 41). To determine if carbonylation of cellular AMPKα/β occurs after 4-HNE treatment, HepG2 cells were subjected to increasing concentrations of 4-HNE followed by biotin hydrazide modification followed by streptavidin capture and Western blotting. As shown in the Western blots depicted in Fig. 5A, 4-HNE treatment resulted in a concentration-dependent increase in carbonylation of AMPKα/β. The quantification of the Western blots demonstrates a 5–10-fold increase in carbonylation after the 25–50 μm 4-HNE treatment (Fig. 5, B--D). These data indicate that intracellular carbonylation of AMPK occurs as a consequence of 4-HNE exposure (8).
FIGURE 5.

AMPKα is modified by reactive aldehydes after 4-HNE treatment in HepG2 cells. A, 4-HNE modified AMPKα, AMPKβ1, and AMPKβ2 from HepG2 cells. Using whole cell extracts, 125 μg of protein from increasing concentrations of 4-HNE (0–50 μm for 60 min in serum-free media) or untreated cells was incubated for 2 h with 2.5 mm biotin hydrazide, purified, and analyzed using SDS-PAGE/Western blotting with rabbit polyclonal anti-AMPKα, AMPKβ1, AMPKβ2. B, quantification of carbonylated AMPKα. C, quantification of carbonylated AMPKβ1. D, quantification of carbonylated AMPKβ2. Statistical analysis was via one-way analysis of variance with Tukey's multiple comparison test. **, p < 0.01; ***, p < 0.001.
Effects of 4-HNE on Lipid Accumulation in HepaRG Cells
We previously demonstrated that the addition of 4-HNE results in increased lipid accumulation in HepG2 cells (11). HepG2 cells are a hepatocarcinoma cell line and may not best represent primary liver cells (42). HepaRG cells were treated with 50 μm 4-HNE or 5% oleic acid (positive control) for 60 min followed by incubation for 24 h, and lipid accumulation was measured using Nile Red. From Fig. 6A, incubation of HepaRG cells with 4-HNE results in a significant 34% increase in lipid accumulation. This is in agreement with data we obtained in HepG2 cells (11).
FIGURE 6.

Effects of 4-HNE on lipid accumulation in HepaRG cells. A, 4-HNE induces lipid accumulation in HepaRG cells. Nile Red fluorescence of HepaRG cells treated with 4-HNE (50 μm 60 min) or oleic acid (5% 60 min-positive control). Cells were washed in PBS followed by incubation for an additional 24 h in serum containing media. B, exposure to 4-HNE does not affect expression of proteins involved in β-oxidation. HepaRG cells were treated with 4-HNE (50 μm for 60 min) or oleic acid (5% 60 min-positive control). Cells were washed in PBS followed by incubation for an additional 24 h in serum containing media. Cells were lysed and examined for expression of ACSL and CPT1α via Western blotting. C, AMPK is carbonylated after 4-HNE exposure in HepaRG cells. HepaRG cells were treated with 50 μm 4-HNE followed by cell lysis and purification of carbonylated proteins using biotin hydrazide capture (BH/SA). Whole cell extracts (WCE) and purified biotinylated proteins were analyzed using SDS-PAGE/Western blotting probing for AMPKα. Experiments was performed in at least triplicate and subjected to one-way ANOVA with Tukey's multiple comparison test. *, p < 0.05; ***, p < 0.001.
The effects of 4-HNE on expression of β-oxidative enzymes in cell culture has not been reported. From Fig. 6B, expression of carnitine palmitoyl transferase (CPT1α) and acyl-CoA synthetase 1 (ACSL1) was not significantly changed after overnight 4-HNE exposure, suggesting that 4-HNE does not have a direct effect on the expression of β-oxidative enzymes.
We sought to determine if AMPK kinases are also carbonylated after HepaRG cell exposure to 4-HNE by using biotin hydrazide modification and streptavidin capture (12). From Fig. 6C, an increase in carbonylation is clearly evident in the 4-HNE treated cells, indicating that AMPK is indeed a target of 4-HNE in HepaRG cells. Combined with these data, we demonstrate that increased 4-HNE concentrations contribute to lipid accumulation as well as increased carbonylation of AMPK.
Effects of Chronic Ethanol on Hepatic Inflammation and Oxidative Stress
We have identified AMPKα as a novel target of reactive aldehydes in hepatocellular cell culture models. Our previous data indicate that ACC phosphorylation is not altered in response to increased AMPK phosphorylation in the liver after chronic ethanol consumption. We hypothesized that AMPK may also be carbonylated in our chronic ETOH consumption model. To determine if AMPK carbonylation was increased after chronic ETOH consumption, mice were fed increasing concentrations of ETOH for 7.5 weeks. As shown in Table 1, chronic ethanol consumption resulted in a significant increase in liver/body weight, serum alanine aminotransferase, hepatic triglycerides, and in overall carbonylation. After ETOH consumption, a significant decrease in GSH and in the ratio of GSH:GSSG indicate an increase in hepatocellular oxidative stress.
TABLE 1.
Liver parameters obtained from pair-fed and ETOH-fed mice
Basic liver parameters were obtained from chronic ETOH consumption for 7.5 weeks. Serum alanine aminotransferase (ALT; units/ml), liver/body weight ratio, hepatic triglycerides (mmol/mg of tissue), percent control carbonylation, GSH (μmol/g of tissue), GSSG (μmol/g of tissue), and GSH:GSSG were all determined as described under “Experimental Procedures.” n = 6 mice/group.
| Parametera | Pair-fed | ETOH | p Value |
|---|---|---|---|
| ALT (units/liter) | 7.810 ± 1.548 | 89.965 ± 8.989b | 0.0003 |
| Liver/body weight | 3.570 ± 0.092 | 4.992 ± 0.153c | 0.0386 |
| Liver triglycerides (μmol/mg tissue) | 0.236 ± 0.028 | 0.349 ± 0.012b | 0.0006 |
| Carbonylation | 100.00 ± 16.056 | 146.639 ± 13.560d | 0.0019 |
| GSH μmol/g tissue | 3.307 ± 0.327 | 2.037 ± 0.067c | 0.0168 |
| GSSG μmol/g tissue | 0.555 ± 0.034 | 0.563 ± 0.032 | 0.8745 |
| GSH:GSSG | 5.935 ± 0.273 | 3.688 ± 0.318b | 0.001 |
a Data are presented as the mean ± S.E. Statistical significance was determined by paired Student's t tests.
b p < 0.001.
c p < 0.05.
d p < 0.01.
In other murine models, chronic ethanol consumption results in an increase in hepatic inflammation (43). The F4/80 antigen is an accepted marker of increased hepatic monocyte/macrophage recruitment during chronic inflammation (6). To verify that this model of ETOH consumption results in an increase in inflammation, immunohistochemical staining of F4/80+ cells was performed using formaldehyde-fixed, paraffin-embedded liver tissue isolated from pair-fed and ETOH-fed mice. As shown in Fig. 7A, chronic ethanol consumption resulted in a marked increase in F4/80+-stained cells in both the periportal and central venous regions of the liver. To determine the percent increase in monocytes/macrophages after ETOH, F4/80+ cells were counted, and the ratio F4/80+ cells:total cells (based on numerical analysis of nuclei) was determined. As shown in Fig. 7B, chronic ethanol consumption significantly increased monocytes/macrophage recruitment by >2-fold in both the periportal and centrilobular hepatic regions but not in the midzonal region.
FIGURE 7.

Characterization of the effects of chronic ETOH administration on hepatic macrophage infiltration. A, immunohistochemical staining of F4/80+ macrophages in liver tissue sections isolated from pair-fed and ETOH-fed mice. PT, periportal; MZ, midzonal; CV, centrilobular. B, quantification of hepatic F4/80+ infiltration in the periportal, midzonal, and centrilobular in pair-fed regions and ETOH-fed mice. NS, not significant. C, time course of serum ETOH concentrations after 7.5 weeks of chronic ETOH consumption (samples were taken once per week at 11:55 p.m. and determined as mg/dl, n = 5 for each time point). Data are the means ± S.E. as analyzed by paired Student's t test. n = 6 mice/group (**, p < 0.01).
We previously determined a blood ethanol concentration of 245 mg/dl after 6 weeks of chronic ethanol consumption (30). As shown in Fig. 7C, after 3 weeks serum ETOH concentrations increase linearly in our ETOH-fed animals, reaching an average concentration of 375 mg/dl at 7.5 weeks. Although high, these concentrations are representative of concentrations found in human alcoholics after hospital admission as well as in forensic analysis of cadaveric alcoholics upon death (44, 45). Although this is higher than levels obtained after 6 weeks of chronic ETOH consumption (30), these concentrations are similar to concentrations found in rodents in other models after chronic ETOH consumption (46, 47).
Although we have previously shown using immunohistochemistry that the reactive aldehydes 4-HNE, malondialdehyde (MDA), and acrolein are all increased by chronic ETOH consumption after 6 weeks, these effects have not been characterized after 7.5 weeks of ETOH consumption (48). From Fig. 8, in the control animals low levels of staining of 4-HNE, MDA, and acrolein occur. Compared with the control animals, 7.5 weeks of ETOH consumption resulted in a marked increase in immunostaining surrounding the portal triad of proteins modified by acrolein, MDA, and 4-HNE.
FIGURE 8.
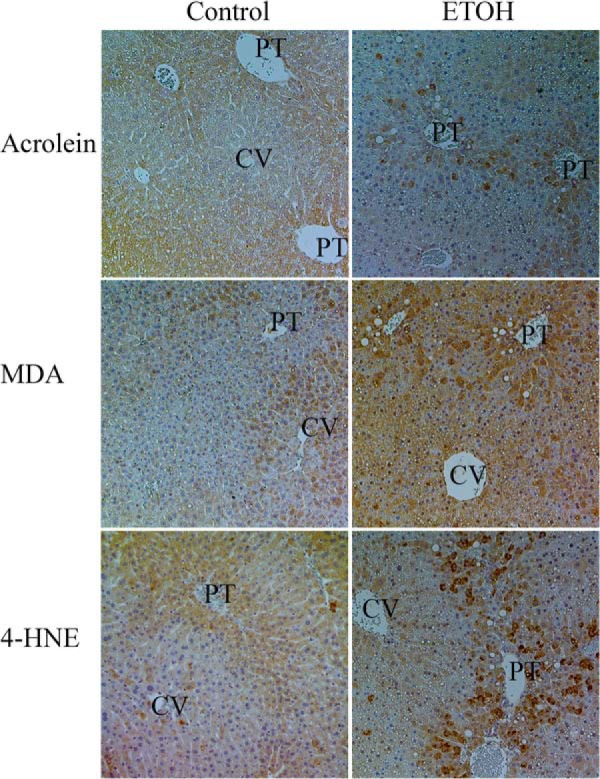
Effects of 7.5 weeks of chronic ETOH consumption on protein carbonylation by acrolein, MDA, and 4-HNE. Formaldehyde-fixed, paraffin-embedded hepatic tissue sections isolated from control and ETOH fed mice were subjected to immunohistochemistry using anti-acrolein, MDA, and 4-HNE as described under methods. PT, portal triad; CV, centrilobular.
In ALD, inflammation is a precursor for fibrosis. To assess if our model exhibited an increase in fibrosis, picrosirius red staining was performed on tissue sections isolated from both pair- and ETOH-fed mice (49). Picrosirius staining of chronic ETOH liver sections did not indicate a substantial increase in hepatic fibrosis in this model (data not shown). This is further demonstrated by using polarized light and the same sections (Data not shown). Nevertheless, these data verify that after ETOH consumption hepatocellular damage, inflammation and carbonylation all are significantly increased in response to ETOH consumption.
In Fig. 5, we identified both AMPKα as a target of carbonylation in 4-HNE-treated HepG2 cells. In our previous report, we determined that although AMPK phosphorylation increased after ETOH consumption, the increase did not correlate with an increase in phosphorylation of ACC (20). In that study, mice were fed ETOH for 6 weeks, and hepatocellular damage as shown by alanine aminotransferase was only increased by 2.5-fold (20). Using whole cell lysates from the current 7.5-week feeding model, phosphorylation of AMPKα and ACC was assessed. As shown in the Western blot, presented in Figs. 9, A--C), AMPK phosphorylation was increased 1.5-fold in ETOH-fed mice, whereas phosphorylation of ACC was not significantly changed. By its ability to convert long-chain fatty acids into long-chain acylcarnitines, CPT1α is the rate-limiting enzyme in β-oxidation. ACSLs activate long-chain fatty acids to form acyl-CoAs, facilitating their transport across membranes for subsequent β-oxidation (50). In a recent report, CPT1 expression was increased after 7 weeks of ETOH consumption in mice (27). The effects of chronic ETOH on expression of CPT1α and ACSL in this study were examined by Western blotting. From Fig. 9A, expression of CPT1α and ACSL was not significantly changed after chronic ETOH consumption. Combined, these data support our hypothesis that expression of proteins associated with β-oxidative processes is not significantly changed in the presence of increased AMPK phosphorylation in our ETOH model. Instead, these data indicate that it may be the lack of increased β-oxidation in the presence of increased AMPK phosphorylation that contributes to lipid accumulation. Furthermore, they also provide additional evidence of regulation of AMPK signaling by carbonylation of AMPK.
FIGURE 9.

Effects of chronic ETOH consumption on hepatic AMPK/ACC phosphorylation. A, Western blotting of Thr(P)172AMPK, total AMPK, Ser(P)79ACC, total ACC, CPT1α, and ACSL in pair-fed and chronic ETOH-fed whole hepatic lysates. B, quantification of normalized Thr(P)172 AMPK. C, quantification of normalized Ser(P)79 ACC. Data are the means ± S.E. as analyzed by paired Student's t test. n = 6 mice/group (**, p < 0.01).
In Figs. 1–5, 4-HNE treatment resulted in an increase in carbonylation and inhibition of AMPK activity in our HepG2 cell culture model. We, therefore, sought to determine if chronic ETOH consumption results in an increase in carbonylation of hepatic AMPK. As shown in Fig. 10, carbonylation of AMPKα is significantly increased in the liver of ETOH-fed mice.
FIGURE 10.

Effects of chronic ETOH consumption on hepatic carbonylation of AMPK. Whole cell hepatic extracts isolated from both pair-fed and ETOH-fed were treated as described under “Experimental Procedures.” Data are the means ± S.E. as analyzed by paired Student's t test. n = 6 mice/group (**, p < 0.01). Human recombinant protein was used.
4-HNE Modifies and Inhibits AMPK Activity in Vitro
Based on data in Figs. 1–5, 4-HNE treatment of HepG2 cells inhibits AMPK activity and modifies AMPKα/β. To further elucidate the effects of 4-HNE on AMPK, recombinant AMPK (rAMPKα/β/γ) was incubated with increasing molar ratios of 4-HNE (51). As shown in Fig. 11A, exposure of rAMPKα/β/γ to increasing molar concentrations of 4-HNE results in increased modification of AMPKα/β but not AMPKγ. We have previously shown that 4-HNE directly inhibits the activity of Akt1 as well as Akt2 kinases. To determine if 4-HNE treatment of rAMPKα/β/γ inhibited activity, rAMPKα/β/γ was treated with increasing molar concentrations of 4-HNE followed by activity assays. As shown in Fig. 11B, rAMPK activity was significantly decreased by 30% at a molar ratio of 5:1. Combined, these data indicate that AMPK is a direct target of reactive aldehydes, and modification of AMPK inhibits activity.
FIGURE 11.

Effects of 4-HNE on recombinant AMPKα/β/γ. A, Western blotting of rAMPKα/β/γ treated with increasing molar concentrations of 4-HNE. Purified rAMPKα/β/γ was incubated with increasing molar ratios of 4-HNE for 30 min at room temperature. Samples were boiled in 5× SDS loading buffer, run on an 8% SDS-PAGE gel, blotted, and probed for 4-HNE using anti-4-HNE polyclonal antibodies. B, inhibition of rAMPKα by 4-HNE. Purified rAMPKα was incubated with increasing ratios of 4-HNE, and activity assays were performed as under “Experimental Procedures.” All samples were performed in at least triplicate. Statistical analysis was via one-way analysis of variance with Tukey's multiple comparison test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Identification of Amino Acid Residues on rAMPKαβ as Targets for 4-HNE Modification
Although 4-HNE inhibited AMPK activity, the mechanism of inhibition is unknown. To identify specific residues in AMPKα modified by 4-HNE, rAMPKα was treated with increasing molar ratios of 4-HNE followed by SDS-PAGE, band excision, tryptic digestion, and LC/MS/MS analysis. The resulting peptides obtained and their respective m/z ratios (mass to charge ratio) are presented in supplemental Table 1. Only peptides with a mascot ion score of >30 were considered as statistically significant and included. From supplemental Table 1, on AMPKα, Cys130, Cys174, Cys227, and Cys304 (all in bold) were identified as targets of 4-HNE modification. On AMPKβ, Cys225 was also identified as a target of 4-HNE. The data in supplemental Fig. 1, A–E show MS/MS ion chromatograms obtained for each 4-HNE-modified peptide on AMPKα/β.
Effects of 4-HNE Modification on AMPK Structure
We examined known crystal structures of AMPK and determined of the identified modified cysteines, only Cys130, Cys174, Cys227(α), and Cys225(β) have known locations in the overall AMPK structure (33). The available crystal structures of AMPK do not indicate the location of Cys304, but based on structural predictions, it is located in an exposed loop between the AMPKα and AMPKγ subunits (data not shown) (33). Computational-based minimization simulations were performed using the crystal structure of AMPK (PDB code 2Y94) combined with each modification. A global view of all the modifications is shown in Fig. 12. Based on the information obtained from the modeling simulation, 4-HNE treatment did not result in any substantial predicted changes to the overall structure of AMPK (data not shown). Examining each adducted residue, 4-HNE Cys225(β) and Cys227(α) were located on the surface and did not appear to significantly alter either the active site or the activation loop (data not shown). As shown in Fig. 13, A and B, the minimized structure of Cys174–4HNE resulted in 4-HNE freely rotating out of the activation loop immediately adjacent to Thr(P)172. This suggests that Cys174–4HNE may decrease the efficiency of phosphorylation and/or dephosphorylation of Thr172 in the activation loop. An examination of the minimized structure of Cys130–4HNE revealed a surprising result. In the crystal structure, the ATP competitive inhibitor staurosporine is shown bound to the active site. As shown in Fig. 13, C and D, in the crystal structure of AMPKα, Cys130–4HNE is buried below the active site under staurosporine. This suggests that the adduction of Cys130 by 4-HNE also has the potential to inhibit ATP binding and or substrate access. Although the specific location of Cys304 is unknown, H2O2 will oxidize Cys304, resulting in an increase in AMPK activity in HEK 293 cells (19). Relative to the location of Lys299 and Arg331, Cys304 is positioned on the opposite face from Cys174 (data not shown). Mutation of Cys304 will inhibit H2O2-induced increases in AMPK activity (19). This suggests that alkylation of Cys304 may also play a role in regulating AMPK activity.
FIGURE 12.
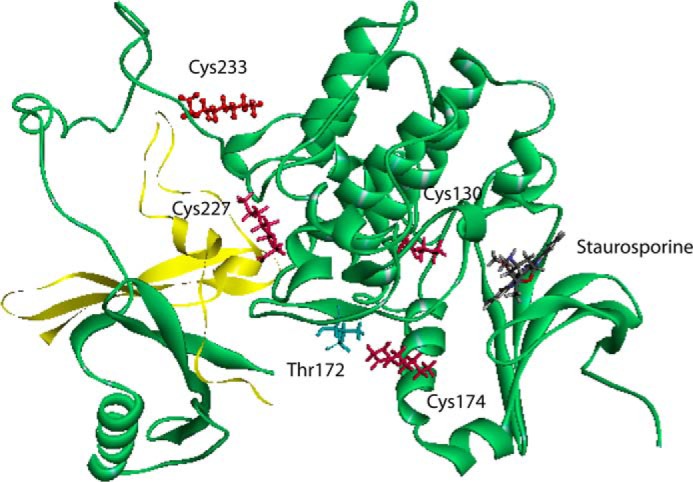
Global in silico molecular modeling of 4-HNE modified human AMPKα/β/γ. Shown is a ribbon diagram demonstrating locations of adducts found on AMPKα/β (AMPKα backbone is in green, AMPKβ backbone is in yellow, and 4-HNE adducted residues are in red). Human recombinant protein was used.
FIGURE 13.
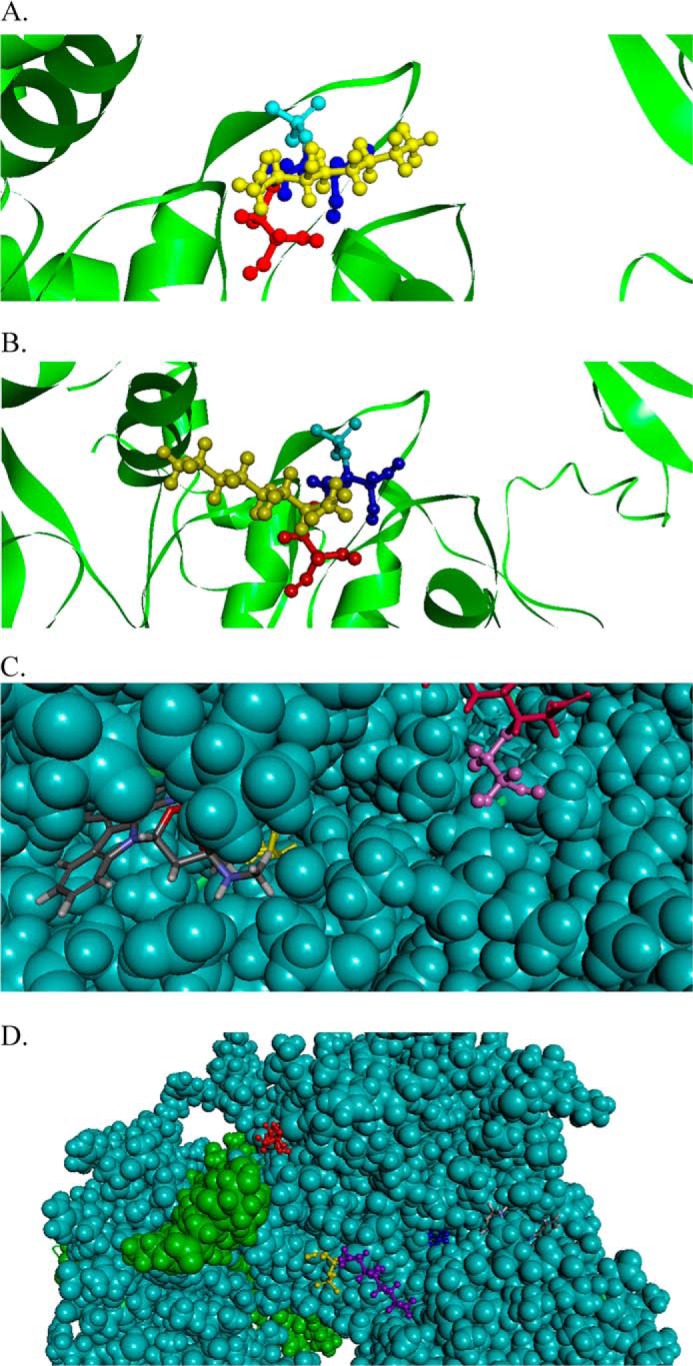
In silico molecular modeling of 4-HNE-modified human AMPKα/β/γ. A, ribbon diagram demonstrating location of Cys174 (backbone, red; 4-HNE, yellow) and The172 (backbone, dark blue; phosphate group, light blue). B, ribbon diagram demonstrating alternative conformation of Cys174 (backbone, red; 4-HNE, yellow) and The172 (backbone, dark blue; phosphate group, light blue). C, ribbon diagram demonstrating location of Cys130–4HNE (yellow) with respect to the competitive ATP inhibitor staurosporine (gray) and Cys174–4HNE (red). D, ribbon diagram demonstrating location of Cys130–4HNE (blue), Cys174–4HNE (purple), and Cys227–4HNE (red). Human AMPK was used.
An attempt was made to immunoprecipitate AMPK followed by detection of direct modification by LC/MS/MS in our animal model as well as our cell culture model. Unfortunately these attempts were not successful (data not shown). Likewise, we were previously unsuccessful in identifying carbonylation of both PTEN as well as Akt2 in our murine models (10, 12). We hypothesize that this is due to the low abundance of AMPK expression in hepatocytes.
DISCUSSION
By its ability to be activated by AMP and subsequently activate β-oxidation, AMPK is a critical cellular energy sensor in hepatocytes. In ethanol models, consumption has been implicated in both activation of AMPK as well as suppression of AMPK (20, 21, 23, 26). These reports, however, utilize different lengths of ETOH consumption, different diets, and different ethanol concentrations. Recent publications have indicated that AMPK is regulated by oxidative stress (52). In this report we characterize the effects of preincubation with 4-HNE on AMPK activation in HepG2 cells. We further translate our cellular results into both in vitro and in vivo models.
The downstream effects of lipid aldehydes such as 4-HNE are multifactorial as well as cell-specific. As a major indicator of oxidative stress and inflammation, 4-HNE is proposed to simultaneously initiate cellular damage as well as promote cellular survival (53). Recent proteomic studies have identified numerous protein targets of 4-HNE in cell culture as well as in a murine model of early ALD (48, 54). In cells, concentrations of 4-HNE are highest within membranes with calculated hepatocellular concentrations in CCl4- and BrCCl3-treated rats of 3.8 and 11.3 mm, respectively (8, 55, 56). These pathophysiological concentrations are significantly higher than concentrations used in this study (0–50 μm) and provide in vivo relevance to these experimental results.
AMPK has been shown to localize to cellular membranes. Localization of AMPK occurs during cellular metabolic stress via myristoylation of the β subunit (57). In MCF-7 cells, another member of the protein A, G, and C kinase family (LKB1) is a known target of 4-HNE (58). Mutational studies identified a critical cysteine residue (Cys210) within the activation loop of LKB1 to be alkylated by 4-HNE. In our study we also identified a critical cysteine residue near the active site. As shown in Figs. 5, 6, and 10, alkylation/carbonylation of AMPKα is significantly increased after 4-HNE treatment of hepatocytes (HepG2 cells as well as HepaRG cells) and in a murine model of ETOH-induced inflammation.
In our cellular model 4-HNE also inhibited AICAR-induced phosphorylation of ACC. This clearly indicates that reactive aldehydes can inhibit AMPK not only under conditions of increased oxidative stress but also during conditions of increased AMP. These conditions occur during chronic ETOH consumption, and based on our cellular data we hypothesize that the inhibition of AMPK by reactive aldehydes occurs not only in conditions of increased oxidative stress but under conditions of increased cellular ATP/AMP ratios (59–61).
In other cell lines, protein phosphatase 2A has been demonstrated to be the primary mechanism involved in the dephosphorylation of Thr172 on AMPK. In HEK293 cells, inhibition of PP2A resulted in increased cell survival after exposure to H2O2 for 12 h (39). In Jurkat cells, 4-HNE has been demonstrated to activate PP2A (62). In our system, preincubation of cells with okadaic acid only partially rescued AMPK phosphorylation in the presence of 4-HNE, indicating a combinatorial effect of 4-HNE in the inhibition of AMPK in HepG2 cells.
Concerning our ETOH model, these data suggest that increasing the length of ETOH consumption from 6 weeks to 7.5 weeks along with the final concentration of ETOH (31.5–34.7%) further increases serum ETOH concentrations as well as hepatocellular damage (12, 20, 30). In our ETOH model, AMPK phosphorylation increased in the ETOH-fed animals, but this increase did not correspond to an increase in Ser(P)79ACC. This is in agreement with our previous six-week study as well as data obtained in an overfeeding ETOH model (21). Concurrently there was not a significant change in expression of CPT1α and ACSL (quantification not shown). This suggests that in the ETOH-fed liver, LKB1 is active as shown by increased AMPK phosphorylation, but decreases in AMPK downstream signaling are in part due to alternative mechanisms such as direct modification by reactive aldehydes. It also indicates that in this model, hepatic fatty acid accumulation may be due to the inability of AMPK to increase β-oxidation.
Examining 4-HNE inhibition of AMPK, the discrepancy between our cell culture model and animal model of ALD is intriguing. In our cell culture model, 4-HNE is administered in the medium and under conditions of short term incubation. This results in a time-dependent inhibition of AMPK phosphorylation. In ALD, there is chronic induction of oxidative stress and activation of Cyp2E1 that contributes to the production of reactive aldehydes in the mitochondria as well as in the endoplasmic reticulum (30, 48, 63). The discrepancy between the intracellular production of reactive aldehydes in ALD and the use of reactive aldehydes as an external cellular stimulus provides a plausible explanation for our cell culture data and our in vivo data.
Using recombinant AMPK and LC/MS/MS, we identify Cys130 (located within the active site of AMPKα), Cys174 (located in the activation loop adjacent to Thr172), Cys227 (located on the external surface of AMPKα), Cys225 (located on AMPKβ), and Cys304 (location not yet determined) as selective targets of 4-HNE Michael addition adducts at molar ratios of 5:1 (Fig. 11, supplemental Table 1 and Fig. 1). The identification of the AMPKα Cys130 and Cys174 as modified amino acids suggests some intriguing possibilities concerning the mechanism of 4-HNE-mediated AMPK inhibition. The presence of a bulky hydrophobic lipid aldehyde group could prevent substrate access and, therefore, phosphorylation.
In vitro, although the specific location of Cys304 is not known, the structure indicates that it is in a loop that is accessible to modification by 4-HNE. In HEK 293 cells, treatment with H2O2 results in glutathionylation of Cys304 and a concomitant increase in AMPK activity (19). If Cys304 is mutated to Ala304, H2O2 activation of AMPK is suppressed. The adduction by 4-HNE of AMPK Cys304 may prevent this mechanism of AMPK activation by H2O2, but it is unknown if adduction would prevent AMPK activation by AMP/AICAR.
In summary, due to its ability to be activated by AMP and subsequently activate β-oxidation, AMPK is a critical cellular energy sensor in hepatocytes. In ethanol models, consumption has been implicated in both activation of AMPK as well as suppression of AMPK/β-oxidation (20, 21, 23, 26). Recent publications have also clearly indicated that AMPK is regulated by oxidative stress (52). As summarized in Fig. 14, we identify AMPK as a direct target of reactive aldehydes in cell culture as well as in a murine chronic ETOH model. Carbonylation of AMPK prevents AMPK activation by H2O2 as well as by AMP by directly inhibiting enzyme activity and thereby preventing phosphorylation of ACC. This marks the first time that AMPKα has been demonstrated to be a target of reactive aldehydes. Using in vitro phosphorylation assays as well as examining downstream targets of AMPK, we further determine that carbonylation of AMPK directly inhibits enzymatic activity both in vitro as well as in the liver of mice chronically fed ETOH. These data provide a plausible mechanism for the occurrence of increased AMPK phosphorylation in the absence of significant changes in phosphorylation of ACC in our ETOH model (20). Furthermore, this also provides a mechanistic explanation for our data indicating no significant change in β-oxidation after chronic ETOH consumption (29).
FIGURE 14.
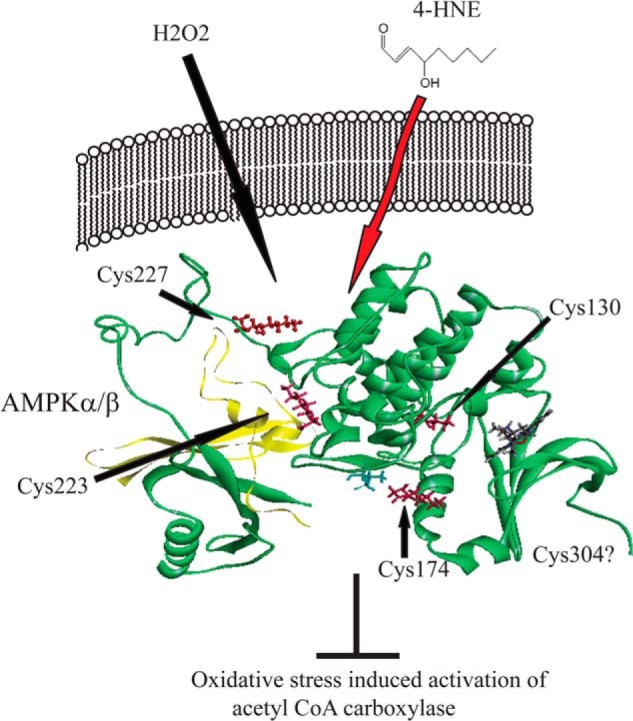
Summary of the effects increased carbonylation of AMPK on cellular processes. Both H2O2 and increasing concentrations of AMP activate AMPK (green ribbon) phosphorylation at Thr172 (blue stick). This leads to increased phosphorylation of downstream targets such as ACC, increased β-oxidation, and repletion of ATP. Under conditions of chronic inflammation and 4-HNE production, AMPKα is carbonylated by 4-HNE on Cys130, Cys174, Cys227, and Cys304, inhibiting activation and subsequent phosphorylation of downstream targets including ACC.
Supplementary Material
Acknowledgments
We thank the Computational Chemistry and Biology Core Facility at the University of Colorado Anschutz Medical Campus and the University of Colorado Denver Cancer Center Research Histology Core (E. Erin Smith, April Otero, and Kathy Lux). The mass spectrometry and histology experiments were supported in part by Center for Research Resources, National Institutes of Health Colorado Clinical and Translational Sciences Institute Grant UL1 RR025780 and the University of Colorado Denver-Anchutz Medical Campus Proteomic Mass Spectrometry Facility, by the Colorado Clinical Translational Science Institute, and University of Colorado Cancer Center Grants Clinical and Translational Science Award UL1 RR025780 and the University of Colorado Cancer Center Grant P30 CA046934.
This work was supported, in whole or in part, by National Institutes of Health Grants F32 AA018613-03 (to C. T. S.) and R37AA009300–17 (to D. R. P.).

This article contains supplemental Fig. 1 and Table 1.
- ALD
- alcoholic liver disease
- 4-HNE
- 4-hydroxy-2-nonenal
- AMPK
- AMP protein kinase
- rAMPK
- recombinant AMPK
- LKB1
- liver kinase B1
- ACC
- acetyl-CoA carboxylase
- AICAR
- 5-aminoimidazole-4-carboxyamide ribonucleoside
- TBST
- TBS-Tween
- ACSL
- acyl-CoA synthetase
- CPT
- carnitine palmitoyl transferase
- MDA
- malondialdehyde
- PP2A
- protein phosphatase 2A.
REFERENCES
- 1. Paradis V., Kollinger M., Fabre M., Holstege A., Poynard T., Bedossa P. (1997) In situ detection of lipid peroxidation by-products in chronic liver diseases. Hepatology 26, 135–142 [DOI] [PubMed] [Google Scholar]
- 2. Paradis V., Mathurin P., Kollinger M., Imbert-Bismut F., Charlotte F., Piton A., Opolon P., Holstege A., Poynard T., Bedossa P. (1997) In situ detection of lipid peroxidation in chronic hepatitis C: correlation with pathological features. J. Clin. Pathol. 50, 401–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roede J. R., Orlicky D. J., Fisher A. B., Petersen D. R. (2009) Overexpression of peroxiredoxin 6 does not prevent ethanol-mediated oxidative stress and may play a role in hepatic lipid accumulation. J. Pharmacol. Exp. Ther. 330, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roede J. R., Stewart B. J., Petersen D. R. (2008) Decreased expression of peroxiredoxin 6 in a mouse model of ethanol consumption. Free Radic. Biol. Med. 45, 1551–1558 [DOI] [PubMed] [Google Scholar]
- 5. Sampey B. P., Korourian S., Ronis M. J., Badger T. M., Petersen D. R. (2003) Immunohistochemical characterization of hepatic malondialdehyde and 4-hydroxynonenal modified proteins during early stages of ethanol-induced liver injury. Alcohol Clin. Exp. Res. 27, 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kohli R., Kirby M., Xanthakos S. A., Softic S., Feldstein A. E., Saxena V., Tang P. H., Miles L., Miles M. V., Balistreri W. F., Woods S. C., Seeley R. J. (2010) High-fructose, medium chain trans fat diet induces liver fibrosis and elevates plasma coenzyme Q9 in a novel murine model of obesity and nonalcoholic steatohepatitis. Hepatology 52, 934–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schaur R. J. (2003) Basic aspects of the biochemical reactivity of 4-hydroxynonenal. Mol. Aspects Med. 24, 149–159 [DOI] [PubMed] [Google Scholar]
- 8. Benedetti A., Comporti M., Fulceri R., Esterbauer H. (1984) Cytotoxic aldehydes originating from the peroxidation of liver microsomal lipids. Identification of 4,5-dihydroxydecenal. Biochim. Biophys. Acta 792, 172–181 [DOI] [PubMed] [Google Scholar]
- 9. Esterbauer H., Schaur R. J., Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde, and related aldehydes. Free Radic. Biol. Med. 11, 81–128 [DOI] [PubMed] [Google Scholar]
- 10. Shearn C. T., Fritz K. S., Reigan P., Petersen D. R. (2011) Modification of Akt2 by 4-hydroxynonenal inhibits insulin-dependent Akt signaling in HepG2 cells. Biochemistry 50, 3984–3996 [DOI] [PubMed] [Google Scholar]
- 11. Shearn C. T., Smathers R. L., Stewart B. J., Fritz K. S., Galligan J. J., Hail N., Jr., Petersen D. R. (2011) Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes. Mol. Pharmacol. 79, 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shearn C. T., Smathers R. L., Backos D. S., Reigan P., Orlicky D. J., Petersen D. R. (2013) Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic. Biol. Med. 65, 680–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stewart B. J., Doorn J. A., Petersen D. R. (2007) Residue-specific adduction of tubulin by 4-hydroxynonenal and 4-oxononenal causes cross-linking and inhibits polymerization. Chem. Res. Toxicol. 20, 1111–1119 [DOI] [PubMed] [Google Scholar]
- 14. Sozio M., Crabb D. W. (2008) Alcohol and lipid metabolism. Am. J. Physiol. Endocrinol. Metab. 295, E10–E16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kohjima M., Higuchi N., Kato M., Kotoh K., Yoshimoto T., Fujino T., Yada M., Yada R., Harada N., Enjoji M., Takayanagi R., Nakamuta M. (2008) SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 21, 507–511 [PubMed] [Google Scholar]
- 16. Harris R. A., Crabb D. W. (1978) Inhibition of hepatic gluconeogenesis by dichloroacetate. Arch. Biochem. Biophys. 189, 364–371 [DOI] [PubMed] [Google Scholar]
- 17. Miller R. A., Birnbaum M. J. (2010) An energetic tale of AMPK-independent effects of metformin. J. Clin. Invest. 120, 2267–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Choi S. L., Kim S. J., Lee K. T., Kim J., Mu J., Birnbaum M. J., Soo Kim S., Ha J. (2001) The regulation of AMP-activated protein kinase by H2O2. Biochem. Biophys. Res. Commun. 287, 92–97 [DOI] [PubMed] [Google Scholar]
- 19. Zmijewski J. W., Banerjee S., Bae H., Friggeri A., Lazarowski E. R., Abraham E. (2010) Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J. Biol. Chem. 285, 33154–33164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shearn C. T., Smathers R. L., Jiang H., Orlicky D. J., Maclean K. N., Petersen D. R. (2013) Increased dietary fat contributes to dysregulation of the LKB1/AMPK pathway and increased damage in a mouse model of early-stage ethanol-mediated steatosis. J. Nutr. Biochem. 24, 1436–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu J., Lai K. K., Verlinsky A., Lugea A., French S. W., Cooper M. P., Ji C., Tsukamoto H. (2011) Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 55, 673–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Everitt H., Hu M., Ajmo J. M., Rogers C. Q., Liang X., Zhang R., Yin H., Choi A., Bennett E. S., You M. (2013) Ethanol administration exacerbates the abnormalities in hepatic lipid oxidation in genetically obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 304, G38–G47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. You M., Matsumoto M., Pacold C. M., Cho W. K., Crabb D. W. (2004) The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 127, 1798–1808 [DOI] [PubMed] [Google Scholar]
- 24. You M., Considine R. V., Leone T. C., Kelly D. P., Crabb D. W. (2005) Role of adiponectin in the protective action of dietary saturated fat against alcoholic fatty liver in mice. Hepatology 42, 568–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen Z., Liang X., Rogers C. Q., Rideout D., You M. (2010) Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G364–G374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. García-Villafranca J., Guillén A., Castro J. (2008) Ethanol consumption impairs regulation of fatty acid metabolism by decreasing the activity of AMP-activated protein kinase in rat liver. Biochimie 90, 460–466 [DOI] [PubMed] [Google Scholar]
- 27. Leung T. M., Lu Y., Yan W., Morón-Concepción J. A., Ward S. C., Ge X., Conde de la Rosa L., Nieto N. (2012) Argininosuccinate synthase conditions the response to acute and chronic ethanol-induced liver injury in mice. Hepatology 55, 1596–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lieber C. S., DeCarli L. M. (1994) Animal models of chronic ethanol toxicity. Methods Enzymol. 233, 585–594 [DOI] [PubMed] [Google Scholar]
- 29. Smathers R. L., Galligan J. J., Shearn C. T., Fritz K. S., Mercer K., Ronis M., Orlicky D. J., Davidson N. O., Petersen D. R. (2013) Susceptibility of L-FABP−/− mice to oxidative stress in early-stage alcoholic liver. J. Lipid Res. 54, 1335–1345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Galligan J. J., Smathers R. L., Shearn C. T., Fritz K. S., Backos D. S., Jiang H., Franklin C. C., Orlicky D. J., Maclean K. N., Petersen D. R. (2012) Oxidative stress and the ER stress response in a murine model for early-stage alcoholic liver disease. J. Toxicol. 2012, 207594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reed D. J., Babson J. R., Beatty P. W., Brodie A. E., Ellis W. W., Potter D. W. (1980) High-performance liquid chromatography analysis of nanomole levels of glutathione, glutathione disulfide, and related thiols and disulfides. Anal. Biochem. 106, 55–62 [DOI] [PubMed] [Google Scholar]
- 32. McMillian M. K., Grant E. R., Zhong Z., Parker J. B., Li L., Zivin R. A., Burczynski M. E., Johnson M. D. (2001) Nile Red binding to HepG2 cells: an improved assay for in vitro studies of hepatosteatosis. In Vitr. Mol. Toxicol. 14, 177–190 [DOI] [PubMed] [Google Scholar]
- 33. Xiao B., Sanders M. J., Underwood E., Heath R., Mayer F. V., Carmena D., Jing C., Walker P. A., Eccleston J. F., Haire L. F., Saiu P., Howell S. A., Aasland R., Martin S. R., Carling D., Gamblin S. J. (2011) Structure of mammalian AMPK and its regulation by ADP. Nature 472, 230–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Onufriev A., Case D. A., Bashford D. (2002) Effective Born radii in the generalized Born approximation: the importance of being perfect. J. Comput. Chem. 23, 1297–1304 [DOI] [PubMed] [Google Scholar]
- 36. Gonzalez E., McGraw T. E. (2009) The Akt kinases: isoform specificity in metabolism and cancer. Cell Cycle 8, 2502–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liangpunsakul S., Wou S. E., Zeng Y., Ross R. A., Jayaram H. N., Crabb D. W. (2008) Effect of ethanol on hydrogen peroxide-induced AMPK phosphorylation. Am. J. Physiol. Gastrointest. Liver Physiol. 295, G1173–G1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shearn C. T., Reigan P., Petersen D. R. (2012) Inhibition of hydrogen peroxide signaling by 4-hydroxynonenal due to differential regulation of Akt1 and Akt2 contributes to decreases in cell survival and proliferation in hepatocellular carcinoma cells. Free Radic. Biol. Med. 53, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li M., Zhao L., Liu J., Liu A., Jia C., Ma D., Jiang Y., Bai X. (2010) Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell. Signal. 22, 1469–1476 [DOI] [PubMed] [Google Scholar]
- 40. Smathers R. L., Fritz K. S., Galligan J. J., Shearn C. T., Reigan P., Marks M. J., Petersen D. R. (2012) Characterization of 4-HNE modified L-FABP reveals alterations in structural and functional dynamics. PLoS ONE 7, e38459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., He L., Zeng N., Sahu D., Cadenas E., Shearn C., Li W., Stiles B. L. (2013) Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen-related receptor α (ERRα). J. Biol. Chem. 288, 25007–25024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Anthérieu S., Rogue A., Fromenty B., Guillouzo A., Robin M. A. (2011) Induction of vesicular steatosis by amiodarone and tetracycline is associated with up-regulation of lipogenic genes in HepaRG cells. Hepatology 53, 1895–1905 [DOI] [PubMed] [Google Scholar]
- 43. Setshedi M., Longato L., Petersen D. R., Ronis M., Chen W. C., Wands J. R., de la Monte S. M. (2011) Limited therapeutic effect of N-acetylcysteine on hepatic insulin resistance in an experimental model of alcohol-induced steatohepatitis. Alcohol Clin. Exp. Res. 35, 2139–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jones A. W., Sternebring B. (1992) Kinetics of ethanol and methanol in alcoholics during detoxification. Alcohol Alcohol 27, 641–647 [PubMed] [Google Scholar]
- 45. Jones A. W., Holmgren P. (2003) Comparison of blood-ethanol concentration in deaths attributed to acute alcohol poisoning and chronic alcoholism. J. Forensic Sci. 48, 874–879 [PubMed] [Google Scholar]
- 46. Badger T. M., Huang J., Ronis M., Lumpkin C. K. (1993) Induction of cytochrome P450 2E1 during chronic ethanol exposure occurs via transcription of the CYP 2E1 gene when blood alcohol concentrations are high. Biochem. Biophys. Res. Commun. 190, 780–785 [DOI] [PubMed] [Google Scholar]
- 47. Ronis M. J., Huang J., Crouch J., Mercado C., Irby D., Valentine C. R., Lumpkin C. K., Ingelman-Sundberg M., Badger T. M. (1993) Cytochrome P450 CYP 2E1 induction during chronic alcohol exposure occurs by a two-step mechanism associated with blood alcohol concentrations in rats. J. Pharmacol. Exp. Ther. 264, 944–950 [PubMed] [Google Scholar]
- 48. Galligan J. J., Smathers R. L., Fritz K. S., Epperson L. E., Hunter L. E., Petersen D. R. (2012) Protein carbonylation in a murine model for early alcoholic liver disease. Chem. Res. Toxicol. 25, 1012–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Deguchi J. O., Huang H., Libby P., Aikawa E., Whittaker P., Sylvan J., Lee R. T., Aikawa M. (2009) Genetically engineered resistance for MMP collagenases promotes abdominal aortic aneurysm formation in mice infused with angiotensin II. Lab. Invest. 89, 315–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li L. O., Ellis J. M., Paich H. A., Wang S., Gong N., Altshuller G., Thresher R. J., Koves T. R., Watkins S. M., Muoio D. M., Cline G. W., Shulman G. I., Coleman R. A. (2009) Liver-specific loss of long chain acyl-CoA synthetase-1 decreases triacylglycerol synthesis and β-oxidation and alters phospholipid fatty acid composition. J. Biol. Chem. 284, 27816–27826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gao X., Yo P., Harris T. K. (2005) Improved yields for baculovirus-mediated expression of human His6-PDK1 and His6-PKBβ/Akt2 and characterization of phospho-specific isoforms for design of inhibitors that stabilize inactive conformations. Protein Expr. Purif. 43, 44–56 [DOI] [PubMed] [Google Scholar]
- 52. Qin S., Rodrigues G. A. (2010) Differential roles of AMPKα1 and AMPKα2 in regulating 4-HNE-induced RPE cell death and permeability. Exp. Eye Res. 91, 818–824 [DOI] [PubMed] [Google Scholar]
- 53. Chaudhary P., Sharma R., Sharma A., Vatsyayan R., Yadav S., Singhal S. S., Rauniyar N., Prokai L., Awasthi S., Awasthi Y. C. (2010) Mechanisms of 4-hydroxy-2-nonenal induced pro- and anti-apoptotic signaling. Biochemistry 49, 6263–6275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Codreanu S. G., Zhang B., Sobecki S. M., Billheimer D. D., Liebler D. C. (2009) Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell Proteomics 8, 670–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Benedetti A., Fulceri R., Ferrali M., Ciccoli L., Esterbauer H., Comporti M. (1982) Detection of carbonyl functions in phospholipids of liver microsomes in CCl4- and BrCCl3-poisoned rats. Biochim. Biophys. Acta 712, 628–638 [DOI] [PubMed] [Google Scholar]
- 56. Benedetti A., Esterbauer H., Ferrali M., Fulceri R., Comporti M. (1982) Evidence for aldehydes bound to liver microsomal protein following CCl4 or BrCCl3 poisoning. Biochim. Biophys. Acta 711, 345–356 [DOI] [PubMed] [Google Scholar]
- 57. Oakhill J. S., Chen Z. P., Scott J. W., Steel R., Castelli L. A., Ling N., Macaulay S. L., Kemp B. E. (2010) β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. U.S.A. 107, 19237–19241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wagner T. M., Mullally J. E., Fitzpatrick F. A. (2006) Reactive lipid species from cyclooxygenase-2 inactivate tumor suppressor LKB1/STK11: cyclopentenone prostaglandins and 4-hydroxy-2-nonenal covalently modify and inhibit the AMP-kinase kinase that modulates cellular energy homeostasis and protein translation. J. Biol. Chem. 281, 2598–2604 [DOI] [PubMed] [Google Scholar]
- 59. Baio D. L., Czyz C. N., Van Horn C. G., Ivester P., Cunningham C. C. (1998) Effect of chronic ethanol consumption on respiratory and glycolytic activities of rat periportal and perivenous hepatocytes. Arch Biochem. Biophys. 350, 193–200 [DOI] [PubMed] [Google Scholar]
- 60. Williams S. P., Newton R. P., Brown E. G. (1987) Analysis of the effects of ethanol, fructose, and nicotinamide on the free nucleotides of rat liver using high performance liquid chromatography. Int. J. Biochem. 19, 879–884 [DOI] [PubMed] [Google Scholar]
- 61. French S. W. (1989) Biochemical basis for alcohol-induced liver injury. Clin. Biochem. 22, 41–49 [DOI] [PubMed] [Google Scholar]
- 62. Liu W., Akhand A. A., Takeda K., Kawamoto Y., Itoigawa M., Kato M., Suzuki H., Ishikawa N., Nakashima I. (2003) Protein phosphatase 2A-linked and -unlinked caspase-dependent pathways for downregulation of Akt kinase triggered by 4-hydroxynonenal. Cell Death Differ. 10, 772–781 [DOI] [PubMed] [Google Scholar]
- 63. Ye Q., Lian F., Chavez P. R., Chung J., Ling W., Qin H., Seitz H. K., Wang X. D. (2012) Cytochrome P450 2E1 inhibition prevents hepatic carcinogenesis induced by diethylnitrosamine in alcohol-fed rats. Hepatobiliary Surg. Nutr. 1, 5–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.