

Background: The accumulation of activated leukocytes correlates with autoimmunity and is regulated by chemokines.
Results: Tpl2-deficient macrophages display impaired chemokine receptor expression and migration under inflammatory conditions.
Conclusion: Tpl2 promotes the induction and maintenance of macrophage chemokine receptor expression and cellular migration in vivo.
Significance: Tpl2 inhibition may represent a treatment for autoimmune disorders by modulating chemokine receptor expression and preventing local accumulation of activated macrophages.
Keywords: Autoimmunity, Chemokines, Inflammation, Lipopolysaccharide (LPS), Macrophages
Abstract
In autoimmune diseases, the accumulation of activated leukocytes correlates with inflammation and disease progression, and, therefore, the disruption of leukocyte trafficking is an active area of research. The serine/threonine protein kinase Tpl2 (MAP3K8) regulates leukocyte inflammatory responses and is also being investigated for therapeutic inhibition during autoimmunity. Here we addressed the contribution of Tpl2 to the regulation of macrophage chemokine receptor expression and migration in vivo using a mouse model of Tpl2 ablation. LPS stimulation of bone marrow-derived macrophages induced early CCR1 chemokine receptor expression but repressed CCR2 and CCR5 expression. Notably, early induction of CCR1 expression by LPS was dependent upon a signaling pathway involving Tpl2, PI3K, and ERK. On the contrary, Tpl2 was required to maintain the basal expression of CCR2 and CCR5 as well as to stabilize CCR5 mRNA expression. Consistent with impairments in chemokine receptor expression, tpl2−/− macrophages were defective in trafficking to the peritoneal cavity following thioglycollate-induced inflammation. Overall, this study demonstrates a Tpl2-dependent mechanism for macrophage expression of select chemokine receptors and provides further insight into how Tpl2 inhibition may be used therapeutically to disrupt inflammatory networks in vivo.
Introduction
The pathologies of a number of autoimmune diseases can be linked to the overabundance of activated cells of the monocyte/macrophage lineage. Accumulation of monocytes and macrophages in the synovium is strongly correlated with disease severity in patients with rheumatoid arthritis (1, 2). CCR1-expressing monocytes have been shown to infiltrate multiple sclerosis plaques, where they are thought to contribute to demyelination (3–5). In addition, infiltration of macrophages at the epidermal/dermal interface is a hallmark of psoriasis, and multiple studies have now implicated macrophage dysregulation in the pathogenesis of the disease (6–8).
Chemokines are small, chemoattractant cytokines that signal through G protein-coupled receptors to initiate leukocyte activation and trafficking (9, 10). They are secreted by a variety of activated cells, including the very cells they recruit, promoting an amplification of cellular recruitment and activation. Therefore, one approach for treating chronic autoimmunity and inflammatory conditions is to control immune cell infiltration into inflamed tissues. Members of the “CC” family of chemokines and chemokine receptors, named for their adjacent conserved cysteine residues, are particularly important for macrophage recruitment. Specifically, CCR1 (ligands CCL3, CCL5, and CCL7), CCR2 (ligand CCL2), and CCR5 (ligands CCL3, CCL4, CCL5, and CCL8) are critical for inflammation-induced macrophage migration and host defense during infection. CCR1 is an important macrophage and neutrophil chemokine receptor, and mice lacking CCR1 expression are more susceptible to parasitic, bacterial, and viral infections (11–13) and display reduced macrophage infiltration and fibrosis in a model of chronic liver injury (14). CCR2-deficient mice have impaired macrophage recruitment in a mouse model of peritonitis and increased susceptibility to the intracellular pathogen Listeria monocytogenes (15, 16). CCR5 has been most intensely studied for its role as a macrophage coreceptor for HIV, but more recent studies have highlighted a role for CCR5 in monocyte entry into atherosclerotic plaques and in the pathology of cardiovascular diseases (17–20).
The specific regulation of the migratory process at the biochemical level is not completely understood, but NF-κB and MAPK activation have been demonstrated to play a role. Induction or repression of chemokine and chemokine receptor expression by macrophages is triggered by cellular recognition of pathogen-associated molecular patterns (i.e. LPS) by conserved pattern recognition receptors (i.e. TLR4) on the macrophage (21–23). This, in turn, triggers the activation of both NF-κB and MAPK, with the ERK MAPK being activated by the serine/threonine kinase tumor progression locus 2 (Tpl2). The importance of Tpl2 in this pathway is evidenced by the fact that Tpl2-deficient mice are resistant to the lethal effects of endotoxin because of a failure in the ERK-dependent secretion of TNF (24). Furthermore, we and others have demonstrated that Tpl2 is important in transducing signals downstream of multiple TLRs, leading to ERK activation and the expression of a subset of proinflammatory mediators, including TNF (24–26). Consequently, Tpl2 small molecule inhibitors are now being developed as a potential treatment for chronic autoimmune conditions, such as rheumatoid arthritis, in which TNF plays a pathologic role (27–30).
Recent studies have directly implicated Tpl2 in the regulation of inflammation-induced cell trafficking. First, Soria-Castro et al. (31) demonstrated that neutrophil chemotaxis toward zymosan in vivo was impaired in tpl2−/− mice. Bandow et al. (32) subsequently described that LPS-induced chemokine ligand expression was regulated by the Cot/Tpl2-ERK axis in macrophages. Although these studies highlight the role of Tpl2 in regulating chemokine expression in inflamed tissues and macrophages, they do not address whether Tpl2 may also regulate chemokine receptor expression and whether this regulation impacts overall macrophage migration in vivo.
In this study, we identified chemokine receptors as being regulated in a Tpl2-dependent manner. First, the maintenance of basal chemokine receptor expression for CCR2 and CCR5 required Tpl2. Second, transcription induction of the macrophage chemokine receptor CCR1 was impaired significantly following LPS stimulation, and this impairment correlated with reduced CCR1 surface protein expression in tpl2−/− macrophages compared with wild-type macrophages. Furthermore, impaired induction of CCR1 expression in Tpl2-deficient BMDMs2 was only slightly diminished by the absence of MyD88 or TRIF, suggesting functional redundancy of those two pathways in the LPS-dependent regulation of chemokine receptor expression. However, both PI3K and ERK were required for the LPS-induced transcription of CCR1 chemokine receptor expression. Consistent with the diminished chemokine receptor expression levels in both resting and activated tpl2−/− macrophages, Tpl2 ablation resulted in impaired macrophage recruitment in vivo to tissue sites of inflammation. The findings presented here provide novel insights into how Tpl2 regulates macrophage homeostasis and further support the targeted inhibition of Tpl2 kinase to disrupt these inflammatory networks in vivo.
EXPERIMENTAL PROCEDURES
Mice
Wild-type C57BL/6, myd88−/−, and ticamlps2/lps2 mice were obtained from The Jackson Laboratory. Tpl2−/− mice backcrossed onto the C57BL/6 background were provided by Dr. Philip Tsichlis and Thomas Jefferson University. Mice used in experiments were 6–12 weeks of age and were age- and sex-matched for individual experiments. Breeding colonies of mice were maintained and housed in sterile caging in a specific pathogen-free facility at the University of Georgia. All animal studies were approved by the Institutional Animal Care and Use Committee at the University of Georgia.
Culture of BMDMs
Bone marrow was harvested from tibiae and femurs of mice by flushing with low-glucose DMEM (HyClone) supplemented with 10% fetal calf serum, 290 μg/ml l-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin (all purchased from Invitrogen) using a 10-ml syringe and 25-gauge needle. Cells were disaggregated by gentle pipetting and centrifuged at 1200 rpm for 10 min at room temperature. The cells were then resuspended in ammonium-chloride-potassium (ACK) lysis buffer (Invitrogen) for 30 s to lyse red blood cells. Cells were washed in PBS (20–30 ml) and centrifuged again at 1200 rpm for 10 min. The cell pellet was resuspended in supplemented DMEM for cell counting. Differentiated macrophages were obtained by culturing bone marrow cells on sterile Petri dishes at 2 × 106/ml in supplemented DMEM with the addition of macrophage colony-stimulating factor (10 ng/ml, Peprotech) for 7 days at 37 °C and 5% CO2. Cells were adherence-purified on day 7 by removing the medium, washing adherent cells with PBS, and then harvesting by incubating in 10 ml of cell dissociation buffer (Invitrogen) for 15 min at 37 °C. Plates were washed with PBS to collect dislodged cells, and cells were centrifuged at 1200 rpm for 10 min. Cell pellets were resuspended in supplemented DMEM for cell counting.
Cell Stimulation
For individual experiments, murine BMDMs were plated at either 0.5 × 106/ml or 1 × 106/ml in supplemented DMEM. Cells were left unstimulated (as a control) or stimulated with ultrapure LPS from Escherichia coli (0111:B4, 1 μg/ml, Invivogen) for 0, 1, 2, 3, or 4 h at 37 °C and 5% CO2. For measurement of mRNA stability, BMDMs were stimulated with 1 μg/ml LPS, and the transcriptional inhibitor actinomycin D (5 μg/ml, Sigma) was added at 1 h of stimulation. Cells were harvested at 1, 2, or 3 h after actinomycin D addition, and mRNA was measured by RT-PCR for CCR1, CCR2, and CCR5. For inhibitor studies, BMDMs were pretreated with the following inhibitors in supplemented DMEM for 30 min at 37 °C and 5% CO2 prior to stimulation with LPS: LY-294,002 hydrochloride, 20 μm (Sigma); rapamycin, 30 nm (Sigma); and U0126 ethanolate, 20 μm (Sigma).
Gene Expression Microarray
For microarray analysis, wild-type or tpl2−/− BMDM were stimulated with 1 μg/ml LPS from E. coli (O111:B4, Sigma-Aldrich) for 4 h. Total cellular RNA was extracted using a mirVana kit (Ambion). Approximately 500 ng of RNA was labeled using a MessageAmpTM II-biotin enhanced kit (Ambion) and hybridized to GeneChip Mouse Genome 430 2.0 arrays (Affymetrix) in accordance with the protocols of the manufacturer. Expression values were determined with GeneChip operating software v1.1.1. All data analyses were performed using GeneSpring software GX 11.0. Expression values for each probe were normalized using the robust multichip average method. The fold changes for each probe were calculated by pairwise comparisons (WT 4 h LPS versus Tpl2 KO 4 h LPS).
RNA Isolation and RT-PCR
Cell pellets were lysed in 350 μl of RNA lysis buffer containing 7 μl of 2-mercaptoethanol, and RNA was isolated using a Total RNA Kit I (Omega Bio-Tek, catalog no. R6834-02) according to the instructions of the manufacturer. RNA was eluted from the columns in 40 μl of diethylpyrocarbonate (DEPC) water and stored at −80 °C. RNA concentration was determined using a NanoDrop spectrophotometer (Thermo Scientific). Conversion of mRNA to cDNA was achieved using a high-capacity cDNA reverse transcription kit (Applied Biosystems).
RT-PCR was performed using the following primer/probe mixes from TaqMan gene expression assays (Applied Biosystems): CCR1 (Mm00438260_s1), CCR2 (Mm01216173_m1), CCR5 (Mm01963251_s1), and actin B (Mm00607939_s1). RT-PCR assays were run in MicroAmp fast optical 96-well reaction plates in a StepOnePlus real-time PCR system (Applied Biosystems). Relative gene expression levels were calculated by normalizing the Ct levels of the target gene to that of an actin B endogenous control using the ΔCt method. Data were further normalized to an experimental control (wild-type, untreated) sample using the ΔΔCt method.
Flow Cytometric Staining
For measurement of CCR1 and CCR5 cell surface protein expression, BMDMs were plated at 0.5 × 106/ml in 0.5 ml on 48-well plates and stimulated with 1 μg/ml LPS for 4 or 24 h. BMDMs were harvested, washed in PBS, and then Fc receptors were blocked by staining with purified anti-mouse CD16/CD32 at 4 °C for 5 min. Cells were subsequently stained with allophycocyanin (APC)-labeled anti-mouse CCR1 (R&D Systems, clone 643854) or phycoerythrin (PE)-labeled anti-mouse CCR5 (eBioscience, clone HM-CCR5) or their appropriate isotype controls for 30 min in PBS containing 0.1% BSA, according to the protocol of the manufacturer. Surface expression was analyzed using a BD LSRII flow cytometer and FlowJo software. For characterization of peritoneal cell populations, 0.5–1.0 × 106 recruited PECs were stained with fluorescently conjugated antibodies to F4/80 and CD11b (eBioscience) for 15 min at 4 °C in PBS and 0.1% BSA. Cells were washed with PBS, fixed in PBS and 1% formalin, and analyzed by flow cytometry as described above.
Peritoneal Exudate Cell (PEC) Isolation
Mice were injected intraperitoneally with 1 ml of 3% Brewer thioglycollate medium to induce local inflammation and recruit effector cells. After 72 h, mice were sacrificed, and the peritoneal cavity was lavaged three times with 3 ml of sterile PBS to collect recruited cells. Cells were centrifuged at 1200 rpm for 10 min at room temperature and were resuspended in supplemented DMEM.
Statistical Analysis
Data are represented as the mean ± S.D. or S.E., as indicated. p values were determined by two-tailed Student's t test or paired Student's t test where indicated.
RESULTS
Chemokine Receptor Expression Is Impaired in tpl2−/− BMDMs
To gain a global view of the early innate functions of Tp12 in macrophages, we analyzed gene expression profiles by microarray in WT versus tpl2−/− BMDMs stimulated with LPS. Consistent with previous reports, Fos, IL-1β, and the chemokines CCL2, CCL7, and CXCL3 were reduced in the absence of Tpl2, whereas IL-12p40 (IL-12B) and IFNβ expression were increased (Fig. 1A) (26, 32–34). These findings served to verify the microarray results. Importantly, in addition to chemokine ligands, we identified the chemokine receptors CCR1 and CCR5 as novel Tpl2-dependent genes (Fig. 1A). To validate the microarray findings, we cultured BMDMs from WT and tpl2−/− mice in medium with or without E. coli LPS for 4 h and measured the mRNA expression levels of multiple chemokine receptors (CCR1, CCR2, and CCR5) clustered on mouse chromosome 9 (human chromosome 3) by quantitative real-time PCR (qRT-PCR). Consistent with the microarray data, we confirmed that CCR1 and CCR5 were decreased in LPS-stimulated macrophages (Fig. 1B). Importantly, LPS stimulation repressed CC chemokine receptor expression in wild-type macrophages, in agreement with previous reports (23, 35). We next performed a time course of stimulations with LPS for up to 4 h to determine the precise mechanism for regulation of each gene by Tpl2. Analysis of CCR1, CCR2 and CCR5 mRNA expression in WT and Tpl2-deficient macrophages following LPS stimulation revealed distinct patterns of regulation. First, there was a dramatic decrease in the basal expression of CCR2 and CCR5 in tpl2−/− macrophages, suggesting that Tpl2 is important for the maintenance of homeostatic levels of these chemokine receptors (Fig. 1C). Of the three receptors, CCR1 expression was uniquely induced at a transcriptional level, with ∼4-fold induction in gene expression induced by LPS, peaking at 2 h, and followed by a rapid decline to near basal expression (Fig. 1C). In contrast, CCR2 and CCR5 mRNA expression was strictly repressed by LPS stimulation (Fig. 1C). Notably, for all three chemokine receptors, expression remained consistently lower in Tpl2-deficient versus WT BMDMs throughout the time course of LPS stimulation. Overall, these results indicate that Tpl2 ablation diminishes chemokine receptor expression in macrophages during inflammation.
FIGURE 1.

Impaired chemokine receptor expression in tpl2−/− BMDMs. A, BMDMs from WT and tpl2−/− mice were stimulated with LPS (1 μg/ml) for 4 h, and mRNA expression was analyzed by gene chip microarray. Data are presented as the fold change in gene expression in WT macrophages compared with tpl2−/− macrophages. B and C, BMDMs from WT and tpl2−/− mice were stimulated in medium with LPS (1 μg/ml) for either 4 h (B) or the indicated time points (C) and were analyzed for mRNA expression of various CC chemokine receptors by qRT-PCR relative to an actin control. Expression levels at each time point were normalized to the endogenous actin control and were expressed relative to the WT unstimulated (Unstim) control. Results are averaged from three independent experiments, and error bars represent mean ± S.E.
The Regulation of CC Chemokine Receptors Is Only Partially MyD88- or TRIF-dependent
LPS signals through TLR4 via either MyD88 or TRIF adaptor proteins (36, 37). Bandow et al. (32) demonstrated that Tpl2 regulates LPS-induced chemokine ligand expression through both pathways. Specifically, Tpl2 promotes CCL2, CCL7, CXCL2, and CXCL3 through the MyD88 pathway, whereas Tpl2 inhibition of CCL5, CXCL10, and CXCL13 was MyD88-independent. To determine the mechanisms regulating CC chemokine receptor expression, WT, MyD88-deficient, or TRIF-mutant (ticamlps/2lps2) BMDMs were stimulated with LPS for up to 4 h, and chemokine receptor expression was determined by qRT-PCR. Signaling deficiencies in either MyD88 or TRIF alone only partially recapitulated the impaired induction of CCR1 expression observed in tpl2−/− BMDMs (Fig. 2, A and B). LPS-mediated repression of CCR2 and CCR5 expression was also not strictly dependent upon MyD88 or TRIF signaling. These data suggest that MyD88 and TRIF are largely redundant for LPS-dependent regulation of CC chemokine receptors by Tpl2 but demonstrate that Tpl2 plays a non-redundant role.
FIGURE 2.

MyD88- and TRIF-dependent signaling are functionally redundant for LPS-mediated chemokine receptor expression. BMDMs from WT and myd88−/− mice (A) or WT and triflps2/lps2 mice (B) were stimulated with LPS for 1, 2, 3, or 4 h. Cells were analyzed for mRNA expression of chemokine receptors by qRT-PCR relative to an actin control. Results are averaged from at least three independent experiments for A and two independent experiments for B. Error bars represent mean ± S.E.
The regulation of mRNA stability is a potential explanation for CCR2 and CCR5 repression. Because Tpl2 has been shown previously to destabilize certain inflammation-related mRNAs, including TNFα, IL-6, and keratinocyte chemoattractant (KC) chemokine mRNAs (38), we investigated whether Tpl2 contributes to chemokine receptor mRNA stability. Addition of the transcriptional inhibitor actinomycin D paradoxically stabilized the mRNAs for all three chemokine receptors (data not shown). Because our time course studies demonstrated no evidence of LPS-induced transcription of CCR2 or CCR5, we calculated the half-life of each mRNA upon LPS stimulation as in Fig. 1C. The half-life of CCR2 mRNA was 2.1 h in WT BMDMs and 2.0 h in tpl2−/− BMDMs, indicating that Tpl2 does not regulate CCR2 via mRNA stability. In contrast, Tpl2 increased the half-life of CCR5 mRNA from 3.0 h in tpl2−/− BMDMs to 4.1 h in WT BMDMs. These data indicate that Tpl2 not only enhances the basal expression of CCR5 but also increases its mRNA half-life by 37%.
CC Chemokine Receptor Expression Is Regulated in a Tpl2-, PI3K-, and ERK-dependent Manner
Tpl2 has been shown to regulate a number of signaling pathways, including ERK, JNK, p38, NF-κB, nuclear factor of activated T-cells (NFAT), PI3K, Akt, and mTOR (38–41). To determine the Tpl2-dependent pathways regulating chemokine receptor expression in response to LPS stimulation, we examined the promoters for CCR1 and CCR5 in both humans and mice. Among the transcription factor binding sites, we observed putative Ets sites in the human and mouse CCR1 promoters and in the human CCR5 promoter (data not shown), indicating that CCR1 and CCR5 are potentially regulated by Map kinase pathways. The presence of putative Ets binding sites suggested that one mechanism by which Tpl2 could be regulating chemokine receptor expression is via ERK-dependent signals. Notably, the murine CCR5 promoter lacked an Ets transcription factor-binding site but, instead, contained an AP-1 site. To test whether CCR1 and CCR5 expression are regulated in an ERK-dependent manner, WT or tpl2−/− BMDMs were pretreated with the MEK inhibitor U0126 or a dimethyl sulfoxide vehicle control 30 min prior to stimulation with LPS. Because Tpl2 has also been shown to regulate PI3K, Akt, and mTOR signaling (41), the PI3K inhibitor LY-294,002 and the mTOR inhibitor rapamycin were also tested for comparison. 1, 2, or 4 h after LPS treatment, cells were harvested, and chemokine receptor expression was quantitated by qRT-PCR. The mTOR inhibitor rapamycin failed to alter chemokine receptor expression in response to LPS (Fig. 3A). In contrast, PI3K inhibition and MEK/ERK inhibition reduced induction of CCR1 expression in LPS-stimulated WT BMDMs, with MEK inhibition approaching levels similar to LPS-stimulated tpl2−/− BMDMs (Fig. 3, A and C). There was no effect of these inhibitors on CCR2 or CCR5 expression levels in LPS-stimulated macrophages. These findings suggest that Tpl2 is required for the PI3K- and ERK-dependent induction and maintenance of CCR1 expression upon LPS stimulation. Tpl2 is also required for the maintenance of CCR2 and CCR5 expression upon LPS stimulation (Fig. 1C), but this apparently occurs through alternate pathways (Fig. 3).
FIGURE 3.

Chemokine receptor 1 expression is regulated in a PI3K- and ERK-dependent fashion. BMDMs from WT and tpl2−/− mice were pretreated in DMEM with the mTOR-specific inhibitor rapamycin (Rapa, 30 nm) (A), the PI3 kinase inhibitor LY-294,002 (LY, 20 μm) (B), or the MEK1/2 inhibitor U0126 ethanolate (ERKi, 20 μm) (C) for 30 min prior to stimulation with 1 μg/ml LPS for 1, 2, or 4 h. Dimethyl sulfoxide-treated (DMSO, vehicle) WT and tpl2−/− BMDMs were included for comparison. After stimulation, cells were lysed and analyzed for mRNA expression of ccr1, ccr2, and ccr5 by qRT-PCR relative to an actin control. Results are expressed as the level of gene expression relative to the unstimulated WT control. Results are mean ± S.D. of two independent experiments.
BMDM mRNA Expression Correlates with Membrane Surface Expression of CCR1 and CCR5 Proteins
Because Tpl2 has been shown to contribute to the posttranscriptional regulation of proinflammatory cytokines, we questioned whether the transcriptional regulation we observed was representative of chemokine receptor expression on the cell membrane. To test this, we performed surface staining and flow cytometry for CCR1 and CCR5 on wild-type and Tpl2-deficient BMDMs that were left unstimulated or stimulated for 4 or 24 h with LPS. In wild-type BMDMs, we observed induction of CCR1 surface expression by 4 h that further increased by 24 h, as seen by an increase in the mean fluorescence intensity of the population (Fig. 4A). This induction was almost completely abrogated in Tpl2-deficient BMDMs. In contrast, CCR5 membrane expression was reduced in both wild-type and Tpl2-deficient BMDMs upon LPS stimulation (Fig. 4B).
FIGURE 4.

Regulation of ccr1 and ccr5 mRNA expression correlates with membrane surface expression of CCR1 and CCR5 proteins. WT and tpl2−/− BMDMs were stimulated with or without LPS (1 μg/ml) for 4 or 24 h, and CCR1 and CCR5 protein expression was determined by surface staining and flow cytometry. A, flow histograms for wild-type (black lines) or Tpl2-deficient (gray lines) BMDMs stained with isotype control antibodies (dashed lines) for CCR1 (top and center panels, bold lines) or CCR5 (bottom panel, bold lines). B, specific mean fluorescence intensities (MFIs) for CCR1 (top and center panel) and CCR5 (bottom panel) were calculated by subtracting the mean fluorescence intensity of the isotype control from the mean fluorescence intensity of the test stain for each condition. Results are representative of three independent experiments for A and are pooled from three independent experiments in B. Error bars represent mean ± S.E. (Student's t test). *, p < 0.05.
Recruitment of Primary Macrophages to the Peritoneal Cavity Is Impaired in tpl2−/− Mice
We next sought to determine whether macrophage migration was altered because of changes in chemokine receptor expression by Tpl2 ablation in an in vivo setting. WT or tpl2−/− mice were injected intraperitoneally with Brewer thioglycollate medium or 1 ml PBS as a control. Thioglycollate medium is a potent inducer of local sterile inflammation and has been shown to cause peritonitis associated with robust monocyte/macrophage recruitment dependent upon monocyte chemokine receptor signaling (15, 43). Thioglycollate-elicited peritoneal macrophages express chemokine receptors CCR1, CCR2, and CCR5 (44), and both CCR2 and CCR5 have been shown to contribute to macrophage recruitment in response to thioglycollate (43). Cells were allowed to migrate to the peritoneal cavity for 72 h before mice were sacrificed and PECs were harvested. The total number of peritoneal cells recovered was increased significantly in thioglycollate-treated mice compared with PBS-treated controls, yet the absolute number of cells recovered from tpl2−/− thioglycollate-treated mice was reduced significantly compared with WT thioglycollate-treated mice (Fig. 5A). Additional characterization of the thioglycollate-recruited cells via flow cytometry staining with F4/80 and CD11b revealed a similarly high proportion of F4/80+CD11b+ primary macrophages recruited to the peritoneal cavity of WT and tpl2−/− mice at this time point, which favors macrophage recruitment (Fig. 5B). However, the total number of macrophages recruited was reduced significantly in tpl2−/− mice (Fig. 5C).
FIGURE 5.
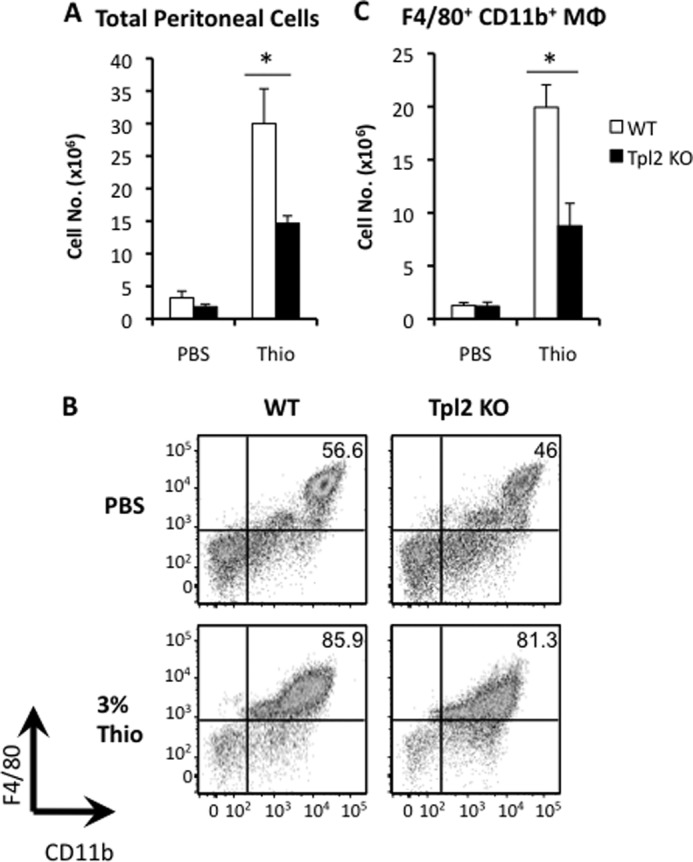
Thioglycollate-elicited recruitment of primary macrophages is impaired in tpl2−/− mice. WT and tpl2−/− mice were injected intraperitoneally with 1 ml of PBS (control) or 3% thioglycollate (Thio) medium to induce sterile peritonitis and macrophage recruitment. Mice were sacrificed 72 h later, and PECs were obtained and analyzed. A, total numbers of PECs obtained from mice 72 h post-injection. B, representative flow cytometric plots showing recruited F4/80+CD11b+ macrophages. C, absolute numbers of recruited macrophages from the peritoneal cavity. Results are averages of three independent experiments, and error bars represent mean ± S.E. (Student's t test). *, p < 0.05.
DISCUSSION
In this study, we demonstrated that macrophage CC chemokine receptors are regulated by Tpl2. By microarray analysis, we identified CCR1 and CCR5, both critical receptors for regulating cellular migration during inflammation (45, 46), as being regulated in a Tpl2-dependent manner. Therefore, we investigated the effects of Tpl2 ablation on the expression of multiple CC chemokine receptors clustered on murine chromosome 9, which includes CCR1, CCR2, and CCR5. We found that CCR2 and CCR5 are expressed at basally decreased levels, whereas LPS-stimulated CCR1 transcription was impaired in tpl2−/− BMDMs. Furthermore, the Tpl2-dependent induction of CCR1 gene expression required both PI3K and ERK activation. To correlate these in vitro chemokine impairments with a possible in vivo functional impairment in macrophage migration, we demonstrated that tpl2−/− mice were defective in the recruitment of primary peritoneal macrophages to the peritoneal cavity following thioglycollate-induced peritonitis. These findings indicate that Tpl2 normally potentiates inflammation by both promoting and stabilizing chemokine receptor expression in activated macrophages. Furthermore, our data suggest that Tpl2 inhibition may help to mitigate excessive cellular infiltration and inflammation in chronic autoimmunity.
LPS stimulation of human monocytes has been shown to inhibit the expression of chemokine receptors, including CCR2, and, to a lesser extent, CCR1 and CCR5 (23, 35). In contrast, a study using mouse dendritic cells demonstrated that CCR1 was induced early by LPS between 30 min and 3 h and was repressed at the mRNA level by 24 h (47). These data demonstrate subtle differences in chemokine receptor regulation that may be related to the host species, cell type, or activation state. Our data in murine BMDMs are consistent with the latter study because we observed LPS-induced induction of CCR1 mRNA as early as 1 h, peaking at 2 h, and showing repression after 4 h. Importantly, CCR1 induction was Tpl2-dependent. We confirmed this CCR1 regulation by measuring CCR1 protein on the cell surface 4 and 24 h after LPS stimulation. On the basis of the fact that CCR1 mRNA was repressed by LPS as early as 4 h after stimulation, it is likely that protein expression would ultimately be reduced at later time points.
The rapid, albeit transient, induction of CCR1 suggests that CCR1 may play a critical early role in monocyte recruitment to inflammatory sites. In laminar flow studies, CCR1 was shown to be responsible for the initial arrest of monocytes to activated endothelium, and both CCR1 and CCR5 subsequently promoted transendothelial migration in response to CCL5 (48). Furthermore, the subsequent repression of CCR1, along with the repression of CCR2 and CCR5, may be necessary for macrophage egress from tissues. In support of this notion, Cao et al. (49) have demonstrated that inflammatory macrophages do not die within tissues but migrate through the lymphatics and into the circulation.
Inhibition of chemokine receptor expression has been reported to occur via multiple mechanisms. CCR2 expression is decreased via mechanisms involving receptor internalization and degradation as well as a reduction in CCR2 mRNA stability (35, 50, 51). Some studies have suggested that LPS represses CCR5 to some extent by transcriptional control (35, 52), whereas others have proposed alternate mechanisms (53, 54). Our data clearly demonstrate a decrease in mRNA expression for CCR1, CCR2 and CCR5 in response to LPS stimulation, although with varying kinetics. Tpl2 deficiency has been shown to result in destabilization of certain inflammation-related mRNAs, including TNFα, IL-6, and KC chemokine mRNAs, in LPS-stimulated macrophages (38). Consistent with this, we observed that Tpl2 increased CCR5 mRNA stability by 37%. Although the focus of this study was the transcriptional regulation of these chemokine receptors, we cannot exclude the possibility of additional mechanisms of regulation. For example, Franchin et al. (54) attributed the reduction in CCR5 surface expression to internalization that is independent of new protein synthesis. Ligand-induced down-regulation of CCR5 by exogenous MIP-1α has also been demonstrated (55). Such a mechanism could contribute to the reduction in CCR5 surface protein expression (Fig. 4). In this regard, Bandow et al. (32) have shown previously that the LPS-induced expression of the CCR1/CCR5 specific chemokine ligand CCL5 (RANTES), was increased in tpl2−/− macrophages. We also observed a time-dependent increase in CCL5 expression levels in tpl2−/− macrophages relative to WT cells stimulated with LPS, although, by 3 h, CCL5 expression was similar in WT and tpl2−/− macrophages (data not shown).
Additional studies revealed that early LPS-induced CCR1 expression was reduced in the absence of either PI3K or ERK. The PI3K/Akt pathway is activated in macrophages by LPS stimulation by a Tpl2-dependent mechanism (42, 56). Furthermore, Akt is one kinase shown to phosphorylate Tpl2 on serine 400 (57). Serine 400 phosphorylation of Tpl2 is required for LPS-stimulated ERK activation in macrophages (58). Therefore, PI3K/Akt, Tpl2, and ERK define the signaling pathway through which LPS induces CCR1 expression in macrophages. Analysis of the CCR1 and CCR5 promoter regions in both humans and mice revealed putative binding sites for known ERK-dependent transcription factors, Ets and/or AP-1. Notably, the murine CCR5 promoter lacked a putative Ets binding site, which could explain the differential transcriptional regulation of CCR1 and CCR5 expression by ERK.
Although this study and others have begun to elucidate the ways in which Tpl2 mediates macrophage chemokine and chemokine receptor expression, currently little is known about the role of Tpl2 in mediating the actual in vivo migration of macrophages during inflammation. To address this issue, we used a thioglycollate model of sterile inflammation to initiate macrophage migration and found that tpl2−/− mice exhibited impaired total cellular and macrophage recruitment to the site of inflammation. On the basis of our findings here, Tpl2 regulates macrophage migration, at least in part, by maintaining appropriate chemokine receptor expression (i.e. CCR1, CCR2, and CCR5) levels in macrophages. Importantly, Tpl2-dependent regulation of chemokine receptors likely extends to other critical innate migratory cells, such as neutrophils and dendritic cells. Future studies targeting specific chemokine receptor/ligand interactions are needed to provide greater insight into the dominant mechanisms by which Tpl2 influences macrophage and/or neutrophil migration.
The selective inhibition of chemokine receptors is emerging as a therapeutic option for the treatment of autoimmune and inflammatory disorders. For example, the selective inhibition of CCR1 has been explored for the treatment of specific autoimmune diseases like rheumatoid arthritis, where CCR1 inhibition correlated with reductions in cellular infiltration and inflammation in the joints of arthritis-induced mice (59). Additionally, CCR1 inhibition has also been investigated in studies for the treatment of tumor-associated diseases such as multiple myeloma, where therapeutic inhibition of CCR1 resulted in a decreased tumor burden in affected mice (60). CCR2 and its ligand CCL2 (MCP-1) have been implicated in the progression of atherosclerosis and multiple sclerosis (19, 61, 62). Mice lacking CCR5 had decreased bacterial burdens and better protection against endotoxin-induced systemic shock (46). Despite these results, the use of CCR1 and CCR5 antagonists in human clinical trials is less prevalent, and the efficacy of these treatments has often been poor. The development of safe CCR1 antagonists for human use has had recent success (63), but few studies have tested the efficacy of these drugs in patients with autoimmune conditions like rheumatoid arthritis. Additionally, CCR5 antagonism has not shown promise in human drug trials for rheumatoid arthritis and, therefore, appears to be a poor target for treatment of this condition on its own (42).
Our findings suggest that Tpl2 inhibition, which can reduce inflammation-associated expression of the macrophage CC chemokine receptors CCR1, CCR2, and CCR5, may provide an alternative treatment for a variety of autoimmune diseases associated with increased inflammatory cell infiltration. Furthermore, CCR1, CCR2, and CCR5 antagonism may be enhanced in combination with selective inhibition of Tpl2 because our findings have suggested that this kinase is responsible for regulating their expression in macrophage populations. Lastly, inhibition of Tpl2 may also help to control in vivo migration of macrophage populations and potentially block the continued accumulation of inflammatory infiltrates in affected tissues. One should note, however, that the physiological effect of Tpl2 ablation (or clinical inhibition) could significantly impair the ability of the host to respond to a variety of bacterial, parasitic, and viral infections as well as weaken host responses to cancers. Clearly, further research into the role of Tpl2 in mediating inflammatory cell trafficking is warranted to evaluate the effects of Tpl2 inhibition in clinical applications.
Acknowledgments
We thank Rebecca Kirkland for technical support throughout this project and the University of Georgia Central Animal Facility staff for animal care. We also thank Julie Nelson and the Center for Tropical and Emerging Global Diseases as well as the Department of Infectious Diseases flow cytometry core facilities for flow cytometry support.
This work was supported by a startup fund from the Office of the Vice President for Research, University of Georgia (to W. T. W.).
The normalized microarray data reported in this paper have been submitted to the Gene Expression Omnibus Repository with accession number GSE48338.
- BMDM
- bone marrow-derived macrophage
- PEC
- peritoneal exudate cell
- qRT-PCR
- quantitative real-time PCR
- mTOR
- mammalian target of rapamycin.
REFERENCES
- 1. Tak P. P., Smeets T. J., Daha M. R., Kluin P. M., Meijers K. A., Brand R., Meinders A. E., Breedveld F. C. (1997) Analysis of the synovial cell infiltrate in early rheumatoid synovial tissue in relation to local disease activity. Arthritis Rheum. 40, 217–225 [DOI] [PubMed] [Google Scholar]
- 2. Davignon J. L., Hayder M., Baron M., Boyer J. F., Constantin A., Apparailly F., Poupot R., Cantagrel A. (2013) Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology 52, 590–598 [DOI] [PubMed] [Google Scholar]
- 3. Barnett M. H., Prineas J. W. (2004) Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 55, 458–468 [DOI] [PubMed] [Google Scholar]
- 4. Huitinga I., van Rooijen N., de Groot C. J., Uitdehaag B. M., Dijkstra C. D. (1990) Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J. Exp. Med. 172, 1025–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Trebst C., Sørensen T. L., Kivisäkk P., Cathcart M. K., Hesselgesser J., Horuk R., Sellebjerg F., Lassmann H., Ransohoff R. M. (2001) CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am. J. Pathol. 159, 1701–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Clark R. A., Kupper T. S. (2006) Misbehaving macrophages in the pathogenesis of psoriasis. J. Clin. Invest. 116, 2084–2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang H., Peters T., Kess D., Sindrilaru A., Oreshkova T., Van Rooijen N., Stratis A., Renkl A. C., Sunderkötter C., Wlaschek M., Haase I., Scharffetter-Kochanek K. (2006) Activated macrophages are essential in a murine model for T cell-mediated chronic psoriasiform skin inflammation. J. Clin. Invest. 116, 2105–2114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stratis A., Pasparakis M., Rupec R. A., Markur D., Hartmann K., Scharffetter-Kochanek K., Peters T., van Rooijen N., Krieg T., Haase I. (2006) Pathogenic role for skin macrophages in a mouse model of keratinocyte-induced psoriasis-like skin inflammation. J. Clin. Invest. 116, 2094–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Olson T. S., Ley K. (2002) Chemokines and chemokine receptors in leukocyte trafficking. Am. J. Physiol. Regul. Integr. Comp. Physiol. 283, R7-R28 [DOI] [PubMed] [Google Scholar]
- 10. García-Ramallo E., Marques T., Prats N., Beleta J., Kunkel S. L., Godessart N. (2002) Resident cell chemokine expression serves as the major mechanism for leukocyte recruitment during local inflammation. J. Immunol. 169, 6467–6473 [DOI] [PubMed] [Google Scholar]
- 11. Khan I. A., Murphy P. M., Casciotti L., Schwartzman J. D., Collins J., Gao J.-L., Yeaman G. R. (2001) Mice lacking the chemokine receptor CCR1 show increased susceptibility to Toxoplasma gondii infection. J. Immunol. 166, 1930–1937 [DOI] [PubMed] [Google Scholar]
- 12. Domachowske J. B., Bonville C. A., Gao J. L., Murphy P. M., Easton A. J., Rosenberg H. F. (2000) The chemokine macrophage-inflammatory protein-1 α and its receptor CCR1 control pulmonary inflammation and antiviral host defense in paramyxovirus infection. J. Immunol. 165, 2677–2682 [DOI] [PubMed] [Google Scholar]
- 13. Rodriguez-Sosa M., Rosas L. E., Terrazas L. I., Lu B., Gerard C., Satoskar A. R. (2003) CC chemokine receptor 1 enhances susceptibility to Leishmania major during early phase of infection. Immunol. Cell Biol. 81, 114–120 [DOI] [PubMed] [Google Scholar]
- 14. Seki E., De Minicis S., Gwak G. Y., Kluwe J., Inokuchi S., Bursill C. A., Llovet J. M., Brenner D. A., Schwabe R. F. (2009) CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Invest. 119, 1858–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurihara T., Warr G., Loy J., Bravo R. (1997) Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 186, 1757–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boring L., Gosling J., Chensue S. W., Kunkel S. L., Farese R. V., Jr., Broxmeyer H. E., Charo I. F. (1997) Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest. 100, 2552–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alkhatib G., Combadiere C., Broder C. C., Feng Y., Kennedy P. E., Murphy P. M., Berger E. A. (1996) CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272, 1955–1958 [DOI] [PubMed] [Google Scholar]
- 18. Jones K. L., Maguire J. J., Davenport A. P. (2011) Chemokine receptor CCR5: from AIDS to atherosclerosis. Br. J. Pharmacol. 162, 1453–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tacke F., Alvarez D., Kaplan T. J., Jakubzick C., Spanbroek R., Llodra J., Garin A., Liu J., Mack M., van Rooijen N., Lira S. A., Habenicht A. J., Randolph G. J. (2007) Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J. Clin. Invest. 117, 185–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gautier E. L., Jakubzick C., Randolph G. J. (2009) Regulation of the migration and survival of monocyte subsets by chemokine receptors and its relevance to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 29, 1412–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ruggiero T., Trabucchi M., De Santa F., Zupo S., Harfe B. D., McManus M. T., Rosenfeld M. G., Briata P., Gherzi R. (2009) LPS induces KH-type splicing regulatory protein-dependent processing of microRNA-155 precursors in macrophages. FASEB J. 23, 2898–2908 [DOI] [PubMed] [Google Scholar]
- 22. Archer K. A., Roy C. R. (2006) MyD88-dependent responses involving Toll-like receptor 2 are important for protection and clearance of Legionella pneumophila in a mouse model of Legionnaires' disease. Infect. Immun. 74, 3325–3333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Parker L. C., Whyte M. K., Vogel S. N., Dower S. K., Sabroe I. (2004) Toll-like receptor (TLR)2 and TLR4 agonists regulate CCR expression in human monocytic cells. J. Immunol. 172, 4977–4986 [DOI] [PubMed] [Google Scholar]
- 24. Dumitru C. D., Ceci J. D., Tsatsanis C., Kontoyiannis D., Stamatakis K., Lin J. H., Patriotis C., Jenkins N. A., Copeland N. G., Kollias G., Tsichlis P. N. (2000) TNF-α induction by LPS is regulated posttranscriptionally via a Tpl2/ERK-dependent pathway. Cell 103, 1071–1083 [DOI] [PubMed] [Google Scholar]
- 25. Banerjee A., Gugasyan R., McMahon M., Gerondakis S. (2006) Diverse Toll-like receptors utilize Tpl2 to activate extracellular signal-regulated kinase (ERK) in hemopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 103, 3274–3279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mielke L. A., Elkins K. L., Wei L., Starr R., Tsichlis P. N., O'Shea J. J., Watford W. T. (2009) Tumor progression locus 2 (Map3k8) is critical for host defense against Listeria monocytogenes and IL-1 β production. J. Immunol. 183, 7984–7993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. George D., Salmeron A. (2009) Cot/Tpl-2 protein kinase as a target for the treatment of inflammatory disease. Curr. Top. Med. Chem. 9, 611–622 [DOI] [PubMed] [Google Scholar]
- 28. Hall J. P., Kurdi Y., Hsu S., Cuozzo J., Liu J., Telliez J.-B., Seidl K. J., Winkler A., Hu Y., Green N., Askew G. R., Tam S., Clark J. D., Lin L.-L. (2007) Pharmacologic inhibition of Tpl2 blocks inflammatory responses in primary human monocytes, synoviocytes, and blood. J. Biol. Chem. 282, 33295–33304 [DOI] [PubMed] [Google Scholar]
- 29. Cusack K., Allen H., Bischoff A., Clabbers A., Dixon R., Fix-Stenzel S., Friedman M., Gaumont Y., George D., Gordon T., Grongsaard P., Janssen B., Jia Y., Moskey M., Quinn C., Salmeron A., Thomas C., Wallace G., Wishart N., Yu Z. (2009) Identification of a selective thieno[2,3-c]pyridine inhibitor of COT kinase and TNF-α production. Bioorg. Med. Chem. Lett. 19, 1722–1725 [DOI] [PubMed] [Google Scholar]
- 30. Green N., Hu Y., Janz K., Li H. Q., Kaila N., Guler S., Thomason J., Joseph-McCarthy D., Tam S. Y., Hotchandani R., Wu J., Huang A., Wang Q., Leung L., Pelker J., Marusic S., Hsu S., Telliez J. B., Hall J. P., Cuozzo J. W., Lin L. L. (2007) Inhibitors of tumor progression loci-2 (Tpl2) kinase and tumor necrosis factor α (TNF-α) production: selectivity and in vivo antiinflammatory activity of novel 8-substituted-4-anilino-6-aminoquinoline-3-carbonitriles. J. Med. Chem. 50, 4728–4745 [DOI] [PubMed] [Google Scholar]
- 31. Soria-Castro I., Krzyzanowska A., Pelaéz M. L., Regadera J., Ferrer G., Montoliu L., Rodríguez-Ramos R., Fernández M., Alemany S. (2010) Cot/tpl2 (MAP3K8) mediates myeloperoxidase activity and hypernociception following peripheral inflammation. J. Biol. Chem. 285, 33805–33815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bandow K., Kusuyama J., Shamoto M., Kakimoto K., Ohnishi T., Matsuguchi T. (2012) LPS-induced chemokine expression in both MyD88-dependent and -independent manners is regulated by Cot/Tpl2-ERK axis in macrophages. FEBS Lett. 586, 1540–1546 [DOI] [PubMed] [Google Scholar]
- 33. Kaiser F., Cook D., Papoutsopoulou S., Rajsbaum R., Wu X., Yang H. T., Grant S., Ricciardi-Castagnoli P., Tsichlis P. N., Ley S. C., O'Garra A. (2009) Tpl-2 negatively regulates interferon-β production in macrophages and myeloid dendritic cells. J. Exp. Med. 206, 1863–1871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sugimoto K., Ohata M., Miyoshi J., Ishizaki H., Tsuboi N., Masuda A., Yoshikai Y., Takamoto M., Sugane K., Matsuo S., Shimada Y., Matsuguchi T. (2004) A serine/threonine kinase, Cot/Tpl2, modulates bacterial DNA-induced IL-12 production and Th cell differentiation. J. Clin. Invest. 114, 857–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sica A., Saccani A., Borsatti A., Power C. A., Wells T. N., Luini W., Polentarutti N., Sozzani S., Mantovani A. (1997) Bacterial lipopolysaccharide rapidly inhibits expression of C-C chemokine receptors in human monocytes. J. Exp. Med. 185, 969–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoebe K., Du X., Georgel P., Janssen E., Tabeta K., Kim S. O., Goode J., Lin P., Mann N., Mudd S., Crozat K., Sovath S., Han J., Beutler B. (2003) Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature 424, 743–748 [DOI] [PubMed] [Google Scholar]
- 37. Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K., Akira S. (2003) Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 301, 640–643 [DOI] [PubMed] [Google Scholar]
- 38. López-Pelaéz M., Fumagalli S., Sanz C., Herrero C., Guerra S., Fernandez M., Alemany S. (2012) Cot/tpl2-MKK1/2-Erk1/2 controls mTORC1-mediated mRNA translation in Toll-like receptor-activated macrophages. Mol. Biol. Cell 23, 2982–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tsatsanis C., Patriotis C., Bear S. E., Tsichlis P. N. (1998) The Tpl-2 protooncoprotein activates the nuclear factor of activated T cells and induces interleukin 2 expression in T cell lines. Proc. Natl. Acad. Sci. U.S.A. 95, 3827–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Das S., Cho J., Lambertz I., Kelliher M. A., Eliopoulos A. G., Du K., Tsichlis P. N. (2005) Tpl2/cot signals activate ERK, JNK, and NF-κB in a cell-type and stimulus-specific manner. J. Biol. Chem. 280, 23748–23757 [DOI] [PubMed] [Google Scholar]
- 41. López-Peláez M., Soria-Castro I., Boscá L., Fernández M., Alemany S. (2011) Cot/tpl2 activity is required for TLR-induced activation of the Akt p70 S6k pathway in macrophages: implications for NO synthase 2 expression. Eur. J. Immunol. 41, 1733–1741 [DOI] [PubMed] [Google Scholar]
- 42. Fleishaker D. L., Garcia Meijide J. A., Petrov A., Kohen M. D., Wang X., Menon S., Stock T. C., Mebus C. A., Goodrich J. M., Mayer H. B., Zeiher B. G. (2012) Maraviroc, a chemokine receptor-5 antagonist, fails to demonstrate efficacy in the treatment of patients with rheumatoid arthritis in a randomized, double-blind placebo-controlled trial. Arthritis Res. Ther. 14, R11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mack M., Cihak J., Simonis C., Luckow B., Proudfoot A. E., Plachý J., Brühl H., Frink M., Anders H.-J., Vielhauer V., Pfirstinger J., Stangassinger M., Schlöndorff D. (2001) Expression and characterization of the chemokine receptors CCR2 and CCR5 in mice. J. Immunol. 166, 4697–4704 [DOI] [PubMed] [Google Scholar]
- 44. Raborn E. S., Marciano-Cabral F., Buckley N. E., Martin B. R., Cabral G. A. (2008) The cannabinoid δ-9-tetrahydrocannabinol mediates inhibition of macrophage chemotaxis to RANTES/CCL5: linkage to the CB2 receptor. J. Neuroimmune Pharmacol. 3, 117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gao J. L., Wynn T. A., Chang Y., Lee E. J., Broxmeyer H. E., Cooper S., Tiffany H. L., Westphal H., Kwon-Chung J., Murphy P. M. (1997) Impaired host defense, hematopoiesis, granulomatous inflammation and type 1-type 2 cytokine balance in mice lacking CC chemokine receptor 1. J. Exp. Med. 185, 1959–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou Y., Kurihara T., Ryseck R.-P., Yang Y., Ryan C., Loy J., Warr G., Bravo R. (1998) Impaired macrophage function and enhanced T cell-dependent immune response in mice lacking CCR5, the mouse homologue of the major HIV-1 coreceptor. J. Immunol. 160, 4018–4025 [PubMed] [Google Scholar]
- 47. Foti M., Granucci F., Aggujaro D., Liboi E., Luini W., Minardi S., Mantovani A., Sozzani S., Ricciardi-Castagnoli P. (1999) Upon dendritic cell (DC) activation chemokines and chemokine receptor expression are rapidly regulated for recruitment and maintenance of DC at the inflammatory site. Int. Immunol. 11, 979–986 [DOI] [PubMed] [Google Scholar]
- 48. Weber C., Weber K. S., Klier C., Gu S., Wank R., Horuk R., Nelson P. J. (2001) Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO+ T cells. Blood 97, 1144–1146 [DOI] [PubMed] [Google Scholar]
- 49. Cao C., Lawrence D. A., Strickland D. K., Zhang L. (2005) A specific role of integrin Mac-1 in accelerated macrophage efflux to the lymphatics. Blood 106, 3234–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Xu L., Khandaker M. H., Barlic J., Ran L., Borja M. L., Madrenas J., Rahimpour R., Chen K., Mitchell G., Tan C. M., DeVries M., Feldman R. D., Kelvin D. J. (2000) Identification of a novel mechanism for endotoxin-mediated down-modulation of CC chemokine receptor expression. Eur. J. Immunol. 30, 227–235 [DOI] [PubMed] [Google Scholar]
- 51. Xu L., Rahimpour R., Ran L., Kong C., Biragyn A., Andrews J., Devries M., Wang J. M., Kelvin D. J. (1997) Regulation of CCR2 chemokine receptor mRNA stability. J. Leukocyte Biol. 62, 653–660 [DOI] [PubMed] [Google Scholar]
- 52. Kuipers H. F., Biesta P. J., Montagne L. J., van Haastert E. S., van der Valk P., van den Elsen P. J. (2008) CC chemokine receptor 5 gene promoter activation by the cyclic AMP response element binding transcription factor. Blood 112, 1610–1619 [DOI] [PubMed] [Google Scholar]
- 53. Sallusto F., Schaerli P., Loetscher P., Schaniel C., Lenig D., Mackay C. R., Qin S., Lanzavecchia A. (1998) Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 28, 2760–2769 [DOI] [PubMed] [Google Scholar]
- 54. Franchin G., Zybarth G., Dai W. W., Dubrovsky L., Reiling N., Schmidtmayerova H., Bukrinsky M., Sherry B. (2000) Lipopolysaccharide inhibits HIV-1 infection of monocyte- derived macrophages through direct and sustained down-regulation of CC chemokine receptor 5. J. Immunol. 164, 2592–2601 [DOI] [PubMed] [Google Scholar]
- 55. Verani A., Scarlatti G., Comar M., Tresoldi E., Polo S., Giacca M., Lusso P., Siccardi A. G, Vercelli D. (1997) C-C chemokines released by lipopolysaccharide (LPS)-stimulated human macrophages suppress HIV-1 infection in both macrophages and T cells. J. Exp. Med. 185, 805–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee J., Mira-Arbibe L., Ulevitch R. J. (2000) TAK1 regulates multiple protein kinase cascades activated by bacterial lipopolysaccharide. J. Leukocyte Biol. 68, 909–915 [PubMed] [Google Scholar]
- 57. Kane L. P., Mollenauer M. N., Xu Z., Turck C. W., Weiss A. (2002) Akt-dependent phosphorylation specifically regulates Cot induction of NF-κB-dependent transcription. Mol. Cell. Biol. 22, 5962–5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Robinson M. J., Beinke S., Kouroumalis A., Tsichlis P. N., Ley S. C. (2007) Phosphorylation of TPL-2 on serine 400 is essential for lipopolysaccharide activation of extracellular signal-regulated kinase in macrophages. Mol. Cell. Biol. 27, 7355–7364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Amat M., Benjamim C. F., Williams L. M., Prats N., Terricabras E., Beleta J., Kunkel S. L., Godessart N. (2006) Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br. J. Pharmacol. 149, 666–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dairaghi D. J., Oyajobi B. O., Gupta A., McCluskey B., Miao S., Powers J. P., Seitz L. C., Wang Y., Zeng Y., Zhang P., Schall T. J., Jaen J. C. (2012) CCR1 blockade reduces tumor burden and osteolysis in vivo in a mouse model of myeloma bone disease. Blood 120, 1449–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Izikson L., Klein R. S., Charo I. F., Weiner H. L., Luster A. D. (2000) Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J. Exp. Med. 192, 1075–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fife B. T., Huffnagle G. B., Kuziel W. A., Karpus W. J. (2000) CC chemokine receptor 2 is critical for induction of experimental autoimmune encephalomyelitis. J. Exp. Med. 192, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dairaghi D. J., Zhang P., Wang Y., Seitz L. C., Johnson D. A., Miao S., Ertl L. S., Zeng Y., Powers J. P., Pennell A. M., Bekker P., Schall T. J., Jaen J. C. (2011) Pharmacokinetic and pharmacodynamic evaluation of the novel CCR1 antagonist CCX354 in healthy human subjects: implications for selection of clinical dose. Clin. Pharmacol. Ther. 89, 726–734 [DOI] [PubMed] [Google Scholar]