

Abstract
Motivation: Modern transcriptomics and proteomics enable us to survey the expression of RNAs and proteins at large scales. While these data are usually generated and analyzed separately, there is an increasing interest in comparing and co-analyzing transcriptome and proteome expression data. A major open question is whether transcriptome and proteome expression is linked and how it is coordinated.
Results: Here we have developed a probabilistic clustering model that permits analysis of the links between transcriptomic and proteomic profiles in a sensible and flexible manner. Our coupled mixture model defines a prior probability distribution over the component to which a protein profile should be assigned conditioned on which component the associated mRNA profile belongs to. We apply this approach to a large dataset of quantitative transcriptomic and proteomic expression data obtained from a human breast epithelial cell line (HMEC). The results reveal a complex relationship between transcriptome and proteome with most mRNA clusters linked to at least two protein clusters, and vice versa. A more detailed analysis incorporating information on gene function from the Gene Ontology database shows that a high correlation of mRNA and protein expression is limited to the components of some molecular machines, such as the ribosome, cell adhesion complexes and the TCP-1 chaperonin involved in protein folding.
Availability: Matlab code is available from the authors on request.
Contact: srogers@dcs.gla.ac.uk
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Cluster analysis is one of the most common techniques in the analysis of mRNA profiles derived from microarray experiments. It can be both a visualization aid and an important tool in exploratory data analysis. For example (Alizadeh et al., 2000), clustered assays and discovered putative new lymphoma subtypes. Gasch et al. (2000) rotated the problem and clustered gene profiles over time to find groups that behaved similarly in response to environmental changes. These are just two examples of the hundreds present in the biological and bioinformatics literature. Many clustering algorithms have been used in and developed for this problem, from hierarchical clustering (Eisen et al., 1998) to more intricate probabilistic models (Lazzeroni and Owen, 2002; Rogers et al., 2004; Segal et al., 2003), each of which is advantageous in certain situations.
Whilst analysis of high throughput mRNA concentration measurements has provided incredible insight into cell operation, there are only limited conclusions that can be drawn from just measuring mRNA. Large-scale measurements of other molecular species, particularly proteins, are becoming more common. Whilst separate analysis of these new datasets is worthwhile, it is particularly interesting to consider how one could create models to jointly analyze these data with data from mRNA assays. Such a combined approach could potentially enable us to make inferences and predictions about how the network of regulatory control varies at the mRNA and protein levels.
If one assumes that there is a high degree of correlation between behaviour at the transcriptomic and proteomic levels, it might appear that the only benefit of combining such data would be a slight reduction in noise brought about by having two independent measurements of the same process. However, several recent investigations have revealed a rather poor correlation between mRNA and protein profiles (Chen et al., 2002; Griffin et al., 2002; Ideker et al., 2001; Tian et al., 2004; Waters et al., 2008), suggesting that different control mechanisms are present at the transcriptomic and proteomic levels
Given mRNA and proteomic data for some set of N genes at T time points, the most obvious way of combining the data is to concatenate them into one vector of length 2T and cluster this vector, as in Waters et al. (2008). This method groups together genes that share similar mRNA and protein profiles. Whilst discovering groups of genes with this property is undoubtedly interesting, the model is rather inflexible. Particularly, genes that share similar mRNA profiles but have very different protein profiles (and vice versa) will remain undiscovered but are still undoubtedly interesting for the very fact that they appear to be regulated differently at the two levels. Additionally, and as we shall demonstrate later, there are a great many more clusters in the concatenated space than in either individual representation, some of which are very small; characteristics of a dataset that make standard cluster analysis (particularly that based on probabilistic models) very challenging. From a purely statistical perspective, we have doubled the size of the feature space (2T rather than T), without increasing the number of data instances, thus significantly increasing the complexity of the problem.
An alternative would be to analyze the two datasets completely independently and there is no doubt that plenty could be learnt from this approach. It overcomes the problem of an increased feature space but we lose the explicit relationship between the two datasets which can surely provide some biological insight.
In this article, we describe a probabilistic clustering model that couples together transcriptomic and proteomic profiles from the same genes in a sensible and flexible manner. The model describes a broad spectrum, of which the two strategies described above (concatenating and clustering independently) are extreme points. At which point on this scale our model naturally sits for a particular dataset is an interesting question in itself; to pose it in a different way—if one is presented with an mRNA profile (or protein profile), how much does this tell us about the shape of the corresponding protein profile (or mRNA profile)? We will see that the answer to this question varies quite considerably between individual genes and groups of genes involved in particuar biological processes.
The model is based on two coupled statistical mixture models and therefore inherits all of the advantages of a probabilistic approach to clustering. For example, posterior probabilities of cluster membership for each gene rather than hard assignments and objective methods for computing the number of clusters present. The two models are coupled through the use of a joint prior distribution on their respective components. This joint distribution is factorized such that the membership of a protein cluster is dependent on the cluster to which the respective mRNA profile was assigned. The use of mixture models at each level provides great flexibility—there are a large number of possible component densities and these need not be the same at the mRNA and protein levels. As well as providing clusterings at the mRNA and protein levels, the approach unravels the links between these clusters. These links are the key to this approach and provide interesting biological insight.
The approach was motivated by and is illustrated on a new dataset describing mRNA and protein evolution in an HMEC cell line stimulated with epidermal growth factor (EGF) (Waters et al., 2008). To our knowledge, this is the first large-scale dataset for which time series data is available for both mRNA and proteins extracted from the same samples. It is likely that more datasets with these characteristics will appear in the future suggesting that the development of bespoke analysis methods is an important area of research.
2 THE COUPLED MIXTURE MODEL
In a standard mixture model, we must assign a prior probability p(k) to each component. Assuming that we have two separate mixture models, one for the mRNA data (with K components) and one for the proteomic data (with J components), we must now define a joint prior distribution over both sets of components, p(k, j). If we have no reason to assume any relationship between the datasets we can assume that k and j are independent and hence p(k, j)= p(k)p(j). At the other extreme, assuming that there is a one to one relationship between mRNA and protein clusters (equivalent to concatenating the data) defines the following joint distribution: p(k,j)=p(k)δkj, where δkj is the Kronecker delta function (δkj=1 if k=j and 0 otherwise).
More generally, we can factorize the joint prior as p(k, j)=p(k)p(j∣k), where p(j∣k)=p(j) in the independent case and δjk in the totally dependent (concatenated) case. We propose treating the components of p(j∣k) as parameters to be inferred in the model, allowing the data to define whereabouts it exists on the spectrum between total independence and total dependence. For our particular application, the components of p(j∣k) provide us with details of the relationship between expression at the mRNA and protein levels.
Formally, defining p(k) as πk, p(j∣k) as θjk and the complete sets of these parameters (over all k and j) as π and θ, respectively, the likelihood of a dataset (X) of G genes is
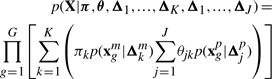 |
(1) |
where Δk and Δj correspond to any parameters unique to the k-th mRNA and j-th protein cluster, respectively.
The only restriction on the component densities p(xgm∣Δk) and p(xgp∣Δj) is that they must be proper probability distributions defined on T-dimensional real vectors. Many suitable choices exist, most of which have been evaluated in the context of biological (particularly mRNA) data. Hand and Heard (2005); Thalamuthu et al. (2006) provide reviews and comparison of several clustering techniques for mRNA data including mixture models. Ouyang et al. (2004), Pan (2006), Medvedovic and Sivaganesan (2002) and Teschendorff et al. (2005) used mixture models with Gaussian components. Alternatively, when clustering time series profiles, one may wish to choose a form of density that explicitly makes allowances for the fact that there is likely to be correlation over time as we might expect concentrations to evolve in a reasonably smooth manner. For example, Chudova et al. (2004) define a functional mixture model where each component is based on an ordinary differential equation model and Luan and Li (2003) use B-splines to define smooth cluster profiles. There are also other approaches based on richer mixture representations developed specifically for microarray data (Rogers et al., 2004) and expanding the proposed model to incorporate such distributions is an interesting avenue for future work. As there is no general consensus on the most appropriate form of noise model, we follow (Medvedovic and Sivaganesan, 2002; Ouyang et al., 2004; Pan, 2006; Teschendorff et al., 2005) and restrict ourselves to Gaussian densities although we stress that the framework presented is not limited to this choice or indeed to the same form of density being used for the two data types.
2.1 Inference and reproducibility
The expectation–maximization (EM) algorithm (Dempster et al., 1977) can be used to find a local maxima of a lower bound on the likelihood function. The required parameter update equations are provided in the supplementary document. One of the drawbacks of such an approach is that different initializations will lead to the algorithm converging to different maxima. To overcome this problem, we ran the algorithm from 100 random initializations and kept the one that gave the highest value of the lower bound. Of course, when using maximum likelihood estimation, one must be careful to avoid overfitting and if suitable prior information was available it would be straightforward to extend this maximum likelihood to a maximum-a-posteriori (MAP) approximation. Such prior information would potentially be of particular use regarding the connection probabilities. For example, a conjugate Dirichlet prior on the connection probabilities could be used to enforce sparsity in connections between clusters, if that were justified for a particular dataset and would take the form of a simple additive factor on both the numerator and denominator of the update equation for the connectivity parameters, θjk. In the current work, we have no prior information regarding the component or connectivity parameters and so work with the maximum likelihood estimation. We deal with the issue of choosing the number of components in the next section.
An important question that arises in this analysis is how reproducible are the results. The symmetry of the likelihood with respect to permutations of the component labels (j and k) makes it very difficult to compare results produced from multiple restarts. However, we can gauge the consistency of the algorithm by comparing the enriched GO terms across multiple restarts. If the results are reproducible, we would expect a significant proportion of GO terms to be enriched over many random initializations. Comparing the enriched terms over 100 random initializations, we found that of the 473 unique terms found to be significant (approximately 50 significant terms in each initialization), 22 where present in at least half of the initializations. Of these, a large proportion (eight) were always present. To place these figures in context, assuming that terms are chosen randomly, the probability of one particular term being present in at least half of the initializations is ∼7×10 −23 and in all of them is 3×10−98 (details in Supplementary Material).
3 RESULTS AND DISCUSSION
After matching the mRNA to their respective protein profiles (more details in Supplementary Material) and removing protein profiles with any missing values, we were left with a dataset consisting of mRNA and protein profiles for approximately ∼500 genes. A comprehensive description of the data generation procedure can be found in Waters et al. (2008). The various pre-processing steps undertaken and further algorithmic details can be found in the Supplementary Material. The number of components K and J were determined individually using the Bayesian Information Criterion (BIC) (K=15, J=20). As well as using BIC, individual Gaussian mixture models were used with Dirichlet process (DP) priors. Such models sample from the posterior distribution of model components as well as the number of components and as such provide a posterior distribution over the number of components. For various choices of the hyper-parameters defining the base measure, the number of components suggested by the DP was in general agreement with that from BIC (more details of both approaches are given in the Supplementary Material). An infinite variant of the full model with DP priors is under development but is non-trivial due to the coupling between the two mixtures.
3.1 Preliminary analysis
Before performing analysis with the coupled model, we did some preliminary experiments to investigate the similarity between the two data sets and what is lost through concatenation. Full details of these investigations are provided in the Supplementary Material. To summarize, we first clustered the two data sets separately and analyzed the similarity between the obtained clusterings, finding that there is a very low (albeit significant) level of agreement. Second, we looked at the number of enriched Gene Ontology (GO) terms found when the two representations are clustered individually and when they are concatenated. Significantly fewer were found when concatenated than when the data sets are analyzed individually. Both of these results provide motivation for the development of a model such as ours.
3.2 High-level observations
We now present some overall observations from the coupled model. The model defines a prior distribution over the component to which a protein profile should be assigned conditioned on which component the associated mRNA profile belongs to—p(j∣k). Hence, the components of this K×J matrix provide us with some insight regarding the level of connectivity between the two representations. We find that p(j∣k) rather than being dominated by a small number of protein clusters (j) for each mRNA cluster (k) is instead very diffuse (a full visualisation of the interactions is provided in the Supplementary Material). Each mRNA cluster is connected to a large number of protein clusters, and vice versa, suggesting that the relationship between transcriptional and translation control is a very complex one. In answer to our original question, knowing the shape of an mRNA profile does not in general tell us much about the associated protein profile. We can quantify the level of complexity by analyzing the entropy of p(j∣k). Figure 1 shows the entropy of p(j∣k) over several algorithm initializations compared to the entropy obtained when the proteins are permuted. We see a small but consistent decrease in entropy (increase in structure) when compared to the value with proteins permuted. This provides an indication of the complexity of the problem—if there was a one-to-one relationship between mRNA and protein clusters, the entropy would be close to 0. The fact that the decrease is so small can be partly explained by the observation that the genes appear to be organized into many small groups with homogenous mRNA and protein profiles. In the following sections we will look at some examples of these small groups in more detail. At this point some of the benefits of using our approach over individual or concatenated clustering become clear. It is obvious that we could make no claim as to the complexity of the relationship between the transciptomic and proteomic profiles by analyzing the data separately but what about concatenation? Of the 15 × 20=300 possible combinations of j and k, we find 191 that are populated with genes. The number of genes in these combined clusters ranges from 1 (60 combinations) to 10 (1 combination). If we attempt to cluster this concatenated data directly into a Gaussian mixture with 191 components, we find only approximately 10 significantly enriched terms—far fewer than the approximately 50 we find with the joint analysis.
Fig. 1.
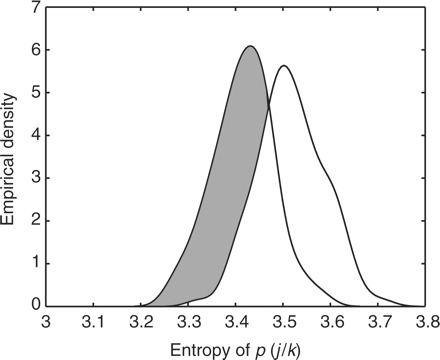
Distribution of mean entropy values of p(j∣k). The left curve gives the true entropy, the right gives the entropy obtained when the proteins are permuted.
3.3 Cluster–cluster relationships
The main benefit of our model is in uncovering relationships between small groups of genes. Particularly, if we take clusters of genes with highly conserved mRNA profiles (i.e. genes that belong to one of the mRNA profiles), it is interesting to look at similarities and differences between their protein profiles. We find that the genes tend to be organized in a modular structure whereby a group of genes with conserved mRNA profiles will have protein profiles belonging to a few, highly conserved clusters of protein profiles. In other words, whilst the mRNA and protein mixture models have reasonably small numbers of components, the diffuse nature of p(j∣k) means that in the joint model we see most possible combinations of mRNA and protein clusters represented. The number of genes in each combination varies from the order of 10 down to 2 or 3. From two reasonably small mixture models, with K and J components, respectively, we have found of the order of K×J=300 concatenated clusters—something that would have been incredibly difficult naively clustering the concatenated data from the approximately 500 genes.
3.3.1 The ribosomes
In the highly complicated network of connections between mRNA and protein clusters, one very strong connection stands out. This is the connection between protein cluster j=4 and mRNA clusters k=3 and k=11. The strength of the (reverse) connection probabilities, p(k=3∣j =4)=0.3653 and p(k=11∣j=4)=0.2316, is clear when it is considered that they are both in the highest 10 values out of the total K×J=300 values. A total of 18 out of the 19 proteins in j=4 are ribosomal (the one exception is SFRS1, a splicing factor) and they exhibit an exceptionally high expression homogeneity (Fig. 2, right heat map). As these proteins must act together to form the large and small ribosomal subunits, this high level of expression similarity at the protein level is no surprise. We might expect this to suggest a strong conservation of mRNA expression but, interestingly, we find that this is not necessarily the case. In the left-hand heat map of Figure 2, we can see the associated mRNA profiles. Note that the mRNA and protein profiles are presented in the same order and have been ordered according to mRNA cluster membership. The two dominant mRNA clusters [the values of p(k∣j=3) can be visualized in the lower chart of Fig. 2], k=3 and k=11 have rather similar expression profiles – initially dropping and then rising towards the end of the experiment) and if one was willing to tolerate noisier clusters there is an argument that these two should be combined. However, the remaining clusters (k=2,4,9,12,14) show quite diverse expression profiles. Particularly the two genes in k=4 and k=14 (both members of the large ribosomal sub-unit) have expression profiles peaking after 13 h rather than 24 h. The ability to be able to observe such diversity of expression and find cases of individual genes that appear to behave abnormally are benefits of the coupled mixture model. Finding these two genes that behave differently would be very difficult in a concatenated model as we would be looking for clusters of size G=1.
Fig. 2.
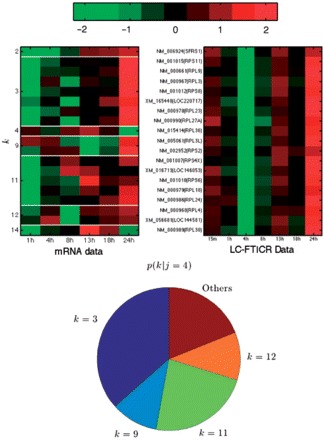
Protein cluster j=4 containing ribosomal proteins. Right-hand heat map shows protein profiles in j=4. Left-hand heat map shows associated mRNA profiles (each row corresponds to the same gene in each side) ordered by the mRNA cluster in which they are placed (i.e. top gene is in k=2, next group are in k=3, etc.). Red corresponds to high, green to low expression. The lower chart shows the probabilities p(k∣j=4) calculated from the conditional prior via Bayes law. Each colored segment corresponds to one mRNA segment and segment size is proportional to probability.
We now change our perspective and concentrate on one of the mRNA clusters which is highly connected to the ribosomal proteins. Isolating mRNA cluster k=3, we can visualize its elements and see which other protein clusters it is connected to. This is shown in Fig. 3. Again, mRNA profiles are shown in the left-hand heat map with protein profiles on the right. Genes are now ordered by their membership on the protein side. The conditional prior values p(j∣k=3) are visualized in the lower chart. We now see enormous diversity of protein profiles for genes whose mRNA profiles are highly conserved. The ribosomal cluster (j=4) stands out, as do various other modules of genes who behave similarly at each level (albeit with no direct correlation between the two). For example, besides all seven of the proteins in k=3, j=7 being ribosomal, all of the proteins in k=3 and j=17 are involved in chromatin structure and nucleo-cytoplasmic transport. Three out of the four proteins in k=3, j=8 are involved in biosynthesis. Two of these four, PAICS (phosphoribosylaminoimidazole succinocarboxamide synthetase) and GSS (glutathione synthetase), are additionally highlighted by GO term GO:0016874 (ligase activity) and two of the four proteins in k=3, j=2 are involved in cell–matrix interaction. Given the diversity of protein profiles and homogeneity of the mRNA profiles, it does not seem unreasonable from these observations to suggest that all of these processes are heavily regulated at the protein level. Thus, the coupled mixture approach can reveal both the complexity of the relationships between transcription and translation while preserving the detection of local, but significant correlations.
Fig. 3.
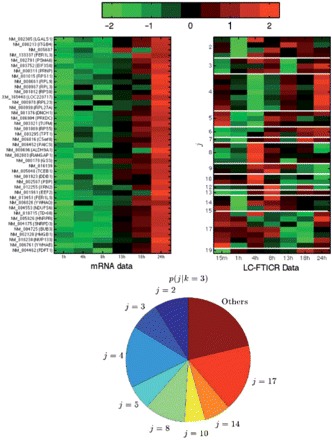
mRNA cluster k=3, containing a large proportion of ribosomal proteins (those in j=4). Left-hand heat map shows mRNA profiles (each row corresponds to the same gene in each side) for genes in k=3, Right-hand heat map shows their associated proteins. mRNA and protein profiles in the two figures are in the same order and are ordered by their membership to the protein clusters (right map). Red corresponds to high and green to low expression. The lower chart shows the conditional prior probabilities p(j∣k=3). Each colored segment corresponds to one protein cluster and size is proportional to probability.
3.3.2 Cell adhesion
We now turn our attention to cluster pair k=6, j=10. Only 3 out of the 542 genes share this clustering. mRNA cluster k=6 is significantly enriched with genes labeled with GO term GO:0005198 (structural molecule activity) whilst those in protein cluster j=10 are significantly enriched with GO term GO:0007155 (cell adhesion). Two genes, JUP (Plakoglobin or γ-catenin) and CTNNA1 (α-catenin), are present in both of these clusters and are labeled with both GO terms. Heat maps for the genes from the two clusters labeled with these GO terms can be seen in Fig. 4. In the left panel, above the white line, we can see genes with mRNA profiles in k=6 and labeled with GO:0005198. Below the line are those genes from j=10 and not k=6, labeled with GO:0007155. The opposite is shown in the right panel. These genes are involved in adherens junctions and desmosomes which are both cell–cell contact structures. The five genes present in j=10 and not k=6 that are labeled with GO:0007155 are also involved in cell to cell adhesion, all being cadherins and catenins as well as a regulator of cadherin adhesion (CD151), and CD166—a gene also involved in cell adhesion, low expression of which has shown to correlate with metastasis and poor prognosis in breast cancer (Tada et al., 2007; Xie et al., 2007), and implicated in the progression of cancers of breast, prostate, colon and in melanoma (Ihnen et al., 2008; Kristiansen et al., 2005; Swart et al., 2005; Weichert et al., 2004). The two genes in k=6 and not in j=10 are tubulin (NM_006009, TUBA3) and septin 7 (NM_001788, CDC10) which is involved in stress fibre regulation and microtubule remodeling (Tada et al., 2007; Xie et al., 2007). Note that these two genes have a reasonably conserved protein profile that is very different from those in j=10. It is striking that genes involved in cell adhesion co-segregate both in certain mRNA and protein clusters, but only two of them show co-regulation at both the transcriptional and translational level. It seems therefore that the two genes, γ-catenin and α-catenin, present in both clusters are more general due to their involvement in different aspects of cell–cell adhesion whilst the others are more specific. In epithelial cells, intercellular adhesion is mediated by the calcium-dependent interaction between the extracellular domains of the cadherins, while the α-, β-, and γ-catenins form a cytoplasmic protein complex that links the intracellular portion of the cadherins with actin filaments and the cytoskeleton. α-Catenin binds to both β- and γ-Catenin linking them to actin filaments. γ-Catenin binds to both cadherin and α-catenin. Thus, in its simplest version a core complex between γ- and α-catenins links cadherins to actin filaments (Kofron et al., 1997). This requires a stoichiometric expression of α- and γ-catenin that may best be afforded by linking mRNA and protein expression. Intriguingly, β-catenin also can double as a transcription factor. Its activity in this role is determined by its levels, as free β-catenin can migrate to the nucleus, associate with LEF/TCF and drive the expression of genes that promote cell cycle progression (Barker et al., 2000). Free β-catenin levels are determined by a balance between production, degradation and association with the cadherin complex. Therefore, it seems plausible that β-catenin expression is uncoupled from the joint regulation of α- and γ-catenin. Extrapolated this could potentially mean that transcriptional and translational co-regulation may be reserved for mono-functional components, while the expression of multi-functional components seems to be regulated more elaborately on different levels.
Fig. 4.
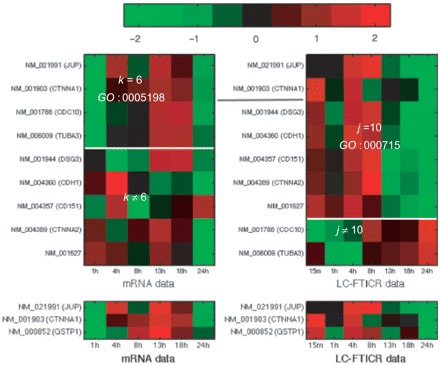
Genes from k=6 and/or j=10 involved in cell adhesion. The top two genes are involved in both clusters and are both tagged with GO:0005198 and GO:0007155. The lower plot shows these two genes and a third (GSTP1) that is present in both k=6 and j=10 but does not have these labels. Red corresponds to high and green low expression.
3.3.3 The chaperonin TCP-1 complex
The genes common to k=2 and j=5 contain almost the entire TCP-1 complex, i.e. seven of eight subunits, with the exception of the CCT1 subunit. TCP-1 is a cylindrical complex made up of stacked rings, which contains a central cavity that binds to unfolded polypeptides, sequesters them from the cellular environment and facilitates folding in an energy (ATP) dependent manner. Thus, the co-regulation of the expression of its components is not unexpected as the proteins need to be in a strict stoichiometric relationship to build a functional TCP-1 complex. Because of the sterical constraints of the cavity, TCP-1 only folds certain substrates (Gomez-Puertas et al., 2004). This function cannot be substituted by other chaperones, and as a result loss of TCP-1 function kills the cell. This specificity distinguishes TCP-1 from other chaperones, and indicates that it may have evolved independently of the chaperone protein folding machinery to serve specialized roles. The classical substrates for TCP-1 are actin and tubulin (Spiess et al., 2004), which are both major constituents of the cytoskeleton. However, the range of substrates is growing. A particular interesting function is the role of TCP-1 in the folding of Huntingtin (Htt), the protein responsible for Huntington disease. This is a major neurodegenerative syndrome due to a progressive expansion of polyglutamine repeats that cause Htt aggregation and cytotoxicity. Htt is folded by TCP-1, but interestingly, the CCT-1 subunit on its own can recognize Htt and ameliorate aggregation and cytotoxicity (Tam et al., 2006). These results suggest that the substrate recognition by CCT-1 may not be strictly dependent on the sterical configuration of the whole TCP-1 complex and could in part be an independent function. This also could explain why CCT-1 is differentially regulated from all the other subunits.
3.4 Summary
Put into a biological context the analysis of these data has several ramifications. First, the correlation between transcription and translation seems to be generally low and diverge with evolution. In lower organisms such as bacteria and yeast, there is a reasonable correlation between transcription and translation (Lu et al., 2007). In bacteria, genes that participate in a particular biological process are often organized in operons that ensure their transcriptional co-regulation and hence may favor the coordinated expression of gene products (Zaslaver et al., 2006). Although in higher organisms the organization in operons gives way to organization of gene clusters and non-random gene orders (Hurst et al., 2004), there still is an appreciable correlation between transcriptome and proteome expression (Lu et al., 2007).
Second, as our data show this correlation becomes very limited in mammals. In fact, it seems to be limited to some molecular machines such as the ribosome and the TCP-1 chaperone, and cell adhesion. The former two cases could be explained by the need to achieve stoichiometric ratios for the successful assembly of molecular machines. However, other molecular machines, such as the proteasome, do not show this correlation suggesting that there may be a fundamental difference at the level of the control that determines the stoichiometry required for the assembly of multi-protein molecular machines.
Third, these results indicate that transcriptional (mRNA) and translational (protein) networks may have evolved independently unless the rare occasions where a strong selection factor in favor of correlation between gene transcription and protein translation was present. In terms of robustness analysis, a system may have increased resilience to perturbations if there are separate levels of control evolving separately, as a failure would not easily propagate through the system (Kitano, 2004). In summary, there seems to be uncoupling of the transcriptome and the proteome in mammalian cells. This will make analysis more complicated but on the other hand enriches research by revealing additional and not always obvious layers of regulation.
Interestingly, systematic comparisons of mRNA and protein expression in different mouse organelles and tissues have suggested that at least in some tissues mRNA and protein expression is well correlated (Kislinger et al., 2006). The reasons for that need to be explored by further large-scale experimentation. A main difference between these and our studies is that we have analyzed a timecourse dataset from an individual cell line stimulated by a distinct growth factor to undergo a round of cell division. In contrast, studies monitoring protein and mRNA expression in tissues are analyzing a heterogenous population consisting of many different cell types, exposed to many different growth factors and hormones in vivo, and presenting a steady state equilibrium of gene expression of cells that due to the low fraction of dividing cells in most normal tissues are mainly in G1 phase. Thus, it is not possible to compare these data directly.
4 CONCLUSIONS
In this article, we have analyzed a new high-throughput trans-criptomic and proteomic dataset using a bespoke probabilistic model. As far as we are aware, these data (Waters et al., 2008) are the first to provide time-series profiles of both mRNA and protein expression levels on such a large scale. The model consists of two Gaussian mixtures coupled through a joint prior on the mixture components and allows us to find clusters of genes similar at the mRNA and protein levels and unravel the links between them.
The mRNA and protein datasets individually do not exhibit many clusters but when they are combined, there are a large number of small modules (approximately 2-10 genes) in which mRNA and protein profile are conserved at the two levels without necessarily any direct correlation between the two levels. The sheer number (approximately 190 in this dataset) of such modules would make attempting to find them with a concatenated clustering approach very cumbersome. Indeed, we have found that performing concatenated clustering with approximately 190 clusters results in an extreme loss of biological information with the number of enriched GO terms dropping from approximately 50 in the joint model to approximately 10 with concatenation. To fit the model, we have used an EM algorithm that finds a local maximum of the log-likelihood. This is computationally very convenient but there are drawbacks, particularly that we are not guaranteed to find the global maxima although we can overcome this to some extent through the use of multiple restarts.
As well as the overall result that the relationship between the mRNA and protein profiles appears highly complex, we have presented three examples of interesting biological phenomena that are uncovered by the model. First, the highly conserved behavior of ribosomes at the protein but not mRNA level. Second, an interesting group of genes involved in cell adhesion and third, the TCP-1 chaperonin which is a specialized protein folding machine. These examples are only a small proportion of those produced by the model and further examining the relationships between clusters is an avenue for future work. Other interesting developments would include experimenting with different forms for the mixture components—we have used spherical Gaussians but there are many alternatives available, some of which explicitly account for the time series nature of the data. It would also be interesting to add genes to both sides of the model for which we only had one representation (i.e. only mRNA profiles or only protein profiles). This would allow us to make predictions as to the time-evolution of the absent profile—our results suggest that the certainty in this prediction would vary greatly from gene to gene depending on the strength of links between clusters, but this is an interesting result in itself.
Matlab code that implements the coupled mixture model algorithm is available from the authors on request.
Supplementary Material
ACKNOWLEDGEMENTS
Portions of this work were conducted at the Environmental Molecular Science Laboratory, a DOE-funded national user facility.
Funding: EPSRC (EP/C010620/1 to S.R. and M.G.); EPSRC Advanced Research Fellow (EP/E052029/1 to M.G.); Interdisciplinary Research Collaboration in Proteomics Technologies RASOR (BBSRC/EPSRC to W.K.); Biomolecular Systems Initiative, part of the Laboratory Directed Research and Development Program at Pacific Northwest National Laboratory (PNNL for data collection); PNNL is operated by Battelle for the US Department of Energy (DE-ACO6-76RLO 1830).
Conflict of Interest: none declared.
REFERENCES
- Alizadeh A, et al. Different types of diffuse large b-cell lymphoma identified by gene expressing profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Barker N, et al. The Yin Yang of TCF/beta-catenin signaling. Adv. Cancer Res. 2000;77:1–24. doi: 10.1016/s0065-230x(08)60783-6. [DOI] [PubMed] [Google Scholar]
- Chen G, et al. Discordant protein and mRNA expression in lung adenocarcinomas. Mol. Cell. Proteomics. 2002;1:304–313. doi: 10.1074/mcp.m200008-mcp200. [DOI] [PubMed] [Google Scholar]
- Chudova D, et al. Gene expression clustering with functional mixture models. In: Thrun S, editor. Advances in Neural Information Processing Systems. Vol. 16 2004. [Google Scholar]
- Dempster A, et al. Maximum likelihood from incomplete data via the em algorithm. J. R. Stat. Soc. B. 1977;39:1–38. [Google Scholar]
- Eisen M, et al. Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch A, et al. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell. 2000;11:4241–4257. doi: 10.1091/mbc.11.12.4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Puertas P, et al. The substrate recognition mechanisms in chaperonins. J. Mol. Recognit. 2004;17:85–94. doi: 10.1002/jmr.654. [DOI] [PubMed] [Google Scholar]
- Griffin TJ, et al. Complementary profiling of gene expression at the transcriptome and proteome levels in s. cerevisiae. Mol. Cell. Proteomics. 2002;1:323–333. doi: 10.1074/mcp.m200001-mcp200. M200001–MCP200–. [DOI] [PubMed] [Google Scholar]
- Hand D, Heard N. Finding groups in gene expression data. J. Biomed. Biotechnol. 2005:215–225. doi: 10.1155/JBB.2005.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst LD, et al. The evolutionary dynamics of eukaryotic gene order. Nat. Rev. Genet. 2004;5:299–310. doi: 10.1038/nrg1319. 2004. 10.1038/nrg1319. [DOI] [PubMed] [Google Scholar]
- Ideker T, et al. Integrated genomic and proteomic analyses of a systematically perturbed metabolic network. Science. 2001;292:929–934. doi: 10.1126/science.292.5518.929. [DOI] [PubMed] [Google Scholar]
- Ihnen M. Breast Cancer Res. Treat. 2008. Predictive impact of activated leukocyte cell adhesion molecule (ALCAM/CD166) in breast cancer. Available at http://www.springerlink.com/content/dm4787127127648p/fulltext.pdf. [DOI] [PubMed] [Google Scholar]
- Kislinger T, et al. Global survey of organ and organelle protein expression in mouse: combined proteomic and transcriptomic profiling. Cell. 2006;125:173–186. doi: 10.1016/j.cell.2006.01.044. [DOI] [PubMed] [Google Scholar]
- Kitano H. Biological robustness. Nat. Rev. Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- Kofron M, et al. The roles of maternal alpha-catenin and plakoglobin in the early xenopus embryo. Development. 1997;124:1553–1560. doi: 10.1242/dev.124.8.1553. [DOI] [PubMed] [Google Scholar]
- Kristiansen G, et al. Expression profiling of microdissected matched prostate cancer samples reveals cd166/memd as nw prognostic markers for patient survival. J. Pathol. 2005;205:359–376. doi: 10.1002/path.1676. [DOI] [PubMed] [Google Scholar]
- Lazzeroni L, Owen A. Plaid models for gene expression data. Stat. Sinica. 2002;12:61–86. [Google Scholar]
- Luan Y, Li H. Clustering of time-course gene expression data using a mixed-effects model with b-splines. Bioinformatics. 2003;19:474–482. doi: 10.1093/bioinformatics/btg014. [DOI] [PubMed] [Google Scholar]
- Lu P, et al. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat. Biotech. 2007;25:117–124. doi: 10.1038/nbt1270. [DOI] [PubMed] [Google Scholar]
- Medvedovic M, Sivaganesan S. Bayesian infinite mixture model based clustering of gene expression profiles. Bioinformatics. 2002;18:1194–1206. doi: 10.1093/bioinformatics/18.9.1194. [DOI] [PubMed] [Google Scholar]
- Ouyang M, et al. Gaussian mixture clustering and imputation of microarray data. Bioinformatics. 2004;20:917–923. doi: 10.1093/bioinformatics/bth007. [DOI] [PubMed] [Google Scholar]
- Pan W. Incorporating gene functions as priors in model-based clustering of microarray gene expression data. Bioinformatics. 2006;22:795–801. doi: 10.1093/bioinformatics/btl011. [DOI] [PubMed] [Google Scholar]
- Rogers S, et al. The latent process decomposition of cDNA microarray datasets. IEEE Trans. Comput. Biol. 2004;2:143–156. doi: 10.1109/TCBB.2005.29. [DOI] [PubMed] [Google Scholar]
- Segal E, et al. Decomposing gene expression into cellular processes. Proceedings of the Pacific Symposium on Biocomputing. 2003:89–100. doi: 10.1142/9789812776303_0009. [DOI] [PubMed] [Google Scholar]
- Spiess C, et al. Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol. 2004;14:598–604. doi: 10.1016/j.tcb.2004.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swart G, et al. Cancer Metastasis Rev. 2005;24:223–236. doi: 10.1007/s10555-005-1573-0. [DOI] [PubMed] [Google Scholar]
- Tada T, et al. Role of septin cytoskeleton in spine morphogenesis and dendrite development in neurons. Curr. Biol. 2007;17:1752–1758. doi: 10.1016/j.cub.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam S, et al. The chaperonin tric controls polyglutamine aggregation and toxicity through subunit-specific interactions. Nat. Cell Biol. 2006;8:155–1162. doi: 10.1038/ncb1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, et al. A variational Bayesian mixture modelling framework for cluster analysis of gene-expression data. Bioinformatics. 2005;21:3025–3033. doi: 10.1093/bioinformatics/bti466. [DOI] [PubMed] [Google Scholar]
- Thalamuthu A, et al. Evaluation and comparison of gene clustering methods in microarray analysis. Bioinformatics. 2006;22:2405–2412. doi: 10.1093/bioinformatics/btl406. [DOI] [PubMed] [Google Scholar]
- Tian Q, et al. Integrated genomic and proteomic analyses of gene expression in mammalian cells. Mol. Cell. Proteom. 2004;3:960–969. doi: 10.1074/mcp.M400055-MCP200. [DOI] [PubMed] [Google Scholar]
- Waters K, et al. Systems analysis of response of human mammary epithelial cells to egf by integration of gene expression and proteomic data. Mol. Syst. Biol. 2008 (under submission) [Google Scholar]
- Weichert W, et al. ALCAM/CD166 is overexpressed in colorectal carcinoma and correlates with shortened patient survival. J. Clin. Pathol. 2004;57:1160–1164. doi: 10.1136/jcp.2004.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, et al. The gtp-binding protein septin 7 is critical for dendrite branching and dendritic-spine morphology. Current Biol. 2007;17:1746–1751. doi: 10.1016/j.cub.2007.08.042. [DOI] [PubMed] [Google Scholar]
- Zaslaver A, et al. Optimal gene partition into operons correlates with gene functional order. Physical Biol. 2006;3:183–189. doi: 10.1088/1478-3975/3/3/003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.