

Abstract
Objective:
To assess efficacy and safety of fulranumab, a fully human monoclonal antibody against nerve growth factor, in patients with diabetic peripheral neuropathic pain (DPNP).
Methods:
In this phase II, double-blind, placebo-controlled trial, patients with moderate to severe DPNP were randomized to treatments with fulranumab (1, 3, or 10 mg) or placebo administered subcutaneously every 4 weeks.
Results:
Because of early study termination (clinical hold) by the US Food and Drug Administration, 77 (intent-to-treat) of the planned 200 patients were enrolled. The primary endpoint, the mean reduction of average daily pain at week 12 compared with baseline, showed a positive dose-response relationship (p = 0.014, 1-sided); the pair-wise comparison between the 10-mg group and placebo was significant (unadjusted p = 0.040, 2-sided). An exploratory responder analysis revealed that a greater proportion of patients in the 10-mg group reported ≥30% reduction in the average DPNP intensity compared with placebo at week 12 (p = 0.006). Although not statistically significant, several secondary endpoints showed directionally similar results to the primary efficacy dose-response relationship. During the combined efficacy and safety extension phases, the top 3 treatment-emergent adverse events in the combined fulranumab group were arthralgia (11%), edema peripheral (11%), and diarrhea (9%). No cases of joint replacement or death were reported.
Conclusion:
Despite early study termination, fulranumab treatment resulted in dose-dependent efficacy and was generally well tolerated.
Classification of evidence:
This study provides Class I evidence that in patients with DPNP, fulranumab 10 mg reduces pain by 1.2 points on an 11-point scale compared with placebo.
Approximately 16% of patients with diabetes have diabetic peripheral neuropathic pain (DPNP), but it is estimated that 2 of 5 such cases do not receive any treatment for the pain.1–3 DPNP seriously affects quality of life and represents an ongoing management challenge for patients and physicians.2,4 Although many pharmacotherapies are available for DPNP, including antidepressants, anticonvulsants, and opioids, the effect size of these treatments is limited.5–7
Nerve growth factor (NGF) contributes to persistent pain in a variety of animal models of inflammatory and neuropathic pain.8–12 Blocking the activity of NGF has shown potential for normalizing neuronal hyperactivity and producing sustained clinical pain relief.13–17 The role of NGF in neuropathic pain and the implications for the development of novel therapeutics are of great interest.18,19
Fulranumab is a fully human recombinant immunoglobulin G2 monoclonal antibody that has a specific capacity to neutralize the biological actions of human NGF. It has shown efficacy in the treatment of moderate to severe knee and hip osteoarthritis pain.20 In the present study, we evaluated the efficacy and safety of fulranumab, compared with placebo, in patients with moderate to severe DPNP that was not adequately controlled by standard pain therapies.
METHODS
Standard protocol approvals, registrations, and patient consents.
The study was approved by an independent ethics committee or an institutional review board. Written informed consent was obtained from all participants at screening. The trial was registered with clinicaltrials.gov (NCT00993018).
Study design.
This was a phase II, randomized, placebo-controlled, double-blind (DB), dose-ranging study conducted at 16 sites from November 9, 2009 through June 23, 2011. The planned study included 5-week screening, 12-week DB efficacy, 40-week DB extension, 52-week open-label extension, and 26-week posttreatment phases. All patients still in the dosing phases stopped treatment on December 23, 2010, when the US Food and Drug Administration (FDA) placed ongoing fulranumab studies on clinical hold because of the concern that the entire class of anti-NGF antibodies may be associated with a condition representing either rapidly progressing osteoarthritis or osteonecrosis.
Patients were randomized 3:2:2:3, via an interactive voice response system, to 1 of 4 treatments (placebo, fulranumab 1, 3, or 10 mg) administered subcutaneously into the thigh or the abdomen every 4 weeks. To maintain the blinding, placebo-treated patients were randomized equally in a 1:1:1 ratio to receive a volume of placebo that matched the volume of the 3 fulranumab groups (1, 3, and 10 mg). Randomization was stratified by current DPNP medication use (yes or no). Patients had clinic visits every 4 weeks (week 1 through week 105).
Patients.
Patients must have been 18 to 80 years of age (inclusive), with chronic DPNP (persistent for >6 months) that was not adequately controlled by standard of care treatment. Patients must have had pain (had to begin in the feet and with relatively symmetrical onset for >6 months) due to bilateral peripheral neuropathy caused by type 1 or type 2 diabetes mellitus, with bilateral decrease or absent reflexes at the ankles, or bilateral decrease of a sensory sign in the distal lower extremities based on medical history, or physical/neurologic examination.
Patients assessed pain using an 11-point numeric rating scale (NRS)21 of 0 to 10 (0 = no pain; 10 = pain as bad as you can imagine), and had an average pain intensity score of ≥5 but <10, over 7 consecutive days before randomization, with a minimum of 5 assessments required. Use of ≤2 of the following medications, each one from a different class, was permitted: anticonvulsants (≤1,800 mg/d gabapentin or ≤300 mg/d pregabalin); opioid analgesics (≤60 mg/d oxycodone equivalent or ≤200 mg/d tramadol); antidepressants (≤75 mg/d amitriptyline equivalent tricyclic antidepressants, ≤60 mg/d duloxetine, or ≤150 mg/d venlafaxine). Patients taking medications for neuropathic pain at screening could choose to continue, reduce the dose or number to within acceptable limits, or discontinue their current pain medications with an appropriate washout period.
Key exclusion criteria included severe autonomic dysfunction or loss of pinprick sensation above the knees or wrists; other chronic, more severe pain conditions than DNPN; and pregnancy or lactation.
Efficacy.
The primary efficacy endpoint was the mean of the daily evening assessment of average pain intensity over the last 24 hours for the last 7 days of the DB efficacy phase minus the mean from the 7-day baseline period. Secondary efficacy outcomes included changes from baseline in (1) the 7-day mean of daily evening assessment of pain at its worst in the last 24 hours to the end of week 12; (2) the monthly Brief Pain Inventory–Short Form22 subscale scores; (3) the monthly Neuropathic Pain Symptom Inventory (NPSI)23 total and subscale scores; and (4) the monthly Patient Global Impression of Change score. An exploratory responder analysis included ≥30% or ≥50% improvement in NRS pain intensity from baseline to the end of week 12.
A post hoc exploratory analysis was conducted to examine the relationship between pain reduction and NPSI baseline subscale scores.
Safety.
Safety assessments included treatment-emergent adverse events (TEAEs), 12-lead ECG, clinical laboratory tests, vital signs, physical examinations, and investigator evaluation of the injection site. Nerve safety evaluation included neurologic examination, total neuropathy scores–nurse (TNSn), skin biopsy,24 and nerve conduction studies.25 We collected 2 adjacent skin biopsies from the distal thigh to determine the intraepidermal nerve fiber density, expressed as the number of fibers per millimeter length of the skin biopsy.26,27 Nerve conduction studies included measures of nerve conduction velocity and amplitude in the distal segments of 2 sensory nerves (ulnar and median) and 2 motor nerves (ulnar and peroneal) using standard surface recording techniques.28 Details of skin biopsy, nerve conduction studies, and TNSn are described in a separate report.
Predefined neurologic treatment-emergent events of interest were significant neurologic signs and symptoms that resulted in discontinuation of treatment, prolonged hypotension, and symptomatic bradycardia. All TEAEs were followed to satisfactory resolution or a clinically stable endpoint. An independent data monitoring committee was established to monitor unblinded safety data, including the protocol predefined events of interest, to ensure the continuing safety of the enrolled patients.
Statistical analysis.
This study was designed to enroll 200 patients to provide ≥85% power at a 1-sided 5% significance level to assess a dose-response relationship, assuming a variance of 2.4 and an effect size of 1.25.
The intent-to-treat population for the efficacy analysis included all randomized patients who received at least 1 dose of fulranumab or placebo. Daily evening assessment of average pain scores for the 7 days before the first dose of study drug were averaged to determine the baseline pain score; those for the last 7 days before the time point of interest (4, 8, and 12 weeks) were averaged to define the pain score for that time point. The last observation carried forward and baseline observation carried forward methods were used for data imputation.
The dose response of the primary efficacy endpoint was evaluated using the MCP-Mod procedure29 at a 1-sided, 5% level of significance. The decision to use a 1-tailed test of significance for the primary endpoint was prespecified. This approach was deemed appropriate because (1) the focus of this phase II pilot study was on a positive dose-response relationship, not an inverse dose-response, and (2) at this early phase, the type 1 error rate is primarily of concern to the sponsor regarding the risk of moving forward into later development stage. The linear, exponential, and Emax models were tested for dose-response relationship using the optimal contrast test for the original fixed allocation ratio of 3:2:2:3 for placebo, fulranumab 1, 3, or 10 mg. The treatment magnitude for each dosage was estimated in 3 pair-wise comparisons of each individual dosage group against placebo using the analysis of covariance model (2-sided at a 5% level). The Cochran-Mantel-Haenszel test and the baseline observation carried forward method were applied to the exploratory responder analysis. All continuous secondary efficacy endpoints were analyzed based on analysis of covariance with treatment and concomitant medication as factors, and baseline score as a covariate. The least squares (LS) means and treatment differences relative to placebo with the corresponding 95% confidence intervals (CIs) and p values were provided. Unadjusted, nominal p values were presented for all secondary efficacy comparisons. Safety results were summarized descriptively.
The relationship between the baseline NPSI score and change from baseline in NRS pain intensity was explored post hoc using a logistic regression model with current baseline pain medication and NPSI score category (below or above a threshold) as factors and baseline NRS pain score as a covariate.
Classification of evidence.
This interventional study provides class I evidence that in patients with DPNP, fulranumab decreased average pain intensity from baseline to the end of the DB efficacy phase in a positive dose-dependent manner (p = 0.014, 1-sided). Fulranumab 10 mg reduced pain by 1.2 points (95% CI −2.44 to −0.06; unadjusted p = 0.040) on an 11-point scale compared with placebo.
RESULTS
Baseline characteristics.
At the time of the clinical hold, 312 patients had been screened and 77 qualified patients had been randomized into 4 treatment groups (figure 1).
Figure 1. Patient flowchart.

*The study was discontinued by the sponsor because of the clinical hold.
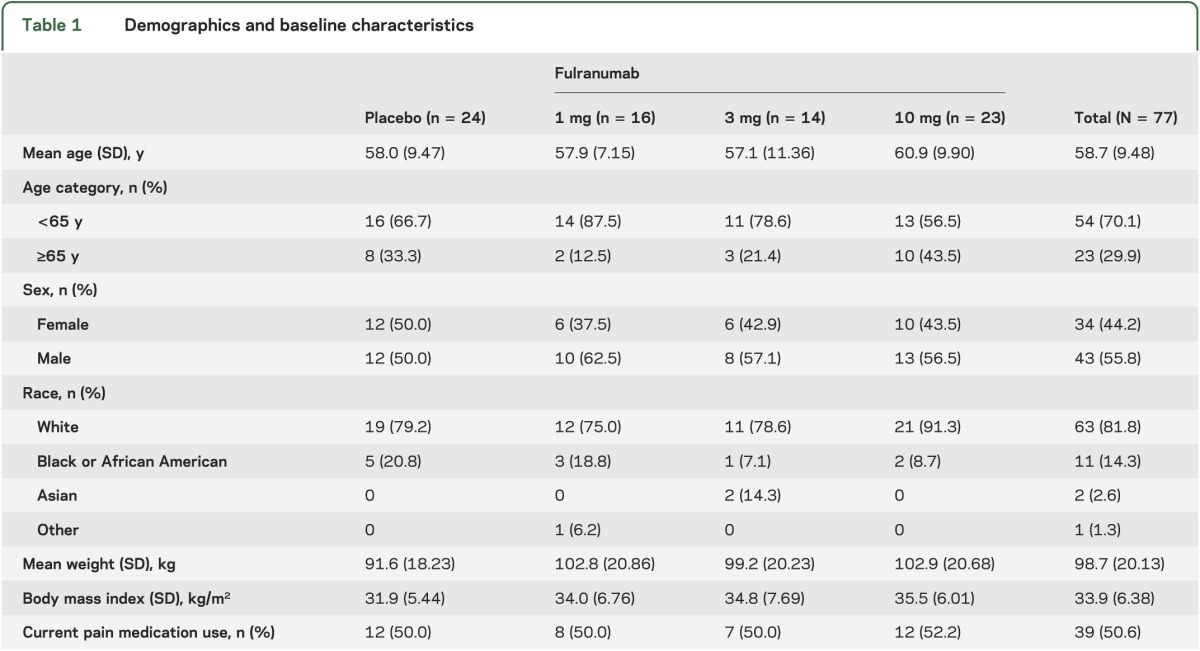
Sixty-two patients (81%) completed the DB efficacy phase, while 13 of 55 patients (24%) completed the DB extension phase, with most patients discontinued by the sponsor because of the clinical hold. All 12 patients entering the open-label phase were withdrawn because of the clinical hold. Patient demographics and baseline characteristics were generally balanced across treatment groups (table 1). During the DB efficacy phase, most patients (62 of 77) received all 3 medication doses. The median treatment exposure was 85 days for the DB efficacy phase, and 194 days for the DB safety phase.
Table 1.
Demographics and baseline characteristics
Efficacy.
The MCP-Mod test results indicated that the linear model was the best fitted one (using the Akaike information criterion) and fulranumab significantly (p = 0.014, 1-sided) decreased average pain intensity from baseline to the end of week 12 in a dose-dependent manner (figure 2A). The p value corresponded to the minimum t statistic (the smaller the more significant, when accounting for the negative sign). At the end of week 12, the reduction of average pain intensity with the fulranumab 10-mg treatment group was significant compared with placebo (LS mean difference −1.2, 95% CI −2.44 to −0.06, unadjusted p = 0.040; figure 2B), and the 10-mg group had significantly more responders with a ≥30% reduction in average pain intensity than placebo (unadjusted p = 0.006; table 2). Although not significant, the improvement from baseline to the end of week 12 in worst pain intensity, NPSI total score and several subscales, and most Brief Pain Inventory–Short Form subscales increased with increasing doses (table 2), consistent with the primary efficacy trend.
Figure 2. Efficacy results.

(A) Estimated linear dose response for the intent-to-treat population using the MCP-Mod procedure with 80% point-wise confidence interval (CI). (B) Mean change from baseline in average pain intensity score to 4, 8, and 12 weeks in the double-blind efficacy phase (intent-to-treat population). Locf = last observation carried forward.
Table 2.
Secondary efficacy outcomes at the end of the 12-week double-blind efficacy phase
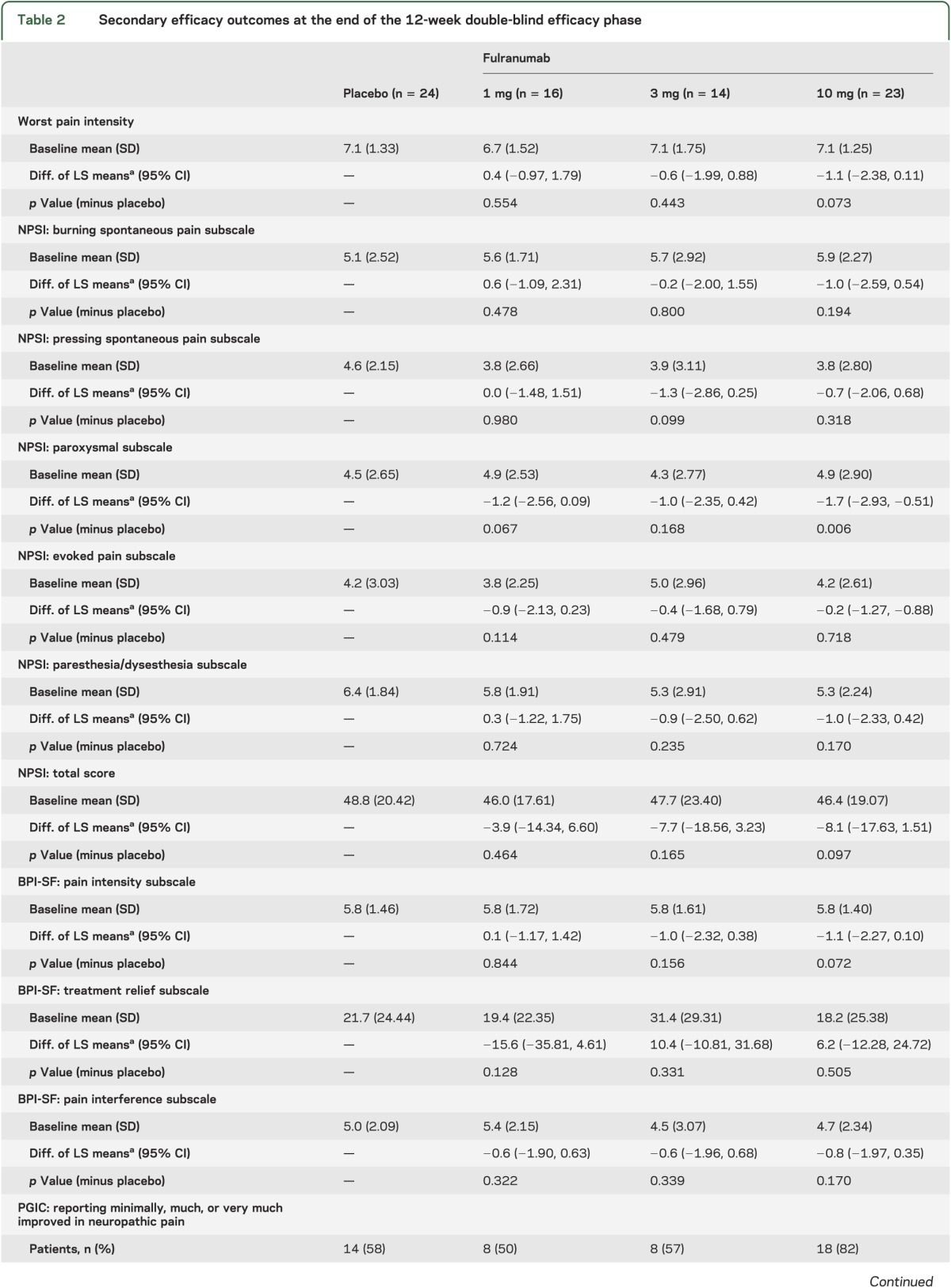

A post hoc exploratory analysis showed that the likelihood of being a responder was related to baseline NPSI levels of burning spontaneous pain (≤5/>5) and pressing spontaneous pain (≤3.5/>3.5) (p = 0.012 and p = 0.045, respectively). Other NPSI baseline subscales were not found to contribute to the responder patient population.
Safety.
During the combined DB efficacy and safety extension phases, the overall percentage of patients with TEAEs was similar among placebo (75%) and fulranumab (81%–86%) groups, with no dose relationship observed among the fulranumab groups (table 3).
Table 3.
TEAEs during the combined double-blind efficacy and safety extension phases
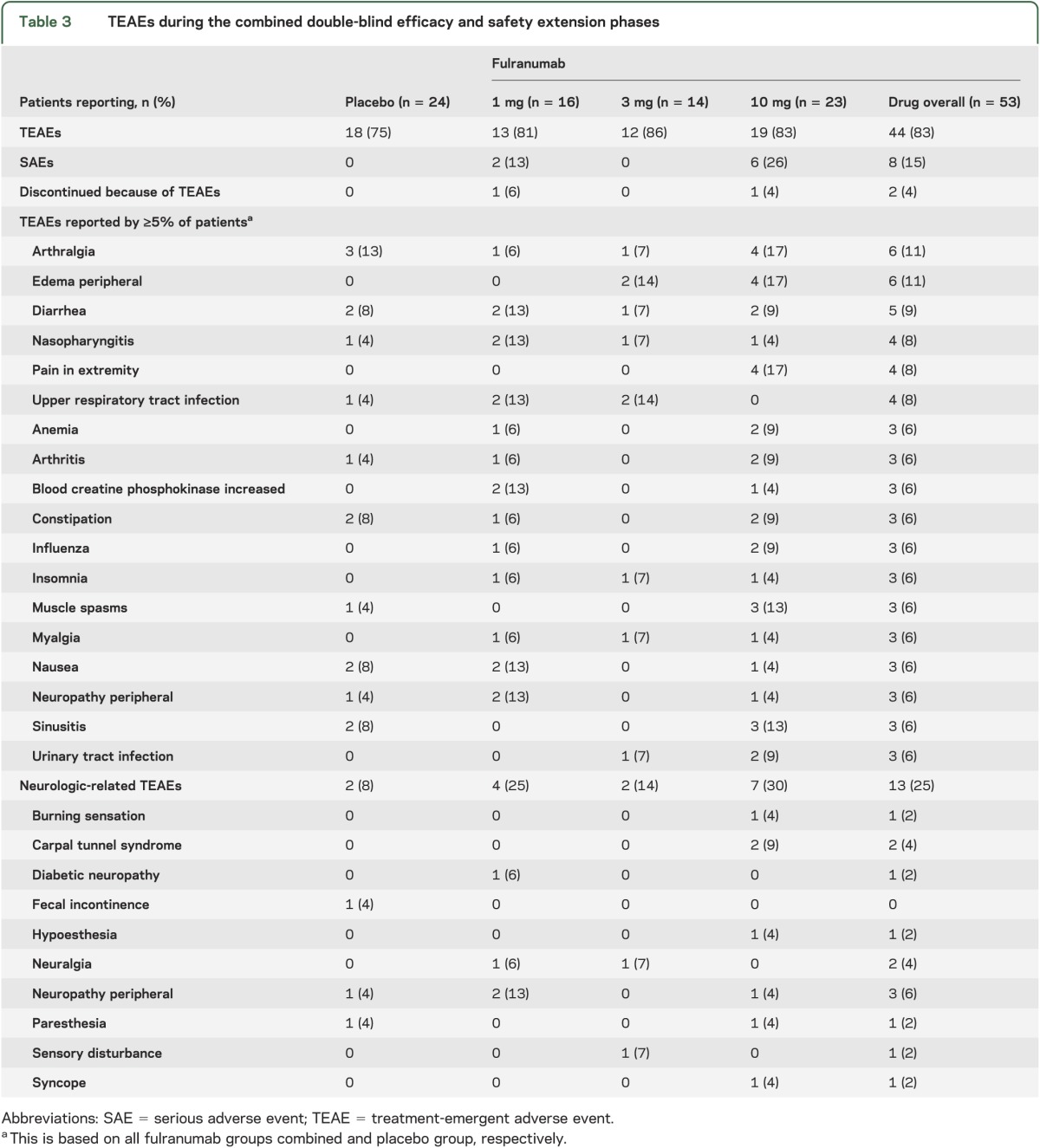
Among fulranumab-treated patients, the 3 most common TEAEs (percentage of patients) were arthralgia (11%), edema peripheral (11%), and diarrhea (9%), compared with arthralgia (13%), constipation (8%), diarrhea (8%), hypertension (8%), nausea (8%), and sinusitis (8%) in the placebo group. Two patients (4%) in the combined fulranumab treatment groups vs none in the placebo group discontinued because of TEAEs: one in the 1-mg group had type III immune complex mediated reaction and the other in the 10-mg group had vesicular lesions in the upper chest. Both TEAEs were moderate in severity and considered by the investigator as probably or possibly related to the study drug. The events resolved after study drug withdrawal.
Neurologic-related TEAEs were reported in 8% (placebo), 25% (1 mg), 14% (3 mg), and 30% (10 mg) of patients during the combined DB efficacy and safety phases (table 3). The incidence of neurologic TEAEs was higher among fulranumab-treated patients (25% overall) than placebo (8%); however, no apparent fulranumab dose relationship was detected. The most common neurologic TEAEs in all fulranumab groups were neuropathic pain (4%), neuropathy (4%), and carpal tunnel syndrome (4%), compared with paresthesia (4%), peripheral neuropathy aggravated (4%), and fecal incontinence (4%) in the placebo group. All neurologic TEAEs were mild or moderate in severity. Twelve of 19 neurologic TEAEs were resolved with durations of 3 to 253 days; 7 of 19 had no end date.
During the combined DB efficacy and safety extension phases, serious adverse events (SAEs) occurred in 2 patients (13%) of the 1-mg group and 6 (26%) of the 10-mg group. These SAEs included hyperglycemia and type III immune complex–mediated reaction (1-mg group), and arthralgia, bacteremia, cellulitis, diverticulitis, localized infection, mental status changes, and urinary retention (10-mg group). There were only 2 patients in the fulranumab 10-mg group who had SAEs (localized infection and urinary retention) during the efficacy phase. At the end of the efficacy phase when efficacy was evaluated, the mean (SD) change in average pain scores from baseline for all 23 patients in the fulranumab 10-mg group was −2.7 (1.8). By comparison, the reductions in average pain intensity from baseline for these 2 patients were respectively −1.3 and 0.4. Thus, these SAEs did not distract those 2 patients from their neuropathic pain during the efficacy phase. All SAEs were resolved within 30 days, except one (right hip joint pain) with no end date.
We observed no clinically important changes in laboratory, ECG, and vital sign findings in any patient during the study. No cases of joint replacement surgery or death were reported during the study. There were no clinically significant changes from baseline in neurologic examination findings, TNSn, or Mini-Mental State Examination score. The assessments of skin biopsy and nerve conduction studies (detailed in a separate report) do not support the hypothesis that fulranumab treatment was associated with improved pain secondary to decreased or damaged nerve function in patients with DPNP.
DISCUSSION
We evaluated the analgesic efficacy and safety of fulranumab (1, 3, and 10 mg administered every 4 weeks) in treating patients with moderate to severe DPNP. Because of the clinical hold by the FDA, only 39% (n = 77) of planned enrollment was achieved; 81% of the study patients (n = 62) received all 3 doses of study medication.
Fulranumab significantly decreased average DPNP intensity in a dose-dependent manner from baseline to the end of the efficacy phase. In addition, a significant pairwise difference from placebo and significantly more responders with a ≥30% reduction in average pain intensity than placebo were observed in the fulranumab 10-mg group. The magnitude of the pain reduction in the 10-mg group could be considered as clinically meaningful pain relief.30,31 Consistent with the primary efficacy dose-response relationship, although not statistically significant, most secondary efficacy outcomes showed similar trends.
It is of scientific interest to understand whether patients with different phenotypic pain respond differently to anti-NGF treatment. NPSI includes 10 descriptors (plus 2 temporal items) that allow discrimination and quantification of 5 distinct clinically relevant dimensions of neuropathic pain syndromes. The psychometric properties of the NPSI suggest that this tool might be used to characterize subgroups of patients with neuropathic pain and verify whether they respond differentially to various pharmacologic agents or therapeutic interventions. The exploratory analysis revealed that patients with a baseline NPSI subscale score of >5 for burning spontaneous pain or >3.5 for pressing spontaneous pain tended to be more responsive to treatment. These findings (need to be verified in an appropriately sized future study) may help inform future research on whether patients with certain baseline phenotypes are likely to be more treatment responsive.
During the combined DB efficacy and safety extension phases, the overall rate of TEAEs was similar among placebo and fulranumab treatment groups, with no dose relationship apparent; however, the rate of neurologic-related TEAEs in the placebo group was approximately one-third of that in the combined fulranumab groups. Based on phase I results, hypoesthesia and paresthesia were expected and were closely monitored using a brief neurologic examination and TNSn assessments. The most common neurologic-related TEAEs in fulranumab-treated patients were those associated with large- and small-fiber sensory function, such as neuropathic pain, neuropathy, paresthesia, and carpal tunnel syndrome. The majority of neurologic TEAEs were mild to moderate in severity and not associated with clinically meaningful neurologic abnormalities. No neurologic-related SAEs were reported during the DB treatment phases. Although not definitive, nerve safety assessments (detailed in a separate report) do not support the hypothesis that fulranumab achieves neuropathic pain relief in DPNP by damaging or otherwise adversely affecting nerves or nerve function.
The FDA has identified rapid joint destruction and/or osteonecrosis as a specific safety concern for the anti-NGF class. No cases of osteonecrosis or joint replacement were reported or adjudicated in this trial, although 13 patients enrolled had a history of osteoarthritis. However, because of the limited sample size due to the clinical hold, we cannot conclude there is no risk. All future trials will incorporate prospective joint surveillance.
The limitations of this study include a small sample size because of the FDA clinical hold. In addition, patients who discontinued the study were often lost to follow-up for safety. However, the discontinuation rate due to TEAEs (4%) in fulranumab treatment groups compares favorably with those reported in other medications (approximately 20%) recommended for treatment of DPNP.32 In addition, neurologic TEAE characteristics of this drug class were typically reported as mild or moderate and investigators opted to continue treatment. Thus, fulranumab showed the potential to address the unmet need for a better tolerated and more effective option compared with currently marketed therapies.33
Despite the limited sample size caused by early study termination, fulranumab treatment resulted in dose-dependent efficacy and was generally well tolerated, with the 10-mg dose demonstrating significant improvement over placebo. Long-term trials involving more patients are needed to fully characterize the efficacy, safety, and tolerability of this potentially new class of analgesic drug for treatment of DPNP.
ACKNOWLEDGMENT
The authors thank David Vitale (Janssen Research & Development, LLC) for outstanding site management, Sue Vallow (former employee of Janssen Global Services, LLC) for advice on patient-reported outcomes, Dr. Didier Bouhassira (CHU Ambroise Paré, APHP, Boulogne-Billancourt, France) for advice on the exploratory analysis, John Ritz (former contractor of Janssen Research & Development, LLC) for the exploratory analysis, Swarna Ramanjulu (Janssen Research & Development, LLC) for confirming and regenerating the plots of the exploratory analysis results, and Dr. Wendy P. Battisti (Janssen Research & Development, LLC) for editorial assistance. The authors thank the study participants without whom this study would never have been accomplished, and also thank the following investigators for their participation in the study: S. Beydoun, B. Beesley, L.H.S. Chuck, T. Heiman-Patterson, J. Hudson, L. Klaff, J. Licht, P. Markovitz, W. McElveen, R. McLean, M. Polydefkis, J. Berg, G. Rederich, A. Runheim, J. Selam, A. Shaibani, M. Tuchman, R. Upender, R. Weinstock, G. Connor, F. Crowell, R. DeGarmo, R. Doty, M. Gurru, L. Harris, O. Omidvar, N. Patel, B. Sorin, M. Warren, E. Wilson, R. Yapundich, L. Zhang, C. Alftine, P. Bhatia, D. Chen, J. Cochran, M. Desantis, G. Haase, B. Horowitz, H. Kerstein, A. Klymiuk, J. Steinberg, and D. Taylor.
GLOSSARY
- CI
confidence interval
- DB
double-blind
- DPNP
diabetic peripheral neuropathic pain
- FDA
US Food and Drug Administration
- LS
least squares
- NGF
nerve growth factor
- NPSI
Neuropathic Pain Symptom Inventory
- NRS
numeric rating scale
- SAE
serious adverse event
- TEAE
treatment-emergent adverse event
- TNSn
total neuropathy score–nurse
AUTHOR CONTRIBUTIONS
Dr. Wang participated in the study design, study governance, supervising study recruitment, monitoring of data quality, and data analysis; contributed to the development of the manuscript and approved the final manuscript for submission. Dr. Romano participated in the study design; contributed to the development of the manuscript and approved the final manuscript for submission. Ms. Frustaci was the study statistician; contributed to the development of the manuscript and approved the final manuscript for submission. Dr. Bohidar participated in the study design; was the study statistician; contributed to the development of the manuscript and approved the final manuscript for submission. Dr. Ma drafted and revised the manuscript for intellectual content; contributed to the development of the manuscript and approved the final manuscript for submission. Dr. Sanga and Dr. Fedgchin participated in the study design, study governance, supervising study recruitment, monitoring of data quality, and data analysis; contributed to the development of the manuscript and approved the final manuscript for submission. Dr. Ness, Dr. Russell, Dr. Kelly, and Dr. Thipphawong participated in study governance, supervising study recruitment, monitoring of data quality, and data analysis; contributed to the development of the manuscript and approved the final manuscript for submission.
STUDY FUNDING
Supported by Janssen Research & Development, LLC.
DISCLOSURE
H. Wang was an employee of Janssen Research & Development, LLC, when this work was performed. The opinions expressed in this article are the author's own and do not reflect the view of the NIH, the Department of Health and Human Services, or the United States government. G. Romano is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. M. Frustaci is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. N. Bohidar is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. H. Ma is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company. P. Sanga is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. S. Ness is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. L. Russell is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. M. Fedgchin is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. K. Kelly is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. J. Thipphawong is an employee of Janssen Research & Development, LLC, a Johnson & Johnson company, and holds Johnson & Johnson stock. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Daousi C, MacFarlane IA, Woodward A, Nurmikko TJ, Bundred PE, Benbow SJ. Chronic painful peripheral neuropathy in an urban community: a controlled comparison of people with and without diabetes. Diabet Med 2004;21:976–982 [DOI] [PubMed] [Google Scholar]
- 2.Bril V, England J, Franklin GM, et al. Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology 2011;76:1758–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tentolouris NK. Painful diabetic neuropathy: new choice of first-line therapy? Nat Rev Endocrinol 2011;7:381–383 [DOI] [PubMed] [Google Scholar]
- 4.Boulton AJ, Vinik AI, Arezzo JC, et al. Diabetic neuropathies: a statement by the American Diabetes Association. Diabetes Care 2005;28:956–962 [DOI] [PubMed] [Google Scholar]
- 5.Cruccu G. Treatment of painful neuropathy. Curr Opin Neurol 2007;20:531–535 [DOI] [PubMed] [Google Scholar]
- 6.Chong MS, Hester J. Diabetic painful neuropathy: current and future treatment options. Drugs 2007;67:569–585 [DOI] [PubMed] [Google Scholar]
- 7.Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ 2007;335:87–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amaya F, Shimosato G, Nagano M, et al. NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci 2004;20:2303–2310 [DOI] [PubMed] [Google Scholar]
- 9.Cirillo G, Cavaliere C, Bianco MR, et al. Intrathecal NGF administration reduces reactive astrocytosis and changes neurotrophin receptors expression pattern in a rat model of neuropathic pain. Cell Mol Neurobiol 2010;30:51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li WW, Sabsovich I, Guo TZ, Zhao R, Kingery WS, Clark JD. The role of enhanced cutaneous IL-1beta signaling in a rat tibia fracture model of complex regional pain syndrome. Pain 2009;144:303–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hefti FF, Rosenthal A, Walicke PA, et al. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol Sci 2006;27:85–91 [DOI] [PubMed] [Google Scholar]
- 12.Hill R. Blocking the effects of NGF as a route to safe and effective pain relief: fact or fancy? Pain 2011;152:2200–2201 [DOI] [PubMed] [Google Scholar]
- 13.Lane NE, Schnitzer TJ, Birbara CA, et al. Tanezumab for the treatment of pain from osteoarthritis of the knee. N Engl J Med 2010;363:1521–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schnitzer TJ, Lane NE, Birbara C, Smith MD, Simpson SL, Brown MT. Long-term open-label study of tanezumab for moderate to severe osteoarthritic knee pain. Osteoarthritis Cartilage 2011;19:639–646 [DOI] [PubMed] [Google Scholar]
- 15.Katz N, Borenstein DG, Birbara C, et al. Efficacy and safety of tanezumab in the treatment of chronic low back pain. Pain 2011;152:2248–2258 [DOI] [PubMed] [Google Scholar]
- 16.Kivitz AJ, Gimbel JS, Bramson C, et al. Efficacy and safety of tanezumab versus naproxen in the treatment of chronic low back pain. Pain 2013;154:1009–1021 [DOI] [PubMed] [Google Scholar]
- 17.Wild KD, Bian D, Zhu D, et al. Antibodies to nerve growth factor reverse established tactile allodynia in rodent models of neuropathic pain without tolerance. J Pharmacol Exp Ther 2007;322:282–287 [DOI] [PubMed] [Google Scholar]
- 18.Ossipov MH. Growth factors and neuropathic pain. Curr Pain Headache Rep 2011;15:185–192 [DOI] [PubMed] [Google Scholar]
- 19.Schmidt RE. The role of nerve growth factor in the pathogenesis and therapy of diabetic neuropathy. Diabet Med 1993;10(suppl 2):10S–13S [DOI] [PubMed] [Google Scholar]
- 20.Sanga P, Katz N, Polverejan E, et al. Efficacy, safety, and tolerability of fulranumab, an anti-nerve growth factor antibody, in the treatment of patients with moderate to severe osteoarthritis pain. Pain 2013;154:1910–1919 [DOI] [PubMed] [Google Scholar]
- 21.Williamson A, Hoggart B. Pain: a review of three commonly used pain rating scales. J Clin Nurs 2005;14:798–804 [DOI] [PubMed] [Google Scholar]
- 22.Cleeland CS, Ryan KM. Pain assessment: global use of the Brief Pain Inventory. Ann Acad Med Singapore 1994;23:129–138 [PubMed] [Google Scholar]
- 23.Bouhassira D, Attal N, Alchaar H, et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain 2005;114:29–36 [DOI] [PubMed] [Google Scholar]
- 24.Polydefkis M, Hauer P, Griffin JW, McArthur JC. Skin biopsy as a tool to assess distal small fiber innervation in diabetic neuropathy. Diabetes Technol Ther 2001;3:23–28 [DOI] [PubMed] [Google Scholar]
- 25.Arezzo JC, Zotova E. Electrophysiologic measures of diabetic neuropathy: mechanism and meaning. Int Rev Neurobiol 2002;50:229–255 [DOI] [PubMed] [Google Scholar]
- 26.McArthur JC, Stocks EA, Hauer P, Cornblath DR, Griffin JW. Epidermal nerve fiber density: normative reference range and diagnostic efficiency. Arch Neurol 1998;55:1513–1520 [DOI] [PubMed] [Google Scholar]
- 27.Stocks EA, McArthur JC, Griffen JW, Mouton PR. An unbiased method for estimation of total epidermal nerve fibre length. J Neurocytol 1996;25:637–644 [DOI] [PubMed] [Google Scholar]
- 28.Kimura J. Electrophysiological diagnosis and therapy of peripheral nerve demyelinating diseases. Nihon Naika Gakkai Zasshi 1996;85:389–392 [PubMed] [Google Scholar]
- 29.Pinheiro J, Bornkamp B, Bretz F. Design and analysis of dose-finding studies combining multiple comparisons and modeling procedures. J Biopharm Stat 2006;16:639–656 [DOI] [PubMed] [Google Scholar]
- 30.Rowbotham MC. What is a “clinically meaningful” reduction in pain? Pain 2001;94:131–132 [DOI] [PubMed] [Google Scholar]
- 31.Jordan K, Dunn KM, Lewis M, Croft P. A minimal clinically important difference was derived for the Roland-Morris Disability Questionnaire for low back pain. J Clin Epidemiol 2006;59:45–52 [DOI] [PubMed] [Google Scholar]
- 32.Attal N, Cruccu G, Baron R, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol 2010;17:1113–e88 [DOI] [PubMed] [Google Scholar]
- 33.Zhang W, Nuki G, Moskowitz RW, et al. OARSI recommendations for the management of hip and knee osteoarthritis: part III: changes in evidence following systematic cumulative update of research published through January 2009. Osteoarthritis Cartilage 2010;18:476–499 [DOI] [PubMed] [Google Scholar]