

Introduction
Asymmetric aldehyde crotylation reactions rank among the most important and widely used methods for non-aromatic polyketide natural product synthesis.1,2 Despite several important conceptual and/or practical advances3–10 since the development of Brown’s crotylboranes11 and Roush’s crotylboronates,12 it remains an unmet but eminently worthy goal to devise an asymmetric aldehyde crotylation method that is characterized by 1) wide scope and generality with respect to the aldehyde substrate, 2) access to either product diastereomer with uniformly very high levels of both diastereoselectivity and enantioselectivity, and 3) sustainable, safe, inexpensive, and otherwise practical and scalable procedures such that the crotylation of a low MW aldehyde on a tens-of-grams or larger scale is a simple undertaking that may be carried out in less than a day by a single chemist. There are only two methods that meet most of these conditions, the Brown protocol11 and our recently reported EZ-CrotylMix methodology.13 The former suffers from being quite labor intensive, requiring the metallation of butene under cryogenic conditions, and quite often from difficulties in product isolation, and it is no accident that this has historically been the method of choice in academic settings where labor is relatively inexpensive. By contrast, the commercially available EZ-CrotylMixes are especially attractive in terms of extraordinary ease of use and otherwise meet every single condition listed above save one: their preparation is involved enough that they cannot be offered at a low enough price for use on larger scales (≥ tens of mmols), a problem which is exacerbated by the fact that the crotylsilane reagents are heavy (569 g/mol) due in large part to the bromine atoms which are necessary for crystallinity. Thus, while the EZ-CrotylMixes remain a highly attractive option for smaller scale reactions and in particular for complex aldehydes, the important goal of devising an equally user-friendly and scalable yet also inexpensive crotylation method remains unmet.
The preparation of the EZ-CrotylMixes involves the reaction of the requisite enantiomer of the diamine ligand with either cis- or trans-crotyltrichlorosilane and DBU, followed by concentration, trituration, and filtration of the DBU·HCl salts with pentane, recrystallization of the crotylsilane reagent, and admixture with the Sc(OTf)3 catalyst, all under strictly anhydrous conditions (Scheme 1A). While this procedure facilitated commercialization of the EZ-CrotylMixes, it seems clear that the ideal approach for larger scale applications would involve skipping all of these technically challenging operations and instead simply adding the Sc(OTf)3 catalyst and the aldehyde to the crotylsilane reagent in situ. Unfortunately, under these conditions no catalysis is observed, a failure which may be attributed to deactivation of the Sc(OTf)3 by the large quantities of chloride ion present in the mixture (Scheme 1B). There thus appeared to be no obvious simple technical solution, and it seemed that the only way to solve this problem would be to devise a new crotylsilane reagent that is as active as the EZ-CrotylMix system, but without requiring the use of a catalyst.
Scheme 1.

(A) The procedure for the synthesis and isolation of the EZ-CrotylMixes. (B) Attempts to form and employ the EZ-CrotylMixes in situ are unsuccessful.
Results and Discussion
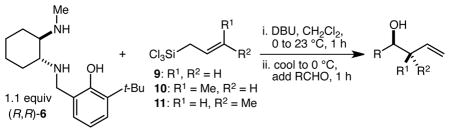
Having few good ideas about how to induce significantly higher levels of reactivity in these crotylsilane reagents without using Lewis acid catalysis, we began instead by focusing on developing derivatives that would display greater hydrolytic stability. The notion was that we might be able to render the isolation of the silane reagent a much simpler and less sensitive process, and in turn that we might then be able to obviate the need for the bromine atoms and arrive at a significantly more air stable and lower MW reagent. Displacement of the chloride of silane 1 with alcohols proved straightforward as outlined in Scheme 2A. Both methoxy and phenoxy silanes 2 and 3 were prepared and their performance in the allylation reactions of several aldehydes was evaluated. Both 2 and 3 were characterized by reactivity that is similar to that of silane 1 (generally poor reactivity in the absence of Sc(OTf)3 e.g.), but with reduced levels of enantioselectivity (Scheme 2A). In addition, silanes 2 and 3 did not appear to be significantly more hydrolytically stable than silane 1, and could not be exposed to air for long periods of time without significant decomposition. The breakthrough came unexpectedly when we decided to explore further the phenoxysilane motif, except with the phenol ring tethered to one of the amino groups. Diaminophenols 5 and 6 were synthesized and used to prepare allylsilanes 7 and 8,14 which for the purposes of initial evaluation were isolated from the DBU salts (Scheme 2B). Noting surprisingly fast reactions with simple aldehydes in the absence of the Sc(OTf)3 catalyst, we interrogated the ability of silanes 7 and 8 to allylate α-methylcinnamaldehyde in the absence of the catalyst. Remarkably, both silanes 7 and 8 reacted smoothly and rapidly with the reactions proceeding to completion in less than 1 h at 0 °C, a level of reactivity that is both dramatically higher than that of silanes 1–3 and at least as high as the combination of silane 1 and 5 mol % Sc(OTf)3. In addition, silane 8 proved highly enantioselective, delivering alcohol 4 in 97% ee. Ligand 6 was selected for further evaluation and much to our delight was found to be quite effective in a one-pot procedure (Scheme 2C). Thus, 6 was treated with allyltrichlorosilane and DBU and after 1 h, α-methylcinnamaldehyde was simply added directly to the mixture. The reaction was complete in less than 1 h at 0 °C, and after isolation alcohol 4 was obtained in 94% yield and 96% ee.
Scheme 2.

(A) Synthesis and allylation reactions of methoxy- and phenoxysilanes 2 and 3. (B) Synthesis and allylation reactions of tethered phenoxysilanes 7 and 8. (C) Optimized one-pot procedure for enantioselective aldehyde allylation reactions with diamine 6 and allyltrichlorosilane.
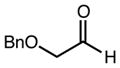
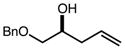
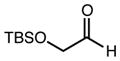
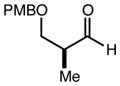
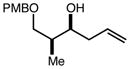
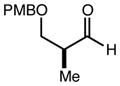
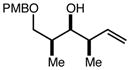
With an effective one-pot procedure established, an exploration of the reaction scope was initiated with an emphasis on the types of aldehydes that are commonly employed in the early stages of polyketide natural product syntheses, including the oft-used aldehydes that are derived from the Roche ester12b (Table 1). Allyltrichlorosilane 9, cis-crotyltrichlorosilane 10,15 and trans-crotyltrichlorosilane 1116 were all employed to great effect, and the reactions were generally highly efficient and diastereoselective and/or enantioselective. The reaction described in entry 12 is especially noteworthy because the alternate procedure using (R,R)-1 and 5 mol % Sc(OTf)3 causes significant decomposition of this sensitive aldehyde.
Table 1.
Highly diastereo- and enantioselective one-pot aldehyde allylation and crotylation reactions with diaminophenol 6
| ||||||
|---|---|---|---|---|---|---|
| entry | silane | RCHO | product | yield (%)a | dr | ee (%) |
| 1 | 9 |
|
|
83 | - | > 99 |
| 2 | 9 |
|
|
81b | - | 98 |
| 3 | 10c |
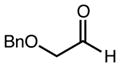
|
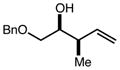
|
94b | 98:2d | 96 |
| 4 | 11e |
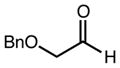
|
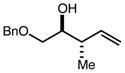
|
88 | 94:6d | 97 |
| 5 | 10c |
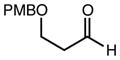
|
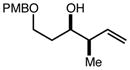
|
92 | > 98:2d | 93 |
| 6 | 9 |
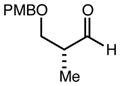
|
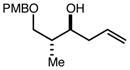
|
82 | 98:2f | - |
| 7 | 10c |
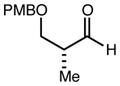
|
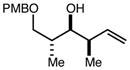
|
82 | 92:8g | - |
| 8 | 11e |
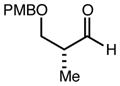
|
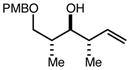
|
80 | 93:7g | - |
| 9 | 9 |
|
|
80 | 98:2f | - |
| 10 | 10c |
|
|
80 | 94:6g | - |
| 11 | 11e |
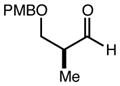
|
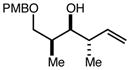
|
80 | 95:5g | - |
| 12 | 9 |
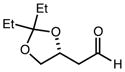
|
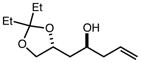
|
87b | 96:4f | - |
Isolated yield of purified products.
A modified workup and isolation procedure was employed.
The geometric purity of the cis-crotyltrichlorosilane employed was measured to be >99:1 by 1H NMR spectroscopic analysis.
Syn:anti diastereoselectivity as measured by 1H NMR spectroscopic analysis.
The geometric purity of the trans-crotyltrichlorosilane employed was measured to be 95:5 by 1H NMR spectroscopic analysis.
Aldehyde diastereoface selectivity as measured by 1H NMR spectroscopic analysis.
Ratio of the major diastereomer to the sum of the minor diastereomers as measured by HPLC and 1H NMR spectroscopic analysis. See the supporting information for details.
We turned next to a demonstration of the practicality and utility of the process with respect particularly to scalability. We selected the crotylation of benzyloxyacetaldehyde with cis-crotyltrichlorosilane 10 as the test reaction, and began with a reaction conducted with 30 mmol (8.71 g) of (R,R)-6 (Scheme 3). The one-pot procedure was employed, and 1 h after the addition of the aldehyde the reaction was quenched by the addition of n-Bu4NF, and after 2 h this was followed by an aqueous workup that effectively separates the diamine 6 from the product. The product 12 was purified by filtration through a small pad of silica, and the diamine 6 was recovered by recrystallization. The recovered 6 was then employed in another run, and then the process was repeated a third time. In total, 14.1 g (68.3 mmol) of 12 was produced starting with 8.71 g (30.0 mmol) of 6, and at the end of this sequence, we still had on hand 6.44 g (22.2 mmol) of 6. Each run may be carried out in ≤ 8 hours, and we noted no other obvious impediments to this chemistry being scaled up further. Because of the experimentally straightforward and efficient recovery of the ligand 6, we believe this methodology has significant potential for commercialization: an initial stock of 20 grams of 6, for example, would be enough to produce ≥ 150 mmol of aldehyde allylation or crotylation products over 3 cycles, with ~15 g of 6 still in hand at the end. Especially for crotylation reactions, we believe this method has the potential to be economically competitive with the relatively inexpensive Brown protocol,11 while being substantially less laborious and less time-consuming to carry out. As for the trichlorosilanes, 9 is commercially available, and we will also be pursuing the commercialization of both 10 and 11. In the meantime, we note that 10, especially, is quite straightforward to prepare on scales as large as 50 g or more, using a modification of the original Tsuji protocol15 (see the supporting information). In terms of significant limitations, we have thus far noted only one: the workup procedure we developed to facilitate the recovery of diamine 6 is not compatible with acid-sensitive protecting groups (see, for example, entries 2 and 12 in Table 1). In these cases an effective (and simpler) alternate workup procedure is used, but as a result the diamine 6 may not easily be recovered.
Scheme 3.
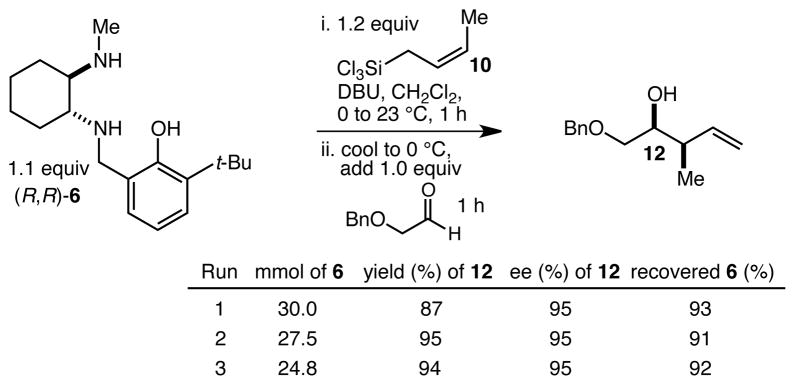
Practical and scalable one-pot enantioselective aldehyde crotylation with recovery and recycling of the activating ligand 6.
The remarkable and dramatic difference in reactivity/Lewis acidity between silanes 3 and 8 is all the more interesting because of their structural similarity. We suggest that it is attributable to stereoelectronic effects that render 8 more acidic than 3, and note that the 29Si NMR chemical shifts of 3 (−25.2 ppm) and 8 (−15.4 ppm) are consistent with this hypothesis. We performed DFT calculations17 on silanes 3 and 8 to gain insight into the nature of these stereoelectronic effects (Fig. 1). In both silanes, the C3-O-Si-N1 torsion angle is close to 90° suggestive of π-type donation from oxygen to the Si-N1 antibonding orbital, which reduces the Lewis acidity of the silicon. Whereas the oxygen in 3 is free to rotate and dynamically interact with both Si-N antibonding orbitals, however, the oxygen in 8 is not, and though the differences are small, the shorter O-Si bond distance and larger C3-O-Si bond angle in 3 are consistent with greater O to Si π-type donation and therefore reduced silane acidity. Transition state effects are almost certainly relevant as well,18 and as the geometry around Si changes along the reaction coordinate for the allylsilylation reaction, the phenoxy group in 3 is more free to rotate than is the phenoxy group in 8. This freer rotation insures that the O to Si π-type donation – and therefore the minimization of the acidity of the silicon center – can be maintained throughout the allylsilylation reaction more fully in 3 than in 8. The substantial planarization of N1 in both 3 and 8 (C1-N1-C2-Si improper torsion angle = 144°) implies that π-type donation from nitrogen to silicon may also be important. In that regard, it is noteworthy that the small but significant planarization observed for N2 in 3 (C4-N2-C5-Si improper torsion angle = 135°) is precluded for N2 in 8 as it occupies a bridgehead position and is forced to remain almost completely pyramidalized (C4-N2-C5-Si improper torsion angle = 123°).
Fig. 1.

Calculated lowest energy structures for (S,S)-3 and (S,S)-8 with selected torsion angles, bond lengths, and bond angles.
Based on this analysis, a model for the reactivity of our family of allylsilane reagents may be advanced. In the chlorosilane series (1, e.g.), donation from nitrogen to the Si-Cl antibonding orbital is the dominant ground state effect that reduces the acidity of the silane, and the only effective way to neutralize this deactivating interaction is the binding of a Lewis acid (i.e. Sc(OTf)3) to the offending nitrogen. In the aryloxysilane series (3, e.g.), donation from nitrogen and oxygen to silicon is similarly responsible for reduced silane acidity, but in addition to the use of Lewis acids, these deactivating interactions may alternatively be counteracted by the imposition of conformational constraints as in silane 8. We note as well that this analysis may facilitate a richer understanding of the origin of activity in our previously reported chiral silane Lewis acid catalyst for Diels-Alder reactions.19
Conclusions
Many non-aromatic polyketide natural products are possessed of truly important and interesting biological activities, while also being both structurally complex and not available in meaningful quantities from natural sources. Having any chance of advancing these compounds or analogs thereof into the clinic will require ever more efficient synthetic methods that are sustainable, safe, inexpensive, technically simple to perform, and readily scalable, all while being possessed of a broad scope. Enantioselective aldehyde crotylation reactions are among the most important methods in this regard, but despite many beautiful conceptual advances over the years, a method that truly satisfies all of these criteria has proved elusive. We believe the method reported here represents a significant advance towards that ultimate aim, and together with the EZ-CrotylMixes, comprises a powerful suite of silane-based reagents for the crotylation of almost any aldehyde, whether simple or complex and whether in the beginning, middle or late stages of a synthesis. Mechanistically, we believe the effect reported here promises to guide the development of a new generation of more powerful silicon based Lewis acidic reagents and catalysts.
Supplementary Material
Acknowledgments
This work was supported by a grant form the National Institute of General Medical Sciences (GM58133). L.M.S. is the recipient of a National Science Foundation Graduate Research Fellowship. M.L.S. is supported by the Center for Re-Defining Photovoltaic Efficiency Through Molecular Scale Control, an Energy Frontier Research Center (EFRC) funded by the Department of Energy (DOE), Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0001085.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures and spectroscopic data; computational procedures and optimized geometries for 3 and 8. See DOI: 10.1039/b000000x/
Notes and references
- 1.(a) Denmark SE, Almstead NG. In: Modern Carbonyl Chemistry. Otera J, editor. Chapter 10 Wiley-VCH; Weinheim: 2000. [Google Scholar]; (b) Chemler SR, Roush WR. In: Modern Carbonyl Chemistry. Otera J, editor. Chapter 11 Wiley-VCH; Weinheim: 2000. [Google Scholar]
- 2.(a) Yamamoto Y, Asao N. Chem Rev. 1993;93:2207–2293. [Google Scholar]; (b) Masse CE, Panek JS. Chem Rev. 1995;95:1293–1316. [Google Scholar]; (c) Denmark SE, Fu J. Chem Rev. 2003;103:2763–2793. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]; (d) Kennedy JWJ, Hall DG. Angew Chem Int Ed. 2003;42:4732–4739. doi: 10.1002/anie.200301632. [DOI] [PubMed] [Google Scholar]; (e) Hall DG. Synlett. 2007:1644–1655. [Google Scholar]
- 3.Garcia J, Kim BM, Masamune S. J Org Chem. 1987;52:4831–4832. [Google Scholar]
- 4.Hafner A, Duthlaer RO, Marti R, Rihs G, Rothe-Streit P, Schwarzenbach F. J Am Chem Soc. 1992;114:2321–2336. [Google Scholar]
- 5.Jain NF, Takenaka N, Panek JS. J Am Chem Soc. 1996;118:12475–12476. [Google Scholar]
- 6.Denmark SE, Fu J. J Am Chem Soc. 2001;123:9488–9489. doi: 10.1021/ja016552e. [DOI] [PubMed] [Google Scholar]
- 7.Lachance H, Lu X, Gravel M, Hall DG. J Am Chem Soc. 2003;125:10160–10161. doi: 10.1021/ja036807j. [DOI] [PubMed] [Google Scholar]
- 8.Hackman BM, Lombardi PJ, Leighton JL. Org Lett. 2004;6:4375–4377. doi: 10.1021/ol0480731. [DOI] [PubMed] [Google Scholar]
- 9.Burgos CH, Canales E, Matos K, Soderquist JA. J Am Chem Soc. 2005;127:8044–8049. doi: 10.1021/ja043612i. [DOI] [PubMed] [Google Scholar]
- 10.(a) Kim IS, Han SB, Krische MJ. J Am Chem Soc. 2009;131:2514–2520. doi: 10.1021/ja808857w. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gao X, Townsend IA, Krische MJ. J Org Chem. 2011;76:2350–2354. doi: 10.1021/jo200068q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Brown HC, Bhat KS. J Am Chem Soc. 1986;108:293–294. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]; (b) Brown HC, Bhat KS. J Am Chem Soc. 1986;108:5919–5923. doi: 10.1021/ja00279a042. [DOI] [PubMed] [Google Scholar]
- 12.(a) Roush WR, Ando K, Powers DB, Palkowitz AD, Halterman RL. J Am Chem Soc. 1990;112:6339–6348. [Google Scholar]; (b) Roush WR, Palkowitz AD, Ando K. J Am Chem Soc. 1990;112:6348–6359. [Google Scholar]; (c) Roush WR, Grover PT. J Org Chem. 1995;60:3806–3813. [Google Scholar]
- 13.Kim H, Ho S, Leighton JL. J Am Chem Soc. 2011;133:6517–6520. doi: 10.1021/ja200712f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silanes 7 and 8 were formed as single diastereomers, and the configuration at silicon was unambiguously established by 1H NMR spectroscopy. See the supporting information for details.
- 15.Tsuji J, Hara M, Ohno K. Tetrahedron. 1974;30:2143–2146. [Google Scholar]
- 16.(a) Furuya N, Sukawa T. J Organomet Chem. 1975;96:C1–C3. [Google Scholar]; (b) Kira M, Hino T, Sakurai H. Tetrahedron Lett. 1989;30:1099–1102. [Google Scholar]; (c) Iseki K, Kuroki Y, Takahashi M, Kishimoto S, Kobayashi Y. Tetrahedron. 1997;53:3513–3526. [Google Scholar]
- 17.Calculations were performed with Jaguar, version 7.8, Schrodinger, LLC, New York, NY, 2010, using the B3LYP functional and the 6–31G**/LACVP basis sets.
- 18.Zhang X, Houk KN, Leighton JL. Angew Chem Int Ed. 2005;44:938–941. doi: 10.1002/anie.200462130. [DOI] [PubMed] [Google Scholar]
- 19.Kubota K, Hamblett CL, Wang X, Leighton JL. Tetrahedron. 2006;62:11397–11401. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.