

Abstract
Reverse thermal gels have numerous biomedical implications, as they undergo physical gelation upon temperature increases and can incorporate biomolecules to promote tissue repair. Such a material is developed for the sustained release of bevacizumab (Avastin), a drug used to treat age-related macular degeneration. The polymer, poly(ethylene glycol)-poly-(serinol hexamethylene urethane) (ESHU), forms a physical gel when heated to 37 °C and shows good cytocompatibility with ocular cells. ESHU is capable of sustaining bevacizumab release over 17 weeks in vitro, and the release kinetics can be altered by changing the drug dose and the ESHU concentration. These results suggest that ESHU is biologically safe, and suitable for ocular drug delivery.
Keywords: age-related wet macular degeneration, compatibility, hydrogels, retinal cells, sustained release
1. Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss in Americans over the age of 50. The wet—or exudative—form is characterized by abnormal blood vessel growth from the choroid, beneath the sensory retina. These vessels are associated with fluid leakage, which over time results in bleeding, retinal detachment, scarring and ultimately, loss of central vision.[1–3]
Intravitreal injection of anti-angiogenic drugs is currently the most widely utilized strategy for preventing choroidal neovascularization, and can decelerate vision loss significantly.[4,5] More specifically, vascular endothelial growth factor (VEGF) is a major target of such treatments as its expression is elevated in patients with AMD, and it is known to promote blood vessel growth, enhance vascular permeability and stimulate inflammatory leukocyte recruitment.[6–9] While effective, the short half-life of VEGF-inhibiting drugs necessitates frequent painful injections, placing an overwhelming burden on patients and their families.[10] Moreover, such frequent injections increase the risk of retinal detachment, endophthalmitis, intraocular hemorrhage, and other ocular complications.[11,12] Therefore, a controlled release delivery system that reduces injection frequency while maintaining the drug’s efficacy would be highly beneficial to those suffering from AMD.
With this goal in mind, we investigated the feasibility of a reverse thermal gel as anintraocular drug-delivery vehicle for AMD. Reverse thermal gels are water-soluble and undergo spontaneous phase transition upon temperature elevation; ideally such a material would be in the solution phase at room temperature and form a physical gel when placed at body temperature.[13–17] When designed in this manner the gel can be loaded with bioactive macromolecules such as proteins, independent of their solubility properties. Since the gel forms rapidly in situ, this system can achieve sustained, localized drug release. Previously we reported synthesis of an ABA type block copolymer, poly(ethylene glycol)-poly(serinol hexamethylene urethane) (ESHU), as a reverse thermal gel in which A is hydrophilic and B is hydrophobic.[18] In this work, we used ESHU to successfully design an anti-angiogenic reverse thermal gel system by combining ESHU with bevacizumab. Bevacizumab was chosen as a model anti-angiogenic agent because it is the most commonly used VEGF inhibitor for treatment of AMD, and is also administered intravitreally for other VEGF-mediated diseases such as central retinal vein occlusion and proliferative diabetic retinopathy.[19–22] We assessed the cytocompatibility of ESHU with bovine and human ocular cells, and studied the release kinetics of the bevacizumab-loaded gel in order to gauge the applicability of ESHU as an intraocular drug-delivery vehicle.
2. Experimental Section
2.1. Materials
N-BOC-serinol, hexamethylene diisocyanate (HDI), tetramethyl benzidine, hyarulonic acid from Steptococcus equi (M̄w: 1.5 million), and bovine serum albumin (BSA) were purchased from Sigma–Aldrich (St. Louis, MO, USA). PEG was obtained from Alfa Aesar (Ward Hill, MA, USA). Anhydrous diethyl ether was obtained from Fisher Scientific (Pittsburgh, PA, USA). Anhydrous chloroform and anhydrous N,N-dimethylformamide (DMF) were purchased from EMD (Gibbstown, NJ, USA). The Spectra/Por dialysis membrane (MWCO: 3500–5000) was bought from Spectrum Laboratories (Rancho Domingues, CA, USA). Recombinant human VEGF 165 was obtained from R&D System (Minneapolis, MN, USA). Horseradish peroxidase-goat anti-human IgG (H + L) was purchased from Invitrogen (Carlsbad, CA, USA). Bevacizumab was purchased from Drugstore (Swedesboro, NJ, USA).
2.2. Equipment
Proton NMR spectra were recorded on a Bruker Avance 600 NMR. The molecular weight was calculated by gel permeation chromatography (GPC) on a Viscotek GPCmax VE2001 system using a Viscotek I-MBMMW-3078 column and a Viscotek 270 dual detector with THF as the eluent. Polyethylene glycol (American Polymer Standard) was used for calibration. Thermal behavior was studied by a thermostatted oscillating rheometer(AR2000, TA Instruments) equipped with 45 mm aluminum parallel plate geometry. 700 μL of the polymer solution was used for the measurements. Data were collected at an angular frequency of 1 rad · s−1 with 0.5% strain. Cell images were obtained on a Nikon TI Fluorescence Microscope. Fluorescence intensity in the images was recorded and analyzed using MetaMorph 7.7.4 software (Molecular Devices Sunnyvale, CA, USA).
2.3. ESHU Synthesis
ESHU was synthesized as described previously with minor modifications.[18] Briefly, the hydrophobic polyurethane block was synthesized by the reaction of N-BOC-serinol (0.5 g, 2.62 mmol) with HDI (0.44 g, 2.62 mmol) at 90 °C under nitrogen atmosphere. After reacting for 3 h, it was dissolved in 10 mL anhydrous DMF with excess HDI (0.88 g, 5.24 mmol) and stirred for 24 h at 85 °C. After cooling to ambient temperature, the mixture was poured into excess anhydrous diethyl ether to precipitate out the polymer. The precipitate was then dissolved in 2 mL anhydrous DMF and the purification process was carried out twice with diethyl ether. Then the precipitates were washed in 100 mL of anhydrous diethyl ether overnight to remove unreacted HDI. A transparent polyurethane middle block was obtained after drying at 45 °C under vacuum (yield: 98%). To conjugate the hydrophilic block, polyurethane (1 g) and PEG (4 g, M̄w: 550) were dissolved in 10 mL anhydrous DMF, and stirred for 24 h at 85 °C. After cooling to ambient temperature, the mixture was poured into excess anhydrous diethyl ether to precipitate out the polymer. The polymer was purified by dialysis in water at room temperature for 3 d. Finally, the dialyzed solution was freeze-dried and a transparent solid, ESHU, was obtained (yield: 95%, M̄w: 7 244, M̄w/M̄n: 1.54). 1H FTNMR (CDCl3, δ): 4.83–5.23 [2H, –NH(CH2)6NH–]; 4.0–4.2 [4H, –CH2(NH–BOC)CH2–]; 3.66 (4H, –OCH2CH2O–); 3.19 [4H, –NHCH2(CH2)4CH2NH–]; 1.41 [9H, –OC(CH3)3]; 1.26 and 1.44 [8H, –NHCH2(CH2)4CH2NH–].
2.4. Cell Culture
To isolate bovine corneal endothelial (CE) cells, fresh whole cow corneas were excised with a scleral rim and then placed endothelium side up in plastic tube caps. The CE cells were enzymatically detached by application of 0.05% trypsin/0.5 mM EDTA with incubation at 37 °C and harvested by gentle scraping. The CE cells were separated from Descemet’s membrane and cell suspensions were broken up by gentle pipetting and sieved through a 70 μm nylon mesh. The suspended CE cells were centrifuged, and pelleted cells to be used for the comet assay were directly re-suspended in 1% low-melting-point (LMP) agarose and used as described below. For culture, CE cells were re-suspended in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), gentamicin, penicillin/streptomycin, and amphotericin B and cultured in 25 cm2 cell culture flasks at 37 °C in 5% CO2 until 90% confluent.
To test human retinal pigment epithelium (RPE), ARPE-19 (American Type Culture Collection) cells were suspended in 1% LMP agarose and centrifuged. The pelleted cells were directly re-suspended in 1% LMP agarose, then DMEM supplemented with 10% fetal bovine serum, gentamicin, penicillin/streptomycin, and amphotericin B and cultured in 25 cm2 cell culture flasks at 37 °C in 5% CO2 until 90% confluent.
To test in vitro cytotoxicity, both primary bovine CE and ARPE-19 cells were incubated with 20 wt% ESHU in glycated human serum albumin (GHSA) medium for 24 and 72 h, respectively, at 37 °C in 5% CO2. Cells cultured in tissue culture polystyrene (TCPS) with no exposure to ESHU gel were used as a positive control.
2.5. Live-dead Cell Counting
After incubation with ESHU, cell viability was assessed using the Live/Dead assay. Cells were washed with DMEM containing 50 μg · mL−1 Calcein AM for 20 min at 37 °C. In the final 5 min of incubation, 5 μg · mL−1 propidium iodide (PI) was added, and then cells were washed with DMEM. Finally, nuclei were counterstained with 1 μg · mL−1 Hoechst 33342 for 5 min. The live and dead cells were analyzed by a fluorescence microscope. Comparative quantification of live cells was assessed using by a cell counting application sensitive to luminescence of Calcein AM, PI, and Hoechst staining in representative photomicrographs.
2.6. Immunoassay of Bevacizumab
The concentration of bevacizumab was measured by an enzyme linked immunosorbent assay (ELISA) as previously described with slight modification.[23] Ninety-six-well plates were coated with 1 μg · mL−1 recombinant human VEGF 165 (100 μL · well−1) and incubated overnight at 4 °C. After washing three times with phosphate-buffered saline (PBS) containing 0.05% Tween-20, wells were blocked with 1% BSA in PBS for one hour at room temperature (200 μL · well−1). Wells were then washed five times with PBS containing 0.05% Tween-20 and stored at 4 °C until use. Samples diluted in PBS containing 0.1% BSA were added to the plates and incubated for 3 h at room temperature. After washing three times with PBS containing 0.05% Tween-20, 1 μg · mL−1 horseradish peroxidase-goat anti-human IgG(H + L) was added (100 μL · well−1) and incubated for 3 h at room temperature. After washing five times with PBS containing 0.05% Tween-20, color development was performed with 100 μL of tetramethyl benzidine, and the reaction was stopped by the addition of 100 μL of 1 M hydrogen chloride. Optical density was measured on a Microplate Reader using Gen5 software (SYNERGY Mx, BioTek) at 450 nm with correction at 570 nm. A standard curve was prepared with bevacizumab concentrations ranging from 0.2 ng · mL−1 to 3.125 μg · mL−1, and the linear region was used for the calculation (Supporting Information, Figure S1). We measured the bevacizumab concentration in each sample thrice.
2.7. Release Profile of Bevacizumab
In vitro bevacizumab release studies were carried out in 1% (w/v) of hyaluronic acid (HA) in PBS (pH 7.4) on a thermal rocker to mimic vitreous fluid composition[24] and eye motion, respectively. Gel solutions (200 μL) of 15 and 20% (w/v) containing 0.5, 1, 3, and 5 mg of bevacizumab were injected into the HA solution through a 27-gauge needle at 37 °C within 5 s. The needle was submerged into the solution completely to mimic clinical injection into the vitreous. The concentrations, 15 and 20%, were chosen in the range that the solution could be easily injected through the 27-gauge needle with a quick sol–gel transition time. At predetermined time points, three samples (20 μL each) were withdrawn from different positions, and the same amounts of fresh HA solution were added. The samples were diluted with PBS containing 0.1% (w/v) BSA, and released bevacizumab was quantified using ELISA. The cumulative release was calculated based on the theoretical loading amount.
3. Results and Discussion
3.1. ESHU Demonstrates Good Cytocompatibility with Ocular Cell Types
In a previous study, we demonstrated that ESHU showed good biocompatibility in vitro with smooth muscle cells as well as in vivo when injected subcutaneously.[18] Here we investigated the suitability of ESHU for intraocular drug delivery. We first tested whether ESHU was compatible with intraocular cell lines. We chose two different lines—bovine CE cells and human ARPE-19 cells—because these cell types are likely to be exposed to ESHU and its degradation products upon intravitreal injection. First we exposed primary bovine CE cells to either control conditions (serum-free DMEM), or ESHU (15% w/v in DMEM). Cells were stained with Calcein AM/PI/Hoechst and viability was evaluated at 1, 12, and 24 h. Representative photomicrographs were taken to quantify cell survival at each time point (Figure 1), and cell morphology was also assessed. Cells exposed to ESHU exhibited typical polygonal morphology that showed no difference from control conditions, suggesting that ESHU did not affect the growth of CE cells (Supporting Information, Figure S2). Cell viability was expressed in terms of percentage cell death (PI nuclei/overall nuclei). There was no significant difference in the survival of CE cells between the control and ESHU groups (p > 0.05, two-way ANOVA) at any time point (Figure 2).
Figure 1.

Representative fluorescence micrograph images of bovine CE cells exposed to control (A, B) and ESHU (C, D) at 24 h. Both groups show comparable intense nuclei staining (A, C) with scarce red stained cells (B, D) indicating little cell death.
Figure 2.

In vitro cytotoxicity of ESHU against bovine CE cells after 1, 12, and 24 h incubation. The number of dead CE cells was determined by counting the number of PI+ cells, and was expressed as a function of the total number of cells labeled by Hoechst staining. Values are expressed as the means ± SD (n = 3).
ESHU showed good cytocompatibility with bovine CE cells, which are likely to be exposed to ESHU degradation products but not necessarily ESHU itself. Therefore, we needed to assess its compatibility with cells from the posterior segment of the human eye, as these cell types will be more directly exposed to the hydrogel. We used human ARPE-19 cells and exposed them to either DMEM (positive control) or ESHU, as described above. We found no evidence of significant cell death as indicated by the small number of PI-dyed dead cells (Figure 3). At all time points, total cell death was less than 1%, and statistically there were no differences between control and ESHU groups (p > 0.05, two-way ANOVA) (Figure 4). Taken together, ESHU shows little to no cytotoxicity when cultured with both bovine and human ARPE-19 cells.
Figure 3.

Representative fluorescence micrograph images of human ARPE-19 cells exposed to control (A, B) and ESHU (C, D) at 72 h. There was no difference between experimental groups in the total number of viable cells (A, C) and dead cells (B, D).
Figure 4.

In vitro cytotoxicity of ESHU against human ARPE-19 cells after 24, 48, and 72 h incubation. The percentage of dead ARPE-19 cells was determined by expressing the number PI+ cells over the total number of cells labeled by Hoechst staining. Values are the means ± SD (n = 3).
3.2. Thermal Behavior of Bevacizumab-loaded ESHU
In order to examine whether bevacizumab-loaded ESHU underwent a phase transition at body temperature, we formulated a 20 wt% ESHU solution containing 1.25 mg of bevacizumab and monitored elastic modulus (G′) changes during heating. We used 1.25 mg because it is the standard dose ophthalmologists inject intravitreally to treat AMD.[25,26] The bevacizumab-loaded ESHU solution underwent phase transition comparable to that of pure ESHU (Figure 5).[18] The elastic modulus, G′, which is indicative of a material’s stiffness and thus its phase, began to increase slightly from 32 to 33 °C, corresponding to the initiation of gelation. It then increased dramatically as temperature continued to rise and reached a maximum stiffness at 39 °C, indicating formation of a complete physical gel. Although G′ steadily increased until 39 °C, it had reached about 90% of its maximum value at 37 °C, at which point the solution had already visibly formed a gel. Given the phase transition behaviors, bevacizumab-loaded ESHU maintained thermal gelling properties as expected.
Figure 5.

Thermal behavior of bevacizumab-loaded ESHU formulated with 20 wt% ESHU and 1.25 mg bevacizumab. The steep increase in G′ from 33 to 39 °C indicated a sol–gel phase transition. The phase transition behavior is similar to pure ESHU.[18]
3.3. In vitro Bevacizumab Release
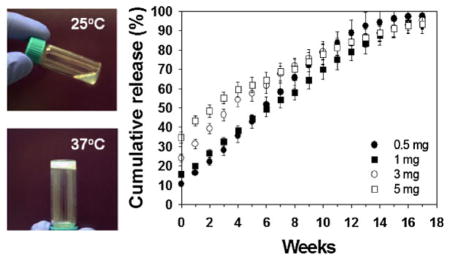
The key challenge of bevacizumab therapy for AMD is to prolong its release because the half-life of free bevacizumab in human eyes after intravitreal injection is on the order of 7–10 d.[27] The intraocular concentration depends on local conditions, such as the presence of blood or fluid under the retina, the state of the vitreous gel and other factors. To evaluate the potential of the ESHU-bevacizumab platform, we performed release tests in 7.5 mL of 1 wt% HA at 37 °C to mimic vitreous fluid, while gyroscopic shaking was conducted to simulate human eye motion. Four formulas, each of 15 and 20 wt% of ESHU containing 0.5, 1, 3, and 5 mg of bevacizumab, gelled immediately upon injection into the 37 °C solution. The resulting sphere quickly sank to the bottom of the vessel, suggesting that the delivery system will sink out of the optical axis of the eye when injected in vivo. In all formulas, release was sustained without reaching plateau during the 17-week observation period (Figure 6). The release was more sustained with 20 wt% ESHU because ESHU formed a more rigid gel at the higher concentration,[18] which presumably affects the diffusion path of bevacizumab. We also observed a small initial burst release for all the formulations. It is well-documented that burst release from injectable systems is higher than from pre-fabricated architectures such as implants;[28] thus we hypothesize that the observed burst release can be attributed to bevacizumab lost during the phase transition. Larger bursts were observed in the 15 wt% system, as well as with higher bevacizumab doses. This observation can be explained by the theory that at higher drug loading more is likely to be lost during the phase transition; additionally the lower ESHU concentration requires a longer gelation time, allowing for more bevacizumab to be released.[18] This burst release might be helpful clinically, however, so as to provide immediate drug exposure in order to maximize efficiency. Moreover, the ability to control this burst by altering the ESHU concentration is attractive in that it can attenuate potential systemic effects bevacizumab absorption, some of which can be serious, while still having an immediate therapeutic effect.[29] Thus, the bevacizumab-ESHU delivery system has three advantages over the current practice of direct bevacizumab injection in that the therapeutic window is much longer, release is sustained over 17 weeks in vitro, and injection frequency can be greatly reduced.
Figure 6.

The release profile of bevacizumab from (A) 15 and (B) 20 wt% ESHU systems. The release was sustained over the 17-weeks period with burst releases at the beginning. The 20 wt% ESHU systems resulted in slower release rates, and smaller burst releases due to its higher concentration.
4. Conclusion
We developed an anti-angiogenic reverse thermal gel system using ESHU and bevacizumab that demonstrates potential as a controlled release drug-delivery system for AMD. ESHU appears to be an excellent material for ocular drug delivery, as it shows good cytocompatibility with bovine CE and human ARPE-19 cells, and bevacizumab release profile extending to 17 weeks. Moreover, ESHU is not limited to the use of bevacizumab; other water-soluble drugs can be easily loaded into the system as well, enabling multifaceted treatment approaches. This preliminary work supports the concept that such a strategy may significantly reduce administration frequency, decrease treatment cost, and improve patient compliance.
Supplementary Material
Acknowledgments
This work was supported by ocular tissue engineering and regenerative ophthalmology (OTERO) Postdoctoral Fellowship provided by the Louis J. Fox Center for Vision Restoration.
Footnotes
Supporting Information for this article is available from the Wiley Online Library or from the author.
Contributor Information
Daewon Park, Department of Bioengineering, University of Colorado Denver Anschutz Medical Campus, Aurora, Colorado 80045, USA.
Veeral Shah, Department of Ophthalmology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15219, USA.
Britta M. Rauck, Department of Bioengineering, Chemical Engineering, Surgery, McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania 15219, USA
Thomas R. Friberg, Department of Ophthalmology, University of Pittsburgh School of Medicine, Pittsburgh, Pennsylvania 15219, USA
Yadong Wang, Email: yaw20@pitt.edu, Department of Bioengineering, Chemical Engineering, Surgery, McGowan Institute for Regenerative Medicine, University of Pittsburgh, Pittsburgh, Pennsylvania 15219, USA.
References
- 1.Couch SM, Bakri SJ. Semin Ophthalmol. 2011;26:114. doi: 10.3109/08820538.2011.577130. [DOI] [PubMed] [Google Scholar]
- 2.El-Mollayess GM, Noureddine BN, Bashshur ZF. Semin Ophthalmol. 2011;26:69. doi: 10.3109/08820538.2010.545100. [DOI] [PubMed] [Google Scholar]
- 3.Prasad PS, Schwartz SD, Hubschman JP. Maturitas. 66:46. doi: 10.1016/j.maturitas.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Anderson OA, Bainbridge JWB, Shima DT. Drug Discovery Today. 2010;15:272. doi: 10.1016/j.drudis.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Do DV. Ophthalmology. 2009;116:S24. doi: 10.1016/j.ophtha.2009.06.049. [DOI] [PubMed] [Google Scholar]
- 6.Adamis AP, Miller JW, Bernal MT, Damico DJ, Folkman J, Yeo TK, Yeo KT. Am J Ophthalmol. 1994;118:445. doi: 10.1016/s0002-9394(14)75794-0. [DOI] [PubMed] [Google Scholar]
- 7.Aiello LP, Avery RL, Arrigg PG, Keyt BA, Jampel HD, Shah ST, Pasquale LR, Thieme H, Iwamoto MA, Park JE, Nguyen HV, Aiello LM, Ferrara N, King GL. New Engl J Med. 1994;331:1480. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 8.Otani A, Takagi H, Oh H, Koyama S, Ogura Y, Matumura M, Honda Y. Microvasc Res. 2002;64:162. doi: 10.1006/mvre.2002.2407. [DOI] [PubMed] [Google Scholar]
- 9.Campa C, Harding SP. Curr Drug Targets. 2011;12:173. doi: 10.2174/138945011794182674. [DOI] [PubMed] [Google Scholar]
- 10.Ciulla TA, Rosenfeld PJ. Curr Opin Ophthalmol. 2009;20:166. doi: 10.1097/ICU.0b013e328329d173. [DOI] [PubMed] [Google Scholar]
- 11.Shah CP, Garg SJ, Vander JF, Brown GC, Kaiser RS, Haller JA. Ophthalmology. 2011;118:2028. doi: 10.1016/j.ophtha.2011.02.034. [DOI] [PubMed] [Google Scholar]
- 12.Day S, Acquah K, Mruthyunjaya P, Grossman DS, Lee PP, Sloan FA. Am J Ophthalmol. 2011;152:266. doi: 10.1016/j.ajo.2011.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Y, Singh J. Int J Pharm. 2009;365:34. doi: 10.1016/j.ijpharm.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 14.Oh HJ, Joo MK, Sohn YS, Jeong B. Macromolecules. 2008;41:8204. [Google Scholar]
- 15.Ogura M, Tokuda H, Imabayashi SI, Watanabe M. Langmuir. 2007;23:9429. doi: 10.1021/la701384q. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen MK, Lee DS. Macromol Biosci. 2010;10:563. doi: 10.1002/mabi.200900402. [DOI] [PubMed] [Google Scholar]
- 17.He CL, Kim SW, Lee DS. J Controlled Release. 2008;127:189. doi: 10.1016/j.jconrel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 18.Park D, Wu W, Wang YD. Biomaterials. 2011;32:777. doi: 10.1016/j.biomaterials.2010.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Julian K, Terrada C, Fardeau C, Cassoux N, Francais C, LeHoang P, Bodaghi B. Acta Ophthalmol. 2011;89:179. doi: 10.1111/j.1755-3768.2010.02046.x. [DOI] [PubMed] [Google Scholar]
- 20.Ehlers JP, Decroos FC, Fekrat S. Retina- J Retinal Vitreous Dis. 2011;31:1856. doi: 10.1097/IAE.0b013e31820d59a5. [DOI] [PubMed] [Google Scholar]
- 21.Hung KH, Lee SM, Lee SY, Lee FL, Yang CS. J Ocular Pharmacol Ther. 2010;26:85. doi: 10.1089/jop.2009.0090. [DOI] [PubMed] [Google Scholar]
- 22.Salam A, Mathew R, Sivaprasad S. Acta Ophthalmol. 2011;89:405. doi: 10.1111/j.1755-3768.2010.02079.x. [DOI] [PubMed] [Google Scholar]
- 23.Krohne TU, Eter N, Holz FG, Meyer CH. Am J Ophthalmol. 2008;146:508. doi: 10.1016/j.ajo.2008.05.036. [DOI] [PubMed] [Google Scholar]
- 24.Swindle-Reilly KE, Shah M, Hamilton PD, Eskin TA, Kaushal S, Ravi N. Invest Ophthalmol Vis Sci. 2009;50:4840. doi: 10.1167/iovs.08-2891. [DOI] [PubMed] [Google Scholar]
- 25.Bakri SJ, Snyder MR, Reid JM, Pulido JS, Singh RJ. Ophthalmology. 2007;114:855. doi: 10.1016/j.ophtha.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Miyake T, Sawada O, Kakinoki M, Sawada T, Kawamura H, Ogasawara K, Ohji M. Invest Ophthalmol Vis Sci. 2009;51:1606. doi: 10.1167/iovs.09-4140. [DOI] [PubMed] [Google Scholar]
- 27.Zhu Q, Ziemssen F, Henke-Fahle S, Tatar O, Szurman P, Aisenbrey S, Schneiderhan-Marra N, Xu X, Grisanti S. Ophthalmology. 2008;115:1750. doi: 10.1016/j.ophtha.2008.04.023. [DOI] [PubMed] [Google Scholar]
- 28.Hacker MC, Haesslein A, Ueda H, Foster WJ, Garcia CA, Ammon DM, Borazjani RN, Kunzler JF, Salamone JC, Mikos AG. J Biomed Mater Res, Part A. 2009;88A:976. doi: 10.1002/jbm.a.31942. [DOI] [PubMed] [Google Scholar]
- 29.Tolentino M. Surv Ophthalmol. 2011;56:95. doi: 10.1016/j.survophthal.2010.08.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.