

Abstract
Occidiofungin is a cyclic nonribosomally synthesized antifungal peptide with submicromolar activity produced by Gram-negative bacterium Burkholderia contaminans. The biosynthetic gene cluster was confirmed to contain two cyclase thioesterases. NMR analysis revealed that the presence of both thioesterases is used to increase the conformational repertoire of the cyclic peptide. The loss of the OcfN cyclic thioesterase by mutagenesis results in a reduction of conformational variants and an appreciable decrease in bioactivity against Candida species. Presumably, the presence of both asparagine and β-hydroxyasparagine variants coordinate the enzymatic function of both of the cyclase thioesterases. OcfN has presumably evolved to be part of the biosynthetic gene cluster due to its ability to produce structural variants that enhance antifungal activity against some fungi. The enhancement of the antifungal activity from the incorporation of an additional cyclase thioesterase into the biosynthetic gene cluster of occidiofungin supports the need to explore new conformational variants of other therapeutic or potentially therapeutic cyclic peptides.
Nonribosomal peptide synthetases (NRPSs) produce a wide array of small and structurally complex peptides that have therapeutic potential. The system enables the incorporation of nonproteinogenic amino acids into the polypeptide. Polyketide synthetases (PKSs) are a family of enzymes or enzyme complexes that produce polyketides. Integration of PKSs into the NRPSs system further increases the variety of polypeptides that can be produced by these systems. Recent studies are aimed at exploiting nonribosomal peptide synthetases (NRPSs) for producing peptide libraries that can be screened for therapeutic applications.1–8
Unlike linear peptides, cyclic peptides are restrained to fewer conformations that facilitate their interaction with their molecular target.9–17 These structural constraints provide resistance to proteases, extreme pH, and temperature.9, 18 These attributes make them one of the most promising scaffolds for pharmacophores. Synthetic approaches of cyclic peptides is hindered by regioselectivity.
Classical total synthesis of peptides by solid phase or solution phase peptide synthesis followed by subsequent cyclization reactions requires the addition and removal of protecting groups at the right stages to drive the cyclization among the correct residues.8 Even with these considerations, proper cyclization is hindered by intermolecular interactions and entropically disfavored pre-cyclization conformations resulting in a vast mixture of compounds or low yields. Microorganisms ensure the formation of a functional cyclic peptide conformation by enzymatically catalyzing the cyclization and release of the peptide with regioselectivity using a cyclase thioesterase.1, 7 The cyclase thioesterase is often located at the C-terminal end of the last NRPS involved in the synthesis of the peptide and is referred to as the TE domain.
The TE domain can hydrolyze the bound peptide as a linear peptide or it can catalyze an intramolecular reaction resulting in the formation of a cyclic peptide. At present, little is known about the cyclization mechanism of peptides. The crystal structure of the surfactin peptide cyclase provided the first basic understanding of its mechanism of action.19, 20 The peptidyl chain bound to 4-phosphopantetheine cofactor (ppan) that is attached to the thiolation (T)-domain is transferred to a serine, which is part of a catalytic triad in the adjacent TE domain. Once the peptide is transferred to the TE domain, the cyclase binding pocket enables proper orientation and cyclization of the peptide substrate. The enzyme was found to share structural homology to the α,β-hydrolase family. The lack of water in the binding cleft of the cyclase, which prevents hydrolysis, is the significant alteration from the hydrolase family that gives the cyclase thioesterase its ability to form cyclic peptides.
Occidiofungin is a broad spectrum nonribosomally synthesized cyclic antifungal peptide that has submicro/nanomolar activity and low toxicity.18, 21–25 ESI-MS and NMR data revealed the existence of four structural variants of the antifungal peptide occidiofungin produced by the soil bacterium Burkholderia contaminans MS14, having a mass [M + 1] of 1,200.39, 1216.41, 1234.17, and 1250.41 Da.21 The mass differences correspond to the addition of oxygen to Asparagine (Asn1) forming β-hydroxyasparagine (BHN1) and/or addition of chlorine to β-hydroxytyrosine (BHY4) forming chlor-BHY4.21 NMR data sets and amino acid analysis revealed that occidiofungin is produced via a hybrid PK-NRPS system and that the antifungal compound is composed of eight amino acids (Figure 1). Using NRPS-PKS web-based software and interProScan software in EMBL-EBI, predicted epimerase domains were identified in the NRPS modules for BHY4, 2,4-diaminobutyric acid (DABA5), and Ser8.21 The peptide is predicted to have L-Asn1(BHN1), L-Ser3, D-BHT4, D-DABA5, L-Asn7, and D-Ser8. An interesting feature in the biosynthetic pathway of occidiofungin is the presence of two putative thioesterases. One is present as an independently expressed thioesterase, OcfN, and the other is a C-terminal TE domain of OcfD. We set out to gain a better understanding of the role of the two putative thioesterases in the biosynthetic gene cluster of occidiofungin.
Figure 1.

Covalent structure of occidiofungin. (R1 = H or OH; R2 = H or Cl)
We have focused our study on the last step in the formation of the cyclic NRP occidiofungin.21 Here we conclusively show that the biosynthesis utilizes two distinct cyclase thioesterases to expand the formation of conformers and that the evolutionary integration of an additional cyclase thioesterase improves its bioactivity against Candida species. Restrictions in the conformational freedom of a peptide may facilitate the interaction of the compound with its molecular target, but there is also an inherent evolutionary constraint using an enzymatic cyclization approach. Our results suggest that when the molecular target is associated with a broad spectrum of microorganisms, the constrained conformers of a cyclic peptide may not always provide the best activity against all organisms. Our study supports the need to investigate new stereoisomers of antimicrobial cyclic peptides in an effort to identify the most effective therapeutic compound. In addition, our study provides further understanding of the function of cyclase thioesterases.
RESULTS AND DISCUSSION
Proportion of Occidiofungin Variants in the Sample
The C-terminal TE domain of OcfD and the OcfN cyclase thioesterase in the occidiofungin biosynthetic gene cluster are both predicted to be involved in the termination of synthesis and formation of the cyclic peptide. Given that the N-terminal of the linear peptide is an Asn or a BHN, we hypothesized that each thioesterase was required for cyclization of the Asn1 and BHN1 variants. The Asn1 and BHN1 variants of occidiofungin are not separable by RP-HPLC, thus, both variants are present in the purified fraction (Figure 2). The final RP-HPLC step in the purification process reveals the presence of three peaks. Occidiofungin samples elute as a double peak before the third peak. Occidiofungin derived from the wild type strain MS14 and the ocfN mutant MS14GG88 have the same chromatographic profile as observed in the last purification step. Occidiofungin peaks were confirmed by MALDI-TOF and bioassays. It is important to note that the presence of the doublet peak is not associated with the presence of Asn1 or BHN1. Each peak of the double peak contains both the Asn1 and BHN1 variants.
Figure 2.

RP-HPLC Chromatograms. A. Chromatogram of the final purification step of the wild-type occidiofungin fraction at 220 nm using a 4.6 × 250 mm C18 column. B. Chromatogram of the final purification step of ocfN mutant occidiofungin fraction at 220 nm using a 4.6 × 250 mm C18 column. C. Overlay of the wild-type (black) and the mutant (grey) fractions of occidiofungin.
The relative proportion of the Asn1 and BHN1 variants could not be directly compared, because direct measurement of the Asn1 peak intensities could not be done due to the peaks overlapping with Asn7. The relative proportion of the Asn1 and BHN1 variants in the wild-type fraction was determined by measuring the 13C NMR HSQC Ha-Ca cross peak intensities of each BHY4 peak in the data set,26, 27 given that each of the BHY4 peaks could be attributed to either the Asn1 or BHN1 variant. Based on the Ha-Ca cross peak intensities for BHY4 in the HSQC spectrum, the Asn1 and BHN1 variants were determined to be approximately 36 and 64% of the total amount of occidiofungin, respectively (Figure 3). The peaks in red and green represent the BHY4 peaks associated with BHN1 and Asn1 variants, respectively. A similar ratio was also observed in the relative abundance of each peak in the ESI-MS spectrum (Figure 4A). Furthermore, the HSQC Ha-Ca cross peak intensities for the BHN1 peaks were determined to be 90.50 and 38.65, which support the intensities measured for BHY4 peaks corresponding to the BHN1conformational variants.
Figure 3.

TOCSY (left panel) and HSQC (right panel) spectra of BHY4 in the wild-type sample. The proportions of Asn1 and BHN1 variants were determined by the measurement of the Ha-Ca cross peak intensities of BHY4 in the HSQC spectra. These values are listed next to their corresponding peaks in the right panel. The peaks in red and green represent the BHY4 peaks associated with BHN1 and Asn1 variants, respectively. Based on the calculation of their relative proportions, i.e. (34.81 + 87.97 for the BHY4 peaks found in the BHN1 conformational variants) and (32.79 + 37.61 for the BHY4 peaks found in the Asn1 conformational variants), the approximate proportion of the Asn1 variants could be calculated as (32.79 + 37.61)/(34.81 + 87.97) + (32.79 + 37.61).
Figure 4.

ESI mass spectrometry. A. ESI-MS data of purified wild-type occidiofungin fraction. B. ESI-MS data of purified ocfN mutant occidiofungin fraction.
Mutagenesis of the ocfN gene was conducted via a marker exchange procedure as described previously,21 to generate the mutant MS14GG88. The percentage of Asn1 to BHN1 variants in the ocfN mutant MS14GG88 fraction could be determined by measuring the proportion of each BHN1 variant using the HSQC data set and by the integration of the HN of Asn1 and BHN1 in the 1H NMR spectra. Asn1 and BHN1 variants are approximately 20 and 80% of the total amount of occidiofungin, respectively. The ESI-MS spectrum also shows a lower relative abundance for the Asn1 variant (1200.39 Da) compared to the BHN1 variant (1216.41 Da) (Figure 4B).
Comparison of Wild-type and ocfN Mutant NMR Spectra
Occidiofungin has a complex spectrum for a peptide of only eight amino acids (Figure 5A, Table 1). The NMR spectrum represents an average of the conformers on the NMR time scale. Conformers in slow exchange on the NMR time scale may result in multiple spin systems for each amino acid. In some situations, multiple conformers are known to arise for cyclic peptides due to slow interconverting conformational families.28, 29 Despite the conformational restrictions brought about by the cyclization, occidiofungin still has a significant amount of conformational freedom. Both Asn1 and BHN1 variants are visibly present in the wild-type fraction, which are colored red in Figure 5A. The TOCSY fingerprint region (NH correlations) is not as complex for the ocfN thioesterase MS14GG88 mutant spectra (Figure 5B). A significant number of spin systems found in the wild-type spectra are absent in the ocfN thioesterase mutant spectra. Our experiments show that the TE domain on the C-terminal region of OcfD is able to perform the peptide macrocyclization of both the Asn1 and BHN1 variants, although there is only one amide spin system for Asn1 produced by OcfD. The loss of OcfN results in the disappearance of the other three Asn1 amide spin systems.
Figure 5.

TOCSY fingerprint region (NH correlations). A. NH correlations in the wild-type sample. The two BHN1 and four Asn1 spin systems present in the wild-type sample are colored red. B. NH correlations in the ocfN mutant sample. C. Overlay of the NH correlations found in the wild-type and ocfN mutant samples. NH correlations that are not present in the ocfN mutant sample are colored green.
Table 1.
Chemical Shift Values for Occidiofungin Derived from the ocfN Mutant MS14GG88a
| Unit | No. | δC | δH (JHNHα in Hz) |
|---|---|---|---|
| 2 | 52.71, CH | 4.59 | |
| Asn1 | 2-NH | 7.75 (12.1) | |
| 3 | 39.91, CH2 | 2.62, 2.41 | |
| 4 | - | ||
| 4-NH2 | 7.39, 6.93 | ||
| 2 | 58.47, CH | 4.66, 4.61 | |
| BHN1 | 2-NH | 7.81, 7.9 (8.1, 12.1) | |
| 3 | 75.01, C | 3.98, 4.02 | |
| 3-OH | 4.66 | ||
| 4 | - | ||
| 4-NH2 | 7.24 | ||
| 2 | 43.88, CH2 | 2.34, 2.36 | |
| NAA2 | 3 | 47.25, CH | 4.23 |
| 3-NH | 7.31, 7.34 (8.4, 11.4) | ||
| 4 | 41.57, CH2 | 1.39, 1.76 | |
| 5 | 66.36, CH | 3.50 | |
| 6 | 76.07, CH | 3.08 | |
| 7 | 79.61, CH | 3.72 | |
| 8 | 33.19, CH2 | 1.54 | |
| 9–17 | 25.14–28.02, CH2 | 1.27 | |
| 18 | 16.94, CH3 | 0.86 | |
| 2 | 58.59, CH | 4.07, 4.15 | |
| Ser3 | 2-NH | 8.11, 8.14 (11.2, 8.0) | |
| 3 | 70.23, 64.29 | 3.49, 3.45 | |
| 3-OH | 4.95 | ||
| 2 | 58.71,CH | 4.06, 4.15 | |
| BHY4 | 2-NH | 7.83, 7.94 (8.7, 11.7) | |
| 3 | 73.75,CH | 4.98, 5.08 | |
| 3-OH | 5.66, 5.73 | ||
| 4 | - | ||
| 5,6 | - | 7.15 | |
| 8,9 | - | 6.67 | |
| 2 | 53.49, CH | 4.43 | |
| DABA5 | 2-NH2 | 7.66 (8.5) | |
| 3 | 32.68, CH2 | 1.88, 2.11 | |
| 4 | 39.17, CH2 | 2.92 | |
| 4, NH | 7.71 | ||
| 2 | 44.76, CH2 | 3.87, 3.58, 3.84, 3.70 | |
| Gly6 | 2-NH | 7.68, 7.85 | |
| 2 | 53.25, CH | 4.51, 4.58 | |
| Asn7 | 2-NH | 8.35, 8.41 (9.3, 8.7) | |
| 3 | 40.03, CH2 | 2.61, 2.38 | |
| 4 | - | ||
| 4-NH2 | 7.39, 6.93 | ||
| 2 | 58.11, CH | 4.33, 4.32 | |
| Ser8 | 2-NH | 7.76, 7.78 (8.2, 12.00) | |
| 3 | 64.59 | 3.61, 3.62 | |
| 3-OH | 4.79 | ||
Proton chemical shift values are from a TOCSY and NOESY experiments. 13C values are from the HSQC experiment.
An overlay of the wild-type and ocfN mutant NMR spectra shows the amino acid spin systems in green that are absent in the mutant spectra (Figure 5C). These spin systems are for Asn7, Ser8, Asn1, Novel Amino Acid 2 (NAA2), Ser3, BHY4, and Gly6. The loss of these spin systems suggests that the complex spin system observed for the wild-type occidiofungin fraction is not only due to interconverting conformational families, but is the result of distinct diastereomers formed by the regiospecific activity of the OcfN cyclase and OcfD TE domain. Dramatic chemical shifts observed, such as the 2 ppm shift for HN of the NAA2, supports the formation of a structurally unique conformer of occidiofungin. A unique conformer is further supported by the subsequent loss of an NAA2 spin system in the NMR spectra of the ocfN mutant. Furthermore, the presence of both Asn1 and BHN1 spin systems in the mutant spectra along with the absence of the amide spin systems shown in green indicate that the additional spin systems are not due to the presence of the β-hydroxy group on Asn1. The additional spin systems are due to the formation of a unique diastereomer produced by OcfN cyclase thioesterase. To further test for the formation of a configurational isomer versus an interchangeable conformer, 1D NMR temperature titrations were performed. Amide and aromatic regions revealed little change in the complexity of peaks present with the occidiofungin derived from ocfN mutant MS14GG88 or wild-type strain MS14 (Figure S5). Given that NAA2 spin systems are a good indicator for the presence of both diastereomers in the wild-type spectrum, we collected TOCSY spectra for occidiofungin derived from ocfN mutant MS14GG88 or wild-type strain at 50°C (Figure S6). There was no loss or addition of a spin system for NAA2 in the mutant spectrum. Furthermore, both spin systems for NAA2 remained in the wild-type spectrum. This data supports that the stereoisomers are non-interchangeable isomers, supporting their classification as diastereomers rather than conformers.
Model for the Coordinated Function of Two Cyclase Thioesterases
There was no loss of an amide spin system for a BHN1 in the ocfN mutant NMR spectra. This suggests that OcfN thioesterase has a substrate requirement for the peptide containing Asn1, since there is no concomitant loss of a BHN1 spin system with the observed loss of the Asn1 spin systems. The C-terminal TE domain of OcfD has a preference for the peptide containing the BHN1, but is capable, albeit at a lower efficiency of cyclizing the Asn1 variant. This provides an interesting scenario for the activity of the two thioesterases (Figure 6). Both thioesterases contain the GXSXG motif, which is important for the catalytic transfer of the peptide from the T domain to the cyclase.30 This suggests that substrate recognition occurs prior to the catalytic transfer of the peptide to the cyclase. Presumably, OcfN cyclase has a higher affinity or better access for the Asn1 peptide product given that the proportion of the Asn1 cyclic peptide produced by OcfD compared to the BHN1 product is reduced in the wild-type fraction. Therefore the biosynthesis of occidiofungin utilizes the structural differences between Asn and BHN to increase the conformational biodiversity of occidiofungin. The increase in conformational diversity is accomplished by the regiospecific activity of each cyclase, presumably by differences in their binding clefts that helps orientate the peptide before cyclization.
Figure 6.

Schematic of occidiofungin ring closure. The completely synthesized eight amino acid linear peptide is bound by a 4-phosphopantetheine cofactor (ppan) linker to the thiolation (T) domain. The peptide varies by the presence or absence of a hydroxy group on the β-carbon of Asn1. The TE domain of OcfD is capable of forming the cyclic peptide of both variants in the absence of a functional OcfN cyclase thioesterase. However, it is not as efficient at producing the Asn1 cyclic peptide variant as OcfN. In the presence of a functional OcfN cyclase thioesterase, novel diastereomers of occidiofungin are formed by the selective ring closure of the Asn1 cyclic peptide. R1 and R2 are BHN1 and Asn1, respectively.
Comparison of the Bioactivity of the Wild-type and ocfN Mutant Product
To determine whether the increase in conformational diversity is important for bioactivity, minimum inhibitory concentrations were determined against medically relevant Candida species (Figure 7A). There was a 2-fold decrease in the MIC with the purified ocfN mutant product with respect to the wild-type product against Candida albicans LL, C. albicans TE, C. glabrata ATCC66032, C. parapsilosis ATCC90018, and C. tropicalis ATCC66029. There was no difference in the MIC for C. albicans ATCC66027. Colony forming units (CFUs/mL) were determined for the MIC wells of wild-type product for each Candida species and compared to the corresponding well containing the same concentration of the ocfN mutant product (Figure 7B). Following exposure to the same concentration of wild-type and ocfN mutant products, these results show a 5 to 7-log decrease in cell density of the Candida species treated with wild-type product. The differences in activity are also visualized by the rate of cell death. Time-kill experiments were performed against C. glabrata ATCC66032. There was a 10-fold difference in yeast present at 4 and 8 hours when cells were treated with 0.5 Og/mL of occidiofungin derived from ocfN mutant MS14GG88 or wild-type strain (Figure S7). Furthermore, a slower rate of cell death was also observed for yeast treated with occidiofungin derived from ocfN mutant MS14GG88 at 1.0 and 2.0 Og/mL. Given that the cyclic occidiofungin variants produced by OcfN constitute less than half of the total structural variants, a 2-fold loss in activity suggests that the diastereomers synthesized by OcfN are 4-fold more active than the isomer produced by OcfD against five of the Candida species tested. Another possible explanation for the observed differences in activity could be attributed to possible synergism between the diastereomers produced by each cyclase thioesterase. Furthermore, the antifungal activity of the ocfN mutant (MS14GG88: 8.79 ± 0.38 mm) was also significantly reduced (P < 0.05) compared to wild-type activity (inhibitory zone radius ± SEM: 13.00 ± 0.58 mm) in an overlay assay against Geotrichum candidum (Figure 8).
Figure 7.

Comparison of the bioactivity from the wild-type and ocfN mutant occidiofungin fractions. A. MICs of wild-type and ocfN mutant fraction determined by CLSI M27-A3 method in RPMI 1640. B. Comparison of the CFUs in the MIC wells of wild-type fraction to the corresponding well having the same concentration of the ocfN mutant occidiofungin fraction. Asterisks represent no detectable colonies in the MIC wells of the wild-type occidiofungin fraction. Black and grey bars are ocfN mutant and wild-type fractions, respectively. Standard deviations for the CFU measurements are presented.
Figure 8.
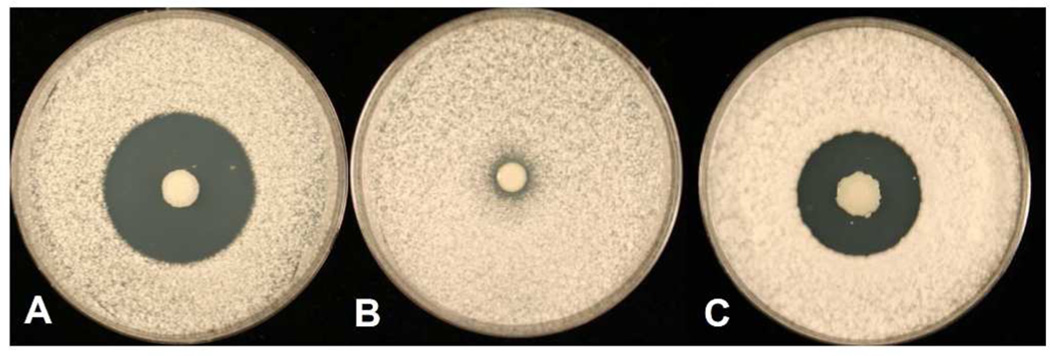
Potato dextrose agar plates were inoculated with each of the strains and incubated for 3 days at 28°C. The plates were oversprayed with the indicator fungus Geotrichum candidum and incubated overnight. A: The wild-type strain MS14; B: Negative control MS14GG78 (ocfJ::nptII); C: MS14GG88 (ocfN::nptII).
General Discussion
The findings from this study include experiments showing the following: the relative proportion of the Asn1 and BHN1 variants in the purified fraction; distinct differences in spin systems for the wild-type and ocfN mutant products; a proposed model for the coordinated function of two cyclase thioesterases; and demonstrated differences in biological activity of wild-type and ocfN mutant products against therapeutically relevant Candida species. Expanding the conformational repertoire of cyclic peptide natural products can be beneficial to microorganisms. These data suggest that the bacterium Burkholderia contaminans MS14 is benefited by maintaining two distinct cyclase thioesterases that improves the spectrum of activity of occidiofungin.
Our data support the observation that cyclase thioesterase substrate recognition occurs prior to the catalytic transfer of the peptide. The presence or absence of a hydroxy group on the β-carbon of the N-terminal amino acid (Asn1) appears to be important for the substrate recognition by the two cyclase thioesterases. It has also been shown that the N-terminal amino acid is important for substrate recognition for other thioesterases.4, 8 It is possible that the presence of the hydroxy group promotes a hydrogen bond with the ocfD cyclase thioesterase domain or more likely promotes an interaction within the T domain of the NRPS. Different bound orientations of the peptide to the T domain would establish a basis for the coordinated function of two cyclase thioesterases.
The presence of the hydroxy group on the β-carbon and the bound orientation of the peptide to the T domain may prevent the interaction of the OcfN cyclase, while enabling the continued substrate recognition by the OcfD TE domain. Conformational diversity of the T domain has been shown to be important for the directed movement of the peptide substrate bound to the ppan cofactor and its interaction with externally acting enzymes.3 More specifically, the active site serine of the cyclase thioesterase needs to attack the linear peptide attached by a thioester linkage to the ppan forming an acyl-O-TE intermediate. The position of the peptide bound to the ppan in the T domain will be important for bringing the peptide substrate in proximity of the appropriate cyclase thioesterase.
Furthermore, some cyclase thioesterases are capable of transacylation of the peptide to the active site serine, when the peptide is bound to a biomimetic prosthetic group.4, 15 However, there are several cyclase thioesterases that will not function when the product is bound to a biomimetic group. These data suggest that the interaction of the peptide with the T domain is important for the enzymatic activity of some thioesterases and this interaction cannot be mimicked using a prosthetic group. It is conceivable that the coordinated function of the two cyclase thioesterases, involved in the synthesis of occidiofungin, utilize differences in the interaction of the ppan bound peptide within the T domain.
Presumably, ocfN was integrated into the occidiofungin biosynthetic gene cluster to improve its spectrum of activity against fungi. Given the broad spectrum of antifungal activity associated with occidiofungin, the molecular target is likely to be highly conserved. However, there must be some variation among fungal species to account for the differences in biological activity. Increasing the conformational repertoire must be a selective advantage to the bacterium for it to maintain the two functional cyclase thioesterases. The microbial environment is considerably different than how we intend to apply the natural products produced by microorganisms. For instance, the bacterium Streptomyces roseosporus is a soil saprotroph responsible for the production of daptomycin.31, 32 The microbial community that this bacterium encounters is far more diverse than the group of bacteria that cause human infection. Thus, evolutionary pressures that selected for the current conformers of daptomycin may not necessarily be the best conformers for treating a Staphylococcus aureus infection. It is very likely that the therapeutic application of daptomycin or other cyclic peptide drugs could be improved by engineering novel conformational or configurational isomers.
Creating novel diastereomers of other cyclic peptide drugs using new or engineered cyclase thioesterases may lead to improvements in their therapeutic activity against clinically relevant pathogens. This is true for occidiofungin produced by the bacterium Burkholderia contaminans MS14, which accomplishes this goal by the evolutionary integration of an additional cyclase thioesterase into the occidiofungin biosynthetic gene cluster.
EXPERIMENTAL SECTION
Materials
Occidiofungin produced by both the wild type strain MS14 and the ocfN mutant MS14GG88 were purified as previously described for the wild-type sample.22 Chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and were the highest grade, unless otherwise stated. Media were purchased from Fisher Scientific, enzymes were purchased from New England BioLabs (Ipswich, MA), and primers were purchased from Integrated DNA Technologies (Coralville, IA) unless otherwise stated. Candida strains used were purchased from the ATCC biological resource center and were a gift from Thomas Edlind (Drexel University College of Medicine).
Site Directed Mutagenesis
A nonpolar mutation was constructed in the open reading frame of wild-type ocfN by the insertion of a kanamycin resistance gene, nptII.33 To mutate ocfN, a 1-kb fragment containing ocfN was obtained by PCR using primers MocfNF (5'-CGCCACCCGTTACGAGGATTC) and MocfNR (5'-ACGCGTCCCCTCTTCCTACG). The 1-kb PCR product was cloned into the pGEM-T Easy Vector System I (Promega Corporation, Madison, WI) resulting in plasmid pGG30. The nptII gene was inserted into the cloned ocfN at SmaI, generating plasmid pGG31. The ~2-kb EcoRI fragment of pGG31 harboring the ocfN gene disrupted by insertion of nptII was cloned into pBR32534 at the EcoRI site to generate pGG32. Mutagenesis of the ocfN gene was conducted via a marker exchange procedure as described previously,35 to generate the mutant MS14GG88. PCR analysis and sequencing were used to verify the double crossover mutants. Production and purification of the antifungal were done as previously described.22
NMR Spectroscopy
A 2 mM sample of ocfN thioesterase mutant fraction of occidiofungin was prepared in DMSO-d6 (Cambridge Isotopes) and data were collected as previously described for the wild-type fraction.21 The NMR data were collected on a Bruker Advance DRX spectrometer, equipped with a CryoProbe, operating at 600 MHz. The 1H NMR resonances were assigned according to standard methods36 using COSY, TOCSY, NOESY and HSQC experiments. NMR data were collected at 25°C. The carrier frequency was centered on the residual water resonance (3.33 ppm), which was suppressed minimally using standard presaturation methods. A 2.0 s relaxation delay was used between scans. The TOCSY experiment was acquired with a 60 ms mixing time using the Bruker DIPSI-2 spinlock sequence. The NOESY experiment was acquired with 400 ms mixing time. The parameters for collecting the HSQC spectrum were optimized to observe aliphatic and aromatic CH groups. The sweep width for the TOCSY and NOESY experiments was 11.35 ppm in both dimensions. The sweep widths for the HSQC experiments were 11.35 ppm in the proton dimensions and 0 and 85 ppm for the carbon dimension. 2D data were collected with 2048 complex points in the acquisition dimension and 256 complex points for the indirect dimensions, except for the HSQC which was collected with 2048 and 128 complex points in the direct and indirect dimension, respectively. Phase sensitive indirect detection for NOESY, TOCSY, and COSY experiments was achieved using the standard Bruker pulse sequences. 1H NMR chemical shifts were referenced to the residual water peak (3.33 ppm). Data were processed with nmrPipe37 by first removing the residual water signal by deconvolution, multiplying the data in both dimensions by a squared sinebell function with 45 or 60 degree shifts (for the 1H NMR dimension of HSQC), zerofilling once, Fourier transformation, and baseline correction. Data were analyzed with the interactive computer program NMRView.38 1D NMR temperature titrations were collected on the wild type and mutant peptides, using a Bruker AVANCE III HD 600 MHz spectrometer equipped with a cryoprobe. Eight scans were collected in each 1D experiment, using 32K points, at 298 K. The experiments were repeated using higher temperatures for both samples in 5 degrees K increments, up to a temperature of 323 K. 2D TOCSY spectra were collected at 323 K, using a mixing time of 60 milliseconds. Eight scans and 256 indirect points were used for both the wild type and mutant peptides. The 2D spectra were processed using NMRPipe, with 45 degree sinebell squared shifts in both dimensions.
Mass Spectrometry
The wild-type occidiofungin and the ocfN mutant sample (10 Og) were evaporated to dryness in a Speed Vac Concentrator (ThermoScientific, San Jose, CA) and the residue was taken up in 50 OL MeOH and analyzed by direct infusion at 3 OL/minutes into an LCQ DecaXP (ThermoScientific, San Jose, CA). Data were acquired over a mass range of m/z 200 to 2000.
In Vitro Susceptibility Testing
Microdilution broth susceptibility testing was performed in triplicate according to the CLSI M27-A3 method in RPMI 1640 [buffered to a pH of 7.0 with MOPS (morpholinepropanesulfonic acid)] growth medium. 100X stock solutions of occidiofungin were prepared in DMSO. MIC endpoints for occidiofungin were determined by visual inspection and were based on the wells that had no visible growth (an optically clear well) after 24 hours of incubation. DMSO containing no antifungal agent was used as a negative control. Colony forming units (CFUs) were determined in triplicate by plating 100 OL from the MIC wells onto a Yeast Peptone Dextrose (YPD) plate as well as plating 100 OL from 10-fold serial dilutions of the cell suspension in Yeast Peptone Dextrose (YPD) Broth. Colony counts were performed and reported as CFUs/mL. Time-kill experiments were performed as previously reported.18 C. glabrata (ATCC 66032) colonies on 24-h-old YPD plates were suspended in 9 mL of sterile H2O. The density was adjusted to a 0.5 McFarland standard and was diluted 10-fold with RPMI 1640 medium to a final volume of 10 mL containing a final concentration of 2, 1, 0.5 and 0 µg/mL of occidiofungin from wild type strain MS14 and the ocfN mutant MS14GG88. The cultures were incubated at 35°C with agitation. Samples were drawn, serially diluted, and plated on YPD medium for colony counts.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to think Ron Shin and Rama Krishna at the UAB High-Field NMR Facility for NMR data collection. This research was supported by funds from Texas A&M University and the Mississippi Agricultural and Forestry Experiment Station.
ABBREVIATIONS
- NRPS
Non-ribosomal Peptide synthetase
- PKS
Polyketide synthetase
- TE domain
Thioesterase domain
- ppan
4-phosphopantetheine cofactor
- ESI-MS
Electrospray ionization mass spectrometry
- NMR
Nuclear magnetic resonance
- DMSO
Dimethyl sulfoxide
- COSY
Correlation spectroscopy
- TOCSY
Total correlation spectroscopy
- NOESY
Nuclear Overhauser Effect Spectroscopy
- HSQC
Heteronuclear Single Quantum Coherence
- RPMI
Roswell Park Memorial Institute
- MOPS
Morpholinepropanesulfonic acid
- CFU
Colony forming units
- MIC
Minimum inhibitory concentration
- YPD
Yeast Peptone Dextrose
- RP-HPLC
Reverse phase High performance liquid phase chromatography
Footnotes
ASSOCIATED CONTENT
Supporting Information
NMR data for occidiofungin from ocfN mutant MS14GG88. Kill kinetics data from the from wild-type and ocfN mutant MS14GG88. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Fischbach MA, Walsh CT. Chem. Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- 2.Koglin A, Löhr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Güntert P, Wagner G, Walsh CT, Marahiel MA, Dotsch V. Nature. 2008;454:907–911. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koglin A, Mofid MR, Löhr F, Schäfer B, Rogov VV, Blum M-M, Mittag T, Marahiel MA, Bernhard F, Dötsch V. Science. 2006;312:273–276. doi: 10.1126/science.1122928. [DOI] [PubMed] [Google Scholar]
- 4.Kohli RM, Takagi J, Walsh CT. PNAS. 2002;99:1247–1252. doi: 10.1073/pnas.251668398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lautru S, Challis GL. Microbiology. 2004;150:1629–1636. doi: 10.1099/mic.0.26837-0. [DOI] [PubMed] [Google Scholar]
- 6.Samel SA, Wagner B, Marahiel MA, Essen L-O. J. Mol. Biol. 2006;359:876–889. doi: 10.1016/j.jmb.2006.03.062. [DOI] [PubMed] [Google Scholar]
- 7.Walsh CT. Science. 2004;303:1805–1810. doi: 10.1126/science.1094318. [DOI] [PubMed] [Google Scholar]
- 8.White CJ, Yudin AK. Nat. Chem. 2011;3:509–524. doi: 10.1038/nchem.1062. [DOI] [PubMed] [Google Scholar]
- 9.Boddy CN. Chem. Biol. 2004;11:1599–1600. doi: 10.1016/j.chembiol.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 10.Boguslavsky V, Hruby VJ, O'Brien DF, Misicka A, Lipkowski AW. J. Pept. Res. 2003;61:287–297. doi: 10.1034/j.1399-3011.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Lopez S, Kim HS, Choi EC, Delgado M, Granja JR, Khasanov A, Kraehenbuehl K, Long G, Weinberger DA, Wilcoxen KM, Ghadiri MR. Nature. 2001;412:452–455. doi: 10.1038/35086601. [DOI] [PubMed] [Google Scholar]
- 12.Fridkin G, Gilon C. J. Pept. Res. 2002;60:104–111. doi: 10.1034/j.1399-3011.2002.02993_1.x. [DOI] [PubMed] [Google Scholar]
- 13.Jelokhani-Niaraki M, Hodges RS, Meissner JE, Hassenstein UE, Wheaton L. Biophys. J. 2008;95:3306–3321. doi: 10.1529/biophysj.108.137471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jelokhani-Niaraki M, Prenner EJ, Kondejewski LH, Kay CM, McElhaney RN, Hodges RS. J. Pept. Res. 2001;58:293–306. doi: 10.1034/j.1399-3011.2001.00893.x. [DOI] [PubMed] [Google Scholar]
- 15.Kohli RM, Walsh CT, Burkart MD. Nature. 2002;418:658–661. doi: 10.1038/nature00907. [DOI] [PubMed] [Google Scholar]
- 16.Rayan A, Senderowitz H, Goldblum A. J. Mol. Graph. Model. 2004;22:319–333. doi: 10.1016/j.jmgm.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 17.Schwarzer D, Mootz HD, Marahiel MA. Chem. Biol. 2001;8:997–1010. doi: 10.1016/s1074-5521(01)00068-0. [DOI] [PubMed] [Google Scholar]
- 18.Ellis D, Gosai J, Emrick C, Heintz R, Romans L, Gordon D, Lu S-E, Austin F, Smith L. Antimicrob. Agents Ch. 2012;56:765–769. doi: 10.1128/AAC.05231-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sieber SA, Marahiel MA. J. Bacteriol. 2003;185:7036–7043. doi: 10.1128/JB.185.24.7036-7043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tseng CC, Bruner SD, Kohli RM, Marahiel MA, Walsh CT, Sieber SA. Biochemistry. 2002;41:13350–13359. doi: 10.1021/bi026592a. [DOI] [PubMed] [Google Scholar]
- 21.Gu G, Smith L, Liu A, Lu S-E. Appl. Environ. Microbiol. 2011;77:6189–6198. doi: 10.1128/AEM.00377-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gu G, Smith L, Wang N, Wang H, Lu S-E. Biochem. Biophys. Res. Commun. 2009;380:328–332. doi: 10.1016/j.bbrc.2009.01.073. [DOI] [PubMed] [Google Scholar]
- 23.Gu G, Wang N, Chaney N, Smith L, Lu S-E. FEMS Microbiol. Lett. 2009;297:54–60. doi: 10.1111/j.1574-6968.2009.01653.x. [DOI] [PubMed] [Google Scholar]
- 24.Lu S-E, Novak J, Austin FW, Gu G, Ellis D, Kirk M, Wilson-Stanford S, Tonelli M, Smith L. Biochemistry. 2009;48:8312–8321. doi: 10.1021/bi900814c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tan W, Cooley J, Austin F, Lu S-E, Smith L, Pruett S. Int. J. Toxicol. 2012;31:326–336. doi: 10.1177/1091581812445185. [DOI] [PubMed] [Google Scholar]
- 26.Heikkinen S, Toikka MM, Karhunen PT, Kilpelainen IA. JACS. 2003;125:4362–4367. doi: 10.1021/ja029035k. [DOI] [PubMed] [Google Scholar]
- 27.Rai RK, Tripathi P, Sinha N. Anal. Chem. 2009;81:10232–10238. doi: 10.1021/ac902405z. [DOI] [PubMed] [Google Scholar]
- 28.Baysal C, Meirovitch H. Biopolymers. 1999;50:329–344. doi: 10.1002/(SICI)1097-0282(199909)50:3<329::AID-BIP8>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 29.Bonmatin J-M, Laprévote O, Peypoux F. Comb. Chem. High Throughput Screen. 2003;6:541–556. doi: 10.2174/138620703106298716. [DOI] [PubMed] [Google Scholar]
- 30.Eys S, Schwartz D, Wohlleben W, Schinko E. Antimicrob. Agents Chemother. 2008;52:1686–1696. doi: 10.1128/AAC.01053-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liao G, Shi T, Xie J. J. Cell. Biochem. 2012;113:735–741. doi: 10.1002/jcb.23414. [DOI] [PubMed] [Google Scholar]
- 32.Vilhena C, Bettencourt A. Mini Rev. Med. Chem. 2012;12:202–209. doi: 10.2174/1389557511209030202. [DOI] [PubMed] [Google Scholar]
- 33.Alexeyev MF. Biotechniques. 1995;18:52. [PubMed] [Google Scholar]
- 34.Prentki P, Karch F, Iida S, Meyer J. Gene. 1981;14:289–299. doi: 10.1016/0378-1119(81)90161-x. [DOI] [PubMed] [Google Scholar]
- 35.Lu S-E, Scholz-Schroeder BK, Gross DC. MPMI. 2002;15:43–53. doi: 10.1094/MPMI.2002.15.1.43. [DOI] [PubMed] [Google Scholar]
- 36.Wüthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 37.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 38.Johnson BA, Blevins RA. J. Biomol. NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.