

Abstract
Fibrils derived from Pmel17 are functional amyloids upon which melanin is deposited. Fibrils of the repeat domain (RPT) from Pmel17 form under strict melanosomal pH (4.5–5.5) and completely dissolve at pH ≥ 6. To determine which Glu residue is responsible for this reversibility, aggregation of single, double, and quadruple Ala- and Gln-mutants were examined by intrinsic Trp fluorescence, circular dichroism spectroscopy, and transmission electron microscopy. Charge neutralization of either E404, E422, E425, or E430, glutamic acids located in the putative amyloid-forming region, modulated aggregation kinetics. Remarkably, the removal of a single negative charge at E422 out of a total of 16 carboxylic acids shifted the pH dependence by a full pH unit. Mutation at E404, E425, or E430 has little to no effect. We suggest that protonation at E422 is essential for initiating amyloid formation while other Glu residues play an allosteric role in fibril stability.
Keywords: Pmel17, repeat domain, melanin, TEM, tryptophan
In the last decade, the emerging concept of “functional” amyloids is challenging the way we view amyloids, which have been previously thought as either a cause or consequence of human diseases as in Alzheimer’s and Parkinson’s.[1] In our work, we have studied a crucial fibril forming domain termed the repeat domain (RPT, residues 315–444) derived from the human functional amyloid, Pmel17, to gain insights into what may differentiate functional from pathological amyloid.[2] Pmel17 is a transmembrane precursor protein that is proteolytically processed to form intralumenal fibrils in melanosomes upon which melanin is deposited.[3] Pmel17 is highly regulated in vivo, undergoing a series of post-translational and proteolytic modifications whereby the timing and sequence of these events permit amyloid formation. RPT is essential for the amyloid structures observed in melanosomes.[4] Fibrils are formed during the early stages of melanosome development and once formed are responsible for the deposition of the pigment melanin. Since melanin precursors are cytotoxic, sequestering their synthesis on fibrils prevents potential detriment to the organelle.[5]
A distinguishing feature that we have discovered is that not only does RPT form amyloid at a mildly acidic, melanosomal pH regime (5±0.5)[2b] but these fibrils completely dissolve at pH ≥ 6.[2d] This reversible polymerization behavior highly contrasts those exhibited by disease-related amyloids, which only upon the harshest treatments will disassemble, e.g. chemical denaturants and non-physiological pH. A potential biological implication for this observed disaggregation process is that if RPT filaments were to escape from the melanosome, they would dissolve under neutral cytosolic pH, and thus remain benign. While this is a compelling hypothesis, there is no current data supporting fibril dissolution in vivo and other domains may be involved.[6] Nevertheless, our results support the requirement of the acidic melanosome pH for amyloid assembly where protonation of specific carboxylic acids promotes key interactions for RPT fibril formation by reducing either intra- or inter-molecular electrostatic repulsion. Here, we sought to pinpoint specific carboxylic acids (protonation sites) that are necessary for aggregation and to assess the role of hydrogen bonding in fibril formation by utilizing Ala- and Gln-mutants, respectively.
Although RPT is rich in carboxylic acid residues (15 Glu and 1 Asp, Figure 1A), several lines of evidence suggest that the amyloid core is located in the C-terminal region. Limited proteinase K (PK) digestion of RPT fibrils showed that peptides corresponding to residues 390–444, 393–444, and 400–444 are PK-resistant (Table S1). This region contains the longest proline-free stretch (403–431) and a predicted amyloidogenic sequence (403–413).[7] Moreover, solid-state nuclear magnetic resonance (ssNMR) data indicate the presence of protonated Glu sidechains in its fibrillar state.[8] Thus, we focused on the effects of mutations at residues, E404, E422, E425 and E430.
Figure 1.
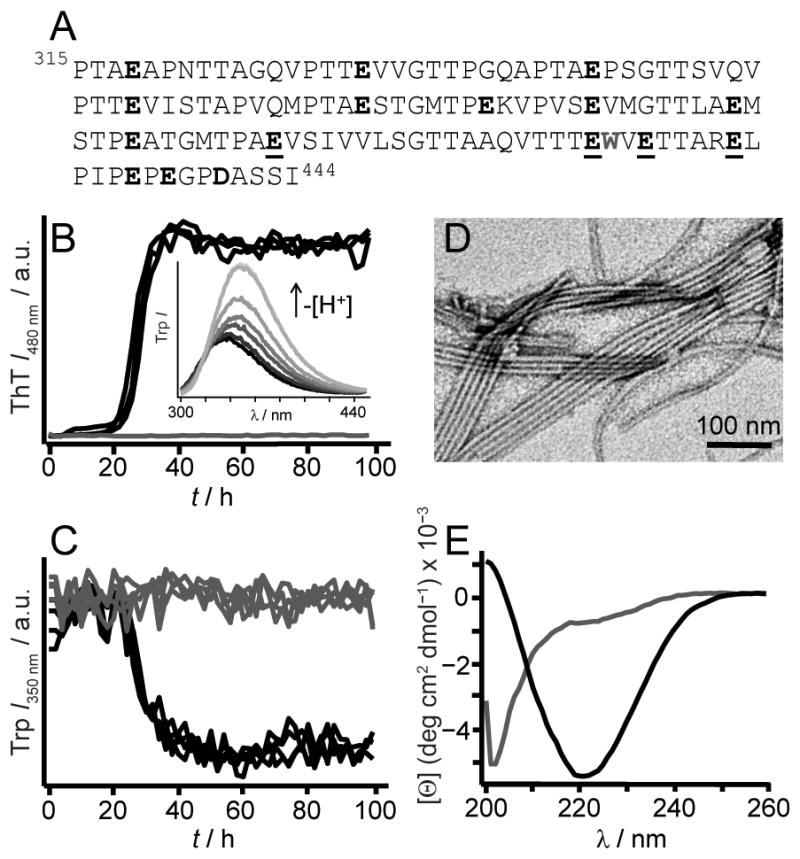
(A) RPT sequence is rich in Pro, Ser, Thr, and Glu (in bold). W423 is colored grey. Mutated residues used in this study, E404, E422, E425, and E430, are underlined. Aggregation kinetics of WT RPT (30 μM, 37 °C) at pH 5 (black) and 6 (grey) monitored simultaneously by ThT (10 μM, B) and W423 (C) fluorescence (n ≥ 3). Inset. W423 emission spectra of preformed fibrils sequentially titrated with NaOH from pH 5 to 7 at 25 °C (black-to-grey, respectively). (D) TEM image of RPT fibrils formed at pH 5. (E) Far-UV CD spectra of RPT samples (pH 5, black and 6, grey) measured at the end of aggregation ([Θ] is mean residue ellipticity).
Aggregation kinetics of single, double, and quadruple Ala/Gln mutants were monitored by thioflavin T (ThT) and Trp fluorescence, with the latter serving as a local probe given the close proximity of W423 to these Glu residues. Samples (200 μL containing 30 μM RPT in 20 mM of appropriate buffers (pH 5–7) with 100 mM NaCl and 10 μM ThT) in a 96-well plate were incubated at 37 °C with orbital shaking using a microplate reader. Fluorescence intensities were recorded every 2 h until completion. Transmission electron microscopy (TEM) and circular dichroism (CD) spectroscopy were used to observe morphological and secondary structural changes, respectively.
In accord with previous data collected in the absence of ThT, [2b] wild-type (WT) RPT aggregated in a pH dependent manner, with fibrillation occurring only below pH 6 (Figure 1). Amyloid formation at pH 5 was verified by (1) the characteristic ThT fluorescence enhancement, (2) a W423 quantum yield decrease, (3) TEM, and (4) a secondary structure change from unstructured to β-sheet as determined by CD spectroscopy, establishing that ThT had little effect on RPT aggregation. Fibril dissolution was also confirmed using both W423 and ThT fluorescence (Figure 1B inset and Figure S1). We next examined aggregation kinetics of single Ala- and Gln-mutants at pH 5.0 (Figure 2). Consistent and reproducible kinetic trends were obtained from multiple independent experiments (n (plates) ≥ 3; n (replicates on each plate) = 12). In comparison to WT protein, either Ala- or Gln-substitution at position E404 accelerated, whereas mutations at E425 and E430 retarded fibril formation, defined here as the total time taken to reach stationary phase. In contrast, E422A and E422Q had opposing behaviors, where Ala slowed and Gln hastened the aggregation time.
Figure 2.
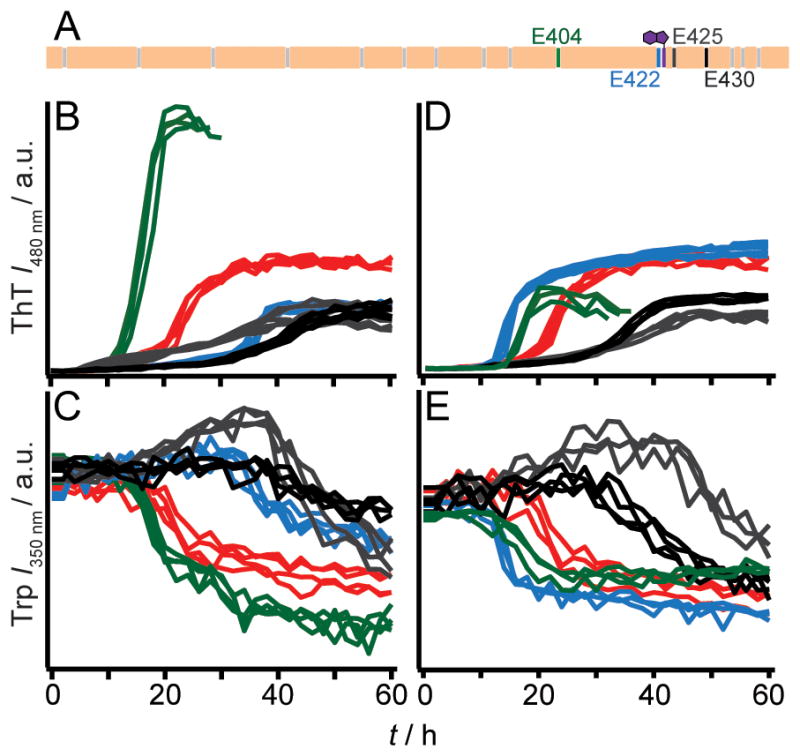
Aggregation kinetics of WT RPT, single Ala- and Gln-mutants at pH 5. (A) Schematic of RPT sequence highlighting positions of residues E404 (green), E422 (blue), E425 (dark grey) and E430 (black). All other carboxylic acid sidechains and W423 are colored grey and purple, respectively. Aggregation kinetics of WT RPT (red), single Ala- (B and C), and Gln-mutants (D and E) monitored by ThT and Trp fluorescence (n = 3). Colored as noted in panel A. Intensity scales are identical for panels B/D and C/E.
Careful scrutiny of the data indicates the presence of multiple kinetic phases. As detected by ThT fluorescence, WT shows two growth transitions, an early (5–20 h) and a later but faster (20–30 h) phase, where the relative amplitudes vary from plate-to-plate (Figure 2B and 2D). Mutations at E425 and E430 exert the greatest effect on their growth phases by significantly lengthening them. Changes in absolute ThT signals were not strictly correlated to the amount of fibrils generated, implying that other factors such as differences in fibril structure may account for this observation.[9] TEM images showed that fibrils were present for all mutants and obvious differences in fibril morphology could not be discerned (Figure S2). Of note, the simultaneously collected Trp data exhibit only a single transition with the exception of E425A/Q (Figure 2C and 2E), which gave an initial quantum yield increase followed by a decrease. Interestingly, Trp kinetics appear to be somewhat slower than those obtained by ThT fluorescence, perhaps indicating subtle, local structural changes within fibrils. Collectively, these data offer mechanistic insights into RPT fibril formation. Protonation of the C-terminal glutamic acids is shown to be vital, likely through the inhibition of intra/intermolecular electrostatic repulsion. Particularly, both charge neutralization and hydrogen bonding play key roles at position E422, where the introduction of an amide (–NH2 vs. – OH) sidechain accelerates aggregation via the increase of hydrogen bonding capability. This is in strong agreement with the inhibitory effect of the Ala mutation where hydrogen bonding donor (–OH) and acceptor (C=O) are removed. However, the difference in residue size, i.e. sidechain packing, cannot be ruled out as a contributing factor in kinetics modulation. By comparison, hydrogen bonding and/or size are not as critical at E404 where Ala/Gln both stimulate aggregation. Mutations at both E425 and E430 have a similar negative effect on aggregation, prolonging fibril growth. Upon protonation, these residues influence the self-assembly process perhaps through the formation of local noncovalent interactions and thus, retarding aggregation.
We next asked if any of these mutated residues could change the pH sensitivity of RPT amyloid formation. Only E422 mutants had a substantial impact where fibrils now form at pH 6.5 (Figure 3 and S3). TEM images taken at pH 6.5 show no significant changes in fibril morphology. Consistently, dissolution experiments conducted on E422Q fibrils verified that it is more stable than WT fibrils and are resistant to disassembly up to pH 7 (Figure S4). We note that E404A/Q occasionally aggregated (6 out of 12 wells) at pH 6 suggesting that it may play an ancillary role (Figure S5). Mutation at either E425 or E430 has no effect on the pH dependent aggregation of RPT (Figure S5).
Figure 3.
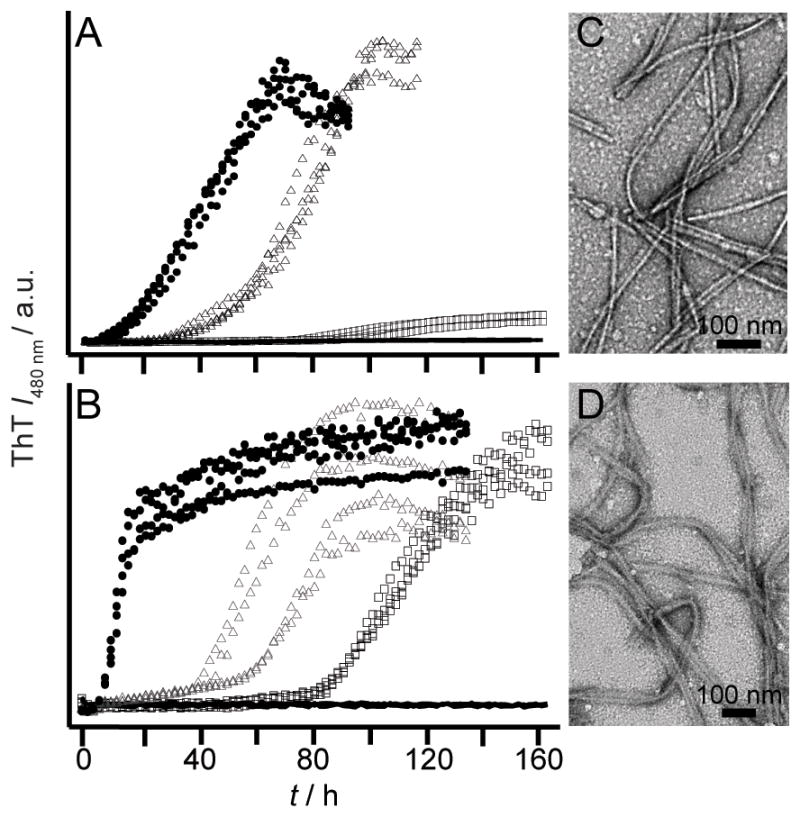
Effect of pH on aggregation kinetics of E422 mutants monitored by ThT fluorescence. E422A (A) and E422Q (B) at pH 5.5 (solid circles), 6.0 (open triangles), 6.5 (open squares), and 7 (black lines) (n ≥ 3). Intensity scales are identical. Representative TEM images for E422A (C) and E422Q (D) fibrils at pH 6.5. Scale bar is 100 nm.
Taken together, our data suggest that residue 422 is the critical sidechain in controlling the pH sensitivity of RPT amyloid formation in accord with ssNMR data where the conserved amyloidogenic region corresponds to residues 405–423 with E422 protonated.[8] Upon protonation of this site, reduced charge repulsion would favor compaction and lead to intermolecular interaction facilitating fibril formation. In the absence of this negative charge, fibril formation is more favorable and thus can occur at a higher pH. However, the observation of E404A/Q sometimes aggregating at pH 6 implied that additional sites may contribute and allosteric effects may be important.
To test this hypothesis of allostery, we studied the double E404/E422 mutant. Indeed, by neutralizing both charges through either Ala or Gln substitutions, fibrils now form at pH 7. A pronounced effect on the kinetics was observed for E404Q/E422Q where the lag phase is abolished and the growth rates attenuated as the solution pH was increased from 5 to 7 (Figure 4B and S6). In contrast, the double Ala mutant showed no changes in either the lag phase or fibril growth rate as the pH is increased (Figure 4A and S6). Again, reiterating an effect of hydrogen bonding and/or sidechain packing. Interestingly, TEM images for E404A/E422A showed differences in fibril morphology for the different pH solution conditions. A twisted ribbon morphology is observed in Figure 4E, which is absent in Figure 4C. CD data also corroborate distinctive fibril structures as spectroscopic differences were observed (Figure S7). Dissolution experiments confirmed the enhanced pH stability of the double mutant fibrils (Figure S8). To rule out the involvement of E425 and E430, the quadruple variant, E404Q/E422Q/E425Q/E430Q, was studied and similar pH dependent aggregation kinetics were observed (Figure S9).
Figure 4.

Effect of pH on aggregation kinetics of E404/E422 mutants monitored by ThT fluorescence. E404A/E422A (A) and E404Q/E422Q (B) at pH 5.0 (black), 6.0 (dark grey), and 7.0 (light grey) (n = 3). Intensity scales are identical. Representative TEM images for E404A/E422A fibrils at pH 5 (C), 6 (D) and 7 (E) and E404Q/E422Q fibrils at pH 5 (F), 6 (G) and 7 (H). Scale bar is 100 nm. (I) Schematic diagram showing a possible conformation of the central core of RPT filaments, composed of two β-strands (403–411) and (415–423), depicting sidechain orientations for E404 (green) and E422 (blue) as outside and inside the β-strands, respectively. Fibril axis is coming out of the page. W423 is colored purple. Removal of the critical amyloidogenic region, VSIVVL (cyan), completely abrogates fibril formation.
From a structural perspective, we propose that E404 and E422 reside within the amyloid-forming region of RPT. In fact, a deletion mutant corresponding to Δ405–410 (Figure S10), failed to aggregate at pH 5, supporting a critical role for this region. A schematic representation for one hypothetical conformation is shown in Figure 4I with two β-strands (indicated by arrows) formed by residues 403–411 and 415–423 aligned perpendicular to the filament axis. A β-turn (not shown) involving G412 links the two strands forming a hairpin-like structure. Here, Glu sidechains are oriented with E404 positioned outside and E422 inside the filament (defined here as individual subunits that make up fibrils).
In this putative model, positioning E422 sidechains within the filament core, suggests that upon protonation, both intra- and inter-sheet contacts are facilitated and essential in stabilizing filament structure. In the absence of a net charge, filaments can form at higher pH. The reduced aggregation rates associated with E422A indicate that either hydrogen bonding or size is involved in inter-sheet packing and stability. Protonation of the outwardly facing E404 would prevent intra-sheet electrostatic repulsion. The increased aggregation propensity associated with E404A also may suggest a role for sidechain interdigitation as the small methyl groups would allow tighter packing between filaments. While this model offers one potential scenario, clearly many other structural polymorphs can exist and may be just as likely (Figure S11).
In summary, these findings offer new insight into the molecular basis that governs the formation of a functional amyloid. In the case of RPT, amyloid regulation is not likely to be controlled via chaperones, but by the pH environment in which it resides. This unique pH switch may be an elegant way for controlling fibril assembly and maintaining other amyloid-like structures their benign nature as in the case of peptide hormones in pituitary secretory granules.[1f] Most recently, it also has been found that a small molecule can trigger the low complexity (LC) domain derived from fused in sarcoma (FUS) RNA-binding protein to form dynamic amyloid-like structures, potentially mimicking how RNA granules are formed.[1e] From an application perspective, a reversible amyloid formation mechanism is highly desirable and could exert its potential in various areas of nanobiotechnology including tissue engineering and drug delivery development.
Experimental Section
Supplementary Material
Acknowledgments
Supported by the Intramural Research Program of the NIH, NHLBI. TEM and CD data were collected in the NHLBI EM and Biophysics Core Facilities.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cbic.20xxxxxxx.
References
- 1.a) Chiti F, Dobson CM. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]; b) Fowler DM, Koulov AV, Balch WE, Kelly JW. Trends Biochem Sci. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]; c) Shewmaker F, McGlinchey RP, Wickner RB. J Biol Chem. 2011;286:16533–16540. doi: 10.1074/jbc.R111.227108. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bayro MJ, Daviso E, Belenky M, Griffin RG, Herzfeld J. J Biol Chem. 2012;287:3479–3484. doi: 10.1074/jbc.M111.313049. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Kato M, Han TNW, Xie SH, Shi K, Du XL, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei JM, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Maji SK, Perrin MH, Sawaya MR, Jessberger S, Vadodaria K, Rissman RA, Singru PS, Nilsson KPR, Simon R, Schubert D, Eisenberg D, Rivier J, Sawchenko P, Vale W, Riek R. Science. 2009;325:328–332. doi: 10.1126/science.1173155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) McGlinchey RP, Shewmaker F, McPhie P, Monterroso B, Thurber K, Wickner RB. Proc Natl Acad Sci U S A. 2009;106:13731–13736. doi: 10.1073/pnas.0906509106. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pfefferkorn CM, McGlinchey RP, Lee JC. Proc Natl Acad Sci U S A. 2010;107:21447–21452. doi: 10.1073/pnas.1006424107. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McGlinchey RP, Shewmaker F, Hu KN, McPhie P, Tycko R, Wickner RB. J Biol Chem. 2011;286:8385–8393. doi: 10.1074/jbc.M110.197152. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) McGlinchey RP, Gruschus JM, Nagy A, Lee JC. Biochemistry. 2011;50:10567–10569. doi: 10.1021/bi201578h. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) McGlinchey RP, Yap TL, Lee JC. Phys Chem Chem Phys. 2011;13:20066–20075. doi: 10.1039/c1cp21376h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raposo G, Marks MS. Nat Rev Mol Cell Biol. 2007;8:786–797. doi: 10.1038/nrm2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoashi T, Muller J, Vieira WD, Rouzaud F, Kikuchi K, Tamaki K, Hearing VJ. J Biol Chem. 2006;281:21198–21208. doi: 10.1074/jbc.M601643200. [DOI] [PubMed] [Google Scholar]
- 5.a) Berson JF, Harper DC, Tenza D, Raposo G, Marks MS. Mol Biol Cell. 2001;12:3451–3464. doi: 10.1091/mbc.12.11.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fowler DM, Koulov AV, Alory-Jost C, Marks MS, Balch WE, Kelly JW. PLoS Biol. 2006;4:100–107. doi: 10.1371/journal.pbio.0040006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Watt B, van Niel G, Fowler DM, Hurbain I, Luk KC, Stayrook SE, Lemmon MA, Raposo G, Shorter J, Kelly JW, Marks MS. J Biol Chem. 2009;284:35543–35555. doi: 10.1074/jbc.M109.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Leonhardt RM, Vigneron N, Hee JS, Graham M, Cresswell P. Mol Biol Cell. 2013;24:964–981. doi: 10.1091/mbc.E12-10-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Maurer-Stroh S, Debulpaep M, Kuemmerer N, de la Paz ML, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, Schymkowitz JWH, Rousseau F. Nat Methods. 2010;7:237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]; b) Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L. Nat Biotechnol. 2004;22:1302–1306. doi: 10.1038/nbt1012. [DOI] [PubMed] [Google Scholar]; c) Trovato A, Chiti F, Maritan A, Seno F. PLoS Comp Biol. 2006;2:1608–1618. doi: 10.1371/journal.pcbi.0020170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu KN, McGlinchey RP, Wickner RB, Tycko R. Biophys J. 2011;101:2242–2250. doi: 10.1016/j.bpj.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiang W, Yau WM, Tycko R. J Am Chem Soc. 2011;133:4018–4029. doi: 10.1021/ja109679q. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.