

Abstract
The transcription factor MYC has fundamental roles in proliferation, apoptosis, tumorigenesis, and stem cell pluripotency. Over the last 30 years extensive information has been gathered on the numerous cofactors that interact with MYC and the target genes that are regulated by MYC as a means of understanding the molecular mechanisms controlling its diverse roles. Despite significant advances and perhaps because the amount of information learned about MYC is overwhelming, there has been little consensus on the molecular functions of MYC that mediate its critical biological roles. In this perspective, the major MYC cofactors that regulate the various transcriptional activities of MYC, including canonical and noncanonical transactivation and transcriptional repression, will be reviewed and a model of how these transcriptional mechanisms control MYC-mediated proliferation, apoptosis, and tumorigenesis will be presented. The basis of the model is that a variety of cofactors form dynamic MYC transcriptional complexes that can switch the molecular and biological functions of MYC to yield a diverse range of outcomes in a cell-type- and context-dependent fashion.
The cancer-associated protein MYC mediates cell growth, proliferation, stem cell self-renewal, and apoptosis. It recruits different interacting proteins (cofactors) to regulate transcription in a context-dependent fashion.
The c-Myc protein (MYC) plays a critical role in cellular proliferation, cell size, differentiation, stem cell self-renewal, and apoptosis. Deregulated MYC expression occurs in many cancers and may be the driving force in up to 70% of human malignancies. MYC is a member of a transcription factor family that includes MYCN and MYCL. MYCN has similar activities compared with MYC, but MYCL is deficient in both transcriptional and transformation activity (Barrett et al. 1992; Oster et al. 2002). There have been tremendous advances in the 30 years since the MYC protein was first identified (Alitalo et al. 1983; Hann et al. 1983). However, the fundamental issue of how MYC mediates its diverse and critical biological roles at the molecular level has not been fully resolved (Meyer and Penn 2008). MYC is involved in many cellular pathways, interacts with dozens of cofactors, and regulates the expression of hundreds of genes, leading to a plethora of models to define the mechanism of MYC action. The prevailing theory is that the activity of MYC stems from its ability to control the expression of large sets of genes involved in cell division, metabolism, protein synthesis, and genomic instability, although few key mediators of MYC-induced proliferation, apoptosis, or tumorigenesis have been agreed on (Oster et al. 2002; Adhikary and Eilers 2005). As with all transcription factors, MYC recruits different sets of interacting proteins (referred to here as cofactors) to engage the transcriptional machinery. One deceptively simple question is how to define an “active” MYC protein complex. Furthermore, are there several different active MYC complexes with distinct molecular and biological roles? There are numerous studies that support a model in which MYC interacts differentially with a variety of cofactors in dynamic complexes that have distinct molecular activities controlling target gene selection and downstream pathways. In this perspective, a comprehensive review of the role of MYC cofactors in the transcriptional functions of MYC will be presented, and then the biological outcomes that are mediated by these interactions will be discussed.
COFACTORS MEDIATING MYC CANONICAL TRANSACTIVATION
Transcriptional activation is a complex process involving multiple steps, including preinitiation, initiation, promoter clearance, elongation, and termination. Typically, transcription factors mediate the binding of RNA polymerase during initiation, forming a transcription initiation complex at promoters. However, before initiation access to condensed chromatin at promoters must occur through chromatin modification, such as acetylation of histones, and chromatin remodeling via ATP-dependent restructuring of nucleosomes. In addition to altering chromatin structure, specific histone modifications or marks can be “read” by other proteins to allow further recruitment of activators or repressors. As a transcription factor, MYC recruits a variety of cofactors to modify or remodel chromatin and to activate RNA polymerase II (Pol II), as well as Pol I and III, at specific promoters (Luscher and Vervoorts 2012). However, as discussed below, the role of MYC and its cofactors in transcriptional activation is complex and unresolved. Although proteomic studies have been performed to identify the most abundant proteins associated with MYC in a particular cell (Koch et al. 2007; Agrawal et al. 2010), few of these protein interactions have been validated or characterized. In contrast, most MYC interacting proteins have been individually identified and characterized for their roles in canonical MYC transcriptional activity. Canonical activity is defined as the binding of MYC to the canonical E-box sequence, CACGTG or “E-box MYC site” (EMS), and subsequent transcriptional activation. MYC is also recruited to several variant (vEMS) E-box sequences (Blackwell et al. 1993; Meyer and Penn 2008). In the past, the term “noncanonical” has been used for any non-EMS site, including vEMS, so to avoid confusion the term noncanonical will only be used for MYC-binding sites that are not E-box sequences, such as those found in MYC-repressed target genes. As discussed in this review, this distinction is important because different MYC cofactor complexes mediate MYC DNA-binding site and target gene selection, as well as different biological outcomes.
The MAX Network
MAX is a basic/helix-loop-helix/leucine zipper (b/HLH/LZ) protein, like MYC, that was initially identified as a ubiquitous cofactor of MYC essential for MYC target gene expression. MAX is critical for canonical target gene induction by MYC because it forms a heterodimer with the MYC b/HLH/LZ domain permitting association with E-box DNA sequences (Grandori and Eisenman 1997; Luscher and Vervoorts 2012). However, MAX may be dispensable for some MYC activities (Gherardi et al. 2013). The members of the MXD (previously known as MAD) protein family compete for MAX binding at EMS and vEMS. MXD/MAX associates with Sin3/histone deacetylase (HDAC) complexes to repress MYC-MAX target gene transcription and act as antagonists of MYC function (see Conacci-Sorrell et al. 2014).
Chromatin Modification
MYC associates with several different chromatin-modifying enzymes, such as JARID1A, an H3K4me3-specific histone demethylase, and Pim1 kinase, which phosphorylates H3S10 (Secombe and Eisenman 2007; Zippo et al. 2007). However, the majority of studies have focused on the association of MYC with the histone acetyltransferases (HATs) GCN5 and TIP60. MYC can influence histone acetylation both globally by inducing GCN5 as a target gene or directly by recruiting GCN5 or TIP60 to promoters (Knoepfler et al. 2006; Murr et al. 2007; Cotterman et al. 2008). HATs are recruited to MYC and other transcription factors by TRRAP, which serves as a scaffold for assembling multiprotein complexes (McMahon et al. 2000; Luscher and Vervoorts 2012). TRRAP is a large (434 kDa) protein having homology with the ATM/PI3-kinase family, but lacks a functional kinase domain (Cowling and Cole 2006). TRRAP complexes have fundamental roles in transcription and have been investigated in several species, including yeast and Drosophila (Murr et al. 2007). The TRRAP/TIP60 complex (NuA4 in yeast) also contains the ATPase/helicase motif-containing cofactors TIP48 and TIP49 and the SWI/SNF-related protein p400 ATPase (McMahon et al. 2000; Frank et al. 2003). TIP48/49 can also bind to MYC independent of TRRAP, but their role in chromatin modification and MYC target gene regulation is unclear (Cowling and Cole 2006). GCN5 is part of a distinct TRRAP complex termed STAGA (SAGA in yeast) (Liu et al. 2003). GCN5 and the closely related PCAF preferentially acetylate H3 (K9/14/18), whereas TIP60 preferentially acetylates H4 (K5/8/12) and H2AK5 (Liu et al. 2003; Ciurciu et al. 2006; Cowling and Cole 2006; Sapountzi et al. 2006). Recruitment of TRRAP/TIP60 and enhanced H4 acetylation appears to predominate at MYC target genes following mitogenic stimulation of rodent fibroblasts (Frank et al. 2001, 2003). However, profiling of 24 lysine-acetylation marks in the human B cell line P493-6, which expresses a tetracycline-repressible MYC transgene, revealed that MYC induces H3 acetylation at K9/14/18 and H4 acetylation at K5/8/12/91 and H2AK5, suggesting that both GCN5 and TIP60 acetylate histones at MYC target genes (Martinato et al. 2008). Distinct TRRAP/HAT complexes may predominate at specific promoters in certain contexts, but the relevance of lysine acetylation by GCN5 compared with TIP60 for Myc function is unclear. Furthermore, although up-regulation of some canonical MYC target genes, such as tert and cyclin D2, is accompanied by both TRRAP recruitment and H3/H4 acetylation (Bouchard et al. 2001; Nikiforov et al. 2002), not all target genes show enhanced acetylation (Nikiforov et al. 2002). A survey of gene expression following a conditional knockout of GCN5 in neural stem cells indicated that one-sixth of the genes affected by loss of GCN5 are also affected in the same manner by loss of MYCN, implicating MYC in histone acetylation at specific promoters (Martinez-Cerdeno et al. 2012).
The discovery of TRRAP/HAT association with MYC led to the logical idea that MYC/MAX/HAT complexes and MXD/MAX/HDAC complexes function as transcriptional switches to regulate the activity of canonical MYC target gene promoters through histone acetylation, which would “open” or “close” the chromatin at MYC targets before Pol II recruitment (Luscher and Vervoorts 2012). However, the numerous findings that MYC binds to sites already possessing active chromatin marks and preloaded Pol II poised for elongation raises questions as to the significance of TRRAP/HAT at target promoters (Bouchard et al. 2004; Guccione et al. 2006; Martinato et al. 2008; Lin et al. 2012; Nie et al. 2012; Sabò and Amati 2014). Interestingly, MYC induces enhanced H4 acetylation at promoters having preloaded RNA Pol II, suggesting a role for TRRAP/HAT complexes in transcriptional elongation (Frank et al. 2003). In fact, TRRAP/HAT complexes have been implicated in pause release and chromatin remodeling, as discussed below, although the possibility remains that there are promoter and/or context-dependent roles of TRRAP/HAT in preinitiation of Pol II transcription. In support of this idea, MYC recruitment of TRRAP/HAT and histone acetylation precedes recruitment of Pol I and III. MYC recruits TRRAP/HAT complexes and the Pol I-specific factor SL1, and enhances histone acetylation at the rRNA promoter before recruitment of Pol I (Arabi et al. 2005; Grandori et al. 2005). MYC recruits TRRAP/GCN5 and TFIIIB to the tRNA and 5S rRNA promoters and stimulates rapid H3 acetylation before Pol III recruitment (Kenneth et al. 2007).
Chromatin Remodeling
MYC has direct roles in chromatin remodeling through several different mechanisms, originally suggested by the finding that MYC interacts with subunits of the multiprotein SWI/SNF complex (BAF in humans), leading to stimulation of reporter genes (Cheng et al. 1999). The SWI/SNF complex has ATPase activity, destabilizes histone-DNA interactions, and regulates transcription through nucleosome repositioning (Cheng et al. 1999; Park et al. 2002; Pal et al. 2003; Vervoorts et al. 2003). MYC also controls chromatin remodeling by inducing the exchange of histone H2A with the variant H2A.Z at target genes mediated by the TRRAP/TIP60 remodeling complex containing p400 ATPase (Martinato et al. 2008; Conerly et al. 2010). The incorporation of H2A.Z in the body of genes is associated with active transcription and reduced DNA methylation (Conerly et al. 2010). Inducible knockout of p400 results in impaired gene expression, including many MYC target genes (Fujii et al. 2010). In addition, knockdown of another ATPase that interacts with MYC, ASH2/ANCCA, causes a reduction in MYC target gene activation (Ciro et al. 2009). ASH2/ANCCA has a bromodomain that binds to acetylated chromatin, suggesting another link between histone acetylation and chromatin remodeling. The role and regulation of chromatin remodeling is evolving, but the impact these various remodeling cofactors have on MYC transcriptional activity at all or subsets of MYC target genes is likely to be significant.
RNA Polymerase Activation
Because MYC interacts with histone modifying and remodeling complexes that typically have roles before Pol II recruitment for transcriptional initiation and elongation, it was surprising to find preloaded Pol II at the CAD promoter and that MYC controls transcriptional elongation (promoter clearance) rather than initiation of CAD transcription (Eberhardy and Farnham 2001, 2002). MYC interacts with the two subunits of P-TEFb, cyclin T1 and CDK9, which control transcriptional elongation through Serine 2 (S2) phosphorylation of the multiple heptapeptide repeats in the carboxy-terminal domain (CTD) of Pol II as well as the negative elongation factors NELF and DSIF, resulting in the release of the paused polymerase (Eberhardy and Farnham 2001, 2002). This finding was later confirmed for many MYC targets (Bouchard et al. 2004). As with chromatin remodeling, transcriptional elongation also appears to be regulated by Myc through the recruitment of chromatin modifiers. Depletion of Pim1 kinase, a Myc cofactor that phosphorylates H3S10, blocks S2 CTD phosphorylation and transcriptional elongation of MYC target genes, suggesting that histone phosphorylation is required for P-TEFb interaction (Zippo et al. 2007). In addition, Brd4—a bromodomain protein that binds highly acetylated chromatin—interacts directly with both MYC and P-TEFb, thus linking MYC and P-TEFb recruitment to histone acetylation at active genes (Brès et al. 2008). In yeast, mutation of HAT activity in TRRAP complexes (SAGA) inhibited elongation, suggesting a direct role of histone acetylation in transcriptional elongation of a subset of genes (Wittschieben et al. 2000). Remodeling of chromatin may also play a role in transcriptional elongation because the FACT complex enhances elongation by remodeling nucleosomes to allow for Pol II transit through a gene (Winkler et al. 2011) and SWI/SNF has been implicated in efficient elongation (Subtil-Rodriguez and Reyes 2011).
Mediator, a multisubunit complex that is essential for transcriptional initiation through phosphorylation of S5 on the CTD of Pol II, also interacts with MYC (Eberhardy and Farnham 2001, 2002) and is recruited by MYC to some target promoters, such as cyclin D2 (Bouchard et al. 2001). Although many MYC target genes have preloaded Pol II, Mediator interaction with MYC may control initiation of transcription in a promoter- or context-dependent fashion. In addition, Mediator has been shown to have a role in transcriptional elongation. The MED26 subunit of Mediator can bind to either the general initiation factor TFIID or P-TEFb and thus contribute to initiation and pause release (Takahashi et al. 2011). Also, TRRAP/GCN5 complex STAGA can recruit Mediator to MYC (Liu et al. 2008). Disruption of the Mediator and STAGA interaction impairs transcription of the MYC target gene telomerase reverse transcriptase (TERT) and inhibits proliferation of MYC-dependent cells, but does not impair recruitment of Pol II (Liu et al. 2008). Thus, rather than playing discrete roles in mediating MYC transcriptional activity, chromatin modifiers and remodelers may interact with Pol II activators to form interdependent complexes to control MYC target gene expression. Although these various complexes, such as TRRAP/HAT, Mediator, and P-TEFb, have been shown to interact with different domains of the MYC protein, the large sizes of the complexes suggest that some cofactors are recruited indirectly, supporting an interdependent association of these cofactors with MYC. In addition, the subunit composition of these large transcriptional complexes is dynamic and can influence the activity of the complex, thus allowing additional regulation in a promoter, cell-type- and/or context-dependent manner. For example, Mediator exists in several different complexes with and without the CDK8 kinase module that controls Mediator function and SWI/SNF exists in distinct complexes with mutually exclusive subunits controlling activation or repression (Knuesel et al. 2009). TRRAP also exists as several different complexes with different activities (Park et al. 2002). In addition to indirect and/or differential composition of the different cofactor complexes there is likely a temporal or serial occupancy of the different complexes at specific promoters.
REGULATORY COFACTORS OF MYC CANONICAL TRANSACTIVATION
In addition to the MYC cofactors that are critical for “basal” transcription of canonical target genes and used by many transcription factors, such as chromatin modifiers and remodelers and activators of RNA polymerase, there is another class of “regulatory” MYC cofactors that control MYC through either controlling the stability of MYC protein or the transcriptional activity of MYC. These regulatory cofactors often modulate the posttranslational modification of MYC. The expression and/or activities of these cofactors are typically regulated by cellular events, such as during the cell cycle, apoptosis, or tumorigenesis. Considering that MYC protein levels can impact MYC-mediated functional outcomes, the regulation of MYC protein stability has been extensively investigated (Hann 2006; Vervoorts et al. 2006; Thomas and Tansey 2011; Farrell and Sears 2014). MYC is a rapidly degraded protein with a half-life of ∼20–30 min and a variety of different proteins interact with MYC to control its stability (Hann and Eisenman, 1984). These include kinases that phosphorylate MYC at T58 and S62 (GSK-3, MAPK, CDK, JNK, and DYRK), E3 ubiquitin ligases (Fbw7, TRUSS/TRPC4AP, and Huwe1), acetyltransferases that block ubiquitylation by directly acetylating MYC (CBP/p300 and TIP60), and cofactors that influence these modifications (Aurora A, Rabring, and SIRT1) (Hann 2006; Vervoorts et al. 2006; Zhao et al. 2008; Choi et al. 2010). In addition to cofactors that control the stability and posttranslational modification of MYC protein, there are other regulatory cofactors that control MYC transcriptional activity through undefined mechanisms, such as Bin1, p107, and nucleophosmin (NPM) (Gu et al. 1994; Sakamuro et al. 1996; Li et al. 2008).
Control of MYC Canonical Transcriptional Activity via Posttranslational Modification
In addition to influencing stability, there is accumulating evidence that posttranslational modifications by regulatory cofactors can control the activity of Myc, with or without affecting stability. Phosphorylation of MYC at S62 by several kinases stimulates MYC transcriptional activity, but the mechanism is unclear (Hann 2006). For example, phosphorylation of S62 by cyclin G1 increased the binding of MYC to the cyclin B1 promoter and transactivation (Seo et al. 2008) and a MYCS62A mutated protein, unlike MYC, fails to be recruited to the γ-glutamyl cysteine synthetase promoter (Benassi et al. 2006). In addition to proteolysis, a different role for ubiquitylation in regulating MYC transcriptional activity was suggested by studies examining the ubiquitin ligase SKP2 (S-phase kinase-associated protein 2). Surprisingly, SKP2 was shown to enhance MYC’s transcriptional activity (Kim et al. 2003; von der Lehr et al. 2003). This led to a model suggesting ubiquitylation plays an important role in transcriptional activity, perhaps by recruiting the proteasome to promoter complexes and activating transcription through clearance of MYC or proteolysis of a specific factor (Muratani and Tansey 2003; Geng et al. 2012). In fact, SKP2 is able to interact with cyclin T1 to induce the ubiquitylation and destruction of CDK9 in the P-TEFb complex (Kiernan et al. 2001). However, the effects of SKP2 on MYC protein stability appear to be variable (Kim et al. 2003; von der Lehr et al. 2003; Zhang et al. 2013). Thus, although Skp2 may influence MYC abundance in some contexts, it is likely to have additional roles that control MYC’s transcriptional activity. Support for an additional role for MYC ubiquitylation in MYC transcriptional activity is suggested by the finding that the loss of amino-terminal domain ubiquitylation through the mutation of six lysines to arginines (MYCN6KR) has a minor on effect on stability, but significantly inhibits canonical MYC target gene induction (Zhang et al. 2013). In addition, interaction of the ARF tumor suppressor with MYC inhibits SKP2 interaction and SKP2-mediated ubiquitylation, leading to inhibition of canonical MYC target gene expression (Datta et al. 2004; Qi et al. 2004; Zhang et al. 2013). It is not known how ubiquitylation controls MYC transcriptional activity, but studies with the viral transcriptional activator protein (VP16) suggest that ubiquitylation of the transactivation domain can enhance the interaction with P-TEFb and stimulate rates of elongation (Kurosu and Peterlin 2004).
COFACTORS CONTROLLING NONCANONICAL MYC TRANSCRIPTIONAL ACTIVITY
MYC Repression of Target Genes
In addition to canonical transactivation of target genes, MYC induction also causes transcriptional repression of target genes. Almost all surveys of MYC target genes using a variety of cells and conditions typically find that about one-third of the putative target genes are repressed (Luscher and Vervoorts 2012). The ability of MYC to repress target genes is controversial (Lin et al. 2012; Nie et al. 2012) and subsets of these putative repressed target genes, as with induced targets, are likely to be regulated indirectly. In fact, MYC represses many genes via the histone-lysine N-methyltransferase (EZH2)-induced genome trimethylation of H3K27. This occurs indirectly through MYC induction of the tumor suppressor PTEN, which then activates EZH2 via AKT phosphorylation in normal cells (Kaur and Cole 2013; Cole 2014). However, there is extensive evidence that MYC is directly recruited to non-E-box sequences at specific promoters through the interaction with other transcription factors, including MIZ-1, SP1/SP3, and NF-YB/NF-YC (Li et al. 1994; Peukert et al. 1997; Schneider et al. 1997; Gartel et al. 2001; Staller et al. 2001). MYC interaction with these factors causes repression by recruiting HDACs (Satou et al. 2001; Jiang et al. 2007; Kurland and Tansey 2008; Matsuoka et al. 2008).
The nature and regulation of these MYC repressive complexes, as well as their competition with canonical MYC activator complexes, is unclear, but may be dependent on the relative abundance of regulatory MYC cofactors under specific conditions. For example, ARF, which is induced by oncogenic MYC or RAS, stabilizes the interaction between MIZ-1 and MYC, displacing the MIZ-1 coactivators NPM and p300 (see Wiese et al. 2013). SP1, MIZ-1, or SP1/MIZ-1 complexes can recruit MYC and HDAC as repressive complexes to noncanonical promoters that are distinct from the MYC/HAT complexes at canonical promoters, suggesting a dynamic exchange of cofactors in a promoter- and context-dependent manner (Gherardi et al. 2013). However, MYC can also interact directly with HDAC3 to repress canonical targets (Kurland and Tansey 2008). This raises an interesting conundrum, because MXD/MAX and MNT/MAX complexes also recruit HDAC to repress canonical promoters, such as cyclin D2 (Popov et al. 2005). What is the purpose of MYC recruiting both HAT and HDAC activities with opposing functions to canonical promoters? One possibility is that the MYC/HDAC repressive complexes are recruited to distinct promoters that are not recognized by MXD-family proteins. For example, the repression of a human miRNA cluster and clusterin occurs through vEMS sequences (Wang et al. 2011b; Corvetta et al. 2013). Another possibility is that MYC may recruit HDAC3 during times when the MXD proteins are not expressed or active.
Noncanonical MYC Transactivation
Because MYC induces canonical target genes and represses target genes at non-E-box sites, it was unexpected to find that ARF is necessary for MYC to induce the expression of the transcription factors EGR-1 and EGR-4, target genes lacking canonical EMS sequences (Boone et al. 2011). This ARF-dependent noncanonical transactivation is in stark contrast to canonical transactivation, because ARF inhibits canonical transactivation (Datta et al. 2004; Qi et al. 2004). Interestingly, the other EGR family members, EGR-2 and EGR-3, are typical canonical MYC target genes (Qi et al. 2004; Boone et al. 2011). Although MYC associates weakly with the EGR-1 promoter without ARF, MYC recruitment is significantly enhanced by ARF (Boone et al. 2011). Because ARF inhibits SKP2-mediated ubiquitylation of the MYC transcriptional domain, the ubiquitylation status of the amino-terminal transcriptional domain of MYC may be a molecular switch controlling ARF-activated noncanonical activity. In support of this idea, inhibiting SKP2 expression or using MYCN6KR also results in the loss of canonical activity and acquisition of noncanonical activity, resulting in EGR-1 induction (Zhang et al. 2013). The control of canonical and noncanonical transcriptional activity by the ubiquitylation status of MYC is intriguing and suggests a novel transcriptional mechanism that remains to be defined.
IS MYC A GENOMIC REGULATOR OR A REGULATOR OF SPECIFIC TARGET GENES?
As discussed above, MYC can be recruited to both canonical and noncanonical target gene promoters for induction and/or repression. The evidence suggests that cofactors control the switch between transactivation and repression, as well as between canonical and noncanonical transactivation. In addition, cofactors may also control different types of canonical or repressive activities. However, recent findings suggest that MYC only binds to E-box sequences to enhance the expression of all active genes in a given cell (Lin et al. 2012; Nie et al. 2012). How can this discrepancy be resolved?
Genomic approaches using ChIP have shown that MYC can be recruited to anywhere from 4000 to 40,000 sites, depending on its level of expression. Normal proliferating cells are estimated to have a few thousand MYC molecules, but tumor cells can have up to 10 times higher levels (Varlakhanova and Knoepfler 2009; Lin et al. 2012; see Sabò and Amati 2014). MYC is not unique in its ability for widespread genomic binding. For example, MYB, Sall4, the estrogen receptor α, and NF-κB can be recruited to 10,000, 3200, 10,000, and 15,000 binding sites, respectively (Yang et al. 2008; Welboren et al. 2009; Kasowski et al. 2010; Quintana et al. 2011). Although some genomic studies show that MYC is recruited to all active promoters at EMS and vEMS sequences (Lin et al. 2012; Nie et al. 2012), other studies show only 60% or less of the binding sites have E-box sequences (Guccione et al. 2006; Kim et al. 2008; Perna et al. 2012). Although many of these widespread interactions may be nonspecific, it is likely that promoter selection is controlled by the expression and activity of specific cofactors, regulated in a cell-type- and context-dependent fashion, which recruit MYC to canonical or noncanonical sites.
Even though MYC binds to thousands of genomic sites that represent potential target genes, the rate-limiting step in target gene expression occurs post recruitment (see Sabò and Amati 2014). Most expression analyses using microarrays, RNAseq, or SAGE (serial analysis of gene expression) show that MYC up-regulates or down-regulates only a few hundred genes, albeit often different genes in different cell types and conditions. For example, in a recent study of genomic MYC binding and expression analysis using fibroblasts having an inducible MYC gene knockout, MYC was found to bind to more than 4000 sites, but only regulated 234 putative genes or 5.5% (Perna et al. 2012). This discordance in genomic binding and expression analyses is also observed for other transcription factors and suggests that target gene expression after binding to DNA is highly regulated by the presence of specific cofactors. To determine the specific role of a cofactor in regulating MYC canonical or noncanonical transcriptional activity it is necessary to examine direct MYC target gene promoters. However, there have been relatively few direct bona fide targets identified and characterized. Target genes can be misidentified owing to improper normalization to total RNA, because MYC increases the levels of both ribosomal RNA and specific mRNAs, and/or setting the threshold for designation too low, because MYC typically regulates endogenous target gene expression by only a few fold (Lin et al. 2012; Levens 2013). In addition, because some expression analyses use constitutively high levels of MYC or examine late time points with inducible MYC, many of the putative targets are likely indirect. Therefore, it is preferable to use low levels of an inducible MYC, such as the dihydroxytamoxifen (OHT)-inducible MYCER fusion protein or inducible knockout/knockdown, and choose early time points to enrich for direct target genes (Littlewood et al. 1995; Lawlor et al. 2006). To verify direct target genes it is preferable to use a combination of approaches, such as luciferase reporter assays to assess promoter activity in addition to chromatin immunoprecipitation (ChIP) assays.
Expression analyses have revealed a core set of signature MYC canonical target genes. This core signature is largely composed of ribosomal, metabolic, and protein synthesis genes, representing a cell-type-independent primordial set of genes involved in biomass accumulation (Ji et al. 2011). These genes may only require a “basal” complex of MYC cofactors, such as the widely expressed MAX and P-TEFb, for their induction. Beyond this core set of target genes, the limited overlap may reflect the cell-type and context-dependent expression of MYC cofactors differentially controlling MYC target gene selection.
Other transcription factors may be one group of targets that may be responsible for the widespread effects of MYC. MYC exerts major effects on target genes and pathways through the control of the expression and/or activities of other transcription factors at specific promoters. For example, MYC regulates the expression of EGR, E2F, ATF3, Foxm1, and AP4 transcription factors (Leone et al. 1997; Jung and Hermeking 2009; Boone et al. 2011). As with the EGR family, MYC also differentially regulates E2F family members to influence cell-cycle progression, apoptosis, and MYC-induced lymphomagenesis (Leone et al. 2001; Rempel et al. 2009). In addition, MYC regulates the activity of other transcription factors through direct interaction, as with SP1 and MIZ-1. Another layer of target gene regulation is through dual site cooperation or competition. Most transcription factors cooperate at target gene promoters and MYC targets are often enriched in binding sites for other factors, including EGR, E2F, Nrf-1, NF-Y, CREB, ELK-1, AhR/Arnt, ETF, and SP1, as well as nuclear hormone receptors ER and AR (Elkon et al. 2004). This cooperation among factors is also illustrated by the observation that serum can induce the immediate early EGR factors in MYC null fibroblasts, but induction is dramatically increased by MYC (Boone et al. 2011). Therefore, these factors can operate as secondary transcriptional nodes for MYC to direct downstream pathways and outcomes dependent on cell-type- and context-dependent cofactors. Furthermore, widespread indirect epigenetic target gene regulation can occur via direct MYC targets, such as GCN5 and HDAC, and indirect targets, such as EZH2, which also may be controlled in a cell-type- and context-dependent fashion. In addition, MYC-induced stimulation of metabolic pathways can generate precursors for histone modification, including methylation and acetylation, which influence epigenetic regulation (Dang 2012).
BIOLOGICAL RELEVANCE OF MYC COFACTOR INTERACTIONS
MYC cofactors have been examined by a variety of molecular and biological assays, but evaluating the influence of a particular cofactor or complex is not always straightforward. First, it is not always clear what molecular activities mediated by the cofactors, such as canonical transactivation or transcriptional repression, are essential for a particular biological function of MYC. Assessing biological relevance is also limited by the assays that are commonly used, which can include in vitro assays for proliferation, differentiation, cell size, apoptosis, transformation and induced pluripotency, as well as in vivo assays for tumorigenesis and embryonic development. Few cofactors have been thoroughly evaluated using a variety of molecular and biological assays in several different cell types and conditions. Common approaches to evaluate cofactors can be difficult to interpret owing to several limitations. Overlapping and multiple cofactor binding sites on MYC, as well as improper folding of MYC having specific mutations or deletions, can make interpretation of structure-function studies difficult. If the cofactor is redundant or nonessential in specific cells or cellular conditions, overexpression or inhibition of expression/activity may have no effects. Conversely, if the cofactor is essential for specific cellular functions, overexpression or inhibition can cause indirect effects. Thus, using the appropriate cell type and cellular condition and examining MYC-inducible functions rather than overall cellular effects is optimal. Despite the limitations and caveats in evaluating the different MYC cofactors, accumulating evidence suggests that specific cofactors can act as molecular switches to mediate MYC-induced proliferation, apoptosis, and tumorigenesis as discussed below.
Proliferation
MYC expression is a hallmark of proliferating cells and thus the ability of MYC to enhance proliferation has been routinely used to determine MYC activity. Proliferation has been assessed by the ability of MYC to induce cell-cycle entry from a quiescent G0 phase into G1, increase cell size in G1 (growth), maintain normal cell-cycle progression through checkpoints, stimulate hyperproliferation, induce loss of contact inhibition, and induce transformation. In addition to how proliferation is assessed, the ability of MYC to impact proliferation is highly dependent on the cell type, as illustrated by studies using different isoforms or mutated forms of MYC. MYC-S is a naturally occurring translational variant of MYC that lacks the amino-terminal 100 amino acids that are necessary to interact with Mediator, P-TEFb, TRRAP, and several other cofactors, and thus MYC-S cannot induce canonical target gene expression (Spotts et al. 1997; Xiao et al. 1998). However, MYC-S can rescue the slow proliferation phenotype of MYC null fibroblasts, promote normal cell-cycle progression in Drosophila, and induce hyperproliferation and anchorage independence in several cell types (Xiao et al. 1998; Hirst and Grandori 2000; Oster et al. 2003; Benassayag et al. 2005; Cowling and Cole 2008). In support of these findings, knockdown of TRRAP does not influence normal cell-cycle progression (Wood et al. 2000). Therefore, these studies suggest that the canonical transcriptional activity of MYC is not required for normal cell-cycle progression or hyperproliferation. In contrast, MYC-S can still repress transcription, suggesting that the repression of noncanonical target genes, such as the cell-cycle inhibitors p21, p27, or gadd45, is sufficient for cell-cycle progression (Xiao et al. 1998). Multiple transcriptional mechanisms have been proposed to mediate MYC repression of cell-cycle inhibitors during normal cell-cycle progression, but it is unclear which mechanism(s) prevail or whether there are different or additional repressive mechanisms in tumor cells as discussed below.
Although canonical MYC transcriptional activity is not essential for cell-cycle progression, it is essential for the ability of cells to exit G0 and increase cell size via biomass accumulation in G1. MYC-S cannot efficiently induce cell-cycle entry of primary quiescent human fibroblasts in the absence of serum or increase cell size, leading to decreased progeny size in Drosophila (Hirst and Grandori 2000; Benassayag et al. 2005). However, the ability of MYC to induce cell-cycle entry appears to be distinct from its ability to induce biomass accumulation, requiring additional molecular activities, cofactors, and canonical target genes. The release of paused Pol II mediated by P-TEFb is observed following activation of MYC in B cells, which respond with a significant increase in cell size (Iritani and Eisenman 1999; Lin et al. 2012). MYC alone can induce cell growth in P493-6 B cells in the absence of serum, but cannot induce proliferation without serum (Schuhmacher et al. 1999), suggesting that cell-cycle entry and progression induced by MYC requires more than P-TEFb recruitment, which may be sufficient for increasing cell size.
Recent studies support the idea that there are different classes of MYC target genes regulated by different molecular mechanisms for cell-cycle entry and cell-cycle progression. On serum induction, only a subset of genes is bound by MYC (Perna et al. 2012). In addition, MYC associates with promoters having paused Pol II that are enriched for genes in ribosomal biogenesis, whereas MYC also associates with a second class of genes having significantly less paused Pol II, which are primarily cell-cycle or cell differentiation genes that also have roles in maintaining pluripotency and reprogramming (Yang et al. 2013). In Drosophila, MYC binding at a subset of this second class of genes overlapped with insulator sites during mitosis that may also be involved in chromatin organization through interaction with the insulator protein Orc2 (Yang et al. 2013). Furthermore, the onconeural antigen cdr2 has also been shown to interact with MYC during mitosis to regulate MYC target gene expression (O’Donovan et al. 2010).
Taken together, these studies suggest that distinct molecular mechanisms mediated by different MYC cofactors control different aspects of proliferation through the differential control of MYC target genes (Fig. 1). Repression of MYC targets, such as p21, p27, and gadd45, via different mechanisms, appears to be sufficient for normal and accelerated cell-cycle progression. Transcriptional elongation mediated by P-TEFb appears sufficient for canonical transactivation of core signature MYC target genes, such as ribosomal, metabolic, and protein synthesis factors, required for rapid biomass accumulation and cell growth. Efficient biomass accumulation also likely needs MYC-induced Pol I-mediated rDNA synthesis and Pol III activation, which requires TRRAP/HAT and NPM (Kenneth et al. 2007; Li and Hann 2013). MYC-induced cell-cycle entry appears to be mediated by another subset of canonical MYC target genes, such as cyclin D2, that additionally requires chromatin modifiers and/or remodeling cofactors, such as TRRAP/HAT, SWI/SNF, TIP60/p400, and/or Mediator. In support of this idea, TRRAP is critical for maintaining the hematopoietic stem cell pool (Loizou et al. 2009) and the TIP60/p400 complex regulates stem cell identity (Fazzio et al. 2008). These studies also highlight the importance of selecting the appropriate cell type and assay when examining the role of cofactors and target genes in proliferation. Canonical MYC activity does not appear to be necessary for normal or accelerated cell-cycle progression in immortalized fibroblasts owing to the deregulation of critical MYC targets. In support of this idea, the majority of canonical MYC target genes have similar expression between immortalized MYC null fibroblasts and parental fibroblasts, whereas a repressed gene was found elevated in MYC null cells (Bush et al. 1998). Other cell types, such as embryonic stem (ES) cells, can function normally without MYC owing to up-regulation of MYCN, requiring a codeletion of MYC and MYCN to assess activity (Davis et al. 1993; Laurenti et al. 2008; Smith et al. 2010).
Figure 1.

Proliferation pathways mediated by MYC and MYC cofactors. MYC drives proliferation through stimulating cell growth (biomass accumulation) and cell-cycle entry from quiescence while inhibiting cell-cycle inhibitors for cell-cycle progression. Cofactors (in red) control the indicated proliferative pathways that can be opposed by other factors, such as TGF-β and MXD/MAX. Repression of cell-cycle inhibitors by MYC, which appears to be sufficient to drive cell-cycle progression, may be mediated by several possible mechanisms, including the recruitment of repressive complexes containing MIZ-1 or SP1 and/or indirect repression caused by EZH2 induction.
Apoptosis
Induction of apoptosis by oncogenic MYC is an essential safeguard to protect cells from uncontrolled proliferation and transformation (see McMahon 2014). MYC plays a critical role in regulating the balance between pro- and antiapoptotic BCL-2 family members, thus modulating the apoptotic threshold. Studies on MYC-mediated apoptosis have focused on both B cells and fibroblasts; however, there appear to be cell-type-dependent differences. In B cells, it appears that MYC induces apoptosis primarily by inhibiting the antiapoptotic factors BCL-XL and BCL-2 via unresolved indirect mechanisms, but up-regulation of proapoptotic factors such as BIM may also be involved (Eischen et al. 2001; Hemann et al. 2005). The ability of MYC to induce apoptosis in fibroblasts is routinely assayed under low serum conditions, which primarily leads to an induction of proapoptotic factors, such as BIM, BAX, and PUMA (Sherr 2006).
As illustrated in Figure 2, MYC-mediated apoptosis can occur through at least three different pathways in fibroblasts, the p53-dependent MYC-ARF-MDM-2-p53 pathway, the p53-dependent/ARF-independent DNA damage pathway, and the p53-independent pathway MYC-ARF-EGR-1 (Pomerantz et al. 1998; Zindy et al. 1998; Boone et al. 2011). Oncogenic levels of MYC induce ARF protein levels by inhibiting the ubiquitin ligase for ARF (ULF) that mediates proteolysis of ARF (Chen et al. 2010, 2013a). Stabilized ARF can then interact with multiple partners to promote apoptosis. ARF binds to and inhibits the E3 ubiquitin ligase MDM-2, leading to stabilization of p53. In addition, ARF can interact directly with MYC, inhibiting SKP2-mediated ubiquitylation, leading to the induction of the proapoptotic EGR-1 noncanonical target gene (Qi et al. 2004; Boone et al. 2011; Zhang et al. 2013).
Figure 2.
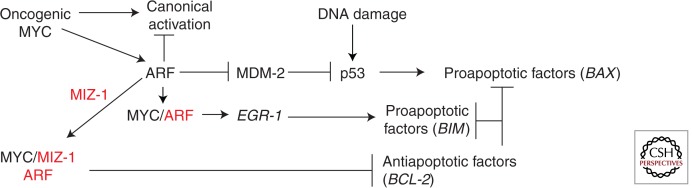
Apoptosis pathways mediated by MYC and MYC cofactors. Overexpressed or deregulated MYC (oncogenic MYC) induces ARF protein levels. ARF can then bind to inactivate MDM2, causing p53 induction, or bind to MYC to induce EGR1 through noncanonical transactivation while inhibiting canonical transactivation or bind to MIZ-1 and MYC to cause repression of BCL2. ARF-independent apoptosis can occur through DNA damage. The relative binding and regulation of ARF to its many partners is unknown. These pathways have primarily been investigated in fibroblasts. ARF and MIZ-1 are shown in red as MYC cofactors.
Although it is clear that MYC can induce both p53-dependent and p53-independent apoptosis in vitro and in vivo, the relative use of these different pathways is not known and may depend on cell type or context (Gregory et al. 2005; Sherr 2006). Interestingly, the major proapoptotic factors implicated in MYC-induced apoptosis are not the targets typically induced by p53, such as PUMA, NOXA, and BAX, but rather the proapoptotic Bim, which is a target of EGR-1 (Egle et al. 2004; Hemann et al. 2005). Overexpressed ARF is often found in nucleoli, sequestered by NPM, but because p53 null fibroblasts with high levels of ARF proliferate normally, nucleolar ARF does not seem to influence cell-cycle progression or apoptosis (Qi et al. 2004). To induce apoptosis, oncogenic MYC releases ARF from nucleolar sequestration, allowing its accumulation in the nucleoplasm (Qi et al. 2004; Li and Hann 2009). In addition to activation of proapoptotic factors, it may be essential to repress antiapoptotic factors in some cell types or contexts (Fig. 2). MIZ-1 was shown to be necessary for MYC repression of BCL-2, which may also require ARF for stabilization of the MYC/MIZ-1 complex (Herold et al. 2002; Patel and McMahon 2006, 2007; Herkert et al. 2010). It is important to note that MYC levels are clearly not the only determinant of apoptosis induction. Although high levels of MYC are required to induce ARF, similar MYC levels in p53/ARF double null mouse embryo fibroblasts do not induce apoptosis (Qi et al. 2004; Boone et al. 2011; Zhang et al. 2013). In addition, two different mutants of MYC, MYCT58A and MYCN6KR, are both more stable than wild-type MYC, but MYCT58A is defective for BIM induction and apoptosis, whereas MYCN6KR induces BIM and apoptosis (Hemann et al. 2005; Zhang et al. 2013).
Transformation/Tumorigenesis
Tumor cells need to maintain high growth and cycling rates, but they also need to disable apoptosis and initiate new transcriptional programs specific for tumor initiation and metastasis. Inducible transgenic mouse studies with MYC and knockdown of MYC in specific tumors show that MYC drives these tumor-promoting cellular processes (Arvanitis and Felsher 2006; see Gabay et al. 2014). A major question that remains unresolved is: How do MYC and its cofactors cause tumorigenesis? Is it just higher levels of MYC that elicit greater target gene expression? Or is it more like apoptosis, in which differential expression of specific cofactors, such as ARF, changes the molecular and biological activities of MYC?
Many of the cofactors mediating canonical MYC activity have been shown to be critical for transformation of primary fibroblasts, including cofactors involved in chromatin modification and remodeling complexes (Wood et al. 2000; Park et al. 2002). However, because many of these cofactors also interact with other transcription factors and have roles in other processes besides transcription, it is difficult to assess their direct role in MYC-induced transformation. Whereas MYC canonical transcriptional activity is likely required to enhance the cycling rate and biomass needed for highly proliferating tumor cells, it is unclear which specific MYC cofactors are driving MYC-induced tumorigenesis. Some MYC cofactors have elevated expression in tumors, suggesting that they may enhance the oncogenic activity of MYC. Two such regulatory cofactors, SKP2 and NPM, are overexpressed in many tumors and are also canonical targets of MYC, thus maintaining high levels in a feed-forward loop (Gstaiger et al. 2001; Grisendi et al. 2006). Elevated SKP2-mediated ubiquitylation enhances canonical transactivation and cell-cycle progression, while also competitively inhibiting the interaction of ARF with MYC to inhibit EGR-1 induction and apoptosis (Kim et al. 2003; Zhang et al. 2013). Overexpression of NPM causes a dramatic increase in MYC-induced anchorage-independent proliferation, but has little effect on normal cell-cycle progression (Li et al. 2008). NPM not only influences both canonical transactivation and repression, it is also essential for translocating a fraction of MYC to nucleoli to activate Pol I for rRNA synthesis and ribosomal biogenesis. High levels of NPM increase rRNA synthesis and can also sequester ARF in nucleoli to inhibit apoptosis (Li et al. 2008; Li and Hann 2009, 2013). Thus, NPM has multiple effects on MYC activities.
Phosphorylation of MYC at S62 is also necessary for efficient MYC-induced transformation and tumorigenesis (Hann 2006). S62 phosphorylation is enhanced in tumor cells through the up-regulation of several different kinases and CIP2A, which inhibits PP2A phosphatase (Hann 2006; Junttila et al. 2007). Loss of T58 phosphorylation, which enhances S62 phosphorylation, or loss of the MYC ubiquitin ligase FBW7 also stimulates MYC-induced transformation (Lutterbach and Hann 1994; Welcker et al. 2004a; Minella and Clurman 2005; Wang et al. 2011a). FBW7 ubiquitylation is dependent on T58 phosphorylation, which is dependent on the prior phosphorylation of S62 (Lutterbach and Hann 1994; Welcker et al. 2004b; Yada et al. 2004). The molecular role of MYC phosphorylation is unresolved, but there is evidence that S62 phosphorylation regulates a subset of both activated and repressed MYC target genes, in addition to influencing MYC protein stability (Hann 2006; Niemela et al. 2012). Interestingly, CDK5, which phosphorylates S62 and is essential for the induction of cyclin B1, is first activated by cyclin G1, which is elevated in cancer tissues (Seo et al. 2008). CDK2 phosphorylation of S62 promotes tumor progression by inhibiting RAS-induced senescence; however, the effect appears to be specific for CDK2, suggesting CDK2 may also regulate other cofactors in the complex (Campaner et al. 2010; Hydbring and Larsson 2010). The MYC target gene AP4 also inhibits senescence via the direct repression of p21 and p16, perhaps controlled by CDK2 phosphorylation of MYC (Jung and Hermeking 2009; Jackstadt et al. 2013). Although SKP2, NPM, and S62 phosphorylation of MYC are all elevated in cancers, it is unclear how they influence the transcriptional activities of MYC or the regulation of transformation-related target genes.
Although it appears there is a shift to higher expression and activity of MYC regulatory cofactors that may influence canonical MYC transcriptional activity and other activities during cellular transformation, is there a fundamental shift in other MYC cofactors during transformation? MYC activation of quiescent fibroblasts or B cells during cell-cycle entry is predominantly correlated with increased histone acetylation and pause release for canonical transactivation (Lin et al. 2012; Perna et al. 2012). Therefore, it might be expected that high levels of MYC would enhance histone acetylation during tumorigenesis, but there is actually a global loss of H4 acetylation in cancer cells and tumors compared with normal cells and tissues (Fraga et al. 2005). In fact, TIP60 is a tumor suppressor that is often found underexpressed in tumors (Lleonart et al. 2006; Gorrini et al. 2007; Sakuraba et al. 2011; Chevillard-Briet et al. 2013). Although GCN5 is highly expressed in non-small-cell lung cancer, NPM, which is also overexpressed in many tumors, binds to GCN5 and inhibits its activity (Zou et al. 2008; Chen et al. 2013a). Interestingly, there may be a shift to enhanced chromatin remodeling, rather than histone acetylation in cancer. There is an increase in the deposition of the variant histone H2A.Z in cancers, which is mediated by the TIP60/p400 complex (Conerly et al. 2010; Rangasamy 2010). However, the ratio of p400 to TIP60 may be important for cancer progression. A greater p400 to TIP60 ratio is pro-oncogenic and may influence target gene selection (Mattera et al. 2009; Chevillard-Briet et al. 2013). In addition, the MYC cofactor BRD4, which is involved in cell-cycle progression through its role in histone acetylation-dependent transcriptional regulation, is down-regulated in cancer (Rodriguez et al. 2012). Therefore, it appears that the mechanisms involved in the regulation of histone acetylation necessary for normal cell-cycle progression are disrupted in cancer.
In concert with an apparent shift away from histone acetylation, there is an induction of HDAC activity in tumor cells, which promotes transformation by repression of tumor suppressors (Huang et al. 2005; Hrzenjak et al. 2006). HDAC overexpression promotes tumor cell proliferation and blocks apoptosis (Huang et al. 2005; Hrzenjak et al. 2006). The significance of HDACs in tumorigenesis is highlighted by studies showing that HDAC inhibitors are effective therapeutic agents to repress tumorigenesis (Gherardi et al. 2013). In addition, HDAC2 is a MYC target gene, resulting in a feed-forward mechanism, as with SKP2 and NPM (Marshall et al. 2010). Therefore, as shown in Figure 3, although it is essential to maintain and enhance proproliferative canonical MYC activity, likely through regulatory MYC cofactor induction and a shift to chromatin remodeling, as well as disable proapoptotic MYC pathways, there also appears to be a shift to “oncogenic” repression of MYC target genes through HDAC recruitment at specific promoters for MYC-induced transformation and tumorigenesis. Oncogenic repressive mechanisms may replace repressive mechanisms that are used during normal cell-cycle progression or may supplement these mechanisms.
Figure 3.

Tumorigenesis pathways mediated by MYC and MYC cofactors. MYC-mediated tumorigenesis is primarily driven by enhanced canonical transactivation caused by overexpressed regulatory cofactors such as NPM, SKP2, and CIP2A and oncogenic MYC repression/silencing of tumor suppressors through cofactor complexes MIZ-1 and/or SP1 with HDACs, DNMTs, and EZH2. Loss of ARF and/or p53 and inhibition of RAS-induced senescence through CDK2 phosphorylation of MYC contributes to MYC-driven tumorigenesis. MYC cofactors are shown in red.
Repression has long been linked to transformation and tumorigenesis (Claassen and Hann 1999; Gherardi et al. 2013). Although direct and indirect repression of cell-cycle inhibitors, including CDK inhibitors, as well as gadd and gas target genes, promotes cell-cycle progression and inhibits senescence in both normal and transformed cells, accumulating evidence suggests tumor-specific or oncogenic repression of target genes is necessary for MYC to mediate tumorigenic functions. This includes loss of cell adhesion (integrins, caveolin-1, N-cadherin), stimulation of angiogenesis (thrombospondin), and metastasis (NDRG1/2) (Claassen and Hann 1999; Gherardi et al. 2013). In addition, comparison of targets regulated by MYCN and HDAC2 knockdown revealed that 13% are coregulated, suggesting that a subset of MYC targets are repressed by HDAC recruitment (Marshall et al. 2010).
Oncogenic repression involves recruitment of HDACs to several different MYC repressive complexes. One type of complex includes SP1, which like MYC is highly expressed in tumors and binds a variety of chromatin-modifying and remodeling complexes (Gherardi et al. 2013). MYC has been shown to recruit HDACs to repress target genes at SP1 sites, such as tissue transglutaminase 2 and cyclin G2, which have roles in tumor cell growth (Liu et al. 2007; Marshall et al. 2010). In addition, MIZ-1 can form a unique complex with SP1 and MYCN to recruit HDAC. This MYCN/SP1/MIZ-1/HDAC1 complex represses the tyrosine kinase receptor TRKA, p75 neurotrophin receptor, and p21 (Iraci et al. 2011). The repression of metastasis-inhibiting NDRG1/2 and other targets has also been linked to MIZ-1 and HDAC recruitment (Gherardi et al. 2013). In addition to SP1 and MIZ-1, YY1 can also recruit MYC and HDACs to YY1 target genes, including α3β1-integrin, in tumor cells (de Nigris et al. 2007). Furthermore, MYC can directly recruit HDAC3 for epigenetic repression of canonical targets, such as Id2, Gadd153, and miRNA-15a/16 (Kurland and Tansey 2008; Zhang et al. 2012). Interestingly, HIF-2α promotes the recruitment of SP1 and MIZ-1 to MYC and enhances neoplastic progression, whereas HIF-1α antagonizes MYC activity by competing for SP1 binding (Gordan et al. 2007; To and Huang 2013).
The MYC repressive complexes can also recruit additional epigenetic repressors, which may be a hallmark of oncogenic repression. MIZ-1 and SP1 associate with DNA cytosine methyltransferase (DNMT1) and DMNT3a, respectively, and HDAC1 can also associate with DNMT1, suggesting that these complexes are involved in gene silencing in tumors at certain promoters, such as p21, which is heavily methylated in leukemias (El-Osta and Wolffe 2000; Herkert and Eilers 2010; Li and Davie 2010). MYC also recruits DNMT3 to SP1 sites in the promoters of CDKN2a, CCND1, and TIMP2 genes (Hervouet et al. 2009). In addition, MYC/HDAC3 can recruit the transcriptional repressor EZH2, which is frequently up-regulated in human cancers (Chase and Cross 2011). MYC/HDAC3/EZH2 represses miR-29 expression, which contributes to aggressive clinical outcome of MYC-driven lymphomas (Zhang et al. 2012) and a MYCN/HDAC/EZH2 complex represses the expression of the tumor suppressor clusterin in neuroblastomas (Corvetta et al. 2013).
Taken together, these studies suggest that rather than an increase in all MYC activities during tumorigenesis there is a shift to enhanced transcriptional activity as a result of induced SKP2, NPM, and S62 MYC phosphorylation, a shift to chromatin remodeling rather than histone acetylation, and a shift to oncogenic repression as a result of HDAC and SP1 induction, as well as a loss of noncanonical transactivation owing to a loss of ARF function. Part of this shift in cofactors could be attributable to high levels of MYC in tumors, because SKP2, NPM, and HDAC2 are target genes of MYC, but it is likely that there are additional influences on other MYC cofactors, such as SP1 and EZH2, from multiple oncogenic signaling pathways. These shifts in MYC cofactors during tumorigenesis result in changes in the nature or magnitude of target gene expression. For example, interaction between MIZ-1 and MYC is enhanced by ARF, so a MYC/MIZ-1/ARF complex has a tumor suppressive role, as suggested by its proapoptotic role in repressing BCL-2, whereas a MYC/SP1/MIZ-1/HDAC complex appears to have a tumorigenic function. MIZ-1 also has little influence on p21 repression during normal proliferation and repression of p21 by MYC during TGF-β-mediated cell-cycle arrest does not appear to be mediated by HDACs (Claassen and Hann 2000; Herkert and Eilers 2010). Furthermore, repression owing to the indirect activation of EZH2 via MYC induction of the tumor suppressor PTEN would be disabled in tumors relative to normal cells, because PTEN is typically lost during tumorigenesis (Kaur and Cole 2013). These changes in transcriptional repressive mechanisms mediated by MYC cofactors during tumorigenesis may also reflect a shift to transcriptional silencing of specific genes, such as cell-cycle inhibitors and other tumor suppressors, either by DNA methylation via DNA methyltransferases (DNMTs) and/or histone methylation via EZH2, rather than transient repression observed during normal cell-cycle progression.
CONCLUSION
Examination of the roles of MYC cofactors suggests that MYC activity cannot be accounted for solely by the recently proposed amplifier model, which suggests that the sole function of MYC is to enhance the expression of all canonical target genes that were initiated by other factors in a given cell (Lin et al. 2012; Nie et al. 2012; Levens 2013; Rahl and Young 2014). The evidence summarized in this review suggests that dynamic complexes of cofactors can differentially regulate the transcriptional activity and target gene selection of MYC to mediate diverse biological outcomes, thus changing the intrinsic activity of MYC. Figure 4 summarizes the active role that cofactors play in controlling various functions of MYC, which is the fundamental basis of the cofactor switch model (Boone and Hann 2011). However, the two models are not mutually exclusive. The amplifier model focuses on the rapid release of paused Pol II in canonical transactivation of the core signature target genes mediating biomass accumulation and cell growth, which is one function of MYC. However, this model fails to consider the role of chromatin modification and remodeling in canonical transactivation involved in cell-cycle entry and Pol I and III activation, transcriptional repression of targets required for cell-cycle progression and tumorigenesis, and ARF-dependent noncanonical transactivation in p53-independent apoptosis. Similar to the amplifier model, the activity of MYC in the cofactor switch model is dependent on cell type and/or context, because many of the cofactors are differentially expressed in different cell types and contexts, such as during the cell cycle, apoptosis, or transformation. Specific thresholds of MYC are also relevant in both models and different MYC thresholds may differentially induce cofactors, such as GCN5, ARF, NPM, SKP2, and HDAC2. However, excessive MYC alone would not be expected to cause a shift in target gene selection or function if critical cofactors are limiting. For example, excessive MYC in B cells is recruited to the same promoters as endogenous MYC (Nie et al. 2012). High levels per se are not essential to cause carcinogenesis, because levels of MYC are often similar in EBV-immortalized nontumorigenic B cells compared with Burkitt lymphoma B cells (Hann and Eisenman 1984).
Figure 4.

Cofactor switch model. A summary of pathways mediated by MYC and MYC cofactors (in red) illustrating how the differential interaction of the various cofactors, which control different transcriptional mechanisms and MYC target genes, mediate distinct biological outcomes. Several pathways can cooperate to achieve optimal outcomes. Cell type and cellular context, as well as the levels of MYC, differentially regulate the expression and activity of the MYC cofactors.
A major unresolved question is what determines and regulates cofactor switching. How do these different cofactors and complexes compete for MYC to determine the cofactor composition at specific promoters to mediate distinct biological outcomes? Several factors likely control the composition of cofactor complexes and several different MYC complexes may exist in the same cell or cell populations. External and internal signals control the relative expression of cooperative or competitive cofactors and posttranslational modifications that influence cofactor activities or localization. For example, SKP2 expression is regulated during the cell cycle. Senescence leads to ARF up-regulation. Excessive ribosomal proteins L11 and S14 compete with TRRAP binding and Aurora A kinase and βTrCP-SCF compete with the ubiquitin ligase Fbw7 binding to MYCN (Dai et al. 2007; Popov et al. 2010; Zhou et al. 2013). NPM and ARF are relocalized from nucleoli to nucleoplasm on DNA damage (Gjerset 2006). PAK2 phosphorylation of the MYC carboxy-terminal domain regulates MAX heterodimerization and differentiation (Uribesalgo et al. 2011). Phosphorylation of MYC at S62 disrupts the interaction of the inhibitor BIN-1 with MYC and phosphorylation of CDCA7 by AKT disrupts its interaction with MYC (Sakamuro et al. 1996; Pineda-Lucena et al. 2005; Gill et al. 2013). Thus a myriad of controls can regulate the activities of the various MYC complexes resulting in specific physiological outcomes.
The models discussed here for MYC transcriptional activities mediating proliferation, apoptosis, and tumorigenesis are meant to be illustrative of the critical molecular and biological functions of MYC cofactors for MYC and do not include all the cofactors that control these processes. There are also other cellular processes controlled by MYC, including replication, genomic stability, α-tubulin acetylation via MYC-NICK, and mRNA cap methylation that likely require differential cofactor interaction with MYC to regulate these disparate activities (Cole and Cowling 2008). These and other cofactors may also be found to control MYC function in development, differentiation, and pluripotency. Researching MYC can often seem like a trip to Wonderland. You are presented with so many contradictory images and unbelievable scenarios that you can feel lost and a little mad. But you are also presented with a rich tapestry of diverse pathways and interesting characters that have a powerful influence on life and death. Understanding the role and regulation of MYC cofactors will certainly allow more effective intervention to control MYC activity in several different contexts, including tumor formation and somatic cell reprogramming.
ACKNOWLEDGMENTS
I thank Qin Zhang for assistance in preparing the manuscript and Erick Spears, Steve Hanks, Bill Tansey, and Mike Cole for critical reading of the manuscript, as well as the editors Chi Dang and Bob Eisenman. I also acknowledge Bob Eisenman, Peter Vogt, and my mother Hilda Passage as sources of inspiration and encouragement for many years.
Footnotes
Editors: Chi V. Dang and Robert N. Eisenman
Additional Perspectives on MYC and the Pathway to Cancer available at www.perspectivesinmedicine.org
REFERENCES
*Reference is also in this collection.
- Adhikary S, Eilers M 2005. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol 6: 635–645 [DOI] [PubMed] [Google Scholar]
- Agrawal P, Yu K, Salomon AR, Sedivy JM 2010. Proteomic profiling of Myc-associated proteins. Cell Cycle 9: 4908–4921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alitalo K, Ramsay G, Bishop JM, Pfeifer SO, Colby WW, Levinson AD 1983. Identification of nuclear proteins encoded by viral and cellular myc oncogenes. Nature 306: 274–277 [DOI] [PubMed] [Google Scholar]
- Arabi A, Wu S, Ridderstrale K, Bierhoff H, Shiue C, Fatyol K, Fahlen S, Hydbring P, Soderberg O, Grummt I, et al. 2005. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol 7: 303–310 [DOI] [PubMed] [Google Scholar]
- Arvanitis C, Felsher DW 2006. Conditional transgenic models define how MYC initiates and maintains tumorigenesis. Semin Cancer Biol 16: 313–317 [DOI] [PubMed] [Google Scholar]
- Barrett J, Birrer MJ, Kato GJ, Dosaka-Akita H, Dang CV 1992. Activation domains of L-Myc and c-Myc determine their transforming potencies in rat embryo cells. Mol Cell Biol 12: 3130–3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benassayag C, Montero L, Colombie N, Gallant P, Cribbs D, Morello D 2005. Human c-Myc isoforms differentially regulate cell growth and apoptosis in Drosophila melanogaster. Mol Cell Biol 25: 9897–9909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, Zupi G, Biroccio A 2006. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell 21: 509–519 [DOI] [PubMed] [Google Scholar]
- Blackwell TK, Huang J, Ma A, Kretzner L, Alt FW, Eisenman RN, Weintraub H 1993. Binding of myc proteins to canonical and noncanonical DNA sequences. Mol Cell Biol 13: 5216–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boone DN, Qi Y, Li Z, Hann SR 2011. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc Natl Acad Sci 108: 632–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B 2001. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev 15: 2042–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Marquardt J, Bras A, Medema RH, Eilers M 2004. Myc-induced proliferation and transformation require Akt-mediated phosphorylation of FoxO proteins. EMBO J 23: 2830–2840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brès V, Yoh SM, Jones KA 2008. The multi-tasking P-TEFb complex. Curr Opin Cell Biol 20: 334–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush A, Mateyak M, Dugan K, Obaya A, Adachi S, Sedivy J, Cole M 1998. c-myc null cells misregulate cad and gadd45 but not other proposed c-Myc targets. Genes Dev 12: 3797–3802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campaner S, Doni M, Hydbring P, Verrecchia A, Bianchi L, Sardella D, Schleker T, Perna D, Tronnersjo S, Murga M, et al. 2010. Cdk2 suppresses cellular senescence induced by the c-myc oncogene. Nat Cell Biol 12: 54–59; Suppl 1–14 [DOI] [PubMed] [Google Scholar]
- Chase A, Cross NC 2011. Aberrations of EZH2 in cancer. Clin Cancer Res 17: 2613–2618 [DOI] [PubMed] [Google Scholar]
- Chen D, Yoon JB, Gu W 2010. Reactivating the ARF-p53 axis in AML cells by targeting ULF. Cell Cycle 9: 2946–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Kon N, Zhong J, Zhang P, Yu L, Gu W 2013a. Differential effects on ARF stability by normal versus oncogenic levels of c-Myc expression. Mol Cell 51: 46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Wei T, Si X, Wang Q, Li Y, Leng Y, Deng A, Chen J, Wang G, Zhu S, et al. 2013b. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J Biol Chem 288: 14510–14521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV 1999. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet 22: 102–105 [DOI] [PubMed] [Google Scholar]
- Chevillard-Briet M, Quaranta M, Grezy A, Mattera L, Courilleau C, Philippe M, Mercier P, Corpet D, Lough J, Ueda T, et al. 2013. Interplay between chromatin-modifying enzymes controls colon cancer progression through Wnt signaling. Hum Mol Genet 10.1093/hmg/ddt604 [DOI] [PubMed] [Google Scholar]
- Choi SH, Wright JB, Gerber SA, Cole MD 2010. Myc protein is stabilized by suppression of a novel E3 ligase complex in cancer cells. Genes Dev 24: 1236–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciro M, Prosperini E, Quarto M, Grazini U, Walfridsson J, McBlane F, Nucifero P, Pacchiana G, Capra M, Christensen J, et al. 2009. ATAD2 is a novel cofactor for MYC, overexpressed and amplified in aggressive tumors. Cancer Res 69: 8491–8498 [DOI] [PubMed] [Google Scholar]
- Ciurciu A, Komonyi O, Pankotai T, Boros IM 2006. The Drosophila histone acetyltransferase Gcn5 and transcriptional adaptor Ada2a are involved in nucleosomal histone H4 acetylation. Mol Cell Biol 26: 9413–9423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claassen GF, Hann SR 1999. Myc-mediated transformation: The repression connection. Oncogene 18: 2925–2933 [DOI] [PubMed] [Google Scholar]
- Claassen GF, Hann SR 2000. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor β-induced cell-cycle arrest. Proc Natl Acad Sci 97: 9498–9503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Cole MD 2014. MYC association with cancer risk and a new model of MYC-mediated repression. Cold Spring Harb Perspect Med 10.1101/cshperspect.a014316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole MD, Cowling VH 2008. Transcription-independent functions of MYC: Regulation of translation and DNA replication. Nat Rev Mol Cell Biol 9: 810–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conacci-Sorrell M, Ngouenet C, Eisenman RN 2010. Myc-nick: A cytoplasmic cleavage product of Myc that promotes α-tubulin acetylation and cell differentiation. Cell 142: 480–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Conacci-Sorrell M, McFerrin L, Eisenman RN 2014. An overview of MYC and its interactome. Cold Spring Harb Perspect Med 4: a014357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN, Henikoff S 2010. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res 20: 1383–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corvetta D, Chayka O, Gherardi S, D’Acunto CW, Cantilena S, Valli E, Piotrowska I, Perini G, Sala A 2013. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma: Functional and therapeutic implications. J Biol Chem 288: 8332–8341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotterman R, Jin VX, Krig SR, Lemen JM, Wey A, Farnham PJ, Knoepfler PS 2008. N-Myc regulates a widespread euchromatic program in the human genome partially independent of its role as a classical transcription factor. Cancer Res 68: 9654–9662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowling VH, Cole MD 2006. Mechanism of transcriptional activation by the Myc oncoproteins. Semin Cancer Biol 16: 242–252 [DOI] [PubMed] [Google Scholar]
- Cowling VH, Cole MD 2008. An N-Myc truncation analogous to c-Myc-S induces cell proliferation independently of transactivation but dependent on Myc homology box II. Oncogene 27: 1327–1332 [DOI] [PubMed] [Google Scholar]
- Dai MS, Sears R, Lu H 2007. Feedback regulation of c-Myc by ribosomal protein L11. Cell Cycle 6: 2735–2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV 2012. MYC on the path to cancer. Cell 149: 22–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A, Nag A, Pan W, Hay N, Gartel AL, Colamonici O, Mori Y, Raychaudhuri P 2004. Myc-ARF (alternate reading frame) interaction inhibits the functions of Myc. J Biol Chem 279: 36698–36707 [DOI] [PubMed] [Google Scholar]
- Davis AC, Wims M, Spotts GD, Hann SR, Bradley A 1993. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev 7: 671–682 [DOI] [PubMed] [Google Scholar]
- de Nigris F, Botti C, Rossiello R, Crimi E, Sica V, Napoli C 2007. Cooperation between Myc and YY1 provides novel silencing transcriptional targets of α3β1-integrin in tumour cells. Oncogene 26: 382–394 [DOI] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ 2001. c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J Biol Chem 276: 48562–48571 [DOI] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ 2002. Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem 277: 40156–40162 [DOI] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S 2004. Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci 101: 6164–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Woo D, Roussel MF, Cleveland JL 2001. Apoptosis triggered by Myc-induced suppression of Bcl-XL or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol 21: 5063–5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Osta A, Wolffe AP 2000. DNA methylation and histone deacetylation in the control of gene expression: Basic biochemistry to human development and disease. Gene Expr 9: 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Zeller KI, Linhart C, Dang CV, Shamir R, Shiloh Y 2004. In silico identification of transcriptional regulators associated with c-Myc. Nucleic Acids Res 32: 4955–4961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Farrell AS, Sears RC 2014. MYC degradation. Cold Spring Harb Perspect Med 4: a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzio TG, Huff JT, Panning B 2008. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 134: 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al. 2005. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 37: 391–400 [DOI] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B 2001. Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev 15: 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B 2003. MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 4: 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Ueda T, Nagata S, Fukunaga R 2010. Essential role of p400/mDomino chromatin-remodeling ATPase in bone marrow hematopoiesis and cell-cycle progression. J Biol Chem 285: 30214–30223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Gabay M, Li Y, Felsher DW 2014. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med 10.1101/cshperspect.a014241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartel AL, Ye X, Goufman E, Shianov P, Hay N, Najmabadi F, Tyner AL 2001. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc Natl Acad Sci 98: 4510–4515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng F, Wenzel S, Tansey WP 2012. Ubiquitin and proteasomes in transcription. Annu Rev Biochem 81: 177–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherardi S, Valli E, Erriquez D, Perini G 2013. MYCN-mediated transcriptional repression in neuroblastoma: The other side of the coin. Front Oncol 3: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill RM, Gabor TV, Couzens AL, Scheid MP 2013. The MYC-associated protein CDCA7 is phosphorylated by AKT to regulate MYC-dependent apoptosis and transformation. Mol Cell Biol 33: 498–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjerset RA 2006. DNA damage, p14ARF, nucleophosmin (NPM/B23), and cancer. J Mol Histol 37: 239–251 [DOI] [PubMed] [Google Scholar]
- Gordan JD, Thompson CB, Simon MC 2007. HIF and c-Myc: Aibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 12: 108–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C, Squatrito M, Luise C, Syed N, Perna D, Wark L, Martinato F, Sardella D, Verrecchia A, Bennett S, et al. 2007. Tip60 is a haplo-insufficient tumour suppressor required for an oncogene-induced DNA damage response. Nature 448: 1063–1067 [DOI] [PubMed] [Google Scholar]
- Grandori C, Eisenman RN 1997. Myc target genes. Trends Biochem Sci 22: 177–181 [DOI] [PubMed] [Google Scholar]
- Grandori C, Gomez-Roman N, Felton-Edkins ZA, Ngouenet C, Galloway DA, Eisenman RN, White RJ 2005. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol 7: 311–318 [DOI] [PubMed] [Google Scholar]
- Gregory MA, Qi Y, Hann SR 2005. The ARF tumor suppressor: Keeping Myc on a leash. Cell Cycle 4: 249–252 [PubMed] [Google Scholar]
- Grisendi S, Mecucci C, Falini B, Pandolfi PP 2006. Nucleophosmin and cancer. Nat Rev Cancer 6: 493–505 [DOI] [PubMed] [Google Scholar]
- Gstaiger M, Jordan R, Lim M, Catzavelos C, Mestan J, Slingerland J, Krek W 2001. Skp2 is oncogenic and overexpressed in human cancers. Proc Natl Acad Sci 98: 5043–5048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Bhatia K, Magrath IT, Dang CV, Dalla-Favera R 1994. Binding and suppression of the Myc transcriptional activation domain by p107. Science 264: 251–254 [DOI] [PubMed] [Google Scholar]
- Guccione E, Martinato F, Finocchiaro G, Luzi L, Tizzoni L, Dall’ Olio V, Zardo G, Nervi C, Bernard L, Amati B 2006. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol 8: 764–770 [DOI] [PubMed] [Google Scholar]
- Hann SR 2006. Role of post-translational modifications in regulating c-Myc proteolysis, transcriptional activity and biological function. Semin Cancer Biol 16: 288–302 [DOI] [PubMed] [Google Scholar]
- Hann SR, Eisenman RN 1984. Proteins encoded by the human c-myc oncogene: Differential expression in neoplastic cells. Mol Cell Biol 4: 2486–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hann SR, Abrams HD, Rohrschneider LR, Eisenman RN 1983. Proteins encoded by v-myc and c-myc oncogenes: Identification and localization in acute leukemia virus transformants and bursal lymphoma cell lines. Cell 34: 789–798 [DOI] [PubMed] [Google Scholar]
- Hemann MT, Bric A, Teruya-Feldstein J, Herbst A, Nilsson JA, Cordon-Cardo C, Cleveland JL, Tansey WP, Lowe SW 2005. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature 436: 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkert B, Eilers M 2010. Transcriptional repression: The dark side of myc. Genes Cancer 1: 580–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkert B, Dwertmann A, Herold S, Abed M, Naud JF, Finkernagel F, Harms GS, Orian A, Wanzel M, Eilers M 2010. The Arf tumor suppressor protein inhibits Miz1 to suppress cell adhesion and induce apoptosis. J Cell Biol 188: 905–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, Syvaoja J, Saluz HP, Haenel F, Eilers M 2002. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell 10: 509–521 [DOI] [PubMed] [Google Scholar]
- Hervouet E, Vallette FM, Cartron PF 2009. Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics 4: 487–499 [DOI] [PubMed] [Google Scholar]
- Hirst SK, Grandori C 2000. Differential activity of conditional MYC and its variant MYC-S in human mortal fibroblasts. Oncogene 19: 5189–5197 [DOI] [PubMed] [Google Scholar]
- Hrzenjak A, Moinfar F, Kremser ML, Strohmeier B, Staber PB, Zatloukal K, Denk H 2006. Valproate inhibition of histone deacetylase 2 affects differentiation and decreases proliferation of endometrial stromal sarcoma cells. Mol Cancer Ther 5: 2203–2210 [DOI] [PubMed] [Google Scholar]
- Huang W, Zhao S, Ammanamanchi S, Brattain M, Venkatasubbarao K, Freeman JW 2005. Trichostatin A induces transforming growth factor β type II receptor promoter activity and acetylation of Sp1 by recruitment of PCAF/p300 to a Sp1.NF-Y complex. J Biol Chem 280: 10047–10054 [DOI] [PubMed] [Google Scholar]
- Hydbring P, Larsson LG 2010. Cdk2: A key regulator of the senescence control function of Myc. Aging (Albany NY) 2: 244–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iraci N, Diolaiti D, Papa A, Porro A, Valli E, Gherardi S, Herold S, Eilers M, Bernardoni R, Della Valle G, et al. 2011. A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res 71: 404–412 [DOI] [PubMed] [Google Scholar]
- Iritani BM, Eisenman RN 1999. c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci 96: 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackstadt R, Jung P, Hermeking H 2013. AP4 directly downregulates p16 and p21 to suppress senescence and mediate transformation. Cell Death Dis 4: e775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Wu G, Zhan X, Nolan A, Koh C, De Marzo A, Doan HM, Fan J, Cheadle C, Fallahi M, et al. 2011. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 6: e26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G, Espeseth A, Hazuda DJ, Margolis DM 2007. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol 81: 10914–10923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung P, Hermeking H 2009. The c-MYC-AP4-p21 cascade. Cell Cycle 8: 982–989 [DOI] [PubMed] [Google Scholar]
- Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, et al. 2007. CIP2A inhibits PP2A in human malignancies. Cell 130: 51–62 [DOI] [PubMed] [Google Scholar]
- Kasowski M, Grubert F, Heffelfinger C, Hariharan M, Asabere A, Waszak SM, Habegger L, Rozowsky J, Shi M, Urban AE, et al. 2010. Variation in transcription factor binding among humans. Science 328: 232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur M, Cole MD 2013. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res 73: 695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenneth NS, Ramsbottom BA, Gomez-Roman N, Marshall L, Cole PA, White RJ 2007. TRRAP and GCN5 are used by c-Myc to activate RNA polymerase III transcription. Proc Natl Acad Sci 104: 14917–14922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiernan RE, Emiliani S, Nakayama K, Castro A, Labbe JC, Lorca T, Nakayama Ki K, Benkirane M 2001. Interaction between cyclin T1 and SCFSKP2 targets CDK9 for ubiquitination and degradation by the proteasome. Mol Cell Biol 21: 7956–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Herbst A, Tworkowski KA, Salghetti SE, Tansey WP 2003. Skp2 regulates Myc protein stability and activity. Mol Cell 11: 1177–1188 [DOI] [PubMed] [Google Scholar]
- Kim J, Lee JH, Iyer VR 2008. Global identification of Myc target genes reveals its direct role in mitochondrial biogenesis and its E-box usage in vivo. PLoS ONE 3: e1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler PS, Zhang XY, Cheng PF, Gafken PR, McMahon SB, Eisenman RN 2006. Myc influences global chromatin structure. EMBO J 25: 2723–2734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knuesel MT, Meyer KD, Bernecky C, Taatjes DJ 2009. The human CDK8 subcomplex is a molecular switch that controls Mediator coactivator function. Genes Dev 23: 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR III, Menssen A, Hermeking H 2007. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle 6: 205–217 [DOI] [PubMed] [Google Scholar]
- Kurland JF, Tansey WP 2008. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res 68: 3624–3629 [DOI] [PubMed] [Google Scholar]
- Kurosu T, Peterlin BM 2004. VP16 and ubiquitin; binding of P-TEFb via its activation domain and ubiquitin facilitates elongation of transcription of target genes. Curr Biol 14: 1112–1116 [DOI] [PubMed] [Google Scholar]
- Laurenti E, Varnum-Finney B, Wilson A, Ferrero I, Blanco-Bose WE, Ehninger A, Knoepfler PS, Cheng PF, MacDonald HR, Eisenman RN, et al. 2008. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 3: 611–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawlor ER, Soucek L, Brown-Swigart L, Shchors K, Bialucha CU, Evan GI 2006. Reversible kinetic analysis of Myc targets in vivo provides novel insights into Myc-mediated tumorigenesis. Cancer Res 66: 4591–4601 [DOI] [PubMed] [Google Scholar]
- Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR 1997. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 387: 422–426 [DOI] [PubMed] [Google Scholar]
- Leone G, Sears R, Huang E, Rempel R, Nuckolls F, Park CH, Giangrande P, Wu L, Saavedra HI, Field SJ, et al. 2001. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol Cell 8: 105–113 [DOI] [PubMed] [Google Scholar]
- *.Levens D 2013. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harb Perspect Med 3: a014233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Davie JR 2010. The role of Sp1 and Sp3 in normal and cancer cell biology. Ann Anat 192: 275–283 [DOI] [PubMed] [Google Scholar]
- Li Z, Hann SR 2009. The Myc-nucleophosmin-ARF network: A complex web unveiled. Cell Cycle 8: 2703–2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hann SR 2013. Nucleophosmin is essential for c-Myc nucleolar localization and c-Myc-mediated rDNA transcription. Oncogene 32: 1988–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LH, Nerlov C, Prendergast G, MacGregor D, Ziff EB 1994. c-Myc represses transcription in vivo by a novel mechanism dependent on the initiator element and Myc box II. EMBO J 13: 4070–4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Boone D, Hann SR 2008. Nucleophosmin interacts directly with c-Myc and controls c-Myc-induced hyperproliferation and transformation. Proc Natl Acad Sci 105: 18794–18799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Loven J, Rahl PB, Paranal RM, Burge CB, Bradner JE, Lee TI, Young RA 2012. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 151: 56–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI 1995. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res 23: 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Tesfai J, Evrard YA, Dent SY, Martinez E 2003. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. Mol Cell Biol 278: 20405–20412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Tee AE, Porro A, Smith SA, Dwarte T, Liu PY, Iraci N, Sekyere E, Haber M, Norris MD, et al. 2007. Activation of tissue transglutaminase transcription by histone deacetylase inhibition as a therapeutic approach for Myc oncogenesis. Proc Natl Acad Sci 104: 18682–18687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Vorontchikhina M, Wang YL, Faiola F, Martinez E 2008. STAGA recruits Mediator to the MYC oncoprotein to stimulate transcription and cell proliferation. Mol Cell Biol 28: 108–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleonart ME, Vidal F, Gallardo D, Diaz-Fuertes M, Rojo F, Cuatrecasas M, Lopez-Vicente L, Kondoh H, Blanco C, Carnero A, et al. 2006. New p53 related genes in human tumors: Significant downregulation in colon and lung carcinomas. Oncol Rep 16: 603–608 [DOI] [PubMed] [Google Scholar]
- Loizou JI, Oser G, Shukla V, Sawan C, Murr R, Wang ZQ, Trumpp A, Herceg Z 2009. Histone acetyltransferase cofactor Trrap is essential for maintaining the hematopoietic stem/progenitor cell pool. Mol Cell Biol 183: 6422–6431 [DOI] [PubMed] [Google Scholar]
- Luscher B, Vervoorts J 2012. Regulation of gene transcription by the oncoprotein MYC. Mol Cell Biol 494: 145–160 [DOI] [PubMed] [Google Scholar]
- Lutterbach B, Hann SR 1994. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol Cell Biol 14: 5510–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall GM, Gherardi S, Xu N, Neiron Z, Trahair T, Scarlett CJ, Chang DK, Liu PY, Jankowski K, Iraci N, et al. 2010. Transcriptional upregulation of histone deacetylase 2 promotes Myc-induced oncogenic effects. Oncogene 29: 5957–5968 [DOI] [PubMed] [Google Scholar]
- Martinato F, Cesaroni M, Amati B, Guccione E 2008. Analysis of Myc-induced histone modifications on target chromatin. PLoS ONE 3: e3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Cerdeno V, Lemen JM, Chan V, Wey A, Lin W, Dent SR, Knoepfler PS 2012. N-Myc and GCN5 regulate significantly overlapping transcriptional programs in neural stem cells. PLoS ONE 7: e39456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Fukamachi K, Uehara N, Tsuda H, Tsubura A 2008. Induction of a novel histone deacetylase 1/c-Myc/Mnt/Max complex formation is implicated in parity-induced refractoriness to mammary carcinogenesis. Cancer Sci 99: 309–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattera L, Escaffit F, Pillaire MJ, Selves J, Tyteca S, Hoffmann JS, Gourraud PA, Chevillard-Briet M, Cazaux C, Trouche D 2009. The p400/Tip60 ratio is critical for colorectal cancer cell proliferation through DNA damage response pathways. Oncogene 28: 1506–1517 [DOI] [PubMed] [Google Scholar]
- *.McMahon SB 2014. MYC and the control of apoptosis. Cold Spring Harb Perspect Med 10.1101/cshperspect.a014407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD 2000. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol 20: 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, Penn LZ 2008. Reflecting on 25 years with MYC. Nat Rev Cancer 8: 976–990 [DOI] [PubMed] [Google Scholar]
- Minella AC, Clurman BE 2005. Mechanisms of tumor suppression by the SCFFbw7. Cell Cycle 4: 1356–1359 [DOI] [PubMed] [Google Scholar]
- Muratani M, Tansey WP 2003. How the ubiquitin-proteasome system controls transcription. Nat Rev Mol Cell Biol 4: 192–201 [DOI] [PubMed] [Google Scholar]
- Murr R, Vaissiere T, Sawan C, Shukla V, Herceg Z 2007. Orchestration of chromatin-based processes: Mind the TRRAP. Oncogene 26: 5358–5372 [DOI] [PubMed] [Google Scholar]
- Nie Z, Hu G, Wei G, Cui K, Yamane A, Resch W, Wang R, Green DR, Tessarollo L, Casellas R, et al. 2012. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 151: 68–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemela M, Kauko O, Sihto H, Mpindi JP, Nicorici D, Pernila P, Kallioniemi OP, Joensuu H, Hautaniemi S, Westermarck J 2012. CIP2A signature reveals the MYC dependency of CIP2A-regulated phenotypes and its clinical association with breast cancer subtypes. Oncogene 31: 4266–4278 [DOI] [PubMed] [Google Scholar]
- Nikiforov MA, Chandriani S, Park J, Kotenko I, Matheos D, Johnsson A, McMahon SB, Cole MD 2002. TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol Cell Biol 22: 5054–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donovan KJ, Diedler J, Couture GC, Fak JJ, Darnell RB 2010. The onconeural antigen cdr2 is a novel APC/C target that acts in mitosis to regulate c-myc target genes in mammalian tumor cells. PLoS ONE 5: e10045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster SK, Ho CS, Soucie EL, Penn LZ 2002. The myc oncogene: MarvelouslY complex. Adv Cancer Res 84: 81–154 [DOI] [PubMed] [Google Scholar]
- Oster SK, Mao DY, Kennedy J, Penn LZ 2003. Functional analysis of the N-terminal domain of the Myc oncoprotein. Oncogene 22: 1998–2010 [DOI] [PubMed] [Google Scholar]
- Pal S, Yun R, Datta A, Lacomis L, Erdjument-Bromage H, Kumar J, Tempst P, Sif S 2003. mSin3A/histone deacetylase 2- and PRMT5-containing Brg1 complex is involved in transcriptional repression of the Myc target gene cad. Mol Cell Biol 23: 7475–7487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Wood MA, Cole MD 2002. BAF53 forms distinct nuclear complexes and functions as a critical c-Myc-interacting nuclear cofactor for oncogenic transformation. Mol Cell Biol 22: 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JH, McMahon SB 2006. Targeting of Miz-1 is essential for Myc-mediated apoptosis. J Biol Chem 281: 3283–3289 [DOI] [PubMed] [Google Scholar]
- Patel JH, McMahon SB 2007. BCL2 is a downstream effector of MIZ-1 essential for blocking c-MYC-induced apoptosis. J Biol Chem 282: 5–13 [DOI] [PubMed] [Google Scholar]
- Perna D, Faga G, Verrecchia A, Gorski MM, Barozzi I, Narang V, Khng J, Lim KC, Sung WK, Sanges R, et al. 2012. Genome-wide mapping of Myc binding and gene regulation in serum-stimulated fibroblasts. Oncogene 31: 1695–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M 1997. An alternative pathway for gene regulation by Myc. EMBO J 16: 5672–5686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Lucena A, Ho CS, Mao DY, Sheng Y, Laister RC, Muhandiram R, Lu Y, Seet BT, Katz S, Szyperski T, et al. 2005. A structure-based model of the c-Myc/Bin1 protein interaction shows alternative splicing of Bin1 and c-Myc phosphorylation are key binding determinants. J Mol Biol 351: 182–194 [DOI] [PubMed] [Google Scholar]
- Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, Potes J, Chen K, Orlow I, Lee HW, et al. 1998. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 92: 713–723 [DOI] [PubMed] [Google Scholar]
- Popov N, Wahlstrom T, Hurlin PJ, Henriksson M 2005. Mnt transcriptional repressor is functionally regulated during cell cycle progression. Oncogene 24: 8326–8337 [DOI] [PubMed] [Google Scholar]
- Popov N, Schulein C, Jaenicke LA, Eilers M 2010. Ubiquitylation of the amino terminus of Myc by SCFβ-TrCP antagonizes SCFFbw7-mediated turnover. Nat Cell Biol 12: 973–981 [DOI] [PubMed] [Google Scholar]
- Qi Y, Gregory MA, Li Z, Brousal JP, West K, Hann SR 2004. p19ARF directly and differentially controls the functions of c-Myc independently of p53. Nature 431: 712–717 [DOI] [PubMed] [Google Scholar]
- Quintana AM, Liu F, O’Rourke JP, Ness SA 2011. Identification and regulation of c-Myb target genes in MCF-7 cells. BMC Cancer 11: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Rahl PB, Young RA 2014. MYC and transcription elongation. Cold Spring Harb Perspect Med 4: a020990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangasamy D 2010. Histone variant H2A.Z can serve as a new target for breast cancer therapy. Curr Med Chem 17: 3155–3161 [DOI] [PubMed] [Google Scholar]
- Rempel RE, Mori S, Gasparetto M, Glozak MA, Andrechek ER, Adler SB, Laakso NM, Lagoo AS, Storms R, Smith C, et al. 2009. A role for E2F activities in determining the fate of Myc-induced lymphomagenesis. PLoS Genet 5: e1000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez RM, Huidobro C, Urdinguio RG, Mangas C, Soldevilla B, Dominguez G, Bonilla F, Fernandez AF, Fraga MF 2012. Aberrant epigenetic regulation of bromodomain BRD4 in human colon cancer. J Mol Med (Berl) 90: 587–595 [DOI] [PubMed] [Google Scholar]
- *.Sabò A, Amati B 2014. Genome recognition by MYC. Cold Spring Harb Perspect Med 4: a014191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamuro D, Elliott KJ, Wechsler-Reya R, Prendergast GC 1996. BIN1 is a novel MYC-interacting protein with features of a tumour suppressor. Nat Genet 14: 69–77 [DOI] [PubMed] [Google Scholar]
- Sakuraba K, Yokomizo K, Shirahata A, Goto T, Saito M, Ishibashi K, Kigawa G, Nemoto H, Hibi K 2011. TIP60 as a potential marker for the malignancy of gastric cancer. Anticancer Res 31: 77–79 [PubMed] [Google Scholar]
- Sapountzi V, Logan IR, Robson CN 2006. Cellular functions of TIP60. Int J Biochem Cell Biol 38: 1496–1509 [DOI] [PubMed] [Google Scholar]
- Satou A, Taira T, Iguchi-Ariga SM, Ariga H 2001. A novel transrepression pathway of c-Myc. Recruitment of a transcriptional corepressor complex to c-Myc by MM-1, a c-Myc-binding protein. J Biol Chem 276: 46562–46567 [DOI] [PubMed] [Google Scholar]
- Schneider A, Peukert K, Eilers M, Hanel F 1997. Association of Myc with the zinc-finger protein Miz-1 defines a novel pathway for gene regulation by Myc. Curr Top Microbiol Immunol 224: 137–146 [DOI] [PubMed] [Google Scholar]
- Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D, Kohlhuber F 1999. Control of cell growth by c-Myc in the absence of cell division. Curr Biol 9: 1255–1258 [DOI] [PubMed] [Google Scholar]
- Secombe J, Eisenman RN 2007. The function and regulation of the JARID1 family of histone H3 lysine 4 demethylases: The Myc connection. Cell Cycle 6: 1324–1328 [DOI] [PubMed] [Google Scholar]
- Seo HR, Kim J, Bae S, Soh JW, Lee YS 2008. Cdk5-mediated phosphorylation of c-Myc on Ser-62 is essential in transcriptional activation of cyclin B1 by cyclin G1. J Biol Chem 283: 15601–15610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ 2006. Divorcing ARF and p53: An unsettled case. Nat Rev Cancer 6: 663–673 [DOI] [PubMed] [Google Scholar]
- Smith KN, Singh AM, Dalton S 2010. Myc represses primitive endoderm differentiation in pluripotent stem cells. Cell Stem Cell 7: 343–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spotts GD, Patel SV, Xiao Q, Hann SR 1997. Identification of downstream-initiated c-Myc proteins which are dominant-negative inhibitors of transactivation by full-length c-Myc proteins. Mol Cell Biol 17: 1459–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, et al. 2001. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol 3: 392–399 [DOI] [PubMed] [Google Scholar]
- Subtil-Rodriguez A, Reyes JC 2011. To cross or not to cross the nucleosome, that is the elongation question. RNA Biol 8: 389–393 [DOI] [PubMed] [Google Scholar]
- Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP, et al. 2011. Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 146: 92–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LR, Tansey WP 2011. Proteolytic control of the oncoprotein transcription factor Myc. Nat Cell Biol 110: 77–106 [DOI] [PubMed] [Google Scholar]
- To KK, Huang LE 2013. An efficient way of studying protein-protein interactions involving HIF-α, c-Myc, and Sp1. Nat Cell Biol 1012: 77–84 [DOI] [PubMed] [Google Scholar]
- Uribesalgo I, Buschbeck M, Gutierrez A, Teichmann S, Demajo S, Kuebler B, Nomdedeu JF, Martin-Caballero J, Roma G, Benitah SA, et al. 2011. E-box-independent regulation of transcription and differentiation by MYC. Nat Cell Biol 13: 1443–1449 [DOI] [PubMed] [Google Scholar]
- Varlakhanova NV, Knoepfler PS 2009. Acting locally and globally: Myc’s ever-expanding roles on chromatin. Nat Cell Biol 69: 7487–7490 [DOI] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff JM, Rottmann S, Lilischkis R, Walsemann G, Dohmann K, Austen M, Luscher B 2003. Stimulation of c-MYC transcriptional activity and acetylation by recruitment of the cofactor CBP. EMBO Rep 4: 484–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vervoorts J, Luscher-Firzlaff J, Luscher B 2006. The ins and outs of MYC regulation by posttranslational mechanisms. J Biol Chem 281: 34725–34729 [DOI] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, et al. 2003. The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 11: 1189–1200 [DOI] [PubMed] [Google Scholar]
- Wang X, Cunningham M, Zhang X, Tokarz S, Laraway B, Troxell M, Sears RC 2011a. Phosphorylation regulates c-Myc’s oncogenic activity in the mammary gland. Cancer Res 71: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Lin S, Li JJ, Xu Z, Yao H, Zhu X, Xie D, Shen Z, Sze J, Li K, et al. 2011b. MYC protein inhibits transcription of the microRNA cluster MC-let-7a-1∼let-7d via noncanonical E-box. J Biol Chem 286: 39703–39714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welboren WJ, Sweep FC, Span PN, Stunnenberg HG 2009. Genomic actions of estrogen receptor α: What are the targets and how are they regulated? Endocr Relat Cancer 16: 1073–1089 [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Grim JE, Eisenman RN, Clurman BE 2004a. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr Biol 14: 1852–1857 [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, Clurman BE 2004b. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci 101: 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Wiese KE, Walz S, von Eyss B, Wolf E, Athineos D, Sansom O, Eilers M 2013. The role of MIZ1 in MYC-dependent tumorigenesis. Cold Spring Harb Perspect Med 3: a014290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DD, Muthurajan UM, Hieb AR, Luger K 2011. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. J Biol Chem 286: 41883–41892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittschieben BO, Fellows J, Du W, Stillman DJ, Svejstrup JQ 2000. Overlapping roles for the histone acetyltransferase activities of SAGA and elongator in vivo. EMBO J 19: 3060–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, McMahon SB, Cole MD 2000. An ATPase/helicase complex is an essential cofactor for oncogenic transformation by c-Myc. Mol Cell 5: 321–330 [DOI] [PubMed] [Google Scholar]
- Xiao Q, Claassen G, Shi J, Adachi S, Sedivy J, Hann SR 1998. Transactivation-defective c-MycS retains the ability to regulate proliferation and apoptosis. Genes Dev 12: 3803–3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI 2004. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J 23: 2116–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Chai L, Fowles TC, Alipio Z, Xu D, Fink LM, Ward DC, Ma Y 2008. Genome-wide analysis reveals Sall4 to be a major regulator of pluripotency in murine-embryonic stem cells. Proc Natl Acad Sci 105: 19756–19761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sung E, Donlin-Asp PG, Corces VG 2013. A subset of Drosophila Myc sites remain associated with mitotic chromosomes colocalized with insulator proteins. Nat Commun 4: 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, et al. 2012. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-cell lymphomas. Cancer Cell 22: 506–523 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhang Q, Spears E, Boone DN, Li Z, Gregory MA, Hann SR 2013. Domain-specific c-Myc ubiquitylation controls c-Myc transcriptional and apoptotic activity. Proc Natl Acad Sci 110: 978–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Heng JI, Guardavaccaro D, Jiang R, Pagano M, Guillemot F, Iavarone A, Lasorella A 2008. The HECT-domain ubiquitin ligase Huwe1 controls neural differentiation and proliferation by destabilizing the N-Myc oncoprotein. Nat Cell Biol 10: 643–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Hao Q, Liao JM, Liao P, Lu H 2013. Ribosomal protein S14 negatively regulates c-Myc activity. J Biol Chem 288: 21793–21801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF 1998. Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev 12: 2424–2433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippo A, De Robertis A, Serafini R, Oliviero S 2007. PIM1-dependent phosphorylation of histone H3 at serine 10 is required for MYC-dependent transcriptional activation and oncogenic transformation. Nat Cell Biol 9: 932–944 [DOI] [PubMed] [Google Scholar]
- Zou Y, Wu J, Giannone RJ, Boucher L, Du H, Huang Y, Johnson DK, Liu Y, Wang Y 2008. Nucleophosmin/B23 negatively regulates GCN5-dependent histone acetylation and transactivation. J Biol Chem 283: 5728–5737 [DOI] [PubMed] [Google Scholar]