

SUMMARY
C3a and C5a receptor (C3aR and C5aR) signaling by dendritic cells and CD4+ cells provides costimulatory and survival signals to T effector cells. Here, we demonstrate that when C3aR and C5aR signals are not transduced into CD4+ cells, PI-3Kγ-AKT-mTOR signaling ceases, PKA activation increases, auto-inductive transforming growth factor- β1 (TGF-β1) signaling initiates, and CD4+ cells become Foxp3+ T regulatory cells (iTregs). Endogenous TGF-β1 suppresses C3aR and C5aR signaling by preventing C3a and C5a production and upregulating C5L2, an alternate C5a receptor. Absent C3aR and C5aR signaling decreases costimulatory molecule and interleukin-6 production and augments interleukin-10 production. The resulting iTregs exert robust suppression, possess enhanced stability, and suppress ongoing autoimmune disease. Human iTregs with potent suppressor activity can be induced exploiting this insight.
T lymphocytes expressing the transcription factor Foxp3 play critical roles in controlling many aspects of immune responses 1. Foxp3+ cells are generated during the normal process of T cell differentiation in the thymus (termed thymic derived T regulatory cells, tTreg cells), but also in peripheral sites during the course of immune responses (termed induced Treg cells, iTreg cells) 1. Although much progress has been made in characterizing the transcriptional machinery involved in the generation of Tregs 2, how iTregs are induced physiologically remains poorly understood. This knowledge is key to understanding how T cell immunity is regulated as well as highly relevant for managing diverse human disorders. Multiple factors 3,4 have been proposed as participating in iTreg generation and much emphasis has been placed on the cytokine microenvironment, in particular the involvement of transforming growth factor-β1 (TGF-β1). Nevertheless, the upstream signals that give rise to iTregs thus far remain poorly characterized.
Previous studies5 showed that an important early event during T effector cell activation is that the cognate interaction of CD4+ T cells with dendritic cells (DCs) amplifies T cell and DC synthesis of the alternative pathway (AP) complement components C3, factor B (fB), factor D (fD) in conjunction with C5 and the G-protein coupled receptors (GPCRs) C3a and C5a receptors (C3aR and C5aR). Concurrent with this, both T cells and DCs downregulate their expression of the cell surface C3 and C5 convertase inhibitor, decay accelerating factor (DAF or CD55). In the absence of the inhibitory effect of DAF, C3 and C5 convertases stably assemble from the locally produced C3, fB, and fD at the adjoining DC-CD4+ T cell surfaces. These enzymes act on the nascent C3 and C5 to generate C3a and C5a. The C3a and C5a anaphylatoxins, like cytokines, engage C3aR and C5aR on both the DCs and CD4+ T cells and transduce GPCR signals into both partners. When C3aR and C5aR signaling were simultaneously disabled, a marked reduction in both costimulatory and survival signals was needed for effector T cell responses5-7. Moreover, C3aR and C5aR signaling was also required for the differentiation of TH1 cells and TH17 cells, as signaling via these receptors mediated the production of interleukin-12 (IL-12) by DCs and expression of the IL-12 receptor (IL-12R) by CD4+ T cells8, as well as the production of IL-6 and IL-238. Some data has suggested that C5aR signaling in DCs is essential for biasing T cell differentiation into a TH17 response8,9, but the role of GPCR signaling within the CD4+ T cells themselves was not investigated. In contrast, other studies indicated that signaling via C3aR and C5aR is important in both responding T cells and DCs 5,7.
The major consequences of C3aR and C5aR signaling are the production of IL-611, IL-12 and the promotion of TH17 differentiation5,7. iTreg induction is suppressed in such a cytokine milieu 3,4,10-12. Therefore, we hypothesized that the absence of C3aR and C5aR signal transduction in both responder CD4+ T cells and DCs might promote the production of iTregs. Here, we demonstrate that the concurrent absence of C3aR and C5aR GPCR signal transduction into CD4+ T cells results in the induction of a high percentage of iTregs in an endogenous TGF-β1-dependent fashion. The iTregs that are generated when CD4+ T cells are devoid of both GPCR signals manifest potent suppressor function and stability both in vitro and in vivo. Human iTregs with potent suppressor activity can be induced exploiting this new insight, a finding that potentially could have therapeutic importance.
RESULTS
Absent C3aR or C5aR signals in CD4+ cells induces iTregs
To determine whether absent C3aR and/or C5aR signaling in naive CD4+ T cells results in the induction of Tregs, we activated sorted Foxp3−CD4+ T cells from Foxp3-GFP reporter mice crossed with C3aR deficient (C3ar1−/−), C5aR deficient (C5ar1−/−) or C3ar1−/−C5ar1−/− double knockout mice with anti-CD3+CD28 activation beads and IL-2 in the absence of DCs. Stimulation of WT Foxp3− CD4+ T cells under these conditions failed to induce Foxp3 expression. However stimulation of C3ar1−/− or C5ar1−/− Foxp3− CD4+ T cells resulted in induction of ~6% Foxp3+ T cells. Furthermore, a much higher percentage of Foxp3+ T cells (~27%) was observed when Foxp3− T cells from C3ar1−/−C5ar1−/− mice were similarly stimulated (Fig. 1a). Incubation for longer times (5 days) did not significantly change the differences between the single and double knockouts (not shown). No Foxp3+ cells were induced in the absence of added IL-2 (not shown). To test whether the induced Foxp3+ cells exerted suppressor activity characteristic of iTregs, we incubated sorted Foxp3+ cells generated from each genotype in decreasing (Teff/Treg) ratios with sorted wild type (WT) Foxp3−CD4+ T cells prelabeled with CellTracker Red and activated with anti-CD3+CD28. Foxp3+ cells derived from the single knockouts exerted suppressor activity similar to that of sorted Foxp3+ cells induced from WT Foxp3− CD4+ T cells with exogenously added TGF-β1 (Fig. 1b). However, sorted Foxp3+ cells derived from the C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells exerted greater suppressor activity (Fig. 1b). Induced C3ar1−/−C5ar1−/− Foxp3+ CD4+ T cells resembled tTregs in that they failed to produce IL-2 following phorbol ester (PMA) and ionomycin treatment 3 and were anergic to anti-CD3+CD28 stimulation 3 (not shown). To verify that the effects of the C3aR and C5aR knockouts apply in WT CD4+ T cells, we stimulated sorted WT Foxp3− CD4+ T cells in the presence of C3aR and C5aR pharmaceutical antagonists (C3aR-A/C5aR-A) or in the presence of anti-C3a and C5a monoclonal antibodies (mAbs) specific for neo-epitopes in the C3a and C5a ligands, in each case individually or together. Few Foxp3+ T cells (6.5-8%) were generated when WT CD4+ T cells were cultured with an individual pharmaceutical receptor antagonist or mAb against C3a or C5a, whereas a much higher percentage (~28%) was induced when both receptors or both ligands were blocked (Fig. 1c). Thus, the simultaneous lack of transmission of C3aR and C5aR GPCR signals into naive CD4+ T cells in the absence of DCs leads to the efficient induction of Foxp3+ cells possessing robust suppressor activity (Fig. 1b). Although these results are consistent with previous findings (Supplementary Fig. 1) 5 that autocrine C3aR and C5aR signaling operates tonically in CD4+ cells 5, these previous data also showed that DCs produce much more C3a and C5a per cell. As the above data show, however, the absence or presence of transmitted C3aR and C5aR GPCR signals in the T cells themselves influences T cell differentiation.
Figure 1. iTregs develop from stimulated CD4+ cells devoid of C3aR and C5aR signaling in the absence of DCs.
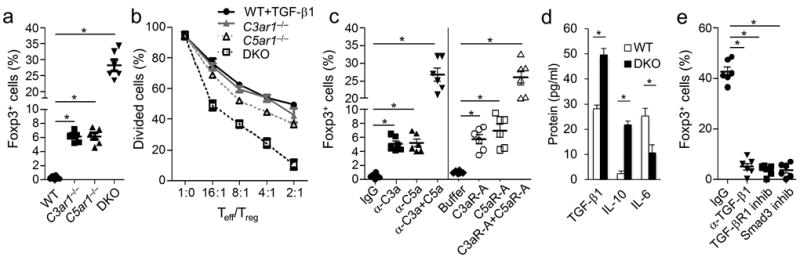
All inductions were performed for 3 days with 1 ×106 CD62LhiFoxp3−CD25− CD4+ T cells, rhIL-2 (5 ng/ml), and anti-CD3 + CD28 beads (per manufacturer’s instructions), experiments were repeated two times. (a) WT, C3ar1−/−, C5ar1−/−, or C3ar1−/− C5ar1−/− Foxp3− cells were activated and assayed for percent Foxp3+CD25+ by flow cytometry (*P<0.001, n=5). (b) Following iTreg induction (as in a), flow sorted WT, C3ar1−/−, C5ar1−/−, or C3ar1−/− C5ar1−/− Foxp3+CD25+ cells were incubated with 106 CellTracker Red™ labeled CD25−Foxp3− CD4+ WT cells in differing Teff/iTreg ratios + anti-CD3 (5 μg/ml) and 2.5×105 CD11c+ DCs. Relative suppressive capacity was determined by percent Red-labeled dividers (n=5). (c) WT Foxp3− CD4+ T cells were induced as in (a) in the absence or presence of anti-C3a (10 μg/ml), anti-C5a (10 μg/ml), or both or in the absence and presence of the antagonists C3aR-A (10 nM), C5aR-A (10 nM), or both. CD4+ T cells then were assayed for Foxp3 expression by flow cytometry (*P<0.001, n=6). (d) Following iTreg induction (as in a), sorted WT and C3ar1−/−C5ar1−/− Foxp3+CD25+ cells were washed and recultured for 24 hr in the absence of further stimulation. Culture supernatants were assayed for TGF-β, IL-6, and IL-10 by ELISA (*P<0.001, n=6). (e) C3ar1−/−C5ar1−/− iTregs were induced (as in a) in the absence and presence of anti-TGF-β mAb (5 μg/ml), TGF-βR1 inhibitor (10 nM), or Smad3 inhibitor (5 μM). Foxp3+ CD25+ Treg percentages were quantified by flow cytometry.
Supernatants from stimulated C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells contained lower levels of IL-6 but higher levels of IL-10 and TGF-β1 than supernatants from stimulated WT Foxp3− WT CD4+ T cells (Fig. 1d). This finding suggested that the absence of C3aR plus C5aR signaling in CD4+ T cells resulted in the endogenous production of TGF-β1 needed for the induction of Foxp3+ cells. This appeared to be the case, as the induction of Foxp3+ T cells from C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells was inhibited when we added anti-TGF-β1 mAb, or specific antagonists of TGF-βR1 or of TGF-βR1 downstream signaling (Smad3 inhibitor) to the induction cultures (Fig. 1e). Thus, analogous to auto-inductive cytokine signaling loops that are mechanistically connected with the induction of TH1, Th2, and TH17 cells 13, TGF-β1 generated from stimulated CD4+ T cells, that are devoid of C3aR and C5aR GPCR signals, amplifies its own production and this auto-inductive signaling loop is essential for iTreg induction (Fig. 1e). Consequently, absent C3aR plus C5aR signaling into naïve CD4+ cells enables auto-inductive TGF-β1 signaling and defines a previously unidentified autocrine signaling loop that is connected with Foxp3+ iTreg lineage commitment (Supplementary Fig. 2).
Absent T cell C3aR and C5aR signals controls DCs
As T cells are activated by DCs physiologically, we next stimulated sorted WT or C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells with anti-CD3 and IL-2 in the presence of WT DCs with or without added TGF-β1. Foxp3+ cells were induced in both cases in the presence of added TGF-β1 (Fig. 2a). Only low numbers of Foxp3+ T cells (~6%) were induced in cultures of C3ar1−/− or C5ar1−/− Foxp3− CD4+ T cells containing WT DCs without added TGF-β1 (Supplementary Fig. 3a). However, almost as many Foxp3+ cells were induced from C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells (45-65%) in the absence of TGF-β1 (Fig. 2a) as in the presence of TGF-β1. Thus, even in the presence of C3aR and C5aR signaling by DCs, the absence of C3aR and C5aR signaling in the responder CD4+ T cells dominates and iTregs are induced.
Figure 2. Absent C3aR and C5aR signaling in CD4+ cells dominates in iTreg induction when WT DCs are present.
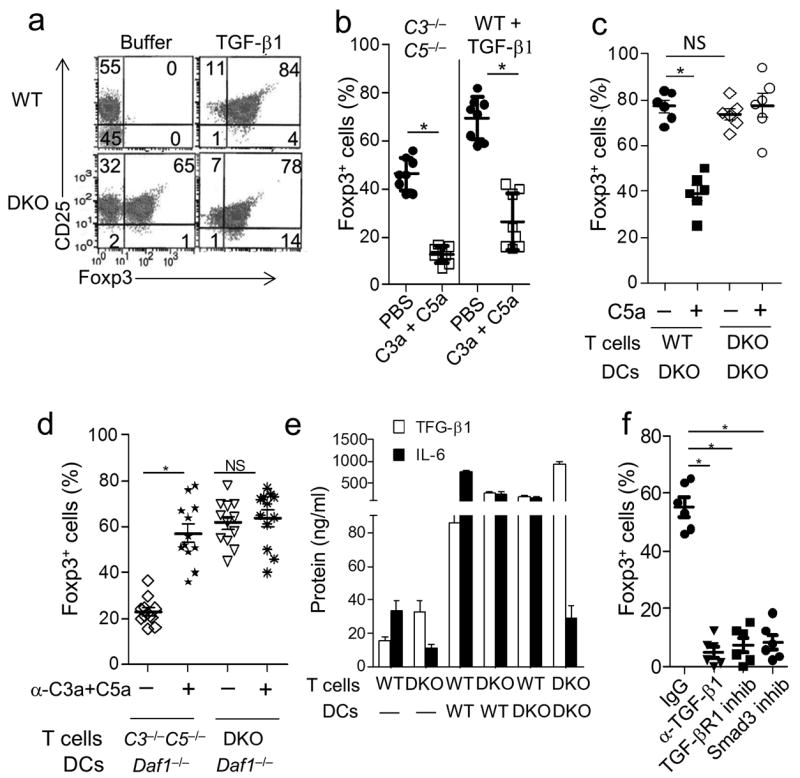
iTreg induction and activation experiments were performed as in Fig 1 with 2.5×105 CD11c+ DCs and anti-CD3 mAb (5 μg/ml) instead of anti-CD3+CD28 beads, experiments were repeated two times. (a) Sorted WT and C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells were incubated with WT DCs ± TGF-β1 (5 ng/ml) and assayed for CD25 and Foxp3 expression by flow cytometry (*P<0.001, n=10). (b) (Left) WT Foxp3− CD4+ T cells were incubated with C3−/−C5−/− DCs without TGF-β1 ± C3a/C5a (100 ng/ml) and Foxp3+CD25+ cells quantified by flow cytometry. (Right) WT Foxp3− CD4+ T cells were incubated in the presence of TGF-β1 (5 ng/ml) ± C3a/C5a (100 ng/ml) and percent Foxp3+CD25+ cells quantified (*P<0.001, n=7). (c) Sorted WT or C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells were incubated with C3ar1−/−C5ar1−/− DCs in the absence or presence of C5a (100 ng/ml) after which percent Foxp3+CD25+ cells was quantified. The % Foxp3+ cells with DKO or WT T cells did not significantly differ (P=0.54). (d) Sorted C3−/−C5−/− or C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells were incubated with Daf1−/− DCs ± anti-C3a and anti-C5a mAbs after which percent Foxp3+CD25+ cells was quantified (*P<0.001; n=6). (e) Sorted WT Foxp3− or C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells were incubated for 24 hr either in the absence of DCs or in the presence of WT DCs or C3ar1−/−C5ar1−/− DCs. Supernatants then were assayed for TGF-β1 and IL-6 by ELISA (*P<0.001, n=6). (f) C3ar1−/−C5ar1−/− Foxp3− cells were incubated for 3 days with anti-CD3 (5 μg/ml), IL-2 (5 ng/ml), and WT DCs ± anti-TGF-β mAb (5 μg/ml), TGF-βR1 inhibitor (10 nM), or Smad3 inhibitor (5 μM). Following the 3 day induction, Foxp3+CD25+ Treg percentages were determined by flow cytometry.
Prior findings that DCs produce up to 1000-fold more copies of C3a and C5a per cell than cognate CD4+ T cells during effector cell activation 5 suggested that absent C3a plus C5a production by the DC and the consequent lack of C3aR plus C5aR signaling into the CD4+ T cell would influence iTreg induction during CD4+ T cell-DC interactions. To test this, we first stimulated WT Foxp3− CD4+ T cells with C3−/−C5−/− DCs (note, C5 is also known as Hc) i.e. antigen presenting cells unable to produce C3 and C5 in the absence of added TGF-β1 (Fig. 2b left). Foxp3+ cells were induced, but the addition of C3a and C5a markedly reduced the induction. Identical results were observed with WT Foxp3− CD4+ T cells and WT DCs in the presence of TGF-β1 (Fig. 2b right). The suppressor activity of the Foxp3+ cells that were induced in the presence of C3a and C5a in both cases also was markedly attenuated (Supplementary Fig. 3b). To test whether absent C3aR and C5aR signaling into the CD4+ T cells is the important condition, we next incubated WT Foxp3− CD4+ T cells (able to receive C3aR and C5aR signals) or C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells (unable to receive C3aR and C5aR signals) with C3ar1−/−C5ar1−/− DCs [unable to receive C3a and C5a signals or produce C3a and C5a 7] (Fig. 2c). High percentages of Foxp3+ cells were induced under both conditions. Added C5a reduced the induction of Foxp3+ cells from WT Foxp3− CD4+ T cells but not from C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells (Fig. 2c). Added C5a similarly reduced the induction of Foxp3+ cells from WT Foxp3− CD4+ T cells and as well as from C3−/−C5−/− T cells and WT DCs in the presence of TGF-β1 (Supplementary Fig. 3c). Lastly, we stimulated mixtures of C3−/−C5−/− CD25− CD4+ T cells (able to receive C3aR and C5aR signals) or C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells (unable to receive C3aR and C5aR signals) with Daf1−/− DCs (in which C3a and C5a production is potentiated 7) (Fig. 2d). Foxp3+ cell induction from C3−/−C5−/− CD25− CD4+ T cells, but not from C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells, was reduced in the presence of Daf1−/− DCs. The addition of anti-C3a and C5a mAbs restored Foxp3+ cell induction in mixtures of Daf1−/− DCs and C3−/−C5−/− CD25− CD4+ T cells and to a lesser extent in mixtures containing WT DCs (Supplementary Fig. 3d) but had no effect in mixtures containing C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells (Fig. 2d). Taken together, these data support the proposition that absent C3a and C5a production by the DC and absent C3aR and C5aR signaling into CD4+ T cells are involved in iTreg induction.
Although absent C3aR and C5aR signaling into CD4+ T cells was necessary and sufficient to evoke iTreg induction in the absence of DCs (Fig. 1a), 10-fold more TGF-β1 was produced (Fig. 2e) when WT DCs were present and a higher percentage of Foxp3+ cells (45-65% [Fig. 2a, and 2b] versus 28% [Fig. 1a]) was induced. More TGF-β1 was produced in mixtures containing WT DCs and WT CD4+ T cells than mixtures containing anti-CD3+CD28 stimulated C3ar1−/−C5ar1−/− CD4+ T cells alone (Fig. 2e). The enhanced TGF-β1 production in WT-WT mixtures, however, was accompanied by markedly enhanced production of IL-6 (Fig. 2e) potentially resulting in inhibition of Treg induction and favoring TH17 cell polarization. The substitution of C3ar1−/−C5ar1−/− DCs for WT DCs in the mixtures with C3ar1−/−C5ar1−/− CD4+ T cells further increased TGF-β1 production and reciprocally decreased IL-6 production compared to the mixtures containing WT DCs and C3ar1−/−C5ar1−/− CD4+ T cells (Fig. 2e).
To understand why greater numbers of iTregs with more potent suppressor activity are generated in the absence of C3aR and C5aR signaling compared to iTregs generated from WT CD4+ T cells with exogenously added TGF-β1, we analyzed DCs in the induction cultures following iTreg generation under the two conditions. Phenotyping of DCs from the cultures of WT DCs and stimulated C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells showed that the lack of C3aR and C5aR signaling only in the CD4+ T cells led to markedly reduced amounts of CD80 (MFI 8±1 vs 79±5), CD86 (MFI 8±1 vs 40±2) and CD40 (MFI 19±1 vs 58±3) on the surface of DC (Supplementary Fig. 4a) compared to those on DCs from cultures in which Foxp3+ cells were induced from WT Foxp3− CD4+ T cells with added TGF-β1, indicating that that iTregs generated from C3ar1−/−C5ar1−/− Foxp3− CD4+ more potently prevented DC maturation. Other studies have demonstrated that TGF-β1 generated by Tregs can exert a feedback effect to downregulate DC costimulatory molecule expression and thereby maintain DC immaturity14. Moreover, iTregs generated from C3ar1−/−C5ar1−/−Foxp3− CD4+ T cells expressed 4.5-fold lower levels of IL-6 receptor (IL-6R) and contained 12-fold less STAT3 phosphorylation than iTregs generated from WT Foxp3− T cells with added TGF-β1 (Supplementary Fig. 4b). The inclusion of anti-C3a plus anti-C5a mAbs or C3aR-A plus C5aR-A in the induction cultures generated from WT Foxp3− T cells with added TGF-β1 reduced p-STAT3 and IL-6R levels on the iTregs to those on iTregs generated from C3ar1−/−C5ar1−/− T cells (Supplementary Fig. 4b and 4c).
Supplementation of the induction culture containing WT DCs and C3ar1−/−C5ar1−/−Foxp3− CD4+ T cells with the specific antagonist of TGF-βR1 or the inhibitor of Smad3 not only blocked Foxp3+ cell induction (Fig. 2f) but the TGF-βR1 antagonist and the Smad3 inhibitor blocked TGF-β1 production. Moreover, the two inhibitors of TGF-βR1 signaling completely blocked IL-10 (Supplementary Fig. 4d) and TGF-β1 (Supplementary Fig. 4e) production, indicative of blockade of auto-inductive signaling induced in the DCs as well as the C3ar1−/−C5ar1−/− CD4+ T cells. Thus, iTregs induced from CD4+ T cells when they do not receive C3ar1 and C5ar1 signals and DCs that do not produce C3a and C5a support the immunosuppressive activities of each other by providing TGF-β1 to each other via auto-inductive signaling. This pathway is analogous to feedback loops involving the induction of TH1 and TH17 cells where CD4+ T cell produced interferon-γ (IFN-γ) feeds back to increase DC class II MHC and costimulatory molecule expression and CD4+ T cell produced IL-17 feeds back to induce DC IL-6 production. Analogously, TGF-β1 that is endogenously produced by iTregs devoid of C3aR and C5aR signals feedback on DCs partners and retain an immature phenotype.
T cell signaling without C3aR and C5aR parallels tTregs
During activation of conventional T cells, protein kinase A (PKA) activation is repressed and AKT phosphorylation (at T308S473) is increased 15. The decrease in PKA activation opposes the phosphorylation and nuclear translocation of the transcription factor CREB and the increase in T308S473 AKT enhances the phosphoinositide-3 kinase (PI-3K)-p-AKT-mammalian target of Rapamycin (mTOR) signaling cascade as well as enabling phosphorylation of p-AKT targets 15. During conventional T cell activation, C3aR and C5aR signaling is mechanistically connected with CD28 and CD40L costimulation 5,7. The C3aR and C5aR GPCRs are Gi coupled GPCRs such that when they are bound by their ligands, the Gαi subunits of their activated G proteins suppress activation of adenylyl cyclase needed for PKA activation (Supplementary Fig. 5). The Gβγ subunits activate PI-3Kγ 16,17 needed for phosphorylating AKT at T308 and S473 5. Downstream T308S473-AKT signaling maintains mTOR complex-1 in an activated state that inhibits TGF-β1 production18,19 and sustains phosphorylation of ribosomal S6 (rbS6) needed for the proliferation of conventional T cells (Supplementary Fig. 5).
As the intracellular signaling in activated tTregs differs from that in activated conventional T cells in that PKA is activated 20,21 and AKT dephosphorylated 20, we examined whether iTregs induced in the absence of C3aR and C5aR signaling resembles the signaling properties of tTregs. We first tested how individually or dually disrupting C3aR and C5aR signal transduction into CD4+ T cells affects the PKA and AKT activation pathways. Consistent with C3aR and C5aR signals synergizing in CD4+ T cells to lower PKA levels during T effector cell responses, adding C3a or C5a individually to anti-CD3 stimulated WT CD4+ T cells reduced adenylyl cyclase activity by 40% and 50%, and adding both in combination reduced adenylyl cyclase activity by >95% (Fig. 3b) thereby repressing PKA activation. In contrast, the lack of transmission of C3aR or C5aR signals individually into CD4+ T cells enhanced PKA activation by 40% and 50% and the concurrent absence of both GPCR signals enhanced PKA activation by 90% (Fig. 3c). C3aR and C5aR signals in CD4+ T cells synergize to activate PI-3Kγ 5. Sorted Foxp3+ cells generated from anti-CD3+CD28 stimulated C3ar1−/−C5ar1−/− Foxp3−CD4+ T cells expressed markedly reduced S473 p-AKT and T308 p-AKT levels (Fig. 3d). The repressed levels of S473 p-AKT and T308 p-AKT inhibit p-AKT dependent regulation of p-AKT targets and mTORC-1 dependent activation rbS6 (Fig. 3e). Anti-CD3+CD28 stimulated C3ar1−/−C5ar1−/− CD4+ T cells expressed p-Smad2 (Fig. 3f), a transcription factor needed for TGF-β1 production and for Foxp3 gene transcription 22-25. Moreover, in contrast to activated conventional WT T cells generated from anti-CD3+CD28 treated WT Foxp3−CD4+ T cells which contained p-STAT3 downstream of IL-6 signaling but not p-STAT5 downstream of CD25 signaling, anti-CD3+CD28 treated C3ar1−/−C5ar1−/− CD4+ T cells contained p-STAT5 but not p-STAT3 (Fig. 3g). Thus, the signaling cascades that operate in iTregs induced from stimulated CD4+ devoid of C3aR and C5aR signals parallel those in activated tTregs.
Figure 3. Absent C3aR and C5aR signaling lifts restraint-on PKA activation and represses PI-3Kγ-AKT- mTOR signaling.
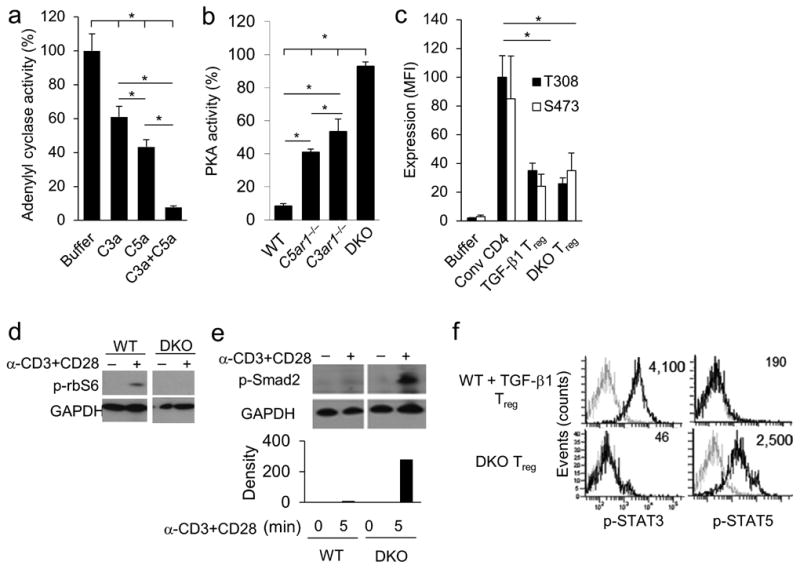
Experiments were repeated two times. (a) WT CD4+ T cells were incubated for 30 min with anti-CD3+CD28 beads after which they were incubated for 10 min with Forskolin (30 μM) ± C3a (100 nM), C5a (100 nM), or both. Levels of cAMP activity were determined by cAMP-Glo assay (*P<0.001). (b) WT, C3ar1−/−, C5ar1−/−, and C3ar1−/−C5ar1−/− CD4+ T cells were stimulated with anti-CD3 mAb for 30 min after which PKA activity was quantified by PepTag assay. (c) iTregs generated from sorted WT Foxp3− CD4+ T cells plus TGF-β1, iTregs derived from C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells, and conventional Foxp3− CD4+ T cells were stimulated with anti-CD3+CD28 beads for 15 min and assayed for S473 and T308 p-AKT by Luminex assay (n=5). (d and e) WT or C3ar1−/−C5ar1−/− CD4+ T cells were incubated for 15 min ± anti-CD3+CD28 beads. Cells were extracted in phospho-lysis buffer and extracts assayed for (d) phospho-rbS6 and (e) phospho-Smad2 by immunoblotting. (f) iTregs induced from WT Foxp3− CD4+ T cells plus TGF-β1 or from C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells were assayed for phospho-STAT3 and phospho-STAT5 by flow cytometry (representative plots of n=6).
TGF-β regulates local complement and C5L2 expression
To understand how absent C3aR and C5aR signaling in CD4+ T cells is mechanistically connected with iTreg induction, we first examined the effect of added TGF-β1 on local complement production by DCs and CD4+ T cells. Adding TGF-β1 to C5a-treated DCs (Supplementary Fig. 6a) or to anti-CD3+CD28 treated CD4+ T cells (Fig. 4a) abolished transcription of all of the mRNA transcripts connected with C3aR and C5aR signaling. Conversely, adding IL-6 to naive CD4+ T cells upregulated expression of C3 mRNA (Fig. 4b), augmented C3a and C5a production (Fig. 4c), and upregulated CD4+ T cell C3aR and C5aR expression (Fig. 4d). IL-6 had a similar upregulatory effect on complement mRNA expression by immature DCs (Supplementary Fig. 6b). The addition of C5a to anti-CD3+CD28-stimulated CD4+ T cells upregulated IL-6 production (Fig. 4e) and down-regulated TGF-β1 production, while blockade of C3aR and C5aR signaling down-regulated IL-6 and up-regulated TGF-β1 production (Fig. 4e). Consistent with the findings in Fig 1e that TGF-β1 enters into an auto-inductive signaling loop in CD4+ cells, blockade of C3aR and C5aR signaling in stimulated CD4+ T cells induced CD4+ T cell TGF-β1 mRNA expression. Anti-TGF-β1 mAb abolished the induction of TGF-β1 mRNA expression elicited by C3aR and C5aR blockade (Supplementary Fig. 6c).
Figure 4. TGF-β1 and C5L2 suppress complement production and enhance iTreg induction.
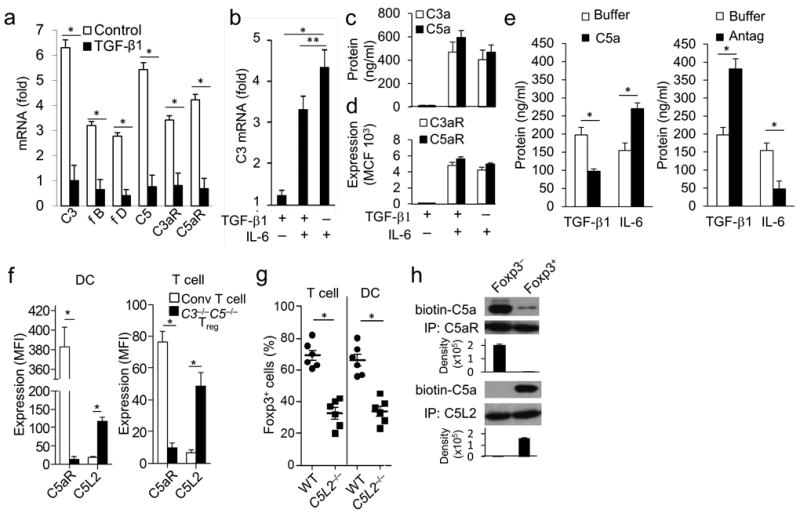
Experiments were repeated two times. (a) Sorted WT Foxp3− cells were incubated for 1 hr with anti-CD3+CD28 beads (as in Fig 1) in the absence or presence of TGF-β1 (5 ng/ml) and then assayed for complement mRNA transcripts by qPCR (*P<0.001, n=5). (b) Sorted WT Foxp3− CD4+ T cells were incubated for 1 hr with anti-CD3+CD28 beads plus TGF-β1 (5 ng/ml) alone, TGF-β1 (5 ng/ml) plus IL-6 (5 ng/ml), or IL-6 (5 ng/ml) alone, after which the cells were assayed for C3 mRNA expression by qPCR (*P<0.001, **P<0.02, n=5). (c and d) Sorted WT Foxp3− CD4+ T cells were incubated for 48 hr with anti-CD3+CD28 beads plus TGF-β1 alone, TGF-β1+IL-6, or IL-6 alone as in (b). Culture supernatants were assayed for (c) C3a and C5a generation by ELISAs, and (d) C3aR and C5aR surface expression by flow cytometry (P<0.01 for TGF-β1 alone vs TGF-β1+IL-6 or IL-6 alone, Mean Fluorescence Intensity (MFI) values; n=5). (e) Sorted WT Foxp3− cells were activated for 48 hr with anti-CD3+CD28 beads ± C5a (100 ng/ml) or C3aR-A/C5aR-A (10 nM), after which culture supernatants were assayed for TGF-β1 or IL-6 by ELISA (P<0.05; n=6). (f) Sorted WT Foxp3− CD4+ T cells were incubated with WT DCs or with C3−/−C5−/− DCs in the absence of TGF-β1 (C3−/−C5−/− iTregs). DCs (left side) were assayed for C5aR and C5L2 expression by gating on CD11c+ cells. Responder T cells (right side) were assayed for C5aR and C5L2 expression by gating on Foxp3− cells in the case of WT CD4+ T cells prepared with WT DCs and on Foxp3+ cells in the case of WT CD4+ prepared with C3−/−C5−/− DCs. (*P < 0.01; n=5). (g) Sorted WT or C5L2−/− (encoded by Gpr77) Foxp3− CD4+ T cells were incubated with WT DCs (Left) and sorted WT Foxp3− CD4+ T cells were incubated with WT or C5L2−/− DCs (right) both in the presence of anti-CD3 and TGF-β1 (5 ng/ml). Percent Foxp3+CD25+ cells were assayed by flow cytometry. (*P<0.001, n=6). (h) WT Foxp3− CD4+ T cells were incubated for 3 days with WT DCs plus TGF-β1 (5 ng/ml) after which Foxp3+ and Foxp3− cells were sorted. Foxp3−CD4+ T cells or Foxp3+ CD4+ iTregs were incubated for 5 min with anti-CD3+CD28 beads + biotin-labeled C5a, the cells chilled to 4°C, and plasma membrane fractions were isolated by ultracentrifugation. Following C5aR and C5L2 IP, bound proteins were eluted with alkaline solution, and equal amounts of protein (determined by A280) were loaded onto SDS PAGE gels. Following electrophoresis, gels were blotted for biotinylated-C5a by adding streptavidin-HRP followed by ECL reagent.
Another factor relevant to the connection of TGF-β1 with suppressed C3aR and C5aR signaling is C5L2 (encoded by Gpr77), an alternative C5aR receptor of incompletely characterized function. While C5L2 has been proposed to scavenge C5a and possibly C3a 26-29, a physiological function has not yet been established. iTregs and DCs derived from mixtures of stimulated WT Foxp3−CD4+ T cells and C3−/−C5−/− DCs showed elevated levels of C5L2 and reduced levels of C5aR compared to conventional CD4+ T cells and DCs derived from cultures of anti-CD3 activated Foxp3− CD4+ T cells and WT DCs (Fig. 4f). iTregs and DCs derived from mixtures of Foxp3− WT CD4+ T cells and WT DCs incubated with TGF-β1 showed a similar but less pronounced increase in C5L2 and decrease in C5aR (Supplementary Fig 7a). The up-regulation of C5L2 was enhanced in the presence of TGF-β1, as anti-CD3+CD28 activation of Foxp3− CD4+ T cells in the presence, but not absence, of added TGF-β1 induced 7- fold up-regulation of C5L2 mRNA transcription at 48 hr (Supplementary Fig. 7b). To study the role C5L2 in iTreg induction, we examined iTregs induced from WT Foxp3− CD4+ T cells (conditions in which C5L2, C3aR, and C5aR are all intact) in the presence of WT DCs and added TGF-β1. Substitution of naive C5L2−/− CD4+ T cells for WT CD4+ T cells or substitution of C5L2−/− DCs for WT DCs markedly reduced iTreg induction (Fig 4g).
To clarify the mechanism by which C5L2 prevents C5aR signaling and thereby diverts CD4+ T cells away from T effector cell induction to iTreg induction, we generated iTregs from WT Foxp3− CD4+ T cells with WT DCs and TGF-β1. We sorted the Foxp3+ and Foxp3− populations and, following resting, we activated the purified Foxp3+ and Foxp3− cells with anti-CD3+CD28 coated beads. We then added biotin-labeled C5a to the activated cells, isolated plasma membranes, and prepared anti-C5aR and anti-C5L2 immunoprecipitates (IPs) of the purified membrane fractions. We added equal amounts (OD280) of the IP purified proteins to gels, and following electrophoresis, we probed immunoblots with streptavidin peroxidase. In the case of sorted Foxp3+ iTregs, the biotin-labeled C5a associated mainly with C5L2. Conversely, in the case of sorted Foxp3− CD4+ T cells activated with anti-CD3+CD28 coated beads, it associated mainly with C5aR (Fig. 4h). In control experiments, no streptavidin signal was observed and no Ig heavy and light chains were detected in anti-C5aR IPs from iTregs generated from C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells confirming specificity (Supplementary Fig. 7c). As recently demonstrated by multi-color confocal microscopy 26, flow cytometry following permeablization showed that most of C5L2 was intracellular consistent wits presence in plasma membrane associated liposomes (Supplementary Fig. 7d). Thus, TGF-β1 prevents C3aR and C5aR signaling in DCs and CD4+ T cells both by inhibiting local complement production and by upregulating C5L2 expression that then sequesters C5a and possibly C3a.
iTregs without C3aR and C5aR signals are stable in vivo
One difficulty with the interpretation of the significance of in vitro generated iTregs is that they have been found to be unstable in vivo 30. To test the stability of iTregs generated in the absence of C3a or C5a signaling, we prepared iTregs in vitro from CD4+ T cells derived from WT (CD45.2) or C3ar1−/−C5ar1−/− (CD45.2) Foxp3-GFP mice bred to OT-II mice expressing a transgenic (Tg) TCR for ovalbumin323-339. Foxp3+ antigen-specific iTregs were then sorted, transferred into CD45.1 recipients, and the recipients immunized with ovalbumin. While five days later only 26% of the Foxp3+ cells generated from WT OT-II cells with TGF-β1 remained Foxp3+ in the draining lymph nodes (dLN) (Fig. 5a), 69% of the Foxp3+ iTregs derived from C3ar1−/−C5ar1−/− OT-II cells remained Foxp3+. No loss of Foxp3 occurred when the mice were immunized irrelevant MOG peptide, consistent with the loss being connected with a specific TCR signal.
Figure 5. iTregs that arise when C3aR and C5aR signaling is disabled are stable and exert robust suppressor activity in vivo.
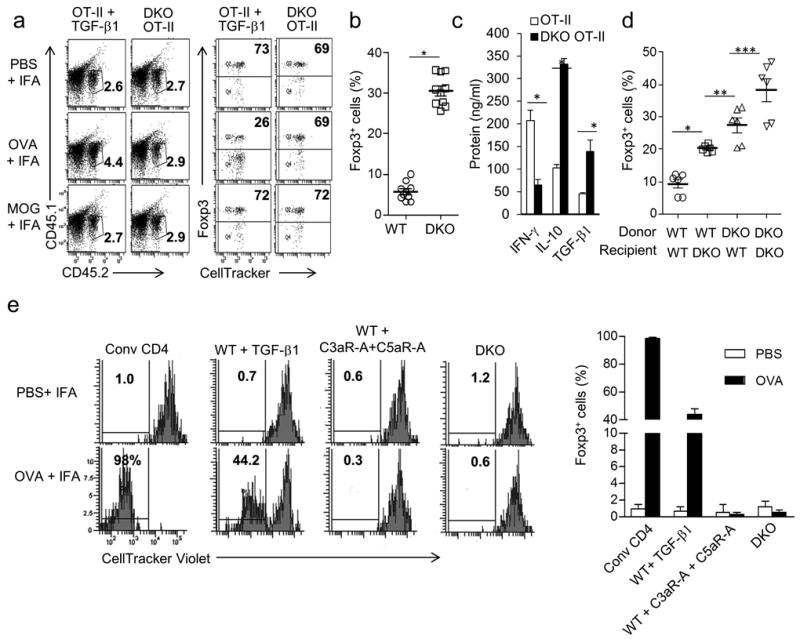
Experiments were repeated two times. (a) iTregs were generated from sorted WT (CD45.2) Foxp3− GFP OT-II cells cultured with WT DCs and TGF-β1 or from sorted WT (CD45.2) Foxp3−C3ar1−/−C5ar1−/− OT-II cells cultured with WT DCs without TGF-β1 as in Fig 2. Sorted (CD45.2) Foxp3+GFP+ cells prepared in each way were labeled with CellTracker Red (for gating to avoid color overlaps), and injected into CD45.1 recipients. Two days later, mice were immunized in hindlimbs with ova323-339 (100 μg) or MOG35-55 (100 μg) in IFA. Five days after immunization, draining inguinal lymph nodes (dLNs) were isolated, and CD45.2 cells were assayed for percent Foxp3+ cells (n=4 each group; representative plots shown). (b) WT and C3ar1−/−C5ar1−/− Foxp3-GFP OT-II mice were immunized with ova323-339 in CFA. Five days following immunization, percent Foxp3+CD25+ cells in the dLN was assessed (*P<0.001, n=10). (c) 1×105 CD4+ T cells (unfractionated) from the dLN (from mice in panel b) were incubated with 1×104 CD11c+ splenic DCs and ova323-339 for 24 hr. Culture supernatants were assayed for IFN-γ, IL-10, and TGF-β1 by ELISA (*P<0.001, n=10). (d) 2×106 WT or C3ar1−/−C5ar1−/− Foxp3−CD25− OT-II cells were adoptively transferred into WT or C3ar1−/−C5ar1−/− mice (non-OT-II). Five days following ovalbumin immunization, Foxp3+CD25+ cells in dLN were assessed by gating on OT-II TCR (*P<0.001, **P<0.02, ***P<0.05, n=5). (e) CellTracker Violet (Invitrogen) labeled (4×106) OT-II cells (Thy-1.2 CD45.1) were transferred into WT Thy-1.1 mice as a source of T effectors. Two days later, 1×106 sorted Foxp3+ CD25+ Tregs (each Thy1.2 CD45.2) prepared from Foxp3− CD4+ T cells and WT DCs with TGF-β1, C3aR/C5aR antagonists, or from C3ar1−/−C5ar1−/− Foxp3− CD4+ T cells and WT DCs were adoptively transferred into the Thy1.1 recipients. Five days following ovalbumin immunization, proliferation of violet-labeled Thy1.2 CD45.1 OT-II cells in the dLN was assessed (values given as percent dividers) (n=8 each group).
Although demethylation of the Treg-specific demethylation region (TSDR) of the Foxp3 locus has been associated with the stability of Foxp3 expression in tTregs compared to iTregs, methylation analyses of the TSDR region of iTregs generated ex vivo in the absence C3aR and C5aR signaling (not shown) showed that it was mostly methylated (88%) similarly to that of iTregs induced with exogenous TGF-β1. This finding suggests that factors other than the methylation status of the TSDR may control stability of Foxp3 expression in vivo as recently reported by other groups 31,32. Insufficient numbers of in vivo generated C3ar1−/−C5ar1−/− Foxp3+ OT-II iTregs could be recovered for analysis of the TSDR.
Disabled C3aR and C5aR signaling induces iTregs in vivo
We next evaluated whether absent C3aR and C5aR signaling in CD4+ T cells in vivo results in the induction of iTregs. Following immunization with ova in CFA, dLNs from WT OT-II mice contained 5.7% CD4+ Foxp3+ T cells while dLNs from C3ar1−/−C5ar1−/− OT-II mice contained 31% Foxp3+ CD4+ T cells (Fig. 5b). Upon in vitro incubation for 24 h with ova and autologous splenic CD11c+ DCs, C3ar1−/−C5ar1−/− OT-II T cells from the dLN produced more TGF-β1 and IL-10, but less IFN-γ than WT OT-II T cells in (Fig. 5c). In a second experiment, we transferred WT Foxp3− OT-II or C3ar1−/−C5ar1−/− Foxp3− OT II cells to WT or C3ar1−/−C5ar1−/− recipients. Two days later, we immunized the mice with whole ovalbumin in IFA. Analyses of the dLNs five days later showed that 9.23 ± 1.34% (n=6) of the transferred cells were Foxp3+ when the donor and host were WT. In contrast, a higher percentage of Foxp3+ cells was seen when the recipient was C3ar1−/−C5ar1−/− and consequently devoid of C3a and C5a production (20.3±0.45%, n=6) or when the donor cells were derived from C3ar1−/−C5ar1−/− mice (27.4±2.2%, n=6). Notably, a much higher percentage (38.4±3.5%, n=6) of Foxp3+ cells was observed when both donor OT-II cells and the recipient mice were C3aR and C5aR deficient (Fig. 5d).
We next compared the in vivo suppressive capacity of iTregs (all CD45.2+Thy-1.2+) prepared in vitro from 1) C3ar1−/−C5ar1−/− Foxp3− OT-II cells in the absence of TGF-β1, 2) WT Foxp3− OT-II cells generated in the presence of C3aR-A + C5aR-A but absence of TGF-β1, or 3) WT Foxp3− OT-II cells conventionally generated in the presence of TGF-β1. We used untreated CD25−Thy-1.2, CD45.2 WT OT-II cells as a control. Sorted Foxp3+ CD4+ T cells from the three iTreg groups or control were co-transferred into WT recipients (Thy-1.1+) together with CellTracker Violet labeled WT OT-II cells (Thy-1.2+ CD45.1+) as the source of T effector cells. Two days later, we immunized all groups with ovalbumin in IFA, and five days thereafter we analyzed dLNs. Almost all (98%) of the CellTracker Violet labeled OT-II cells proliferated when co-transferred with the conventional WT Foxp3− OT-II cells. iTregs conventionally generated in the presence of TGF-β1 inhibited T effector cell proliferation by 44%, while iTregs generated from C3ar1−/−C5ar1−/− Foxp3− OT-II cells or C3aR-A + C5aR-A treated WT Foxp3− OT-II cells completely inhibited T effector cell proliferation (Fig. 5e). Given the above findings that iTregs induced in the absence of C3aR and C5aR signals are stable in vivo whereas those induced with exogenous TGF-β1 are not, further studies are needed to determine whether the enhanced suppressor capacity of the latter groups was secondary to a true increase in suppressive function or secondary to enhanced stability in vivo. Nevertheless, the data show that C3ar1−/−C5ar1−/− iTregs suppress antigen specific T effector cells with high efficiency.
iTregs lacking C3aR and C5aR suppress autoimmune disease
We next evaluated the suppressive capacity of iTregs induced in the presence of C3aR and C5aR antagonists for the prevention and treatment of experimental autoimmune encephalomyelitis (EAE). We immunized Foxp3-GFP mice with MOG35-55 in CFA. When the clinical scores were > 2, we isolated CD4+ T cells from LNs and the spleen and purified Foxp3− CD4+ T cells by cell sorting. We kept half of the Foxp3- cells as a source of T effectors and stimulated the other half in the presence of C3aR-A plus C5aR-A to produce iTregs. We then transferred Foxp3− T cells (4×106) alone or together with iTregs (1×106) into Rag2−/− mice. While the Rag2−/− recipients that received T effectors alone showed progressive weakness and weight loss, the recipients that also received C3aR-A + C5aR-A generated iTregs showed lesser disease with gradual improvement and recovery of their weights (Fig. 6a and 6b). At day 7 (Fig. 6c) post adoptive transfer, Foxp3+ cells were detectable in LNs, spleens, and spinal cords from these mice, but not from mice that received the T effector population alone, indicating stability of Foxp3 expression on the transferred iTregs.
Figure 6. iTregs induced when C3aR and C5aR signaling is disabled in CD4+ cells inhibit autoimmune disease.
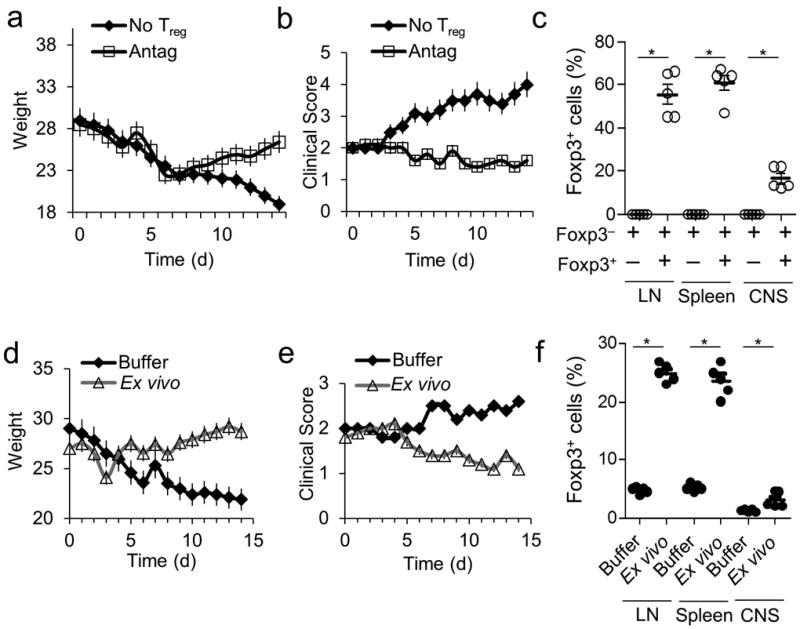
Experiments were repeated two times. (a-c) EAE was induced in Foxp3-GFP mice. Ten days after disease onset, CD4+ T cells were isolated from spleens and LNs and separated into GFP+ and GFP− populations by cell sorting. GFP− cells (4×106) were adoptively transferred into Rag2−/− mice. The mice were subsequently injected with 1×106 sorted Foxp3-GFP+ cells generated ex vivo with C3aR-A/C5aR-A (10 nM) (control is no Tregs) after which (a) weight change, (b) clinical scores, and (c) percent Foxp3+CD25+ cells in the LNs, spleen, and CNS were assayed. (d-f) EAE was induced in Foxp3-GFP mice. Ten days after induction, mice were treated with PBS or with 1×106 sorted Foxp3+ Tregs (generated ex vivo from Foxp3-GFP mice with C3aR-A/C5aR-A). (d) Clinical scores, (e) weights were assessed daily and (f) percent Foxp3+CD25+ cells in dLNs, spleen, and CNS were assessed at day 7 post treatment.
To evaluate whether iTregs generated ex vivo by C3aR + C5aR antagonism can modulate established EAE, we induced EAE in WT mice. We treated sick mice (scores >2) with Foxp3+ cells generated by stimulating naive CD4+ T cells from Foxp3-GFP mice in the presence of C3aR-A + C5aR-A. Following adoptive transfer of sorted Foxp3+ cells (1 × 106), clinical scores decreased and weights increased (Fig. 6d and 6e). At days 7 and 14 (Fig. 6f) post adoptive transfer, Foxp3+ cells were detectable in LNs and spinal cords of the iTreg treated mice. Thus, the administration of a single dose iTregs induced ex vivo in the absence C3aR and C5aR signals can suppress ongoing EAE as well as prevent EAE induction.
C3aR and C5aR antagonism induces functional human iTregs
To date, it has been very difficult to generate functional human Tregs in vitro by stimulating naive CD4+ T cells in the presence of TGF-β1 despite the fact that the cells express high levels of Foxp3 33-36. To test whether C3aR + C5aR antagonism is effective in generating functional human iTregs, we incubated naive human CD45RA+CD25−CD4+ T cells for 3 days with anti-CD3, IL-2, and DCs together with a) TGF-β1, b) C3aR-A + C5aR-A, or c) anti-C3a + anti-C5a mAbs. More than 95% of the cells generated in all three ways became Foxp3+ and were also CD25+ (Fig. 7a). iTregs generated with C3aR-A plus C5aR-A or anti-C3a plus C5a mAbs did not produce IL-2 in response to PMA plus ionomycin treatment, and did not proliferate when re-stimulated with anti-CD3 (Supplementary Fig. 8a and 8b). This contrasted with iTregs cells generated with TGF-β1 which, as reported by others 33, produced IL-2, and proliferated upon re-stimulation. The CD25+ (Foxp3+) cells prepared by C3aR and C5aR antagonism either with the antagonists or mAbs exerted robust suppression (Fig. 7b), whereas those prepared with TGF-β1 had little suppressive activity. Comparable results were obtained with iTregs generated from circulating CD45RA+ CD25− CD4+ T cells from 7 other individuals, three with asymptomatic multiple sclerosis (Fig. 7c). Thus, in the absence of C3aR and C5aR signaling we were able to generate fully functional, bona-fide human iTregs.
Figure 7. Human iTregs with robust suppressor activity can be induced by C3aR and C5aR antagonism.
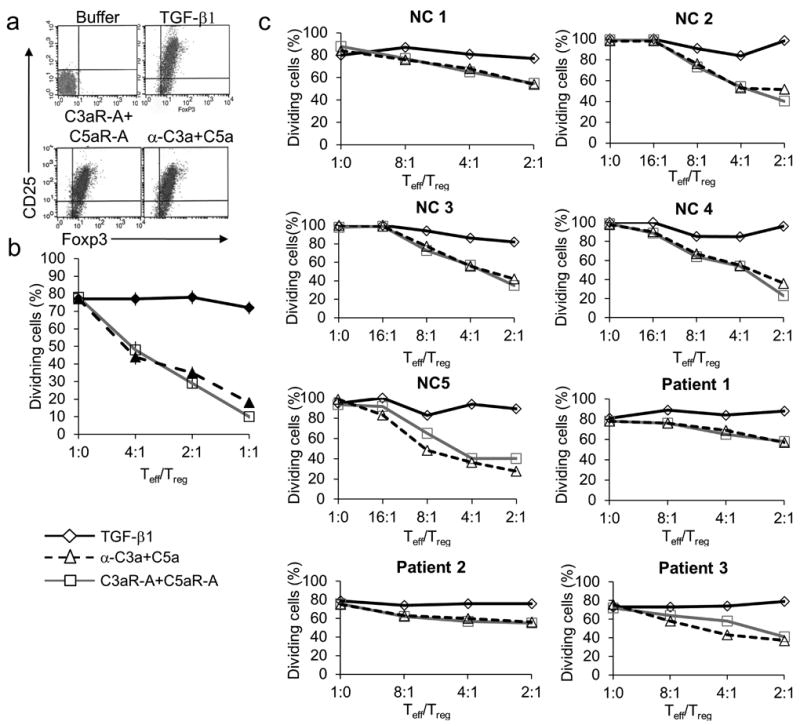
Experiments were repeated two times. (a) Flow sorted human CD45RA+CD25−CD4+ T cells (1×106) were incubated for 3 days with soluble anti-CD3 mAb (3 μg/ml), rhIL-2 (5 ng/ml), and 2.5×105 autologous CD11c+ DCs in the absence or presence of 1) TGF-β1 (5 ng/ml), 2) each of C3aR-A/C5aR-A (10 nM), or 3) each of anti-C3a/C5a mAbs (10 μg/ml). Percent Foxp3+CD25+ CD4+ T cells then were determined by flow cytometry. (b) Flow sorted CD25+ cells from (a) were incubated for 3 days with in differing Teff/iTreg ratios with 1×106 CFSE labeled autologous naive CD25− cells, anti-CD3 mAb (3 μg/ml), and 1×104 autologous CD11c+ DCs. Percent dividers was determined by CFSE dilution. (c) CD45RA+CD25−CD4+ T cells (1×106) were isolated from 5 healthy controls (NC) and 3 MS patients by flow sorting. The cells were incubated for 3 days with soluble anti-CD3 mAb (3 μg/ml), rhIL-2 (5 ng/ml), and 2.5×105 autologous DCs in the absence or presence of 1) rhTGF-β1 (5 ng/ml), 2) C3aR-A/C5aR-A (10 nM), or 3) anti-C3a/C5a mAbs (5 μg/ml). Cells were washed and sorted on CD25. After sorting, CD25+ (Treg) were incubated for 3 days with anti-CD3 mAb, 2.5×105 autologous DCs, and 106 CD25− (Effector) cells prelabeled with CFSE. Percent dividers was determined by CFSE dilution.
DISCUSSION
The studies presented in this report highlight that complement, widely associated with innate immunity, plays an integral role in modulating Treg function/induction and dominant immunologic tolerance Previous studies 5 demonstrated that C3aR and C5aR signaling by cognate CD4+ T cells and DCs up-regulates costimulatory molecule expression on both partners, enhances survival of T effector cells 6,7, and increases TH1 and TH17 T effector function 5,7. Herein, we have examined in depth, the reverse situation. The major consequence of absent transmission C3aR and C5aR signals into CD4+ T cells resulting from absent C3a and C5a production of by antigen presenting cells is the induction of Foxp3+ iTregs without the addition TGF-β1. Thus, signaling in CD4+ T cells themselves via the C3aR and C5aR on the one hand potentiates immune responses, while the absence of this signaling diverts naive T cells to an iTreg response. The data support the concept that the differential regulation of C3a and C5a production and C3aR and C5aR signaling by TGF-β1 and IL-6 is a critical component of the induction of iTregs versus TH17 effector cells37.
Collectively, these studies demonstrate that forward and backward feedback loops are involved in the induction of iTregs. First, the lack of C3aR and C5aR signaling in DCs results in enhanced TGF-β1 production by the DC and the consequent suppression of DC C3a and C5a production 5,7 which deprives the responder T cells of their major source of C3a and C5a. The DC production of TGF-β1 prevents C3a and C5a production by the responder CD4+ T cells (or any other nearby cells) and evokes auto-inductive TGF-β1 signaling in the CD4+ T cell and its conversion to an iTreg. The iTreg produced TGF-β1 feeds back to induce auto-inductive TGF-β1 signaling in the DC and maintain the DC in an immature state. When C5aR signaling is disabled solely in the DC and the delivery of C3aR and C5aR signals into CD4+ T cells is not prevented, much lower percentages of Foxp3+ cells are induced (~5%) 8. Thus, it is the dual absence of C3aR and C5aR signaling into the CD4+ T cells that is important, a concept that has not been previously explored.
Previous studies 5 showed that in addition to abolishing S473 AKT phosphorylation, disrupting C3aR and C5aR signaling abolishes IL-2 and IFN-γ production. The analyses herein show that absent C3aR and C5aR signaling not only leads to disabled AKT phosphorylation, but also to disabled mTORC-1 dependent phosphorylation of rbS6 and disabled mTORC-2 (PDK2) dependent phosphorylation of the S473 residue on AKT. We also demonstrate that absent C3aR and C5aR signaling in stimulated CD25− CD4+ T cells is associated with STAT5 phosphorylation downstream of CD25 and not STAT3 phosphorylation as previously reported for tTregs 38.
The iTregs induced by C3aR and C5aR blockade are both similar and different from iTregs induced by exogenous TGF-β1. While both populations are anergic and suppressive, the DCs isolated from co-cultures of C3ar1−/−C5ar1−/− T cells and WT DCs have markedly reduced levels of CD80, CD86, and CD40 compared to DCs derived from cultures of iTregs prepared with TGF-β1. Similarly, iTregs induced from C3ar1−/−C5ar1−/− T cells express lower levels of IL-6R and p-STAT3, but higher levels of p-STAT5 than iTregs generated with TGF-β1.
A marked up-regulation of C5L2 was observed on iTregs generated with DCs devoid of C3a and C5a, while lower levels were seen on iTregs generated with exogenous TGF-β1. Although C5L2 was described more than 10 years ago 28,39, a physiological function has remained obscure. While C5L2 initially was hypothesized to function as a decoy receptor 29, subsequent data concerning its specificity for C5a alone 40,41 versus both C3a and C5a 42, as well as its downstream signaling 26,43,44 have been conflicting. Recent studies26, however, have provided evidence that it scavenges C5a from C5aR by a β-arrestin dependent mechanism. Our studies demonstrate the capacity of C5L2 to scavenge C5a and thereby circumvent the transmission of C5aR signals. While others 8 concluded that there was no effect of C5L2 deficiency on Treg induction, they did not conduct their testing under conditions in which Tregs were actually induced (i.e., in the presence of added TGF-β1). More in depth studies will be needed to determine the precise relationship between C5L2 expression and Treg suppressor function and stability.
We also compared TGF-β1 iTregs to C3aR and C5aR disabled iTregs in an in vivo assay 30 that measures iTreg stability. When antigen-specific iTregs prepared with TGF-β1 were transferred to a recipient challenged with their cognate antigen, they rapidly lost Foxp3 expression. In contrast, iTregs generated by C3aR and C5aR deficiency retained their Foxp3 expression. In addition, these iTregs were much more potent at inhibiting the expansion of co-transferred naive Foxp3− T cells upon antigen challenge and could be used in a therapeutic mode to reverse the clinical course of established EAE. The molecular signals that underlie the enhanced stability of C3aR and C5aR disabled iTregs are not clear, as the TSDR region of the Foxp3 locus remained mostly methylated following induction in vitro. We have not yet analyzed methylation of their TSDR following in vivo transfer, but the data argue that factors other than the methylation status of the TSDR may also regulate iTreg stability in vivo 31,32.
Our findings that human iTregs with robust suppressor function can be prepared from circulating human CD45RA+CD25−CD4+ T cells by C3aR and C5aR antagonism could have value clinically in disorders in which suppression of T effector responses would be beneficial. Our previous studies of T effector responses5,7 showed that an advantage of this approach would be the concurrent suppression of TH1 and TH17 effector cell responses. Based on this study, a combination regimen incorporating an agent that blocks C3aR as well as C5aR signaling would constitute the optimal approach. It might also be of interest to incorporate these antagonists into regimens now used to expand human tTregs for cellular biotherapy where loss of Foxp3 expression remains a problem.
Many studies 45-48 have implicated TLR signaling in DCs as triggering C3a and C5a production that would induce CD4+ T effector cell polarization. However, in the absence of external activation, there is no evidence that signaling through C3aR or C5aR at threshold levels needed for T effector responses occurs tonically in DCs or CD4+ T cells. Consequently, according to the linkage in this paper that absent C3aR and C5aR GPCR signaling leads to endogenous TGF-β1 production, the default state should be associated with TGF-β1 production and the induction of Tregs. In principle, this pathway should be primarily operative during the generation of tolerogenic responses that naturally give rise to Foxp3+ iTregs to self-antigens or to iTregs to oral or airway antigens.
Taken together, the studies herein thus demonstrate a critical role for C3a and C5a in the differentiation of T regulatory cells as well as T effectors in vivo. Further studies using CD4+ T cell and DC specific C3 and C5 reporter mice guided by this work could enable detailed characterization of this important issue.
Materials and Methods
Reagents and antibodies
Murine C5a was from Cell Sciences. C3aR-A was purchased from Calbiochem (now EMD Biochemicals). C5aR-A synthetic peptide was kindly provided by John Lambris (Univ of Penn). Anti-mouse C3a (I87-1162) and C5a (I52-1486) mAbs were from BD Biosciences. Mouse IL-2, IL-10, IFN-γ, human IL-2, IL-10, IFN-γ and human TGF-β1 were from Prospec Bio. Anti-murine C5aR1 (20/70) was purchased from Abcam. Antibodies against mouse B7-1 (16-10A1), and B7-2 (GL1), were from BD Biosciences. Anti-CD40L mAb was from BioExpress. Anti-murine C3aR (sc-20138) was purchased from Santa Cruz Biotech. Anti-murine C5L2 (HP8015) was purchased from Hylcult Biotech. Anti-Human C3a (20C-CR6032RP) was purchased from Fitzgerald and anti-human C5a (AF2037) was purchased from R&D Systems. Mouse and Human anti-FoxP3 mAbs and Treg staining kits were purchased from eBiosciences. Anti-CD4 (RM-4-5, FITC), anti-Thy1.1 (HIS51, PE-Cy7), anti-Thy1.2 (53-2.1, APC), anti-CD45.1 (A20, PE), and anti-CD45.2 (104, APC-eFluor 780) were purchased from eBiosciences. CFSE, CellTracker Red™, and CellTracker Violet™ were purchased from Invitrogen and used per the manufacturer’s instructions. The TGF-βR1 inhibitor, anti-TGF-β1 mAb, and Smad3 inhibitor were kindly provided by John Letterio (CWRU).
Animals
Thy1.1, C57BL/6, OT-II, Rag2−/−, C3−/−, and C5 deficient mice were from Jackson labs. C3−/− mice and C3ar1−/− and C5ar1−/− were gifts of Dr. Michael Carroll and Dr. Craig Gerard (Harvard). Foxp3-GFP Tg mice were a gift from Vijay Kuchroo (Harvard). C5−/−C3−/− mice were generated by crossing C5 deficient B10.D2 mice with C57BL/6 congenic C3−/− mice. B6.SJL (CD45.1) and OT-II CD45.1 Rag−/− were obtained from Taconic Farms. All studies were approved by the Case Western Reserve University Institutional Animal Care and Use Center (IACUC).
RNA purification, cDNA synthesis, and qPCR
mRNA expression was performed as previously described 5.
Murine DCs and T cell isolations
CD11c+ cells and CD4+ T cells were isolated from spleens and lymph nodes using the CD4+ negative selection cocktail or CD11c positive selection beads from Miltenyi per the manufacturer’s instructions. Both were purified using the Automacs Pro. Foxp3− and Foxp3+ CD4+ T cells were further purified via flow sorting gating on GFP+ and GFP− populations.
T cell activation
1×106 CD62LhiCD25−Foxp3− CD4+ T cells were activated with anti-CD3 (3 μg/ml, or alternatively with anti-CD3+CD28 Dynabeads [Invitrogen] per the manufacturer’s suggestion), 2.5×105 CD11c+ DCs and rhIL-2 (5 ng/ml) with either rhTGF-β1 (5 ng/ml, Prospecbio), C3aR-A+C5aR-A (10 nM, Calbiochem and John Lambris as detailed above), or anti-C3a and anti-C5a mAbs (10 μg/ml, as detailed above).
Immunizations
Mice were immunized s.c. with OVA323-339 or MOG35-55 peptide as described 49.
In vivo stability
Treg stability was measured as previously described 30.
In vivo suppression
4×106 Thy1.2 CD45.1 CD4+ OT-II cells were labeled with CellTracker Violet (Invitrogen) per the manufacturer’s recommendation and adoptively transferred into WT Thy1.1 mice via tail vein injection (in 200 μl RPMI). Two days later, 1×106 in vitro generated Treg from naive Thy1.2 CD45.2 FoxP3-GFP CD4+ T cells with anti-CD3 (5 μg/ml), rhIL-2 (5 ng/ml), and 2.5×105 CD11c+ WT DCs for 5 days ± rhTGF-β1 (5 ng/ml) or C3aR-A+C5aR-A (10 nM) or from Thy1.2 CD45.2 C3ar1−/−C5ar1−/− mice with anti-CD3, rhIL-2 (5 ng/ml), and 2.5×105 CD11c+ WT DCs were adoptively transferred into the Thy1.1 recipients via tail vein injection (in 200 μl RPMI). Mice were immunized s.c. with OVA323-339 (100 μg) in IFA in the hindflank. Five days following immunization, draining LNs were harvested, cells were stained with anti-CD4 (FITC), anti-Thy1.1 (PE-Cy7), anti-Thy1.2 (APC), anti-CD45.1 (PE), and anti-CD45.2 (APC-eFluor 780), and proliferation of draining LN violet-labeled Thy1.2 CD45.1 OT-II cells was assessed.
Anti-CD3 and anti-CD28 stimulations
Cells were stimulated three different ways: 1) anti-CD3 (5 μg/ml) and/or anti-CD28 (BD Biosciences, San Diego, CA) in complete RPMI 1640 + 10% FBS; 2) anti-CD3 (5 μg/ml) and CD11c+ DCs in complete RPMI 1640; or 3) with 1:1 ratio of T cell Dynabeads (Invitrogen) per the manufacturer’s instructions.
FoxP3+ Staining Assays
For both Human and Murine Treg staining assays, the relevant FoxP3+ T regulatory staining kits (236A/E7 for Human and FJK-16s for mouse) were purchased from eBiosciences and used per the manufacturer’s instructions.
Murine Treg Suppression Assays
1×106 CellTracker Red-labeled CD4+Foxp3− T cells (Teff) sorted by FACS were stimulated with 2.5×105 autologous CD11c+ DCs and anti-CD3 mAb (5 μg/mL) alone or with various numbers of suppressor cells (iTreg). The cells were cultured for 3 days in 96-wellflat-bottom plates and CellTracker Red dilution was analyzed by FACS.
ELISAs
For C5a, C3a, IL-2, IL-10, and IL-6, capture and biotinylated detection Ab pairs were purchased from BD Biosciences. For TGF-β1, the human Ab pair (cross reacts with mouse) was purchased from eBiosciences. ELISAs were performed as previously described 5.
Intracellular cAMP and Intracellular PKA Activity Assays
Intracellular cAMP levels were measured using cAMP-Glo Assay (Promega) and PepTag Assay (Promega) respectively.
Luminex Assay
Cells were restimulated with anti-CD3+anti-CD28 mAbs (5 μg/ml) and assayed for pAKT and tAKT using Millipore’s Luminex assay according to the manufacturer’s instructions.
Induction and Evaluation of EAE
EAE was induced and evaluated as previously described 7.
Human Treg Isolation
The acquisition of blood products was approved according to the policies of the University Hospitals Cleveland Case Medical Center in accordance with the Declaration of Helsinki. Peripheral blood mononuclear cells (PBMCs) were obtained from 5 to 10 mL blood from healthy donors. Written informed consent was obtained from all donors in accordance with the Declaration of Helsinki. CD4+ T cells were enriched over the AutoMACS Pro Separator by positive selection with human CD4 microbeads (Miltenyi Biotec) and DCs were isolated by positive selection using the Blood Dendritic Cell Isolation Kit II (Miltenyi Biotec). The cells were labeled with CD4 FITC, CD25 PE, CD45RA PE-Cy5.5 (Invitrogen) and CD127 Alexa Fluor 647 (BD Biosciences).
Human Treg assays
CFSE-labeled CD4+CD25− T cells (1×106) sorted by FACS were stimulated with 2.5×105 autologous DCs and anti-CD3 mAb (3 μg/mL, BD Biosciences) alone or with various numbers of suppressor cells. The cells were cultured for 3 days in 96-well flat-bottom plates and CFSE dilution was analyzed by FACS.
Statistics
Statistical significance for all experimental data was determined by Student’s t test (unpaired, two-tailed) performed in Microsoft Excel or GraphPad Prizm 5. All experiments were repeated at least 3 times.
Supplementary Material
Acknowledgments
We thank Craig Gerard for providing C3ar1−/−, C3ar1−/−, Gpr77−/− (C5L2 knockout) mice, V.J. Kuchroo for FoxP3-GFP mice, B. Cobb (CWRU), Sathyamangla v naga Prasad (CCF), and George Dubyak CWRU) for critical reading, Michael Sramkoski for help with 6-color flow cytometry in vivo suppression assays, and Hae Suk Kim (CWRU) for performing immunoblots. We give special thanks to John Letterio (CWRU) both for providing TGF-β1 inhibitors and helpful discussion. This work was supported by NIH grants R0123598 (MEM), EY011288 (MEM), T32 HL083823-03 (MS), DOD grant BC085077 (MEM), and by NIH grant AI071125 (Peter Heeger). We thank Peter Heeger for ongoing collaborative discussion.
Footnotes
Author contributions
Michael Strainic performed and analyzed the experiments as well as participated in their design and the writing of the manuscript.
Ethan M Shevach consulted in the design and analysis of the experiments as well as participated in the writing of the manuscript.
Fengqi An performed some of the experiments and prepared the knockout mice.
Feng Lin consulted in the design and analysis of the experiments.
M. Edward Medof designed and analyzed the experiments as well as wrote of the manuscript. All work was performed in his lab under his direction.
Image integrity
None of the manuscript data images were edited or adjusted in any manner.
References
- 1.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 2.Josefowicz SZ, et al. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Josefowicz SZ, Rudensky A. Control of regulatory T cell lineage commitment and maintenance. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strainic MG, et al. Locally produced complement fragments C5a and C3a provide both costimulatory and survival signals to naive CD4+ T cells. Immunity. 2008;28:425–435. doi: 10.1016/j.immuni.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lalli PN, et al. Locally produced C5a binds to T cell-expressed C5aR to enhance effector T-cell expansion by limiting antigen-induced apoptosis. Blood. 2008;112:1759–1766. doi: 10.1182/blood-2008-04-151068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, et al. IFN-gamma and IL-17 production in experimental autoimmune encephalomyelitis depends on local APC-T cell complement production. J Immunol. 2008;180:5882–5889. doi: 10.4049/jimmunol.180.9.5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weaver DJ, Jr, et al. C5a receptor-deficient dendritic cells promote induction of Treg and TH17 cells. Eur J Immunol. 2010;40:710–721. doi: 10.1002/eji.200939333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hashimoto M, et al. Complement drives TH17 cell differentiation and triggers autoimmune arthritis. J Exp Med. 2010;207:1135–1143. doi: 10.1084/jem.20092301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shevach EM, et al. The lifestyle of naturally occurring CD4+ CD25+ Foxp3+ regulatory T cells. Immunol Rev. 2006;212:60–73. doi: 10.1111/j.0105-2896.2006.00415.x. [DOI] [PubMed] [Google Scholar]
- 11.Wei J, et al. Antagonistic nature of T helper 1/2 developmental programs in opposing peripheral induction of Foxp3+ regulatory T cells. Proc Natl Acad Sci U S A. 2007;104:18169–18174. doi: 10.1073/pnas.0703642104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itoh S, et al. Interleukin-17 accelerates allograft rejection by suppressing regulatory T cell expansion. Circulation. 2011;124:S187–196. doi: 10.1161/CIRCULATIONAHA.110.014852. [DOI] [PubMed] [Google Scholar]
- 13.Paul WE, Zhu J. How are TH2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–235. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laouar Y, et al. TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A. 2008;105:10865–10870. doi: 10.1073/pnas.0805058105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chi H. Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol. 2012;12:325–338. doi: 10.1038/nri3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schraufstatter IU, Trieu K, Sikora L, Sriramarao P, DiScipio R. Complement c3a and c5a induce different signal transduction cascades in endothelial cells. J Immunol. 2002;169:2102–2110. doi: 10.4049/jimmunol.169.4.2102. [DOI] [PubMed] [Google Scholar]
- 17.Alcazar I, et al. Phosphoinositide 3-kinase gamma participates in T cell receptor-induced T cell activation. J Exp Med. 2007;204:2977–2987. doi: 10.1084/jem.20070366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ching CB, Hansel DE. Expanding therapeutic targets in bladder cancer: the PI3K/Akt/mTOR pathway. Lab Invest. 2010;90:1406–1414. doi: 10.1038/labinvest.2010.133. [DOI] [PubMed] [Google Scholar]
- 20.Sauer S, et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A. 2008;105:7797–7802. doi: 10.1073/pnas.0800928105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yaqub S, Tasken K. Role for the cAMP-protein kinase A signaling pathway in suppression of antitumor immune responses by regulatory T cells. Crit Rev Oncog. 2008;14:57–77. doi: 10.1615/critrevoncog.v14.i1.40. [DOI] [PubMed] [Google Scholar]
- 22.Merkenschlager M, von Boehmer H. PI3 kinase signalling blocks Foxp3 expression by sequestering Foxo factors. J Exp Med. 2010;207:1347–1350. doi: 10.1084/jem.20101156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang M, Fraser D, Phillips A. ERK, p38, and Smad signaling pathways differentially regulate transforming growth factor-beta1 autoinduction in proximal tubular epithelial cells. Am J Pathol. 2006;169:1282–1293. doi: 10.2353/ajpath.2006.050921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Flanders KC, Holder MG, Winokur TS. Autoinduction of mRNA and protein expression for transforming growth factor-beta S in cultured cardiac cells. J Mol Cell Cardiol. 1995;27:805–812. doi: 10.1016/0022-2828(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 25.Kim SJ, Jeang KT, Glick AB, Sporn MB, Roberts AB. Promoter sequences of the human transforming growth factor-beta 1 gene responsive to transforming growth factor-beta 1 autoinduction. The Journal of biological chemistry. 1989;264:7041–7045. [PubMed] [Google Scholar]
- 26.Bamberg CE, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. The Journal of biological chemistry. 2010;285:7633–7644. doi: 10.1074/jbc.M109.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao H, et al. Evidence for a functional role of the second C5a receptor C5L2. FASEB J. 2005;19:1003–1005. doi: 10.1096/fj.04-3424fje. [DOI] [PubMed] [Google Scholar]
- 28.Cain SA, Monk PN. The orphan receptor C5L2 has high affinity binding sites for complement fragments C5a and C5a des-Arg(74) The Journal of biological chemistry. 2002;277:7165–7169. doi: 10.1074/jbc.C100714200. [DOI] [PubMed] [Google Scholar]
- 29.Okinaga S, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry. 2003;42:9406–9415. doi: 10.1021/bi034489v. [DOI] [PubMed] [Google Scholar]
- 30.Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM. IL-2 Controls the Stability of Foxp3 Expression in TGF-{beta}-Induced Foxp3+ T Cells In Vivo. J Immunol. 2011 doi: 10.4049/jimmunol.1100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haribhai D, et al. A Requisite Role for Induced Regulatory T Cells in Tolerance Based on Expanding Antigen Receptor Diversity. Immunity. 2011;35:109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pothoven KL, et al. Rapamycin-conditioned donor dendritic cells differentiate CD4CD25Foxp3 T cells in vitro with TGF-beta1 for islet transplantation. Am J Transplant. 2010;10:1774–1784. doi: 10.1111/j.1600-6143.2010.03199.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allan SE, et al. Activation-induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol. 2007;19:345–354. doi: 10.1093/intimm/dxm014. [DOI] [PubMed] [Google Scholar]
- 35.Wang J, Ioan-Facsinay A, van der Voort EI, Huizinga TW, Toes RE. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol. 2007;37:129–138. doi: 10.1002/eji.200636435. [DOI] [PubMed] [Google Scholar]
- 36.Gavin MA, et al. Single-cell analysis of normal and FOXP3-mutant human T cells: FOXP3 expression without regulatory T cell development. Proc Natl Acad Sci U S A. 2006;103:6659–6664. doi: 10.1073/pnas.0509484103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bettelli E, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 38.O’Gorman WE, et al. The initial phase of an immune response functions to activate regulatory T cells. J Immunol. 2009;183:332–339. doi: 10.4049/jimmunol.0900691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ohno M, et al. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Mol Immunol. 2000;37:407–412. doi: 10.1016/s0161-5890(00)00067-5. [DOI] [PubMed] [Google Scholar]
- 40.Honczarenko M, et al. Leukemia : official journal of the Leukemia Society of America. Vol. 19. Leukemia Research Fund; U.K.: 2005. C5L2 receptor is not involved in C3a / C3a-desArg-mediated enhancement of bone marrow hematopoietic cell migration to CXCL12; pp. 1682–1683. [DOI] [PubMed] [Google Scholar]
- 41.Lee H, Whitfeld PL, Mackay CR. Receptors for complement C5a. The importance of C5aR and the enigmatic role of C5L2. Immunol Cell Biol. 2008;86:153–160. doi: 10.1038/sj.icb.7100166. [DOI] [PubMed] [Google Scholar]
- 42.Chen NJ, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature. 2007 doi: 10.1038/nature05559. [DOI] [PubMed] [Google Scholar]
- 43.Gerard NP, et al. An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. The Journal of biological chemistry. 2005;280:39677–39680. doi: 10.1074/jbc.C500287200. [DOI] [PubMed] [Google Scholar]
- 44.Rittirsch D, et al. Functional roles for C5a receptors in sepsis. Nat Med. 2008;14:551–557. doi: 10.1038/nm1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fang C, Zhang X, Miwa T, Song WC. Complement promotes the development of inflammatory T-helper 17 cells through synergistic interaction with Toll-like receptor signaling and interleukin-6 production. Blood. 2009;114:1005–1015. doi: 10.1182/blood-2009-01-198283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hajishengallis G, Lambris JD. Crosstalk pathways between Toll-like receptors and the complement system. Trends Immunol. 2010;31:154–163. doi: 10.1016/j.it.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hawlisch H, Kohl J. Complement and Toll-like receptors: key regulators of adaptive immune responses. Mol Immunol. 2006;43:13–21. doi: 10.1016/j.molimm.2005.06.028. [DOI] [PubMed] [Google Scholar]
- 48.Zhang X, et al. Regulation of Toll-like receptor-mediated inflammatory response by complement in vivo. Blood. 2007;110:228–236. doi: 10.1182/blood-2006-12-063636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.