

Summary
During maize anther development, somatic locular cells differentiate to support meiosis in the pollen mother cells. Meiosis is an important event during anther growth and is essential for plant fertility as pollen contains the haploid sperm. A subset of maize male sterile mutants exhibit meiotic failure, including ms8 (male sterile 8) in which meiocytes arrest as dyads and the locular somatic cells exhibit multiple defects. Systematic proteomic profiles were analysed in biological triplicates plus technical triplicates comparing ms8 anthers with fertile sibling samples at both the premeiotic and meiotic stages; proteins from 3.5 to 20 kDa were fractionated by 1-D PAGE, cleaved with Lys-C and then sequenced using a LTQ Orbitrap Velos MS paradigm. Three hundred and 59proteins were identified with two or more assigned peptides in which each of those peptides were counted at least two or more times (0.4% peptide false discovery rate (FDR) and 0.2% protein FDR); 2761 proteins were identified with one or more assigned peptides (0.4% peptide FDR and 7.6% protein FDR). Stage-specific protein expression provides candidate stage markers for early anther development, and proteins specifically expressed in fertile compared to sterile anthers provide important clues about the regulation of meiosis. 49% of the proteins detected by this study are new to an independent whole anther proteome, and many small proteins missed by automated maize genome annotation were validated; these outcomes indicate the value of focusing on low molecular weight proteins. The roles of distinctive expressed proteins and methods for mass spectrometry of low molecular weight proteins are discussed.
Keywords: anther development, male sterility, Zea mays L., ms8, proteomics, LTQ Orbitrap tandem mass spectrometry
Introduction
Maize is a major crop worldwide used for both direct human consumption and as animal feed. Hybrid seed is produced by cross-pollinating two lines, a methodology that is tedious by hand and greatly facilitated by using one male sterile parent (Laughnan and Gabay-Laughnan, 1983). Consequently, understanding male fertility has been a major research focus for maize flower development as a strategy to improve grain yield (Duvick, 2001). Maize male reproductive development requires approximately 30 days from differentiation of stamen primordia through pollen shed at anthesis; the complex developmental programme is highly regular permitting accurate staging of key developmental events based on anther length (Kelliher and Walbot, 2011). The stamen is a compound organ with a slender filament connecting the anther to the vegetative plant. At the start of meiosis, the maize anther has four lobes each containing five cell types plus a single central vascular bundle and the connective parenchyma tissue surrounding the vasculature (Figure 1a,b; Wang et al., 2010, 2011). These features facilitate the acquisition of sufficient carefully staged anthers for biochemical analysis.
Figure 1.
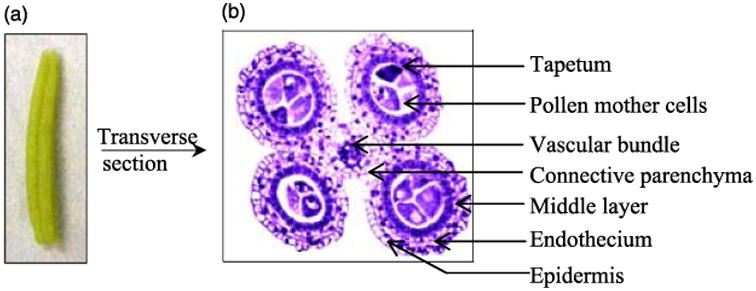
Maize anther morphology and anatomy. (a) Photograph of a 1.5-mm maize anther when the PMC is ready for meiosis; (b) In transverse view (safranine and fast green staining, Wang et al., 2009), each anther has four locules, a single vascular bundle and surrounding connective parenchyma. The anther lobe somatic tissue is composed of four wall layers: epidermis, endothecium, middle layer and tapetum. PMC is located centrally. Meiosis, from the 1.5–2.5-mm anther stages, results in four microspores from each PMC. These separate from the tetrad and mature into pollen.
Transcriptome profiling has established that more than 33 000 maize genes are expressed during anther development, with many examples of stage-specific expression (Ma et al., 2008). As rapid cell proliferation ceases (anther length of 1.0 mm) and three days later at the start of meiosis (1.5 mm), more than 30 000 genes are expressed (Ma et al., 2008; Wang et al., 2010). Maize male sterile mutants with defects during this progression exhibit diverse phenotypes: excess Pollen Mother Cell (PMC) in mac1 (multiple archesporial cells 1) anthers (Sheridan et al., 1999); ms23 (male sterile 23), ms32 (male sterile 32) and ocl4 (outer cell layer 4) have excess somatic cells in one layer (Chaubal et al., 2000; Vernoud et al., 2009); ms8 (male sterile 8) has somatic cell division and expansion defects (Wang et al., 2010); in am1 (ameiotic 1), PMCs conduct mitosis instead of meiosis (Pawlowski et al., 2009); and msca1 (male sterile converted anther1) anthers have normal external morphology but contain leaf cell types (Chaubal et al., 2003). Transcriptome profiling of mac1, ms23 and msca1 (Ma et al., 2007), ms8 (Wang et al., 2010), and am1 (Nan et al., 2011) demonstrate that mutants with more severe phenotypes exhibit a greater disruption in the transcriptome and that detection of transcriptome deviation occurs before anatomical defects are visualized.
Protein evaluation has lagged behind transcriptome profiling in maize and other flowering plants, largely because near complete gene sequence information is required. Detection of protein confirms that transcripts are translated and can quantify post-translational modifications, two rationales for conducting proteomic investigations. Several recent studies have investigated rice anther protein profiles: 40 proteins were identified as consistently differentially expressed during anther development (Kerim et al., 2003); energy production–related proteins were down-regulated at the postmeiotic uninucleate stage in CMS-HL (Honglian Cytoplasmic Male Sterility) lines, which might explain male sterility in this genotype (Sun et al., 2009). Similarly, post-meiotic rice anthers from plants exposed to drought (Liu and Bennett, 2011), cold (Imin et al., 2004) or heat (Jagadish et al., 2010) treatments have been profiled, because these abiotic conditions can limit fertility. To date, there are no proteomic studies of Arabidopsis anthers, because they are extremely small and difficult to dissect from flowers. A key advantage of maize is that the tassel flowers are initially perfect, but the carpels abort early in floral development resulting in a male-only flower. Additionally, maize anthers are exceptionally large when key developmental stages are reached and are highly canalized in their development (Bedinger and Fowler, 2009); these factors facilitate accurate staging for pooling anthers in proteomic analysis. To date in maize, identification of proteins differentially expressed in ms8 compared to fertile siblings (Wang et al., 2010) and in mutator transposon–laden anthers compared to a sibling line with silenced Mu elements (Skibbe et al., 2009) has relied on identifying differentially accumulated proteins visualized by 2D-DIGE followed by LC/MS/MS identification of proteins in individual spots excised from gels. Generally, there is a strong positive correlation between novel protein detection and significant up-regulation of the corresponding transcript; however, the magnitude of differential expression is often different (Skibbe et al., 2009).
The primary aims of this study were to provide a detailed proteome profile of the low molecular weight proteins expressed premeiotically and during meiosis and to compare sibling fertile and ms8 anthers. Proteins expressed prior to and during meiosis are largely unknown in plants, with individual proteins implicated in meiosis defined by loss-of-function alleles and by antibody detection of proteins in situ. The low molecular weight proteome was selected because small secreted proteins are proposed to coordinate anther growth and cell fate setting (Yang et al., 2003; Zhao et al., 2008). Additionally, the pathogen Ustilago maydis causes anthers to switch to tumour production (Skibbe et al., 2010) by expressing many likely secreted low molecular weight proteins, a strategy that implicates small proteins as potent developmental signalling molecules.
The maize ms8 mutant was selected, because it has multiple, discrete phenotypes. It was discovered because anthers do not exert and meiosis fails (Beadle, 1931; Albertsen and Phillips, 1981). Our transcriptome profiling indicated that more than 11% of the gene expression programme is aberrant in ms8 anthers at the 1.0 mm premeiotic, 1.5-mm entry into meiosis and 2.0-mm mid-meiosis stages; a striking feature is that there is precocious expression of many transcripts indicating acceleration of some developmental programmes as well as ectopic expression of genes and the absence of some normal gene expression (Wang et al., 2010). In ms8 anthers, there are more but shorter epidermal cells, while the tapetal layer has too few but larger than normal cells. Thus, ms8 affects cell expansion in one locular somatic cell type and cell proliferation in a different cell type. At the 1.5-mm stage, ms8 PMCs are more widely separated than normal because of excess callose accumulation. Although the mutant meiocytes complete the first meiotic division to form dyads, these structures are defective and the cells collapse; concomitantly, the ms8 anthers stop growing and then senesce (Wang et al., 2010). Using two-dimensional fluorescence difference gel electrophoresis (2D-DIGE) and LC/MS/MS, 63 differentially expressed proteins were identified previously; the largest representation was in metabolism; however, proteins involved in plasmodesmatal remodelling and cell division were also differentially expressed (Wang et al., 2010). In the current study, we have greatly expanded the assessment of protein changes in ms8 compared to fertile siblings by size-fractionating total proteins by 1-D PAGE, digesting with Lys-C to favour recovery of large peptides from small proteins and sequencing peptides with an LTQ Orbitrap MS to acquire accurate mass and high-resolution measurements (Wong et al., 2009).
Results
High reproducibility: intra- and inter-sample correlation (R2) tests
To further explore the phenotypic consequences of the ms8 defect, carefully staged 1.5 mm (entry into meiosis, PMC fully mature) and 2.0 mm (mid-meiosis stage) sterile and corresponding fertile anthers in a primarily A619 inbred background were dissected for protein extraction; three biological replicates were prepared for each sample type. 25 micrograms of each protein extract was size-separated by NuPAGE (Invitrogen, http://www.invitrogen.com) 1-D gel electrophoresis; there was high reproducibility in the visible staining patterns of three technical replicates of each sample (Figure S1). Using alignment with molecular size markers, the regions corresponding to 3.5–10, 10–15 and 15–20 kDa were excised from individual gel lanes and then subjected to in-gel digestion using Lys-C (Figure S1). There were three biological replicates of each sample type. The resulting peptides were then sequenced in technical triplicates (yielding a total of nine data sets for each sample type) using an LTQ Orbitrap Velos mass spectrometer. Among biological triplicates at the 2.0-mm stage when meiosis is well underway, both fertile (0.94) and sterile (0.95) samples showed excellent inter-sample correlations (Table S1). Meiosis is synchronous in maize, and the very large PMC/meiocytes contain ∼20% of anther RNA (Nan et al., 2011) and likely a similar fraction of total protein, two factors that should contribute to exceptional similarity among samples. At the 1.5-mm stage, the correlation among biological triplicates is very high: 0.81 for fertile samples and 0.89 for sterile (Table S1). The 1.5-mm stage includes anthers in which the PMC is still in interphase between the end of mitosis and the start of meiosis, and anthers in which the meiocytes are in the early steps of prophase I; the asynchrony in cell events is the likely cause of the slightly lower correlation (Ma et al., 2008). As shown in Table S2, intra-correlation coefficients were investigated among technical triplicates of each sample type; these averaged 0.89, proving exceptionally high reproducibility; this experimental workflow has been proven reproducible in previous applications (Hack, 2004).
Proteomic profile and identification of proteins with altered abundance by Orbitrap sequencing
Filtering the combined data set of proteins from all four sample types can be done in myriad ways. Here, we report peptide probability (Nesvizhskii and Aebersold, 2005), protein probability (Nesvizhskii and Aebersold, 2005; Nesvizhskii et al., 2007), number of peptides indicative of a single protein (Bantscheff et al., 2007) and finally the number of times peptides were counted; these data are presented in Table S3 (Old et al., 2005). In using a minimum peptide identity probability of 95%, a minimum protein identity probability of 90% and a minimum of two assigned peptides, counted at least twice resulted in 359 proteins with a 0.3% peptide false discovery rate (FDR) and 0.2% protein FDR. Applying the less stringent criteria of only one assigned peptide, requiring only one count gives 2761 unique proteins with a 0.4% peptide FDR and a 7.6% protein FDR (Table S3). As expected, the criterion of two assigned peptides dramatically improves the FDR with the cost of a much smaller data set. Because we are only focusing on proteins smaller than 20 kDa, it is important to note that some very small proteins may only have a single Lys-C cleavage site and therefore generate only a single peptide amenable to a MSMS shotgun proteomics regimen. These proteins are not included in the highest confidence data set, but are undoubtedly present. To increase the robustness of the semi-quantitative measurement of spectral counting (Old et al., 2005), an additional criterion of a minimum of two spectra counted was applied with a minimum of one peptide assignment, which resulted in a total of 672 proteins (Table S3). Because the FDR for peptide assignment is 0.4%, we are confident the majority of the protein assignments based on these peptides are accurate. Molecular weight distribution analysis of the protein models demonstrated that 68% of them were <25 kDa as expected, and the remaining predicted high molecular weight proteins might be processed or truncated products; some protein fragments could be low molecular weight signalling proteins (Figure S2).
Distinctive and shared features of proteome patterns of ms8 and fertile anthers were explored with the 359 highly confident set. As shown in Figure 2a, 93 proteins were specifically expressed at the 1.5-mm stage, and 36 proteins are exclusive to the 2.0-mm stage in fertile anthers. These proteins are valuable stage markers for maize anther development. Proteins involved in translation and catalytic activity are the largest component of these stage-specific proteins; there are 27 proteins in these two categories specifically expressed in 1.5-mm fertile anthers, and 12 proteins in these categories are specific to 2.0-mm anthers (Figure 3). Proteins involved in protein binding and transport are also represented in the stage-specific proteins: seven transport and five protein-binding proteins are found only in the 1.5-mm stage fertile anthers, while nine transport proteins and seven protein-binding proteins are found only in the 2.0-mm stage fertile anthers (Figure 3). These data indicate that execution of meiosis results in obvious changes in the protein profile, especially for translation- and catalytic-activity-related proteins.
Figure 2.
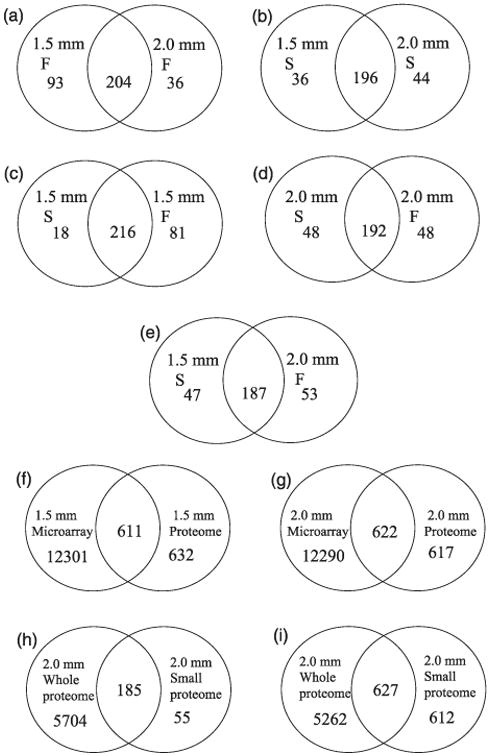
Venn diagrams of proteome content and overlap in various comparisons. (a) Comparison of fertile 1.5- and 2.0-mm stages; (b) Comparison of sterile 1.5- and 2.0-mm stages; (c) Comparison of sterile and fertile anthers at the 1.5-mm stage; (d) Comparison of sterile and fertile anthers at the 2.0-mm stage; (e) Comparison of 1.5-mm sterile anthers with 2.0-mm fertile anthers protein profile to test for precocious protein accumulation in ms8 anthers; (f) Comparison of transcriptome (transcripts with unique protein ID) and proteome data sets at the 1.5-mm stage; (g) Comparison of transcriptome (transcripts with unique protein ID) and proteome data sets at the 2.0-mm stage; (h) and (i) Comparison of a deep-sequenced whole anther proteome (unpublished data from Zhouxin Shen and S. Briggs utilizing staged W23 anther samples dissected at Stanford) and the reported proteome focused on low molecular weight proteins; (h) comparison of the whole anther proteome with 240 highly secure identified proteins in 2-mm fertile anthers addressed here; (i) comparison of whole anther proteome with 1239 proteins with at least one peptide hit identified in 2-mm fertile anthers in our study.
Figure 3.

Gene ontology annotation. Percentage of each gene ontology category of proteins expressed in various comparisons. (a–d): stage-specific proteins; (a) compared with 2.0-mm fertile anthers, specifically expressed proteins in 1.5-mm fertile anthers; (b) compared with 2.0-mm sterile anthers, specifically expressed proteins in 1.5-mm sterile anthers; (c) compared with 1.5-mm fertile anthers, specifically expressed proteins in 2.0-mm fertile anthers; and (d) compared with 1.5-mm sterile anthers, specifically expressed proteins in 2.0-mm sterile anthers; (e–h): fertile or sterile specific proteins; (e) compared with 1.5-mm sterile anthers, specifically expressed proteins in 1.5-mm fertile anthers; (f) compared with 2-mm sterile anthers, specifically expressed proteins in 2.0-mm fertile anthers; (g) compared with 1.5-mm fertile anthers, specifically expressed proteins in 1.5-mm sterile anthers; and (h) compared with 2.0-mm fertile anthers, specifically expressed proteins in 2.0-mm sterile anthers. Categories are in alphabetical order, starting from the top of the list.
Protein profiles overlap substantially between ms8 and fertile siblings (Figure 2c,d). At the 1.5-mm stage, 92% of ms8 anther proteins were shared with fertile anthers; for the 2.0-mm stage, the overlap is 80% (Figure 2c,d). As for the 18 proteins detected in 1.5-mm sterile but not found in 1.5-mm fertile anthers, the top two protein categories are transport and nucleotide binding. For the 81 proteins found in fertile anthers but missing in ms8 anthers, catalytic activity, translation and transport are the top three categories (Figures 2c and 3). These data indicate that ms8 mutants are expressing many proteins in the same categories as fertile siblings; however, different genes are expressed within each category. For the 48 proteins found in 2.0-mm sterile samples but not detected in the fertile counterparts, catalytic activity, DNA binding and translation are predominate. Transport, protein binding and translation are the most abundant classes for the 48 proteins found in 2.0-mm fertile and missing from the sterile data set (Figures 2d and 3); these proteins highlight processes facilitating normal meiosis and should be of interest in future studies of the progression of meiotic stages in plants.
A developmental analysis was conducted for ms8: when comparing proteins identified in two stages of sterile anthers, 36 proteins were detected only at the 1.5-mm stage and 44 proteins only at 2.0 mm (Figure 2b). The Gene Ontology (GO) terms associated with the proteins are slightly different from the pattern in fertile anthers at both stages (Figure 3). Proteins involved in transport, translation and nucleotide binding are the major categories among the 1.5-mm stage-specific proteins; catalytic activity, DNA binding and protein metabolic process are the three main contributors for 2.0-mm stage-specific proteins (Figure 3).
Proteins differentially expressed between ms8 and fertile anthers at two stages were analysed by spectral count, when comparing profiles, a Fisher's exact score (P-value, Zhang et al., 2006) was generated; the twenty top statistically significant differentially expressed proteins are shown in Figure 4a,b. Expression of many nucleic acid–binding proteins changed dramatically before and during meiosis (Figure 4a,b); although these proteins do not show an absolute on/off pattern, quantitative mis-expression is likely to contribute to the many defects in ms8 anthers.
Figure 4.

Differentially expressed protein between ms8 and fertile at the 1.5 and 2.0-mm stages. (a) 1.5 mm differentially expressed proteins; (b) 2.0 mm differentially expressed proteins. The statistical significance was ranked by a Fisher's exact score (P-value). The protein lists were produced from spectral count data of proteins that were identified in both sterile and wild type fractions. All data were manually validated to ensure proteins corresponding to low spectral count (<3) were true positives. The data plotted represent the log of P-value in which a negative value indicates a lower quantitative response in the sterile form and a positive value a higher quantitative response in the sterile form.
Gene Ontology analysis for all proteins identified in fertile, sterile and at both anther developmental stages was considered (Figure S3). In all four groups, proteins involved in translation, transport and catalytic activity are among the top three categories. We conclude that translational capacity and protein transport are key cell processes in both premeiotic and meiotic maize anthers.
ms8 transcriptome profiles at the 1.0, 1.5 and 2.0 mm stages were analysed using custom-designed Agilent 4 × 44 K microarrays containing 42034 probes (excluding controls) to 39612 genes. Major shifts in the transcriptome were detected between sterile and fertile anthers from sibling plants in the same family (Wang et al., 2010). One notable finding was that ms8 anthers achieve some developmental landmarks at a smaller size than fertile anthers; reflecting this precocious development, more than 41% of the 1.5-mm stage-specific ms8 transcripts are expressed at the 2.0-mm stage in fertile siblings (Wang et al., 2010). The current proteomic data substantiate the precocious phenotype first detected in the transcriptome pattern: 80% of the proteins expressed in ms8 anthers at the 1.5-mm stage are also expressed by 2.0-mm fertile anthers (Figure 2e).
Furthermore, the proteomic data set provided a significant expansion of the expressed gene set (Figure 2f,g). When comparing 1.5-mm fertile microarray data (among the 39 612 gene probes, 18 730 transcripts with unique protein ID) with the same stage proteomic data with the less strict criterion of only one or more assigned peptides, 632 proteins were identified only in the protein data set (Figure 2f). At 2.0 mm, 617 proteins were identified only in proteomic data (Figure 2g). A total of 742 small proteins were found in the proteomic analysis for which either transcripts were not detected or no probe was present on the microarray. Maize encodes many small proteins: 8.6% of the 27 500 maize full-length cDNAs encode ORFs of <100 amino acids, with nearly half of these encoding proteins <50 amino acids (Soderlund et al., 2009). Such small proteins are often missed in genome annotation (Silverstein et al., 2007; Lease and Walker, 2006), because only open reading frames encoding more than 100 amino acids are accepted in most automated genome annotations. Consequently, the present study with a focus on small proteins can be used to improve genome annotation as well as highlight the dynamic expression of small proteins during anther development.
Search for processed proteins
To screen for proteins that may have been processed to a shorter length, particularly the class of proteins with transit peptides, the small proteome data set for maize anthers was analysed further to detect peptides not predicted to arise from Lys-C cleavage, an enzyme that cleaves after lysine residues. Peptides that map to the amino terminal region of a protein model but for which the preceding amino acid is not lysine are candidates cases of signal peptide cleavage (Figures S4 and S5); signal peptides average 22.6 amino acids in eukaryotes and have defined features (http://www.cbs.dtu.dk/services/SignalP-1.1/sp_lengths.html). Several protein models match these criteria. One is a 60 S ribosomal protein encoded in the nucleus but with protein function in an organelle; two other models correspond to different isoforms of heat shock factor-binding protein 1 (Table S4). Many heat shock proteins are mitochondrial or Golgi proteins (Lindquist and Craig, 1988), and in plants, additional types are plastid-localized (Nguyen et al., 1993; Lenne and Douce, 1994; Suzuki et al., 1998; Lee et al., 2000; Wang et al., 2004), with the requirement of signal peptide cleavage for correct localization. A high complexity of heat shock proteins was detected when isolated maize microspores were subjected to thermal stress, suggesting that heat shock proteins are inducible during pollen development around the first mitosis stage (Magnard et al., 1996). This corresponds to a stage about 1 week later than the 1.5 and 2.0-mm stages examined in our study. Heat shock proteins are also constitutively expressed as part of the cellular protein folding machinery. To date, 22 maize heat shock protein isoforms have been identified, and individual heat shock binding proteins have specific interactions with them during plant development in the absence of heat shock (Fu et al., 2006). Our data set contains heat shock binding proteins present in anthers before and during meiosis, implying that these factors are critical in early anther development at optimal growth temperature conditions (∼28 °C day/20 °C night).
Comparison of the small anther proteome to a whole proteomic data set
Fertile maize anthers at the 2.0-mm stage were dissected from five plants in the W23 inbred background as a contribution to assembly of a whole protein expression atlas for maize. There are 5889 proteins identified in this global proteome data set in fertile 2.0-mm anthers; this is 24 times the diversity of the focused small proteome data set from this stage identified in a 1 h Orbitrap analysis with at least two peptide hits, and nearly five times higher compared to the small protein data set based on a single peptide hit (Figure 2h,i). Although the small protein data set had a short LTQ Orbitrap operating time and only a small portion of the sizing gel (Figure S1) was sampled, the small proteome data set contains a substantial number of unique proteins (Figure 2h,i). Among the 359 high confidence identified proteins with at least two peptide hits, there are 55 proteins that were not detected in the whole anther analysis; as for proteins with at least one peptide hit, 612 proteins were acquired only in the small protein data set (Figure 2h,i). Functional annotation of these proteins indicates a high diversity of GO representation, indicating that the small protein data set is an important addition to the anther proteome, totalling more than 10% of the combined data sets (Tables S5 and S6). Table S7 provides the GO annotation of the high confidence proteins.
The small protein class is not abundant, based on the paucity of gel staining in the regions sampled (Figure S1); there could be an extraction bias against recovery of small proteins from whole anthers or unexpected migration behaviour as well as the intrinsic inability to reliably identify small proteins after trypsin digestion. Because 49% of the total small proteome found in the restricted analysis was not detected in the whole proteome, our results indicate the value of focused proteome analysis, particularly for small proteins. Validation that specific small proteins exist will be extremely useful in updating the annotation of the proposed gene models for small proteins and in identifying a class of short open reading frames as authentic protein-coding maize genes.
Discussion
As a major crop in terms of economic value and a primary food source for hundreds of millions of people, corn has been improved through intensive breeding. Increasing grain yield is the main target of selection, and the most substantial progress has come from use of high-quality hybrid seed (Duvick, 2001). Mature maize male and female flowers are located in the tassel and ear, respectively, allowing mechanical de-tasselling to permit pollination of one row only by a neighbouring pollen-shedding inbred line (Duvick, 2001). From the mid-1950s until 1970, CMS was used to circumvent detasseling of the ear parent; however, the resulting monocultures with regard to the CMS T (Texas) mitochondrial genome resulted in massive crop failures after emergence of race T of the fungal pathogen Helminthosporium maydis (Levings and Siedow, 1992). A return to male sterility to simplify and lower the cost of hybrid corn seed production will require the use of diverse germplasm to prevent genetic vulnerability in the resulting crop. There are many spontaneous and induced male sterile mutants caused by defects in maize nuclear genes. If deployed appropriately, nuclear male sterility could be as effective as CMS employed previously. Assessment of protein profiles between male sterile mutants and fertile sibling anthers before and during meiosis could clarify the mechanisms of this important progression, and eventually result in the establishment of male sterile inbred lines useful in hybrid seed production.
In this study, differential quantitative proteomic profiling was used to assess developmental changes in maize anthers. The first observation is that maize anthers staged by length have remarkably consistent proteomes, illustrating the appropriateness of length staging within an inbred line, a notable advantage for the investigation of male reproductive development. On average, the protein yield is 3.76 μg per 1.5-mm anther and 7.76 μg per 2.0-mm anther (D. Wang, unpublished data); therefore, samples as small as seven anthers suffice for one replicate in maize. In contrast, about 3000 meiotic stage anthers were needed to produce enough rice protein for analysis (Kerim et al., 2003); even at later stages, such as the uninucleate pollen stage, 40-50 individual rice plants were needed for dissection to produce one biological replicate (Sun et al., 2009). Given the effort involved in obtaining carefully staged anthers, some rice anther proteomic analyses have pooled anthers at different developmental stages (Liu and Bennett, 2011). To date, no proteomic profiles are available for Arabidopsis anthers, given the difficulty of dissecting and collecting tiny anthers. Therefore, the maize anther proteome will serve as the model for flowering plants; because there is high conservation of anther gene expression programmes (Ma et al., 2008), many discoveries in maize will be of general utility.
In maize, male gametophyte development is a tightly programmed and elaborate process central to crop productivity. Therefore, improving our understanding of protein expression in male sterile plants with defects prior to meiosis is a major research priority. We have previously used a 2-D DIGE and LC/MS/MS approach to analyse ms8 and its fertile siblings at three early developmental stages. Sixty-three differentially expressed proteins were identified, with metabolism being the most abundant GO category (Wang et al., 2010). Here, we have expanded this analysis to focus on a profile of small proteins (3.5–20 kDa) profile via LTQ Orbitrap MS analysis. In the set of 359 proteins identified with high confidence, differentially expressed proteins were identified by spectral count (Figure 4a,b). Hundreds of proteins are expressed in a stage-specific manner in both fertile and ms8 sterile anthers, providing new insights about entry into and progression through meiosis; additionally, the hundreds of proteins differentially expressed between fertile and sterile anthers provide new insights into the aberrant phenotypes of ms8 (Figure 2). In addition to the metabolism GO category, proteins involved in transport, translation and catalytic activity showed distinctive expression patterns (Figure 3). The translation category is the major category in 1.5- and 2.0-mm fertile maize anthers, with catalytic activity second (Figure S3). During the ∼3 days separating these two stages, 36 proteins appear and 93 disappear from the proteome lists. Data from a later stage of fertile rice anther development are available: at the uninucleate pollen stage in rice, 97 proteins were identified, the most abundant GO designation was catalytic activity (34%), and 27% of the rice proteins had an unknown molecular function (Sun et al., 2009).
An important contribution of this study is identification of hundreds of distinctively expressed proteins in the premeiotic and mid-meiosis stages as well as markers for male sterility in the ms8 mutant. For example, ADP ribosylation factors (Table S7) exist in functionally redundant plant gene families; they are low molecular weight GTPases required for vesicle trafficking and plasma membrane expansion in eukaryotes. In plants, ARFs play critical roles in secretion and intracellular molecular distribution and are required for the acquisition of epidermal cell polarity (Xu and Scheres, 2005), secretion of callose (Böhlenius et al., 2010) and polar auxin trafficking (Kleine-Vehn et al., 2009). Six maize ADP ribosylation factor proteins were quantified in the high confidence protein data set, and they show complex regulation. Sterile 1.5-mm anthers contain three ADP ribosylation factor proteins (GRMZM2G044368, GRMZM2G075719 and GRMZM2G061912), none of which were found in their 1.5 mm fertile counterparts. Interestingly, all three proteins are detected in 2.0-mm fertile anthers. This protein accumulation pattern confirms that ms8 anthers exhibit precocious expression of some developmental programmes, first evidenced as a substantial proportion of shared transcripts between the fertile 2.0 and ms8 1.5-mm anthers (Wang et al., 2010). Fertile anthers at both developmental stages express GRMZM2G106960, and this protein is present in ms8 1.5-mm anthers but was not detected at 2.0 mm nor were the three precociously expressed ADP ribosylation factors; consequently, at 2.0 mm, ms8 anthers are missing (or have below detectable amounts) of four ADP ribosylation factors accumulating in fertile siblings.
Fertile 1.5-mm anthers express two ADP ribosylation factor genes (GRMZM2G113995 and GRMZM2G086636) that were not detected in the sibling ms8 sterile anthers; however, they were detected in 2.0-mm sterile anthers, somewhat of a surprise because the general pattern is that ms8 mutant anthers show acceleration of numerous developmental programmes (Wang et al., 2010). Given the diverse roles of ADP ribosylation factors in differentiation, mis-regulation of individual isoforms could contribute to ms8 defects in specific cell types such as epidermal expansion, tapetal cell division and callose secretion. Future studies of individual anther cell types could clarify this hypothesis.
GRMZM2G030523 encodes proliferating cell nuclear antigen, an accessory protein for DNA polymerase (Nagar et al., 1995). It is a marker for 1.5-mm fertile anthers compared to the 2.0-mm meiotic stage. Most anther cell division is completed by 1.0 mm (Kelliher and Walbot, 2011); however, a low frequency of division continues over the following 3 days leading up to meiosis, while there is only rare somatic cell division during meiosis. The ADP ribosylation factor GRMZM2G086636 is also a 1.5-mm stage marker, because the protein was not detected in 2.0-mm fertile anthers. Several proteins involved in managing reactive oxygen species are also differentially accumulated in 1.5-mm fertile compared to 2.0-mm fertile anthers: two thioredoxins (GRMZM2G436199 and GRMZM2G300066) and one glutaredoxin (GRMZM2G148387). Glutaredoxins play stage-specific roles in anther development, because knockout mutations confer striking defects (Xing and Zachgo, 2008; Hong et al., 2012). Additionally, two glutathione S-transferases (GRMZM2G031545 and GRMZM2G146246) are stage markers characteristic of 2-mm fertile anthers and are also absent in both stages of ms8 sterile anthers.
Four differentially expressed proteins (GRMZM2G009048, GRMZM2G012479, GRMZM2G075655 and GRMZM2G157443) in the data set were annotated as antifreeze proteins. Antifreeze proteins are usually 12-35 kDa with hydrophilic interfaces that bind to ice crystals in intercellular spaces, altering crystal patterns to reduce cellular dehydration and damage (Jia and Davies, 2002; Pearce, 2001; Griffith et al., 1992; Hon et al., 1994). These proteins are expressed in cold-tolerant species in response to temperature and day-length stimuli; however, maize is not among these cold-tolerant species (Antikainen and Griffith, 1997). Antifreeze proteins are closely related to pathogen response proteins such as β-1, 3-glucanases and chitinases, and plants expressing these proteins have increased resistance to pathogen attack (Huang and Duman, 2001; Hon et al., 1995). It is likely that the proteins detected in maize anthers serve stress response or antipathogenic roles, rather than cold tolerance. The four proteins identified in this data set each have a different pattern of abundance; two of the proteins are more abundant in 1.5-mm fertile anthers compared to 2-mm fertile anthers, and the other two proteins are more abundant in 2-mm anthers compared to 1.5 mm. For all four proteins, when a difference between sterile and fertile comparisons was observed, the proteins were found to be more abundant in the sterile samples. Therefore, the antifreeze proteins show stage-specific accumulation patterns in 1.5 and 2-mm anthers, and the ms8 mutant has increased levels of these proteins. We speculate that the absence of the ms8 protein increases the stress response in sterile plants leading to an overall increase in classes of stress response proteins expressed in anthers. Finally, using the larger proteome list with 1 or more assigned peptides, a cell death protein (protein ID: GRMZM2G021710_P01, death effector) is detected only in 1.5-mm ms8 anthers, and this protein may contribute to the lethal defects of the mutant, a subject for future investigation of the mechanisms underlying male sterility in ms8 and other maize mutants.
The small molecular weight proteome analysis also provides information on an often neglected class of proteins, some of which may serve as signalling molecules during anther development. In a shotgun proteomics approach, the ideal peptide is of a molecular weight larger than 1000 Da, this corresponds to a 9–10 mer using an averaging model. Our data were generated from gel regions corresponding to 3.5–20 kDa, and Lys-C was used for protein digestion. In studying the low molecular weight proteome, particularly using an initial SDS-PAGE size fractionation, there are inherent advantages to using this enzyme. Trypsin is the standard protease used in proteomics research; it cleaves C-terminal to both lysine and arginine residues. For small proteins with multiple lysine and arginine residues, the multiple small peptides are unlikely to be assigned successfully to protein models. Therefore, we used Lys-C to increase average peptide length.
On the basis of full-length cDNA sequencing, about 10% of transcripts encode proteins <100 amino acids. Given that maize anthers at 1.5 and 2.0 mm express about 30 000 transcripts, we might expect up to 3000 small proteins. Based on the intensity of gel staining, however, the low molecular weight region of the anther proteome contains very little protein. In this study, 359 proteins were identified with high confidence, perhaps 12% of the total small proteome; at the more relaxed criterion of one peptide assignment, 2761 proteins were identified, or about 90% of the expectation. Because the overall protein complexity is so reduced compared to the whole anther proteome, a much shorter LC/MS run time should be sufficient to identify most proteins. At the chosen time of 1 h analysis per molecular weight range, the instrument was matched very well with the level of protein complexity, because the maximum duty cycle (the number of peptides sequenced for every survey scan) was rarely pushed to its limitations.
Experimental procedures
Plant material and anther dissection
ms8 and fertile sibling planting and anther dissection were performed as previously described (Wang et al., 2010); cytological study and anther sampling were carried out according to recent publications (Wang et al., 2009, 2010). For the deep whole proteome, fertile maize plants in the W23 inbred background were planted in the field during summer 2009; 300 anthers at the 2.0-mm stage were dissected from each individual and five biological replicates were used for analysis.
SDS-PAGE and in-gel digestion
Anther protein extraction was performed as reported previously (Skibbe et al., 2009). Three biological replicates of each stage were analysed to identify differentially expressed proteins between ms8 and fertile siblings. Total proteins were separated by the NuPAGE electrophoresis system (Invitrogen, http://www.invitrogen.com/site/us/en/home.html), with three technical replicates to improve reliability. Gels were stained with Simply-Blue SafeStain (Invitrogen); three zones (20-15, 15-10 and 10-3.5 kDa) were excised per lane using the Sharp Protein Standard (Invitrogen) as a guide. An example of sample reproducibility and gel cutting is shown in Figure S1. In-gel digestion was performed as previously described (Shevchenko et al., 1996, 2007), but using Lys-C as the protease. In brief, excised gel slices were reduced with DTT and free cysteines alkylated using propionamide. Each gel slice was destained using an acetonitrile/water solution and dried in a speed vacuum. The slices were reconstituted in 100 nanogram of Lys-C, 50 mM Ammonium bicarbonate, pH 8.0. Gel slices were digested overnight at 37 °C in the presence of 0.02% protease max (Promega, Madison, Wl). Peptides were extracted, and each extract was taken to dryness in a speed vacuum centrifuge. Each sample was reconstituted with the same sample buffer (0.1% formic acid, 2% acetonitrile and 97.9% water) and injected onto an HPLC column, as described in the next section.
LTQ Orbitrap Velos sequencing and protein data analysis
The HPLC was an Eksigent (Dublin, CA) nano2D LC run at 600 nL/min with a 60 min linear gradient where solvent B (acetonitrile) started at 2% ramping to 35% over 45 min, followed by column washing and re-equilibration; a Michrom Advance source (Michrom Advance, Auburn, CA) was used in conjunction with a in-house packed fused silica C18 reversed phase column (150 μM inner diameter, 12 cm length). The mass spectrometer was a LTQ Orbitrap Velos, controlled using Xcalibur 2.0 software (Thermo Scientific, San Jose, CA) and run in data-dependent acquisition mode, in which the mass spectrometer was instructed to sequence the top 12 most intense peptide precursor ions. The precursor scan occurred in the Orbitrap at a 60 000 resolution, where each of the ms/ms scans occurred in the LTQ Velos ion trap; this workflow is commonly referred to as a high-low protocol.
Each Thermo (Thermo Scientific, San Jose, CA) RAW file was converted to mzXML format using an msconvert script, after which the file was submitted to database searches using Sequest on a Sorcerer platform. The database was downloaded from the NCBI as Zea mays. Variable modifications of serine, threonine, tyrosine phosphorylation, lysine methylation, demethylation and acetylation were allowed in the analysis. The precursor ions were searched with a 20 ppm mass tolerance and 2 mis-cleavages. Each search result was combined into a master scaffold file for which protein ID, statistical analysis (Fisher's Exact score) and spectrum counts were reported. The data presented as high confidence identification represent proteins that were identified with at least two peptides; this greatly reduced the size of our data set but significantly increased the confidence in protein identification as well as relative quantification using spectral counting. Whole anther proteome analysis was performed in the laboratory of Steven P. Briggs at UCSD (http://briggs.ucsd.edu/publications.html).
Venn diagram and GO chart construction
Comparisons between microarray and proteome results were done by assigning maize protein IDs to each microarray probe using the BLASTx version and BLAST program. Protein sequences were downloaded from version 2 of MaizeGDB's (http://www.maizegdb.org) filtered gene set. Protein assignment for the proteome spectra utilized the same MaizeGDB data set. GO terms for the filtered gene set were also downloaded from MaizeGDB.
Whole anther total protein sequencing analysis
Protein and spectra counts were extracted from the whole anther total protein mass spectrometry data set for each sample type using Scaffold 3 software (http://www.proteomesoftware.com) with the following criteria: minimum number of peptides =1, minimum peptide percent = 95% and minimum protein per cent = 90%. For certain charts, a stricter criterion was applied where the minimum number of peptides = 2.
Supplementary Material
Figure S1 Gel image for three samples of 1.5-mm anther proteins. Each lane is one of three technical replicates of sample 293_1F_1.5 mm. The boxes indicate the gel regions recovered for the 3.5–10, 10–15 and 15–20 kDa samples. Size markers in kDa are shown on the right.
Figure S2 68% of all proteins were <25 kDa, as expected. The x-axis is molecular weight, the y-axis is protein count.
Figure S3 Gene ontology annotation for identified proteins in all four sample types. Percentage of each gene ontology category of expressed proteins in fertile or sterile anthers at different developmental stages. Categories are in alphabetical order, starting from the top of the list.
Figure S4 Protein models for the trans-membrane peptide search.
Figure S5 Peptide start index count.
Table S1 Inter-correlation coefficients between biological triplicates.
Table S2 Intra-correlation coefficients among technical triplicates.
Table S3 Criteria for data filtration.
Table S4 Analysis of potential trans-membrane proteins.
Table S5 GO analysis for 55 proteins that were only identified in the low molecular proteome with more than two assigned peptide, 0.3% peptide FDR and 0.1% protein FDR.
Table S6 GO analysis for 612 proteins that were only identified in the low molecular proteome with at least one assigned peptide, 0.3% peptide FDR and 5.6% protein FDR.
Table S7 GO annotation for the high confidence set of proteins to identify stage-specific and fertile or sterile restricted expression. Note that some proteins are represented in more than one GO category; some proteins identified in this study lacked any GO annotation.
Acknowledgments
We thank Dr. Guo-ling Nan for anther sample dissection for the deep, whole proteome analysis conducted by Dr. Zhouxin Shen and Dr. Steven P. Briggs of UC-SD. We thank Dr. Allis Chien at the SUMS facility and Dr. David S. Skibbe for guidance on the NuPAGE. This research was supported by the National Science Foundation (07-01880) and Award Number S10RR027425 from the National Center for Research Resources. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health.
Footnotes
Supporting information: Additional Supporting information may be found in the online version of this article:
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Albertsen MC, Phillips RL. Developmental cytology of 13 genetic male sterile loci in maize. Genome. 1981;23:195–208. [Google Scholar]
- Antikainen M, Griffith M. Antifreeze protein accumulation in freezing-tolerant cereals. Physiol Plant. 1997;99:423–432. [Google Scholar]
- Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389:1017–1031. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- Beadle GW. Genes in maize for pollen sterility. Genetics. 1931;17:413–431. doi: 10.1093/genetics/17.4.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedinger PA, Fowler JE. The maize male gametophyte. In: Bennetzen JL, Hake SC, editors. Handbook of Maize: Its Biology. New York: Springer; 2009. pp. 57–77. [Google Scholar]
- Böhlenius HJ, Mørch SM, Godfrey D, Nielsen ME, Thordal-Christensen H. The multivesicular body-localized GTPase ARFA1b/1c is important for callose deposition and ROR2 syntaxin-dependent preinvasive basal defense in barley. Plant Cell. 2010;22:3831–3844. doi: 10.1105/tpc.110.078063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaubal R, Zanella C, Trimnell MR, Fox TW, Albertsen MC, Bedinger P. Two male-sterile mutants of Zea mays (Poaceae) with an extra cell division in the anther wall. Am J Bot. 2000;87:1193–1201. [PubMed] [Google Scholar]
- Chaubal R, Anderson JR, Trimnell MR, Fox TW, Albertsen MC, Bedinger P. The transformation of anthers in the msca1 mutant of maize. Planta. 2003;216:778–788. doi: 10.1007/s00425-002-0929-8. [DOI] [PubMed] [Google Scholar]
- Duvick DN. Biotechnology in the 1930s: the development of hybrid maize. Nat Genet. 2001;2:69–74. doi: 10.1038/35047587. [DOI] [PubMed] [Google Scholar]
- Fu S, Rogowsky P, Nover L, Scanlon MJ. The maize heat shock factor-binding protein paralogs EMP2 and HSBP2 interact non-redundantly with specific heat shock factors. Planta. 2006;224:42–52. doi: 10.1007/s00425-005-0191-y. [DOI] [PubMed] [Google Scholar]
- Griffith M, Ala P, Yang D, Hon WC, Moffatt B. Antifreeze protein produced endogenously in winter rye leaves. Plant Physiol. 1992;100:593–596. doi: 10.1104/pp.100.2.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hack CJ. Integrated transcriptome and proteome data: the challenges ahead. Briefings in Funct. Genomics Proteomics. 2004;3:212–219. doi: 10.1093/bfgp/3.3.212. [DOI] [PubMed] [Google Scholar]
- Hon WC, Griffith M, Chang P, Yang D. Extraction and isolation of antifreeze proteins from winter rye leaves. Plant Physiol. 1994;104:879–889. doi: 10.1104/pp.104.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon WC, Griffith M, Mlynarz A, Kwok YC, Yang DS. Antifreeze proteins in winter rye are similar to pathogenesis-related proteins. Plant Physiol. 1995;109:879–889. doi: 10.1104/pp.109.3.879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong L, Tang D, Zhu K, Wang K, Li M, Cheng Z. Somatic and reproductive cell development in rice anther is regulated by a putative glutaredoxin. Plant Cell. 2012 doi: 10.1105/tpc.111.093740. doi. org/10.1105/tpc.111.093740 Online Publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang T, Duman J. Cloning and characterization of a thermal hysteresis (antifreeze) protein with DNA-binding activity from winter bittersweet nightshade, Solanum dulcamara. Plant Mol Biol. 2001;447:339–350. doi: 10.1023/a:1014062714786. [DOI] [PubMed] [Google Scholar]
- Imin N, Kerim T, Rolfe BG, Weinman JJ. Effect of early cold stress on the maturation of rice anthers. Proteomics. 2004;4:1873–1882. doi: 10.1002/pmic.200300738. [DOI] [PubMed] [Google Scholar]
- Jagadish SVK, Muthurajan R, Oane R, Wheeler TR, Heuer S, Bennett J, Craufurd PQ. Physiological and proteomic approaches to address heat tolerance during anthesis in rice (Oryza sativa L.) J Exp Bot. 2010;61:143–156. doi: 10.1093/jxb/erp289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Z, Davies P. Antifreeze proteins: an unusual receptor-ligand interaction. Trends Biochem Sci. 2002;27:101–106. doi: 10.1016/s0968-0004(01)02028-x. [DOI] [PubMed] [Google Scholar]
- Kelliher T, Walbot V. Emergence and patterning of the five cell types of the Zea mays anther locule. Dev Biol. 2011;350:32–49. doi: 10.1016/j.ydbio.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerim T, Imin N, Weinman JJ, Rolffe BG. Proteome analysis of male gametophyte development in rice anthers. Proteomics. 2003;3:738–751. doi: 10.1002/pmic.200300424. [DOI] [PubMed] [Google Scholar]
- Kleine-Vehn J, Huang F, Naramoto S, Zhang J, Michniewicz M, Offringa R, Friml J. PIN auxin efflux carrier polarity is regulated by PINOID kinase-mediated recruitment into GNOM-independent trafficking in Arabidopsis. Plant Cell. 2009;21:3839–3849. doi: 10.1105/tpc.109.071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laughnan JR, Gabay-Laughnan S. Cytoplasmic male sterility in maize. Annu Rev Genet. 1983;17:27–48. doi: 10.1146/annurev.ge.17.120183.000331. [DOI] [PubMed] [Google Scholar]
- Lease KA, Walker JC. The Arabidopsis unannotated secreted peptide database, a resource for plant peptidomics. Plant Physiol. 2006;142:831–838. doi: 10.1104/pp.106.086041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Won SH, Lee HS, Miyao M, Chung WI, Kim IJ, Jo J. Expression of the chloroplast-localized small heat shock protein by oxidative stress in rice. Gene. 2000;245:283–290. doi: 10.1016/s0378-1119(00)00043-3. [DOI] [PubMed] [Google Scholar]
- Lenne C, Douce R. A low molecular mass heat-shock protein is localized to higher plant mitochondria. Plant Physiol. 1994;105:1255–1261. doi: 10.1104/pp.105.4.1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levings CS, Siedow JN. Molecular basis of disease susceptibility in the Texas cytoplasm of maize. Plant Mol Biol. 1992;19:135–147. doi: 10.1007/BF00015611. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Liu JX, Bennett J. Reversible and irreversible drought-induced changes in the anther proteome of rice (Oryza sativa L.) genotypes IR64 and Moroberekan. Mol Plant. 2011;4:59–69. doi: 10.1093/mp/ssq039. [DOI] [PubMed] [Google Scholar]
- Ma J, Duncan D, Morrow DJ, Fernandes J, Walbot V. Transcriptome profiling of maize anthers using genetic ablation to analyze pre-meiotic and tapetal cell types. Plant J. 2007;50:637–648. doi: 10.1111/j.1365-313X.2007.03074.x. [DOI] [PubMed] [Google Scholar]
- Ma J, Skibbe DS, Fernandes J, Walbot V. Male reproductive development: gene expression profiling of maize anther and pollen ontogeny. Genome Biol. 2008;9:R181, 1–17. doi: 10.1186/gb-2008-9-12-r181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnard JL, Vergne P, Dumas C. Complexity and genetic variability of heat-shock protein expression in isolated maize microscopes. Plant Physiol. 1996;111:1085–1096. doi: 10.1104/pp.111.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar S, Pedersen TJ, Carrick KM, Hanley-Bowdoin L, Robinson D. A geminivirus induces expression of a host DNA synthesis protein in terminally differentiated plant cells. Plant Cell. 1995;7:705–719. doi: 10.1105/tpc.7.6.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan GL, Ronceret A, Wang RC, Fernandes JF, Cande WZ, Walbot V. Global transcriptome analysis of two ameiotic1 alleles in maize anthers: defining steps in meiotic entry and progression through prophase I. BMC Plant Biol. 2011;11:1–20. doi: 10.1186/1471-2229-11-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Aebersold R. Interpretation of shotgun proteomic data. Mol Cell Proteomics. 2005;4:1419–1440. doi: 10.1074/mcp.R500012-MCP200. [DOI] [PubMed] [Google Scholar]
- Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;10:787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- Nguyen HT, Weng J, Joshi CP. A wheat (Triticum aestivum) cDNA clone encoding a plastid-localized heat-shock protein. Plant Physiol. 1993;103:675–676. doi: 10.1104/pp.103.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Old WM, Meyer-Arendt K, Aveline-Wolf L, Pierce KG, Mendoza A, Sevinsky JR, Resing KA, Ahn NG. Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol Cell Proteomics. 2005;4:1487–1502. doi: 10.1074/mcp.M500084-MCP200. [DOI] [PubMed] [Google Scholar]
- Pawlowski WP, Wang CR, Golubovskaya IN, Szymaniak JM, Shi L, Hamant O, Zhu T, Harper L, Sheridan WF, Cande WZ. Maize AMEOTIC1 is essential for multiple early meiotic processes and likely required for the initiation of meiosis. Proc Natl Acad Sci USA. 2009;106:3603–3608. doi: 10.1073/pnas.0810115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce R. Plant freezing and damage. Ann Bot. 2001;87:417–424. [Google Scholar]
- Sheridan WF, Golubeva EA, Abrhamova LI, Golubovskaya IN. The mac1 mutation alters the developmental fate of the hypodermal cells and their cellular progeny in the maize anthers. Genetics. 1999;153:933–941. doi: 10.1093/genetics/153.2.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometry sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2007;6:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Silverstein KAT, Moskal JWA, Wu HC, Underwood BA, Graham MA, Town CD, VandenBosch KA. Small cysteine-rich peptides resembling antimicrobial peptides have been under-predicted in plants. Plant J. 2007;51:262–280. doi: 10.1111/j.1365-313X.2007.03136.x. [DOI] [PubMed] [Google Scholar]
- Skibbe DS, Fernandes JF, Medzihradszky KF, Burlingame AL, Walbot V. Mutator transposon activity reprograms the transcriptomes and proteomes of developing maize anthers. Plant J. 2009;59:622–633. doi: 10.1111/j.1365-313X.2009.03901.x. [DOI] [PubMed] [Google Scholar]
- Skibbe DS, Doehlemann G, Fernandes JF, Walbot V. Maize tumor formation after Ustilago maydis infection requires organ-specific gene expression by both partners. Science. 2010;328:89–92. doi: 10.1126/science.1185775. [DOI] [PubMed] [Google Scholar]
- Soderlund C, Descour A, Kudrna D, Bomhoff M, Boyd L, Currie J, Angelova A, Collura K, Wissotski M, Ashley E, Morrow DJ, Fernandes JF, Walbot V, Yu Y. Sequencing, mapping and analysis of 27,455 maize full-length cDNAs. PLoS Genet. 2009;5:e1000740. doi: 10.1371/journal.pgen.1000740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Hu C, Hu J, Li S, Zhu Y. Quantitative proteomic analysis of CMS-related changes in Honglian CMS rice anther. Protein J. 2009;28:341–348. doi: 10.1007/s10930-009-9199-7. [DOI] [PubMed] [Google Scholar]
- Suzuki TC, Krawitz DC, Vierling E. The chloroplast small heat-shock protein oligomer is not phosphorylated and does not dissociate during heat stress in vivo. Plant Physiol. 1998;116:1151–1161. doi: 10.1104/pp.116.3.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernoud V, Laigle G, Rozier F, Meeley RB, Perez P, Rogowsky PM. The HD-ZIP IV transcription factor OCL4 is necessary for trichome patterning and anther development in maize. Plant J. 2009;59:883–894. doi: 10.1111/j.1365-313X.2009.03916.x. [DOI] [PubMed] [Google Scholar]
- Wang W, Vinocur B, Shoseyov O, Altman A. Role of plant heat-shock proteins and molecular chaperones in the abiotic stress response. Trends Plant Sci. 2004;9:244–252. doi: 10.1016/j.tplants.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Wang D, Li C, Zhao Q, Zhao L, Wang M, Zhu D, Ao G, Yu J. Zm401p10, encoded by an anther-specific gene with short open reading frames, is essential for tapetum degeneration and anther development in maize. Fund Plant Biol. 2009;36:73–85. doi: 10.1071/FP08154. [DOI] [PubMed] [Google Scholar]
- Wang D, Oses-Prieto JA, Li KH, Fernandes JF, Burlingame AL, Walbot V. The male sterile 8 mutation of maize disrupts the temporal progression of the transcriptome and results in the mis-regulation of metabolic functions. Plant J. 2010;63:939–951. doi: 10.1111/j.1365-313X.2010.04294.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Skibbe DS, Walbot V. Maize csmd1 exhibits pre-meiotic somatic and post-meiotic microspore and somatic defects but sustains anther growth. Sex Plant Reprod. 2011;24:297–306. doi: 10.1007/s00497-011-0167-y. [DOI] [PubMed] [Google Scholar]
- Wong CCL, Cociorva D, Venable JD, Xu T, Yates JR., III Comparison of different signal thresholds on data dependent sampling in orbitrap and LTQ mass spectrometry for the identification of peptides and proteins in complex mixtures. J Am Soc Mass Spectrom. 2009;20:1405–1414. doi: 10.1016/j.jasms.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing S, Zachgo S. ROXY1 and ROXY2, two Arabidopsis glutaredoxin genes, are required for anther development. Plant J. 2008;5:790–801. doi: 10.1111/j.1365-313X.2007.03375.x. [DOI] [PubMed] [Google Scholar]
- Xu J, Scheres B. Dissection of Arabidopsis ADP-RIBOSYLATION FACTOR 1 function in epidermal cell polarity. Plant Cell. 2005;17:525–536. doi: 10.1105/tpc.104.028449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SL, Xie LF, Mao HZ, Puah CS, Yang WC, Jiang L, Sundaresan V, Ye D. TAPETUM DETERMINANT1 is required for cell specialization in the Arabidopsis anther. Plant Cell. 2003;15:2792–2804. doi: 10.1105/tpc.016618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, VerBerkmoes NC, Langston MA, Uberbacher E, Hettich RL, Samatova NF. Detecting differential and correlated protein expression in label-free shotgun proteomics. J Proteom Res. 2006;5:2909–2918. doi: 10.1021/pr0600273. [DOI] [PubMed] [Google Scholar]
- Zhao X, Palma JD, Oane R, Gamuyao R, Luo M, Chaudhury A, Herve P, Xue Q, Bennett J. OsTDL1A binds to the LRR domain of rice receptor kinase MSP1, and is required to limit sporocyte numbers. Plant J. 2008;54:375–387. doi: 10.1111/j.1365-313X.2008.03426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Gel image for three samples of 1.5-mm anther proteins. Each lane is one of three technical replicates of sample 293_1F_1.5 mm. The boxes indicate the gel regions recovered for the 3.5–10, 10–15 and 15–20 kDa samples. Size markers in kDa are shown on the right.
Figure S2 68% of all proteins were <25 kDa, as expected. The x-axis is molecular weight, the y-axis is protein count.
Figure S3 Gene ontology annotation for identified proteins in all four sample types. Percentage of each gene ontology category of expressed proteins in fertile or sterile anthers at different developmental stages. Categories are in alphabetical order, starting from the top of the list.
Figure S4 Protein models for the trans-membrane peptide search.
Figure S5 Peptide start index count.
Table S1 Inter-correlation coefficients between biological triplicates.
Table S2 Intra-correlation coefficients among technical triplicates.
Table S3 Criteria for data filtration.
Table S4 Analysis of potential trans-membrane proteins.
Table S5 GO analysis for 55 proteins that were only identified in the low molecular proteome with more than two assigned peptide, 0.3% peptide FDR and 0.1% protein FDR.
Table S6 GO analysis for 612 proteins that were only identified in the low molecular proteome with at least one assigned peptide, 0.3% peptide FDR and 5.6% protein FDR.
Table S7 GO annotation for the high confidence set of proteins to identify stage-specific and fertile or sterile restricted expression. Note that some proteins are represented in more than one GO category; some proteins identified in this study lacked any GO annotation.