

Abstract
Prodrugs are widely used in the targeted delivery of cytotoxic compounds to cancer cells. To date, targeted prodrugs for cancer therapy have achieved great diversity in terms of target selection, activation chemistry, as well as size and physicochemical nature of the prodrug. Macromolecular prodrugs such as antibody-drug conjugates, targeted polymer-drug conjugates and other conjugates that self-assemble to form liposomal and micellar nanoparticles currently represent a major trend in prodrug development for cancer therapy. In this review, we explore a unified view of cancer-targeted prodrugs and highlight several examples from recombinant technology that exemplify the prodrug concept but are not identified as such. Recombinant “prodrugs” such as engineered anthrax toxin show promise in biological specificity through the conditionally targeting of multiple cellular markers. Conditional targeting is achieved by structural complementation, the spontaneous assembly of engineered inactive subunits or fragments to reconstitute functional activity. These complementing systems can be readily adapted to achieve conditionally bispecific targeting of enzymes that are used to activate low-molecular weight prodrugs. By leveraging strengths from medicinal chemistry, polymer science, and recombinant technology, prodrugs are poised to remain a core component of highly focused and tailored strategies aimed at conditionally attacking complex molecular phenotypes in clinically relevant cancer.
KEY WORDS: cancer, complementation, conditional bispecificity, prodrugs, targeted therapy
INTRODUCTION
Targeted therapy is the cornerstone of contemporary cancer treatment. For cancers that are completely characterized by uniquely aberrant markers, agents that inhibit or target these markers are excellent therapeutics with minimal adverse effects on normal tissues. The clinical success of imatinib (Gleevec®), for example, which targets the Bcr-Abl tyrosine kinase, is directly due to the unique and casual role of the Bcr-Abl oncoprotein in chronic myeloid leukemia and related “Philadelphia chromosome” leukemias (1). Unfortunately, specific agents are not yet available for the majority of cancers, or even the most common cancers. Prodrugs, which are inactive or less active derivatives of drug molecules and undergo enzymatic or chemical transformation to regenerate the active forms, have been a major strategy in meeting this challenge. By modifying physicochemical characteristics of drugs (e.g., shielding of charges or protection of ionization groups) prior to reaching their sites of action, prodrugs have a long history of overcoming physiologic barriers such as the GI tract. In targeted cancer therapy, conventional chemotherapeutic agents, which lack intrinsic target specificity, are rationally modified to focus and redirect their cytotoxicity to tumor cells. The usefulness of many conventional, nonspecific chemotherapeutic agents, such as doxorubicin, paclitaxel, camptothecan, cisplatin, and their derivatives have been significantly extended by modification into prodrugs, particularly those harboring cell-targeting moieties.
During the past decades, the development of novel approaches to targeting the delivery and activation of prodrugs has been rapid, varied, and impressive. The details of these advances have been documented in several recent reviews (2–6). Here, our aim is to present a unified view of the prodrug concept in targeted cancer therapy. Specifically, we will highlight the functional equivalence of seemingly disparate cell-targeting schemes, and how such schemes may complement each other in targeting complex cancer phenotypes based on two or more molecular markers.
Passively Activated Cancer-Targeted Prodrugs
A diverse range of chemistry has been developed to trigger the activation of cancer-targeted prodrugs to their cytotoxic counterparts in situ (Fig. 1a). Broadly, these strategies can be described as passive or active. Passive strategies make use of aberrant local physicochemical (e.g., reduced pH, hypoxia) or physiologic changes (e.g., overexpression of surface receptors) in tumor tissue to deliver or bioactivate prodrugs via a single step. Active strategies use prodrugs with specialized activation chemistry that must be proffered by a separate, target-directed exogenous enzyme (directed enzyme/prodrug therapy).
Fig. 1.
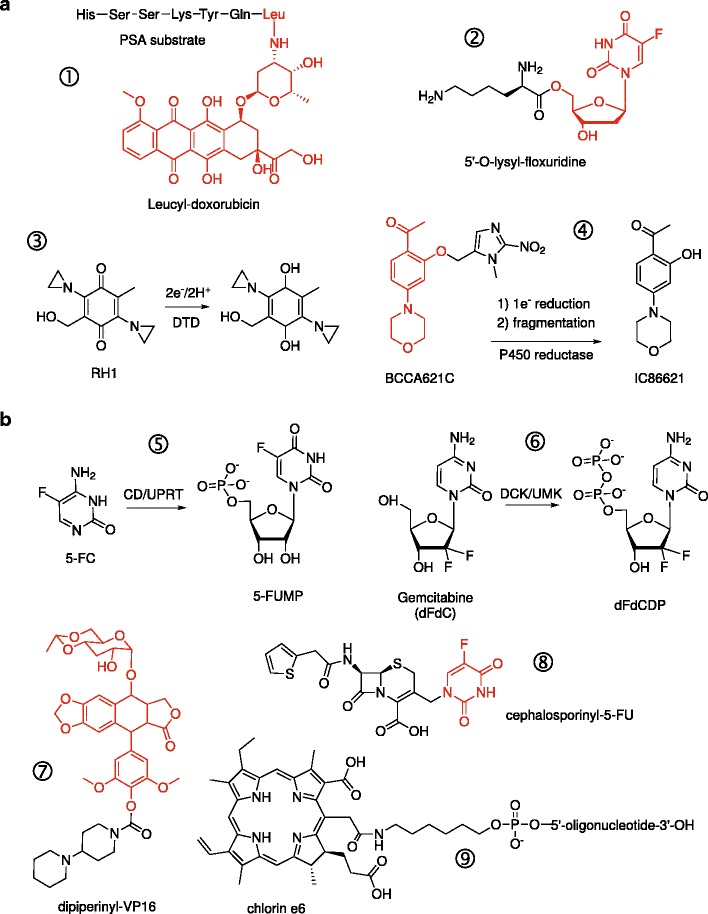
Passive and active conversion of prodrugs. Shown are illustrative examples of prodrugs that are activated by endogenous (passively) or exogenous (actively) enzymes, proteins, or conditions. In the case of conjugates, the active drug moiety is colored in red. a Examples of prodrugs that are substrates for endogenous proteases (① prostate-specific antigen, PSA) (8), membrane transporters (② PEPT1 oligopeptide transporter in pancreatic carcinomas) (14), or intracellular reductases (③ DT-diaphorase and ④ NADPH:cytochrome P450 reductase). b Prodrugs requiring exogenously administered enzymes or energy for activation. Activation of 5-fluorocytosine (⑤ 5-FC) and gemcitabine (⑥ dFdC) by engineered chimeric enzymes to their first cytotoxic antimetabolites. “Designer” conjugates of cytotoxic compounds as substrates for specific exogenous enzymes: ⑦ a recombinant carboxylesterase for dipiperinyl-VP-16 (40) and ⑧ β-lactamase for cephalosporinyl-5-FU (41). ⑨ A conjugate of the photosensitizer chlorin e6 with a single-stranded DNA aptamer that targets epithelial cancers presenting hypo-glycosylated MUC1 antigens (20). Irradiation at 664 nm generates cytotoxic singlet oxygen
Studies in cancer biology have revealed a wide range of enzymes that are aberrantly upregulated in cancer cells. Many proteases are now known to be overexpressed in tumors and contribute to an aggressive or metastatic phenotype. These enzymes can be targeted by incorporating appropriate substrates into the prodrug structure. The most common targets include lysosomal proteases such as the cathepsins and legumain, as well as proteases found in the extracellular matrix (ECM) such as the matrix metalloproteases (MMPs) and urokinase-type plasminogen activator (uPA). Targeting is achieved by incorporating a sequence-specific peptide linker as a “trigger” moiety that prevent free diffusion of the prodrug into cells but, upon cleavage, releases the cytotoxic agent (7). In the case of ECM proteases, tropism for tumors is conferred by proximity of the enzymes near the targeted cell’s surface. For example, a tissue-specific protease in the ECM is prostate-specific antigen (PSA), which has been targeted in prostate cancer by conjugating doxorubicin (8) or with L12ADT (9), a thapsigargin analog (a disruptor of intracellular Ca2+ homeostasis), to the PSA-specific peptide substrate HSSKLQ. Similarly, doxorubicin has been targeted at MMP-expressing fibrosarcoma cells by conjugation with a synthetic MMP-selective peptide substrate (10).
Non-proteolytic targets, such as cell-surface receptors that are aberrantly overexpressed on cancer cells, can also be targeted by prodrug conjugates harboring ligands for these receptors. Many of these receptors undergo endocytosis or transport substrates and therefore act as specific portals into cells. Targeting ligands range widely in size and chemistry. Due to their availability or ease of synthesis, folic acid and short peptides are two of the most common ligands in targeted prodrug conjugates. Folic acid conjugates target the folate receptor (FR) which is differentially overexpressed on many cancer cells and accessible from systemic circulation (11). Among peptides, the tripeptide RGD and their cyclic derivatives are widely used to target integrins and surface protein aminopeptidase N (APN, also known as CD13), both of which are highly expressed in tumor-induced angiogenesis (12,13). Mono- and di-amino acid prodrugs of floxuridine have also been reported to target the PEPT1 transporter, which is highly expressed in some pancreatic adenocarcinomas (14). Bulkier ligands include peptide hormones such as somatostatin and vasoactive intestinal peptide; growth factors/cytokines such as epidermal growth factor (EGF) and various interleukins; as well as antibodies (antibody-drug conjugate, or ADC) (15). The anti-microtubule agent mertansine (DM1) has been used in several clinically successful ADCs, including a conjugate with trastuzumab (T-DM1) to target the HER2 receptor (16), a well-known cell-surface target in metastatic breast cancer. Saccharides represent another class of targeting ligands, ranging from galactose to target the asialoglycoprotein receptor expressed selectively in hepatomas (17), to the polysaccharide hyaluronic acid that binds CD44 on many epithelial cancers (18). A distinct class of ligands used in targeted prodrug conjugates consists of agents derived entirely from in vitro selection, such as affibodies (compact folded proteins) and nucleic acid aptamers (folded single-stranded DNA or RNA). These “artificial” ligands are particularly valuable for targeting disease-related targets for which no endogenous ligand exists. Examples include affibodies for the HER2 receptor (19), and DNA aptamers for the hypo-glycosylated MUC1 antigen (20).
Finally, intracellular targets can be utilized as well. DT-diaphorase (DTD) is a cytosolic enzyme that mediates the two-electron reductase of quinone substrates. DTD levels are elevated in a number of tumor types, including non-small cell lung carcinoma, colorectal carcinoma, liver cancer, and breast carcinoma (21). DTD can bioreductively activate a wide range of quinones, notably the classic DNA cross-linker mitomycin C. Designer alkylating agents such as RH1 (currently in clinical trials in breast cancer) make use of the bioreduction of an attached quinone to selectively activate aziridine-based mustards in cancer cells (22). Another intracellular cancer target is telomerase, a normally repressed enzyme that is active in pancreatic and other cancers (23). Telomerase hydrolyzes acycloguanosyl 5′-thymidyltriphosphate, a thymidine analog prodrug, to acyclovir diphosphate (the active form of acyclovir) (24).
An alternative approach to targeting specific cellular targets is to target aberrant physicochemical features of the tumor microenvironment. For example, deregulated cell growth and poor vascularization of solid tumors lead to severely hypoxic interior environments that promote metastatic dissemination. Hypoxia can be passively targeted with prodrugs that are activated by metabolic reduction (25). A major group of hypoxia-activated prodrugs is represented by nitroheterocycles, which exhibit a range of bioreductive chemistry, including activation of nitro substituents to various labile species or fragmentation to release active moieties (26). For instance, 2-methylimidazole has been incorporated as a trigger moiety in several prodrugs targeting hypoxic cancer cells. BCCA621C is one such example that targets chronically hypoxic cancer cells in which DNA-dependent protein kinase (DNA-PK), a major component of the repair pathway for double-strand breaks, is induced (27). One-electron reduction (for example, by NADPH-dependent P450 reductase) leads to an unstable radical anion that releases the DNA-PK inhibitor IC86621 and enhances the radiosensitivity of hypoxic lung cancer cells (28). Similarly, leukemic cells in hypoxic bone marrow can be targeted with a 2-methylimidazole conjugate of bromo-isophosphoramide mustard termed TH-302 (29).
Coordination complexes containing metal centers that are capable of redox chemistry also represent bioreductively activated prodrugs. Oxidized analogs of cisplatin and other platinating agents, in which the platinum center exists as Pt(IV), are kinetically less reactive cross-linkers relative to Pt(II) and are used in many prodrug designs. Satraplatin, a Pt(IV) analog of cisplatin, is currently under investigation as an orally bioavailable platinating agent (30). Importantly, the oxidized Pt(IV) state stably coordinates two axial ligands that are absent in the more biologically active Pt(II) state. Short peptides containing RGD and NGR motifs have been conjugated to Pt(IV)(NH3)2Cl2 to target integrins and APN (31). Reduction by intracellular thiols rapidly generates cisplatin. A reversed role for the metal center is found in cobalt(III) coordination complexes which act as prodrug “chaperones” by releasing their therapeutic ligands upon reduction to cobalt(II) (32). Targeting can be additionally refined by manipulating the net charge on a prodrug, to take advantage of the relatively acidic (0.5 to 1 pH unit lower than physiologic) of the tumor microenvironment.
Active Conversion of Cancer-Targeted Prodrugs
To expand the target repertoire beyond endogenous activators or conditions, exogenous enzymes can be targeted at cancer cells to activate a specific inactive substrate (prodrug), which is administered separately, to a cytotoxic product (Fig. 1b). In these applications, cytosine deaminase (CD) is historically the most commonly used enzyme. Recombinant CD, cloned from a bacterial, yeast, or fungal source, converts the prodrug 5-fluorocytosine (5-FC) to 5-fluorouracil (5-FU), whose downstream antimetabolites lead to a so-called “thymineless death.” More recently, a yeast cytosine deaminase/uracil phospho-ribosyltransferase fusion (CD/UPRT; encoded by the Fcy::Fur gene) has been introduced as a more efficient alternative to generate the 5-FU-based antimetabolites (33). Other antimetabolite prodrugs include the nucleoside analogs such as acyclovir and ganciclovir, which are activated to their active triphosphate using recombinant thymidylate kinase from herpes simplex virus (34), as well as 6-methyl-2′-deoxyriboside and 2-fluoro-2′-deoxyadenosine, which are converted by Escherichia coli purine nucleoside phosphorylase to 6′-methylpurine and 2-fluoroadenine, respectively (35). Concerns with immunogenicity due to the non-human origin of the activating enzymes can be addressed by the use of engineered human deoxycytidine kinase (DCK) and thymidylate kinase (tmpk) capable of mono-phosphorylating a range of (non-physiologic) prodrugs such as gemcitabine (dFdC), bromovinyl-deoxyuridine (BVdU), cytarabine (AraC), and 3′-azido-3′-deoxythymidine (AZT) monophosphate (36,37). A chimeric fusion of DCK with uridine monophosphate kinase (DCK::UMK) has also been developed to directly activate gemcitabine to its cytotoxic diphosphate metabolite (dFdCDP) in pancreatic carcinoma (38). There are also “designer” prodrugs in which a chemotherapeutic agent is derivatized to a substrate for a specific activating enzyme. Examples include phenoxyacetamide conjugates of doxorubicin and melphalan that are hydrolyzed by penicillin-V amidase (39), a dipiperidinyl conjugate of etoposide (VP-16) that is hydrolyzed by a recombinant carboxylesterase (40), and a cephalosporin conjugate of 5-FU designed for hydrolysis by β-lactamase (41).
In enzyme-activated prodrug therapy, many targeted approaches for delivery of the activating enzymes now exist. The enzyme may be directed to cancer cells as a conjugate with an antibody (antibody-directed enzyme/prodrug therapy, ADEPT), as polymer-based nanoparticles (PDEPT), genetically using engineered non-replicative viruses (GDEPT) or even whole cells (42,43). In particular, the hypoxic tumor microenvironment, which hinders passive drug distribution, can be targeted using engineered bacterial spores of the anaerobic Clostridium sp. harboring genes that encode prodrug-activating enzymes (CDEPT) (44). This approach combines the targeting and transducing capability of Clostridia with their intrinsic bacteriolytic properties. Recently, engineered tumor-tropic Salmonella typhimurium has also been reported (45).
In addition to chemical triggers, physical triggers can be employed to activate cytotoxic prodrugs externally. Photodynamic therapy (PDT) has established itself as a useful treatment for many solid tumors. In PDT, visible or near-infrared light is used to photo-activate inert prodrugs to cytotoxic agents. Examples include the photo-activation of chlorin e6 (46) to generate highly reactive singlet oxygen species, and platinum(IV)-based prodrugs to active platinum(II) species (47). PDT shares some similarities with radiosensitizing prodrugs, and the loco-regional irradiation per se may be considered as targeted therapy. However, PDT has the advantage of using far less damaging radiation and lacking the immunosuppressive toxicity of radiotherapy.
Trageted Macromolecular Prodrugs
Historically, prodrug approaches in targeted therapies have been associated with chemical or enzymatic activation of low-molecular-weight compounds. More recently, the clinical success of the liposomal doxorubicin (DOX) product Doxil® in achieving equivalent anticancer efficacy as free DOX, while significantly reducing off-target toxicity (48), has stimulated the formulation of many nonspecific chemotherapeutics as nanoparticles. Nanoparticles up to ∼100 nm in size can extravasate efficiently through endothelial gaps in tumor vasculature and preferentially accumulate in solid tumors, a phenomenon termed the enhanced permeability and retention (EPR) effect (49). EPR therefore serves as a passive cancer-targeting mechanism. In addition, surface functionalization with high-MW polyethylene glycol (PEGylation) confers prolonged circulation time and avoid uptake by the reticuloendothelial system. To take advantage of these features, the prodrug concept has been incorporated extensively in nanoparticle-based therapeutics, as polymer-drug conjugates or encapsulated nanoparticles. Combined with ligands such as antibodies, peptides, nucleic acid aptamers, and folic acid, nanoparticle-based prodrugs can also be targeted at specific cell-surface receptors, further refining their specificity. To date, a plethora of “nano-formulated” prodrugs of conventional chemotherapeutics have appeared, including liposomes, micelles, and polymer-drug conjugates. Here, we will focus on prodrugs that incorporate additional, specific targeting mechanisms.
Small molecules within a broad range of physicochemical parameters experience relatively few impediments in distribution into cells and subcellular compartments. Compared to low-MW counterparts, macromolecules must overcome specific barriers to reach their final site of action (e.g., nucleus for platinating agents). Unlike small molecules that can freely diffuse across biological membranes, macromolecular agents can only enter live cells via endocytic pathways. Many cell-surface receptors and antigens serve as regulated portals for cell entry if targeted with ligands (50). Once endocytosed, contents are routed into endosomes and subject to acidification, proteolytic enzymes, and exocytosis. Upon reaching the cytosol, it may be additionally necessary to route the drug to specific compartments, such as the nucleus and mitochondria, where they interact with their molecular targets. Thus, the cell biology of regulated entry presents both challenges and opportunities for macromolecular prodrugs to target cancer cells.
In response to these opportunities, a broad range of conjugate strategies has been developed. Many of these approaches extend directly from low-molecular weight conjugates. Antibody-drug conjugates (ADC), for example, are macromolecular analogs of low-MW prodrug conjugates in which a chemotherapeutic is linked to an antibody via engineered linkers (Fig. 2a). These linkers are cleavable under specific physicochemical conditions (low pH, reducing environment) or by specific enzymes (such as esterases or specific ECM or lysosomal proteases), releasing the active drug when the conjugate encounters the targeted endogenous enzyme or condition. Another major class of macromolecular prodrugs is represented by polymer-drug conjugates (Fig. 2b), in which low-molecular weight cytotoxins are conjugated via labile linkers to hydrophilic, biocompatible polymers such as N-(2-hydroxypropyl)methacrylamide copolymers (HPMA) (51). In addition to the cytotoxic agent, cell-targeting moieties (such as receptor ligands, antibodies, or aptamers) can also be anchored to the polymeric backbone, maintaining a unitary targeted polymer-drug conjugate. In addition, many such systems incorporate self-immolative linkers (or spacers) that spontaneously fragment when triggered by a primary targeted event (52). This technology allows the simultaneous release of multiple equivalents of drug from a single triggering event to amplify the cytotoxic effect upon bioactivation.
Fig. 2.
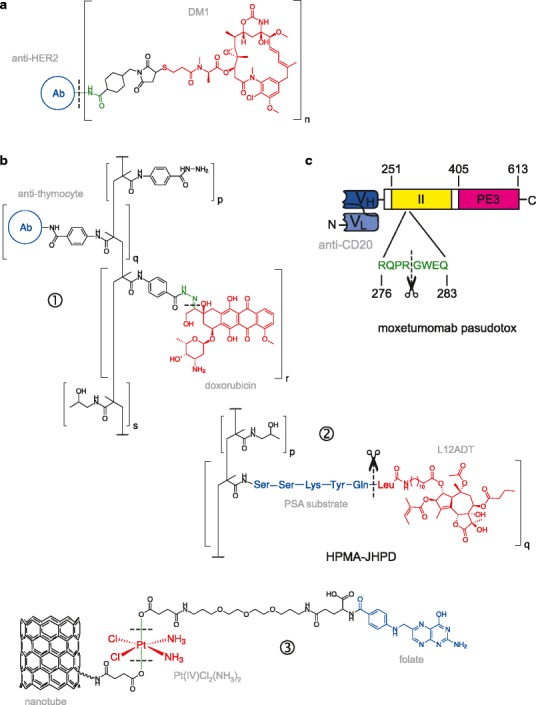
Diversity of targeted macromolecular prodrug conjugates. Shown are illustrative examples of cytotoxic agents (drawn in red) that are covalently attached to targeting moieties (blue) to form macromolecular prodrug conjugates. In some cases, a specifically cleavable linker (or spacer; green) connects the drug and targeting moiety. Cleavage sites are marked with a dashed line; enzyme-mediated cleavage is denoted with a scissors symbol. a Antibody-drug conjugates (ADCs), such as trastuzumab emtansine (T-DM1), in which the anti-tubular agent DM1 is conjugated to trastuzumab that targets HER2-positive metastatic breast cancer (16). In T-DM1, there are on average n = 3.5 equivalents of DM1 per antibody. b Targeted polymer-drug conjugates. HPMA-based copolymers are frequently used as a biocompatible polymeric scaffold to form polymeric nanoparticles. ① A pH-sensitive HPMA-doxorubicin conjugate in which the drug and anti-thymocyte globulin are linked at different HPMA units (94). Release of doxorubicin is triggered by hydrolysis of a hydrazone linker at endosomal pH (5 to 6). ② In HPMA-JHPD, L12ADT (an alkylated thapsigargin analog) targets prostate cancer cells via a sequence-specific peptide linker that is cleaved by prostate-specific antigen (95). ③ Carbon nanotube as a novel macromolecular carrier for Pt(IV)-based prodrugs. The targeting moiety (folic acid) and “longboat” carrier are anchored to the two axial positions present in Pt(IV), which are eliminated when the metal center is reduced to Pt(II), generating cisplatin, under intracellular conditions (53). c Redirected toxins, exemplified by the immunotoxin moxetumomab pasudotox, which is a recombinant conjugate of an anti-CD22 single-chain variable fragment (scFv) to residues 251 to 613 of Pseudomonas exotoxin A. Cytotoxicity encoded in domain III (PE3) is conditionally activated by furin-mediated cleavage between residues 279 and 280 in domain II
In addition to conventional biocompatible polymers, novel materials such as carbon nanotubes are increasingly used to construct macromolecular prodrugs. In the case of platinating agents, the axial positions of Pt(IV) prodrugs have been used to attach them to nanotubes as well as cell-targeting ligands (53,54). As with their low-molecular weight Pt(IV) counterparts, intracellular bioreduction releases the reactive Pt(II) species. Carbon nanotubes exhibit photo-excitation properties and can also act as substrate for PDT to produce reactive singlet oxygen (46). In this way, the nano-carrier can itself exert a therapeutic effect by serving a second, light-activating prodrug.
Besides direct conjugation to a polymeric scaffold, prodrug nanoparticles can also be attained through noncovalent assembly. Pt(IV)(NH3)Cl2 has been derivatized at the axial positions with hexyl chains to impart solubility in the interior of a PLGA-PEG copolymer, to which an aptamer targeting prostate-specific membrane antigen (PSMA) is conjugated (55). Since the interior of nanoparticles is more hydrophobic than bulk solution, the design objective of such a prodrug is to maximize its solubility for encapsulation, rather than to attach the promoiety directly. In the case of highly hydrophobic drugs, they have been incorporated directly as a component of amphiphilic polymers to form micelles or liposomes. For example, paclitaxel has been conjugated with poly(ethyl ethylene phosphate) and folic acid (56). Similarly, phytosphingosine (an anticancer sphingolipid metabolite) has been conjugated with poly(2-hydroxyethyl l-aspartamide) and folic acid (57). In both cases, the resultant amphiphilic conjugates self-assemble to form FR-targeting polymeric micelles capable of loading a second drug (such as doxorubicin), forming two-drug nanoparticles. Hydrolytic cleavage of the polymer at endosomal pH releases both the conjugated drug in the polymer carrier as well as the cargo drug. Finally, hydrophobic anticancer ether lipids have been conjugated as phosphoglycerol and phosphocholine analogs that self-assemble into stable liposomes (58). Cleavage of the phospholipids by secretory phospholipase PLA2 (sPLA2), which is elevated in a variety of epithelial cancers, releases the toxic ether lipids.
In summary, these examples highlight the trend in targeted cancer prodrugs towards macromolecular platforms, which embody the prodrug concept through a broader range of approaches relative to their low-molecular weight counterparts.
Protein Therapuetics As Targeted Cancer Prodrugs
At its core, targeted cancer therapy is concerned with the selective delivery of biologically active payloads to the tumor cells in vivo. It is clear from the foregoing and other reviews (2–6) that conjugates of cytotoxic agents to the full gamut of targeting carriers (ligand-, antibody-, polymer-drug conjugates) constitute a major swath of targeted cancer prodrugs, which release the cytotoxic moiety upon reaching their intended site of action. The diversity of targeted prodrug conjugates per se underscores the general applicability of this approach. In this light, it is helpful to include certain protein therapeutics in this discussion. Protein toxins from a variety of bacterial, fungal, and plant sources are highly potent cytotoxins whose potential as ablative therapeutic agents has been investigated for many decades. Like chemotherapeutics, many (though not all) targeted toxin conjugates employing the same strategies as prodrug conjugates have been reported, although unlike their non-protein counterparts, these targeted toxin conjugates are generally not identified as prodrugs. In our view, there is little reason against a more unified view of targeted prodrug conjugates. Like prodrug conjugates, many targeted toxins consist of a targeting moiety (e.g., an antibody, in the case of immunotoxins), a cleavable linker, and a drug (cytotoxic enzyme). Moxetumomab pasudotox, the most clinically advanced immunotoxin currently in clinical trials for several leukemias and lymphomas, is a bona fide example of this concept (Fig. 2c). It consists of a truncated exotoxin A from Pseudomonas aeruginosa in which the native receptor-binding domain (located in the N-terminal 250 residues) has been replaced with a single-chain variable fragment targeting the cell-surface CD22 antigen (59). Cytotoxic activity is conferred entirely by the C-terminal segment (residues 405 to 613, termed PE3). As is, this conjugate is an inactive toxin: cytotoxic activation requires cleavage by the protease furin during endocytosis between residues 279 and 280 (60). Thus, moxetumomab pasudotox is functionally a targeted prodrug conjugate in which residues 251 to 364 from exotoxin A (domain II) serves as a linker whose cleavage releases the cytotoxic PE3.
In addition to accounting for targeted cancer prodrugs more completely, the inclusion of protein therapeutics in this discussion offers other benefits as well. While conjugates represent the most common type of targeted protein toxins, protein-based prodrugs that incorporate other schemes for targeting cancer cells have also proven promising. The remainder of this review will describe and analyze these schemes with respect to targeting complex cancer phenotypes based on two or more molecular targets. Recent advances in protein engineering present several forward-looking opportunities for adoption by existing prodrug strategies and can potentially refine the specificity of targeted cancer therapeutics.
Beyond Targeting Single Phenotypes: Conditional Multi-specific Targeting
Currently, the majority of targeted therapeutic approaches, including prodrugs, target a single molecular target or condition. These prodrugs are therefore monospecific with respect to their mode of activation, and are generally highly pharmacologically specific. While some cancers can be uniquely differentiated by a single target, most diseases present more complex cellular phenotypes consisting of multiple molecular targets that are mutated or overexpressed. As a result, monospecific agents may be highly selective pharmacologically (for the marker) but be inadequately selective biologically. For example, because the HER2 receptor, a major marker in metastatic breast cancer, is also found on myocardial tissues, trastuzumab and its conjugates that target HER2 alone lead to cardiotoxicity in vivo (61). Despite these apparent limitations, monospecific therapeutics have shown widespread clinical utility. This suggests that if their selectivity profiles could be further refined, significant improvements in their clinical profiles might be attained. Specifically, a reduction in toxicity to normal tissues widens the therapeutic window, thus permitting larger tolerated doses, increased targeted cell killing, and reduced risk of acquired (secondary) resistance. This promise has spurred the development of new therapeutics that incorporates specificities for two or more molecular markers.
Dual- and triple-targeting therapeutics have stirred considerable interest, from simple co-administration of monospecific agents to complex molecular designs. Currently, bi- and tri-specific oncolytic viruses, monoclonal antibodies, immunotoxins, and other protein toxin conjugates comprise emerging classes of targeted therapeutics. Broadly, these multi-specific agents may be divided into those that target a single marker on different cells or multiple targets on a single cell. On the one hand, agents that target markers on different cells are typically intended to elicit cytotoxic immune responses (62) or redirect the tropism of oncolytic viruses (63). On the other hand, bispecific toxin conjugates transductionally target two surface markers (such as receptor tyrosine kinases, interleukin receptors, and surface antigens) on the same cell. Importantly, targeting does not usually depend conditionally on both markers since each marker can initiate endocytosis independently of the other. Thus, these agents typically exhibit enhanced biological activity but not enhanced specificity, since activity on cells that express only one or the other marker (and are therefore “off-target”) remains unaltered. For highly cytotoxic therapeutics, reduced off-target activity is as important a consideration as increased activity on targeted cells. On this front, bispecific conjugates have encountered mixed successes, due to toxicity in off-target cells expressing only one of the two receptors (Fig. 3). For example, a bispecific diphtheria toxin-based conjugate (DTEGF13) targeting both EGFR and the IL-13 receptor (IL13R) in pancreatic adenocarcinoma (64) requires a toxicity blocking or “ToxBloc” protocol (65) to pretreat the host with EGF before administering DTEGF13 to suppress off-target EGFR-mediated toxicity. Physically, bispecific ligands that target cell-surface markers can discriminate cells conditionally on both targets only if the markers are found within the same macromolecule or if the markers associate at least noncovalently (66), a criterion that is infrequently met. Thus, significant challenges remain in engineering refined, conditional multi-specificity into targeted therapeutics.
Fig. 3.
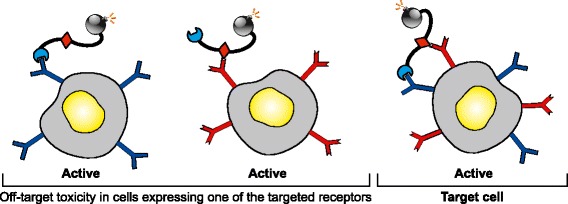
Off-target effects of transductionally bispecific toxin conjugates. A cartoon showing the various cellular interactions of bispecific toxin conjugates harboring two receptor-targeting ligands (blue and red). Since binding by each ligand to its targeted receptor is independent, and each receptor is capable is endocytosis, intoxication ensues in all cells harboring one or both of the targeted cell-surface receptors. Activity in normal cells harboring only one of the targeted receptor leads to off-target, dose-limiting toxicity
Role for the Prodrug Concept in Conditional Multi-specific Targeting
A more tractable route to achieving conditional multi-specificity is to incorporate orthogonal mechanisms of targeting. One approach is to combine transductional targeting of cell-surface receptors with targeting of surface-associated or lysosomal proteases in a trigger moiety consisting of a cell-targeting ligand and protease-specific linker. If access to the protease is consequent to, or is highly localized to the vicinity of the targeted surface receptor, then the free drug would be liberated in a conditionally bispecific manner in the desired cell. When these criteria are satisfied, activation of the prodrug release of cytotoxic drug nominally occurs only in cells presenting both the targeted surface receptor and overexpressed protease. This approach has been adopted by a range of conditionally bispecific prodrugs, including low-MW conjugates, ADCs, polymer-drug conjugates, and nano-carrier systems (Fig. 4). By sparing cells in which one or the other marker is absent, these prodrugs are expected to be more biologically specific than counterparts harboring only one targeting mode.
Fig. 4.
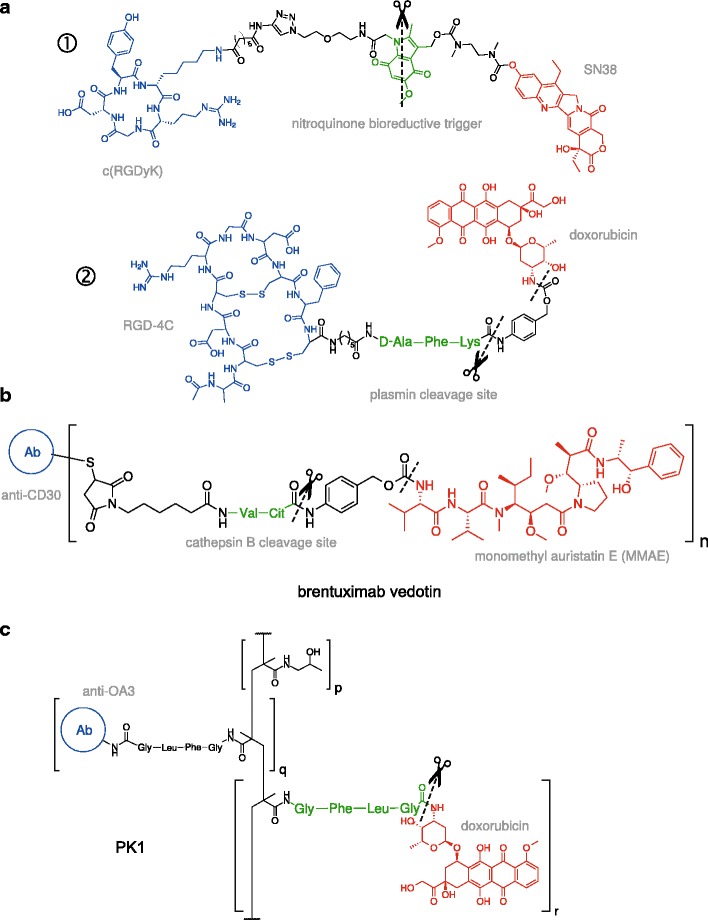
Conditionally bispecific prodrug conjugates. Shown are illustrative examples of cytotoxic agents (red) that are covalently linked to a promoiety that targets two independent markers in a sequential, conditional manner. Receptor-targeting ligands are colored in blue and substrates for target-specific cleavage in green. a “Low-MW” prodrug conjugates targeting cell-surface integrins with cyclic RGD motifs. ① A prodrug conjugate of SN38 (the active form of irinotecan) linked by a nitroquinone trigger. Specific two-electron reduction of the indole nitrogen by intracellular DT-diaphorase (DTD) leads to fragmentation of the linker and drug release (96). ② A doxorubicin conjugate linked by a substrate for plasmin (97). Plasmin cleavage conditionally triggers 1,6-elimination of the adjacent p-aminobenzyl alcohol (PABOH), releasing the free drug. b Brentuximab vedotin is a conjugate of monomethyl auristatin E (MMAE), an anti-mitotic agent, with the anti-CD30 antibody brentuximab (98). Upon endocytosis, cleavage of the valine-citrulline linker by lysosomal cathepsin B, followed by decomposition of the adjacent PABOH moiety, releases MMAE (99). This ADC (Adcetris®) is currently approved for use or in clinical trials for several lymphomas. c A targeted HPMA-doxorubicin (termed PK1) in which DOX and an antibody targeting the surface antigen OA3 on ovarian cancer cells are attached to polymeric HPMA via a peptide substrate (GFLG) for lysosomal cathepsin B (100)
In the biotechnology realm, significant progress has been achieved in transducing proteins in a conditionally multi-specific manner. Currently, engineered anthrax toxins represent the most advanced example of a conditionally bispecific cancer therapeutic that embodies the prodrug concept (Fig. 5). Anthrax toxin (ATx) (from Bacillus anthracis) is an ensemble of three large proteins that represent a highly specialized system for protein transduction: protective antigen (PrAg or PA) (83 kDa), lethal factor (LF) (90 kDa), and edema factor (EF) (89 kDa) (67,68). LF and EF are effector enzymes that target cytosolic substrates in mammalian cells: LF is a zinc metalloprotease that cleaves members of the mitogen-activated protein kinase kinase (MAP2K) family, while EF is a calmodulin- and calcium-dependent adenylyl cyclase. Individually, PA, LF, and EF are nontoxic but together constitute two toxic binary toxin complexes: lethal toxin (LF+PA) and edema toxin (EF+PA). PA is a receptor-binding transporter that specifically transduces LF and EF into the cytosol via a multi-step process. Native PA binds to the surface host receptors ANTXR1 (also called TEM8) and ANTXR2 (also called CMG2), both of which are widespread in human tissues. Receptor binding is coupled to cleavage by a furin protease to a truncated species, PA63, which oligomerizes to form a heptameric or octameric receptor-bound “pre-pore.” (69) Binding of LF or EF to the pre-pore stimulates endocytosis into endosomes (70). Acidification within endosomes rapidly triggers conversion of pre-pores to transmembrane pores that translocate the effector cytotoxic enzymes into the cytosol (71). Thus, effector translocation and cytotoxicity are strictly activated by furin cleavage of PA to PA63. If receptor binding and/or proteolytic activation can be redirected to other targets, ATx may serve as a useful targeted therapeutic.
Fig. 5.
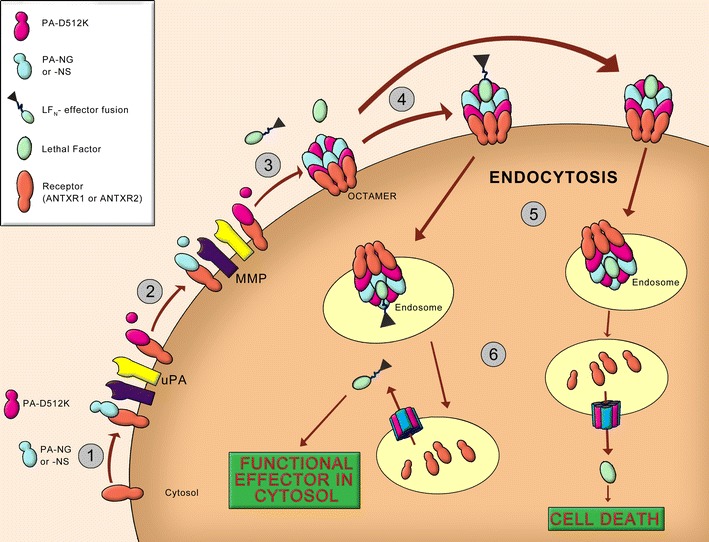
Conditionally bispecific intoxication of cells by engineered anthrax toxin targeted at two ECM proteases: uPA and MMP. Shown is the implementation as reported by Phillips et al. (84). (1) Anthrax protective antigen (PA) recognizes the two receptors ANTXR1 and ANTXR2. Wildtype PA undergoes proteolytic cleavage by furin to a 63-kDa truncated form (PA63) that self-associates in the receptor-bound state. Two variants of protective antigen (PA) were engineered to redirect wildtype PA’s specificity for furin to uPA (2) and MMP (3). (4) Oligomeric PA63 (pre-pore) binds lethal factor (LF) or engineered fusions of its N-terminal domain (LF N) with a cargo effector. (5) The complex is internalized into endosomes. (6) Acidification within the endosomes triggers a transition of the pre-pore to a pore that translocates LF or the LFN-based fusion into the cytosol. Wildtype LF leads to cell death by activating the mitogen-activated protein kinase kinase (MAP2K) pathway. An alternative cytotoxin is a LFN conjugate with Pseudomonas exotoxin A (FP59) that induces apoptosis through inhibition of protein synthesis (72)
The potential for recombinant ATx as a delivery platform is based on two adaptable features of its translocation mechanism. First, PA is able to transport a wide range of protein cargo as simple fusions to the N-terminus of LF (LFN). For example, fusions of LFN with the catalytic domains of diphtheria toxin (DTA), Pseudomonas exotoxin A (PE3), and Shiga toxin have been used as cytotoxic payloads (72,73). Even fusion with polycationic peptides (composed of Lys, Arg, His) can trigger PA-dependent delivery (74). The second adaptable feature of ATx is the ability to redirect the specificity of PA, in two possible ways. One is by mutating the native toxin receptor-binding domain and genetically linking ligands for other cell surface receptors. This has been established for the EGF and HER2 receptors (75–77). The other is by replacing the native furin cleavage site with recognition sequences for other proteases. Such substitutions have been made with cleavage sites for the ECM proteases uPA and MMP (78–82). Thus, engineered ATx with transductionally or proteolytically redirected PA acts functionally as a targeted cancer prodrug. More precisely, targeted receptor binding and/or cleavage of oligomerization-deficient, full-length PA (prodrug) is bioactivated to generate the active moiety (PA63) that oligomerizes to act as a functional translocase.
The native PA oligomer is composed of a single species of subunit. To render pore formation by PA63 conditional on two target proteases, it is necessary to change PA oligomerization from a homotypic transition to a heterotypic one (Fig. 5). This has been achieved by engineering the PA sequence to generate two complementing variants (83,84). For each PA variant, the native furin cleavage site (RXXR) is additionally mutated to recognition sites for uPA (PGSGR↓SA) or MMP (GPLG↓MLSQ). As a result, pore formation occurs only in cells expressing both uPA and MMP, as processing only by both proteases conditionally triggers the formation of a hetero-heptameric or octameric pore that mediates cell entry of the cytotoxic components. Using LF as effector, the engineered toxin system is conditionally toxic to lung tumor cells in vitro and in murine xenograft transplant models when the two variants are administered together (84). The cytotoxic potency of LF using conditionally bispecific ATx (∼10−11 M in vitro) is within 10-fold of its wildtype counterpart. Since transductionally targeted PA still requires furin cleavage for pre-pore formation (75–77), it is likely that conditionally multi-specific PA can also be designed by combining transductional and proteolytic targeting (e.g. EGFR + MMP), although such constructs have not yet been reported.
It is important to note that the use of engineered ATx in cancer targeting involves only the recombinants proteins (PA variants, and LF-based effectors). No microbiological agents, such as the anthrax bacterium or its spore, are involved. Surviving animals at the maximum tolerated dose show no acute nephro- or hepatotoxicity as judged by clinical biochemistry parameters (84).
Incorporation of Conditional Multi-specificity into Directed Enzyme-Prodrug Therapy
Engineered anthrax PA represents a distinctive embodiment of a macromolecular prodrug (as a translocase of LFN-based cargos) when activation is made conditional on targeted proteolysis. Given an apparently broad range of protein cargos that are efficiently transported as LFN-based fusions (85,86), one may envision many applications for the targeted delivery of toxic and nontoxic proteins, as suggested by Collier and co-workers (77). Recent advances in the use of recombinant sortase A, a transpeptidase, to ligate protein and peptides bearing the recognition sequence (LBXT↓G) have made the production of chimeric protein conjugates a relatively efficient task. Indeed, sortase technology has been recently adapted to prepare recombinant affibody ligands for conjugation with a variety of cargo including transductionally targeted PA variants (75).
The feasibility of engineering protein conjugates using sortase and other technologies suggests that, in addition to direct-acting toxins (such as LF or LFN fusions with cytotoxic payloads), complementing PAs present a ready route to incorporating conditional bispecific targeting for enzymes that activate low-MW prodrugs (Fig. 6a). This approach would have the advantage over direct-acting toxins in that it adds a local bystander effect typically associated with prodrug-enzyme approaches. Indeed, low-MW prodrug activation has been incorporated in other recombinant targeted therapeutics to augment cell killing. For example, novel oncolytic viruses, which kill cells through their replication, have been engineered to also transduce genes encoding 5-FC activating enzymes to enhance their antitumor activities and add a bystander effect (87,88). These engineered viruses represent an extension of the GDEPT approach in which the viral vector target as well as directly participate in cell killing.
Fig. 6.
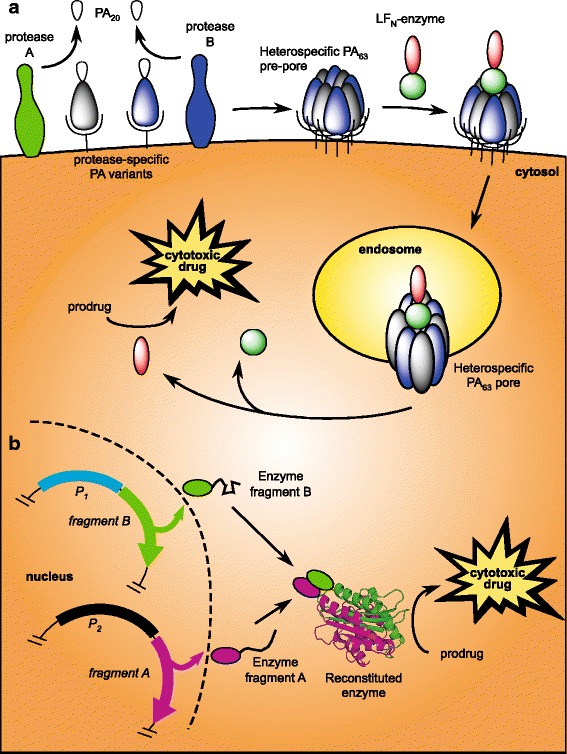
Two schemes for conditionally bispecific enzyme activators for low-MW prodrugs. a Enzymes can be constructed as fusions with the N-terminus of LF to be translocated by conditional bispecific anthrax toxins (ATx). Proteolytic activation of PA and translocation of LFN-based cargos are as described in Fig. 5. In the cytosol of cells expressing both markers, the enzyme would catalyze the bioactivation of a low-MW prodrug. b Alternatively, inactive fragments or subunits of an enzyme can be targeted transcriptionally as transgenes. By placing each transgene under the control of a cell- or disease-specific different promoter (P1 and P2), functional reconstitution becomes conditional within target cells in which both promoters are active. For natively monomeric enzymes, this requires splitting of the primary sequence and likely fusion to a high-affinity oligomerization domain to drive re-assembly. Complementing split enzymes have been demonstrated for a number of systems, including β-lactamase (93), which can be used to bioactivate cephalosporin conjugates of 5-FU (41)
Conditional multi-specific targeting can also be achieved outside the context of anthrax toxin (ATx). Bispecific ATx is one example of a well-established biochemical paradigm known as structural complementation in which protein activity is controlled through the availability of all required subunits. Proteins with intrinsic quaternary structure such as ATx are therefore well-suited for conditional multi-specific targeting by complementation. Functional proteins that are monomeric can also be engineered for complementation, but they must be split into two (or more) inactive fragments. Short of an exhaustive scan of the primary sequence, a site for dissection in the primary sequence must usually be determined from structural data (if available). Since covalent dissection of a folded protein will likely lead to unfolding due to a major loss of the intramolecular interactions that maintain the folded state, useable dissection sites are generally found at minimally folded sites (loops) in the structure. If crystallographic data are available, a convenient parameter is the B (or temperature) factor for the backbone carbon atoms, which is a quantitative indicator of the disorder along the peptide backbone. The order parameter (S2) provides an analog of the crystallographic B-factor in the case of structures solved by solution nuclear magnetic resonance (NMR).
Using these principles, our laboratory has recently developed split fragments of the monomeric PE3 that are capable of structural complementation (89). More precisely, we have engineered fragments that are individually inactive (unfolded) but restore cytotoxicity in combination. Although complementing split PE3 is less potent than wildtype PE3, it maintains similar efficacy in inducing apoptotic cell death. Thus, these inactive fragments may serve as conditionally bispecific toxins if they are targeted individually at different molecular markers. Targeting may be accomplished either by direct transduction of the polypeptide fragments via cell-surface receptors, or genetically by placing transgenes encoding each fragment under the control of independent promoters. With respect to the latter approach, an array of tissue- (90,91) and cancer-specific (92) transcriptional targets have been identified and can be readily incorporated into viral or non-viral gene vectors. Beyond direct-acting toxins, one may envision a similar approach for incorporating conditional bispecificity into enzyme activators of low-MW prodrugs (Fig. 6b). For example, Michnick and co-workers have described a complementing split β-lactamase (93), which can be used to bioactivate cephalosporin conjugates of 5-FU (41).
CONCLUSION
The prodrug concept is utilized in targeted cancer therapy more pervasively than the use of the term presently indicates. In addition to small-molecule and polymer-based approaches, recombinant approaches are properly covered under the prodrug umbrella. In this review, we have adopted an inclusive view of the prodrug concept and highlighted examples of recombinant technology that suggest a promising role for complementation as a strategy to significantly extend the prodrug approach to target complex cancer phenotypes. Certainly, these are forward-thinking concepts that will require extensive additional characterization to establish their therapeutic potential relative to current agents. Recombinant approaches leverage a vast body of pharmaceutical know-how from protein- and gene-based biotherapeutics and are already advancing GDEPT through the combination of prodrug activation and viral oncolysis. We anticipate that medicinal chemistry, polymer science, and recombinant approaches will continue to achieve new synergy in targeting specificity, reduction of host toxicity, and pharmacokinetic optimization.
ACKNOWLEDGMENTS
G.M.K.P. received financial support from Grant Number UL1RR025014 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research, and the College of Pharmacy, Washington State University.
REFERENCES
- 1.Kurzrock R, Kantarjian HM, Druker BJ, Talpaz M. Philadelphia chromosome-positive leukemias: from basic mechanisms to molecular therapeutics. Ann Intern Med. 2003;138(10):819–830. doi: 10.7326/0003-4819-138-10-200305200-00010. [DOI] [PubMed] [Google Scholar]
- 2.Singh Y, Palombo M, Sinko PJ. Recent trends in targeted anticancer prodrug and conjugate design. Curr Med Chem. 2008;15(18):1802–1826. doi: 10.2174/092986708785132997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Han HK, Amidon GL. Targeted prodrug design to optimize drug delivery. AAPS Pharmsci. 2000;2(1). [DOI] [PMC free article] [PubMed]
- 4.Silva AT, Chung MC, Castro LF, Guido RV, Ferreira EI. Advances in prodrug design. Mini Rev Med Chem. 2005;5(10):893–914. doi: 10.2174/138955705774329528. [DOI] [PubMed] [Google Scholar]
- 5.Kratz F, Müller IA, Ryppa C, Warnecke A. Prodrug strategies in anticancer chemotherapy. ChemMedChem. 2008;3(1):20–53. doi: 10.1002/cmdc.200700159. [DOI] [PubMed] [Google Scholar]
- 6.Mahato R, Tai W, Cheng K. Prodrugs for improving tumor targetability and efficiency. Adv Drug Deliv Rev. 2011;63(8):659–670. doi: 10.1016/j.addr.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi KY, Swierczewska M, Lee S, Chen X. Protease-activated drug development. Theranostics. 2012;2(2):156–178. doi: 10.7150/thno.4068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denmeade SR, Nagy A, Gao J, Lilja H, Schally AV, Isaacs JT. Enzymatic activation of a doxorubicin-peptide prodrug by prostate-specific antigen. Cancer Res. 1998;58(12):2537–2540. [PubMed] [Google Scholar]
- 9.Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, et al. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst. 2003;95(13):990–1000. doi: 10.1093/jnci/95.13.990. [DOI] [PubMed] [Google Scholar]
- 10.Albright CF, Graciani N, Han W, Yue E, Stein R, Lai Z, et al. Matrix metalloproteinase-activated doxorubicin prodrugs inhibit HT1080 xenograft growth better than doxorubicin with less toxicity. Mol Cancer Ther. 2005;4(5):751–760. doi: 10.1158/1535-7163.MCT-05-0006. [DOI] [PubMed] [Google Scholar]
- 11.Low PS, Antony AC. Folate receptor-targeted drugs for cancer and inflammatory diseases. Adv Drug Deliv Rev. 2004;56(8):1055–1058. doi: 10.1016/j.addr.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 12.Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, et al. Aminopeptidase N is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 2000;60(3):722–727. [PMC free article] [PubMed] [Google Scholar]
- 13.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2(2):91. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 14.Landowski CP, Vig BS, Song X, Amidon GL. Targeted delivery to PEPT1-overexpressing cells: acidic, basic, and secondary floxuridine amino acid ester prodrugs. Mol Cancer Ther. 2005;4(4):659–667. doi: 10.1158/1535-7163.MCT-04-0290. [DOI] [PubMed] [Google Scholar]
- 15.Ducry L, Stump B. Antibody-drug conjugates: linking cytotoxic payloads to monoclonal antibodies. Bioconjug Chem. 2010;21(1):5–13. doi: 10.1021/bc9002019. [DOI] [PubMed] [Google Scholar]
- 16.Lewis Phillips GD, Li G, Dugger DL, Crocker LM, Parsons KL, Mai E, et al. Targeting HER2-positive breast cancer with Trastuzumab-DM1, an antibody-cytotoxic drug conjugate. Cancer Res. 2008;68(22):9280–9290. doi: 10.1158/0008-5472.CAN-08-1776. [DOI] [PubMed] [Google Scholar]
- 17.Seymour LW, Ulbrich K, Wedge SR, Hume IC, Strohalm J, Duncan R. N-(2-hydroxypropyl)methacrylamide copolymers targeted to the hepatocyte galactose-receptor: pharmacokinetics in DBA2 mice. Br J Cancer. 1991;63(6):859–866. doi: 10.1038/bjc.1991.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oh EJ, Park K, Kim KS, Kim J, Yang JA, Kong JH, et al. Target specific and long-acting delivery of protein, peptide, and nucleotide therapeutics using hyaluronic acid derivatives. J Control Release. 2010;141(1):2–12. doi: 10.1016/j.jconrel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 19.Orlova A, Magnusson M, Eriksson TLJ, Nilsson M, Larsson B, Höidén-Guthenberg I, et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006;66(8):4339–4348. doi: 10.1158/0008-5472.CAN-05-3521. [DOI] [PubMed] [Google Scholar]
- 20.Ferreira CS, Cheung MC, Missailidis S, Bisland S, Gariepy J. Phototoxic aptamers selectively enter and kill epithelial cancer cells. Nucleic Acids Res. 2009;37(3):866–876. doi: 10.1093/nar/gkn967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Danson S, Ward TH, Butler J, Ranson M. DT-diaphorase: a target for new anticancer drugs. Cancer Treat Rev. 2004;30(5):437–449. doi: 10.1016/j.ctrv.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Hargreaves RH, Hartley JA, Butler J. Mechanisms of action of quinone-containing alkylating agents: DNA alkylation by aziridinylquinones. Front Biosci. 2000;5:E172–E180. doi: 10.2741/hargreav. [DOI] [PubMed] [Google Scholar]
- 23.Shay JW, Zou Y, Hiyama E, Wright WE. Telomerase and cancer. Hum Mol Genet. 2001;10(7):677–685. doi: 10.1093/hmg/10.7.677. [DOI] [PubMed] [Google Scholar]
- 24.Polvani S, Calamante M, Foresta V, Ceni E, Mordini A, Quattrone A, et al. Acycloguanosyl 5′-thymidyltriphosphate, a thymidine analogue prodrug activated by telomerase, reduces pancreatic tumor growth in mice. Gastroenterology. 2011;140(2):709–20.e9. doi: 10.1053/j.gastro.2010.10.050. [DOI] [PubMed] [Google Scholar]
- 25.Bennewith KL, Dedhar S. Targeting hypoxic tumour cells to overcome metastasis. BMC Cancer. 2011;11:504. doi: 10.1186/1471-2407-11-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer. 2011;11(6):393–410. http://www.nature.com/nrc/journal/v11/n6/suppinfo/nrc3064_S1.html. [DOI] [PubMed]
- 27.Bouquet F, Ousset M, Biard D, Fallone F, Dauvillier S, Frit P, et al. A DNA-dependent stress response involving DNA-PK occurs in hypoxic cells and contributes to cellular adaptation to hypoxia. J Cell Sci. 2011;124(Pt 11):1943–1951. doi: 10.1242/jcs.078030. [DOI] [PubMed] [Google Scholar]
- 28.Lindquist Kirstin E, Cran Jordan D, Kordic K, Chua Peter C, Winters Geoffrey C, Tan Jason S, et al. Selective radiosensitization of hypoxic cells using BCCA621C: a novel hypoxia activated prodrug targeting DNA-dependent protein kinase. Tumor Microenviron Ther. 2013;1:46. [Google Scholar]
- 29.Portwood S, Lal D, Hsu Y-C, Vargas R, Johnson MK, Wetzler M, et al. Activity of the hypoxia-activated prodrug, TH-302, in preclinical human acute myeloid leukemia models. Clin Cancer Res. 2013 doi: 10.1158/1078-0432.CCR-13-0674. [DOI] [PubMed] [Google Scholar]
- 30.Bhargava A, Vaishampayan UN. Satraplatin: leading the new generation of oral platinum agents. Expert Opin Investig Drugs. 2009;18(11):1787–1797. doi: 10.1517/13543780903362437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mukhopadhyay S, Barnés CM, Haskel A, Short SM, Barnes KR, Lippard SJ. Conjugated platinum(IV) − peptide complexes for targeting angiogenic tumor vasculature. Bioconjug Chem. 2007;19(1):39–49. doi: 10.1021/bc070031k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto N, Renfrew AK, Kim BJ, Bryce NS, Hambley TW. Dual targeting of hypoxic and acidic tumor environments with a cobalt(III) chaperone complex. J Med Chem. 2012;55(24):11013–11021. doi: 10.1021/jm3014713. [DOI] [PubMed] [Google Scholar]
- 33.Erbs P, Regulier E, Kintz J, Leroy P, Poitevin Y, Exinger F, et al. In vivo cancer gene therapy by adenovirus-mediated transfer of a bifunctional yeast cytosine deaminase/uracil phosphoribosyltransferase fusion gene. Cancer Res. 2000;60(14):3813–3822. [PubMed] [Google Scholar]
- 34.Moolten FL. Tumor chemosensitivity conferred by inserted herpes thymidine kinase genes: paradigm for a prospective cancer control strategy. Cancer Res. 1986;46(10):5276–5281. [PubMed] [Google Scholar]
- 35.Parker WB, Allan PW, Shaddix SC, Rose LM, Speegle HF, Gillespie GY, et al. Metabolism and metabolic actions of 6-methylpurine and 2-fluoroadenine in human cells. Biochem Pharmacol. 1998;55(10):1673–1681. doi: 10.1016/S0006-2952(98)00034-3. [DOI] [PubMed] [Google Scholar]
- 36.Hazra S, Sabini E, Ort S, Konrad M, Lavie A. Extending thymidine kinase activity to the catalytic repertoire of human deoxycytidine kinase. Biochemistry. 2009;48(6):1256–1263. doi: 10.1021/bi802062w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato T, Neschadim A, Konrad M, Fowler DH, Lavie A, Medin JA. Engineered human tmpk/AZT as a novel enzyme/prodrug axis for suicide gene therapy. Mol Ther. 2007;15(5):962–970. doi: 10.1038/mt.sj.6300122. [DOI] [PubMed] [Google Scholar]
- 38.Vernejoul F, Ghenassia L, Souque A, Lulka H, Drocourt D, Cordelier P, et al. Gene therapy based on gemcitabine chemosensitization suppresses pancreatic tumor growth. Mol Ther. 2006;14(6):758–767. doi: 10.1016/j.ymthe.2006.07.010. [DOI] [PubMed] [Google Scholar]
- 39.Kerr DE, Senter PD, Burnett WV, Hirschberg DL, Hellstrom I, Hellstrom KE. Antibodypenicillin-V-amidase conjugates kill antigen-positive tumor cells when combined with doxorubicin phenoxyacetamide. Cancer Immunol Immunother. 1990;31(4):202–206. doi: 10.1007/BF01789169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yoon KJ, Qi J, Remack JS, Virga KG, Hatfield MJ, Potter PM, et al. Development of an etoposide prodrug for dual prodrug-enzyme antitumor therapy. Mol Cancer Ther. 2006;5(6):1577–1584. doi: 10.1158/1535-7163.MCT-06-0090. [DOI] [PubMed] [Google Scholar]
- 41.Phelan RM, Ostermeier M, Townsend CA. Design and synthesis of a β-lactamase activated 5-fluorouracil prodrug. Bioorg Med Chem Lett. 2009;19(4):1261–1263. doi: 10.1016/j.bmcl.2008.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tietze LF, Schmuck K. Prodrugs for targeted tumor therapies: recent developments in ADEPT, GDEPT and PMT. Curr Pharm Des. 2011;17(32):3527–3547. doi: 10.2174/138161211798194459. [DOI] [PubMed] [Google Scholar]
- 43.Aboody KS, Najbauer J, Metz MZ, D’Apuzzo M, Gutova M, Annala AJ, et al. Neural stem cell-mediated enzyme/prodrug therapy for glioma: preclinical studies. Sci Transl Med. 2013;5(184):184ra59. doi: 10.1126/scitranslmed.3005365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Umer B, Good D, Anne J, Duan W, Wei MQ. Clostridial spores for cancer therapy: targeting solid tumour microenvironment. J Toxicol. 2012;2012:862764. doi: 10.1155/2012/862764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Massa PE, Paniccia A, Monegal A, de Marco A, Rescigno M. Salmonella engineered to express CD20-targeting antibodies and a drug-converting enzyme can eradicate human lymphomas. Blood. 2013;122(5):705–714. doi: 10.1182/blood-2012-12-474098. [DOI] [PubMed] [Google Scholar]
- 46.Fan J, Zeng F, Xu J, Wu S. Targeted anti-cancer prodrug based on carbon nanotube with photodynamic therapeutic effect and pH-triggered drug release. J Nanoparticle Res. 2013;15(9):1–15. doi: 10.1007/s11051-013-1911-z. [DOI] [Google Scholar]
- 47.Min Y, Li J, Liu F, Yeow EK, Xing B. Near-infrared light-mediated photoactivation of a platinum antitumor prodrug and simultaneous cellular apoptosis imaging by upconversion-luminescent nanoparticles. Angew Chem Int Ed Engl. 2013 doi: 10.1002/anie.201308834. [DOI] [PubMed] [Google Scholar]
- 48.Barenholz Y. Doxil®—the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 49.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Pt 1):6387–6392. [PubMed] [Google Scholar]
- 50.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422(6927):37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 51.Li C, Wallace S. Polymer-drug conjugates: recent development in clinical oncology. Adv Drug Deliv Rev. 2008;60(8):886–898. doi: 10.1016/j.addr.2007.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wong AD, DeWit MA, Gillies ER. Amplified release through the stimulus triggered degradation of self-immolative oligomers, dendrimers, and linear polymers. Adv Drug Deliv Rev. 2012;64(11):1031–1045. doi: 10.1016/j.addr.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 53.Dhar S, Liu Z, Thomale J, Dai H, Lippard SJ. Targeted single-wall carbon nanotube mediated Pt(IV) prodrug delivery using folate as a homing device. J Am Chem Soc. 2008;130(34):11467–11476. doi: 10.1021/ja803036e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ. Soluble single-walled carbon nanotubes as longboat delivery systems for platinum(IV) anticancer drug design. J Am Chem Soc. 2007;129(27):8438–8439. doi: 10.1021/ja073231f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhar S, Gu FX, Langer R, Farokhzad OC, Lippard SJ. Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A. 2008;105(45):17356–17361. doi: 10.1073/pnas.0809154105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang G, Zhang M, He J, Ni P. Synthesis and characterization of a new multifunctional polymeric prodrug paclitaxel-polyphosphoester-folic acid for targeted drug delivery. Polym Chem. 2013;4(16):4515–4525. doi: 10.1039/c3py00419h. [DOI] [Google Scholar]
- 57.Jung BY, Jeong YC, Min JH, Kim JE, Song YJ, Park JK, et al. Tumor-binding prodrug micelles of polymer-drug conjugates for anticancer therapy in HeLa cells. J Mater Chem. 2012;22(18):9385–9394. doi: 10.1039/c2jm30534h. [DOI] [Google Scholar]
- 58.Jensen SS, Andresen TL, Davidsen J, Høyrup P, Shnyder SD, Bibby MC, et al. Secretory phospholipase A2 as a tumor-specific trigger for targeted delivery of a novel class of liposomal prodrug anticancer etherlipids. Mol Cancer Ther. 2004;3(11):1451–1458. [PubMed] [Google Scholar]
- 59.Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res. 2011;17(20):6398–6405. doi: 10.1158/1078-0432.CCR-11-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ogata M, Fryling CM, Pastan I, FitzGerald DJ. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J Biol Chem. 1992;267(35):25396–25401. [PubMed] [Google Scholar]
- 61.Hervent AS, De Keulenaer GW. Molecular mechanisms of cardiotoxicity induced by ErbB receptor inhibitor cancer therapeutics. Int J Mol Sci. 2012;13(10):12268–12286. doi: 10.3390/ijms131012268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chames P, Baty D. Bispecific antibodies for cancer therapy: the light at the end of the tunnel? mAbs. 2009;1(6):539–547. doi: 10.4161/mabs.1.6.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Verheije MH, Rottier PJ. Retargeting of viruses to generate oncolytic agents. Adv Virol. 2012;2012:798526. doi: 10.1155/2012/798526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stish BJ, Chen H, Shu Y, Panoskaltsis-Mortari A, Vallera DA. A bispecific recombinant cytotoxin (DTEGF13) targeting human interleukin-13 and epidermal growth factor receptors in a mouse xenograft model of prostate cancer. Clin Cancer Res. 2007;13(21):6486–6493. doi: 10.1158/1078-0432.CCR-07-0938. [DOI] [PubMed] [Google Scholar]
- 65.Oh S, Stish BJ, Vickers SM, Buchsbaum DJ, Saluja AK, Vallera DA. A new drug delivery method of bispecific ligand-directed toxins, which reduces toxicity and promotes efficacy in a model of orthotopic pancreatic cancer. Pancreas. 2010;39(6):913–922. doi: 10.1097/MPA.0b013e3181cbd908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poon GM. Quantitative analysis of affinity enhancement by noncovalently oligomeric ligands. Anal Biochem. 2013;433(1):19–27. doi: 10.1016/j.ab.2012.10.005. [DOI] [PubMed] [Google Scholar]
- 67.Young JA, Collier RJ. Anthrax toxin: receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 68.Ascenzi P, Visca P, Ippolito G, Spallarossa A, Bolognesi M, Montecucco C. Anthrax toxin: a tripartite lethal combination. FEBS Lett. 2002;531(3):384–388. doi: 10.1016/S0014-5793(02)03609-8. [DOI] [PubMed] [Google Scholar]
- 69.Kintzer AF, Thoren KL, Sterling HJ, Dong KC, Feld GK, Tang II, et al. The protective antigen component of anthrax toxin forms functional octameric complexes. J Mol Biol. 2009;392(3):614–629. doi: 10.1016/j.jmb.2009.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160(3):321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: prepore-to-pore conversion. Biochemistry. 1999;38(32):10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 72.Arora N, Klimpel KR, Singh Y, Leppla SH. Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J Biol Chem. 1992;267(22):15542–15548. [PubMed] [Google Scholar]
- 73.Arora N, Leppla SH. Fusions of anthrax toxin lethal factor with shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect Immun. 1994;62(11):4955–4961. doi: 10.1128/iai.62.11.4955-4961.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Blanke SR, Milne JC, Benson EL, Collier RJ. Fused polycationic peptide mediates delivery of diphtheria toxin A chain to the cytosol in the presence of anthrax protective antigen. Proc Natl Acad Sci U S A. 1996;93(16):8437–8442. doi: 10.1073/pnas.93.16.8437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.McCluskey AJ, Collier RJ. Receptor-directed chimeric toxins created by sortase-mediated protein fusion. Mol Cancer Ther. 2013;12(10):2273–2281. doi: 10.1158/1535-7163.MCT-13-0358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McCluskey AJ, Olive AJ, Starnbach MN, Collier RJ. Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol Oncol. 2013;7(3):440–451. doi: 10.1016/j.molonc.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mechaly A, McCluskey AJ, Collier RJ. Changing the receptor specificity of anthrax toxin. MBio. 2012;3(3):e00088-12. doi: 10.1128/mBio.00088-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu S, Aaronson H, Mitola DJ, Leppla SH, Bugge TH. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc Natl Acad Sci U S A. 2003;100(2):657–662. doi: 10.1073/pnas.0236849100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu S, Bugge TH, Leppla SH. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J Biol Chem. 2001;276(21):17976–17984. doi: 10.1074/jbc.M011085200. [DOI] [PubMed] [Google Scholar]
- 80.Liu S, Netzel-Arnett S, Birkedal-Hansen H, Leppla SH. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000;60(21):6061–6067. [PubMed] [Google Scholar]
- 81.Liu S, Wang H, Currie BM, Molinolo A, Leung HJ, Moayeri M, et al. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J Biol Chem. 2008;283(1):529–540. doi: 10.1074/jbc.M707419200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abi-Habib RJ, Singh R, Liu S, Bugge TH, Leppla SH, Frankel AE. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol Cancer Ther. 2006;5(10):2556–2562. doi: 10.1158/1535-7163.MCT-06-0315. [DOI] [PubMed] [Google Scholar]
- 83.Liu S, Redeye V, Kuremsky JG, Kuhnen M, Molinolo A, Bugge TH, et al. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat Biotechnol. 2005;23(6):725–730. doi: 10.1038/nbt1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Phillips DD, Fattah RJ, Crown D, Zhang Y, Liu S, Moayeri M, et al. Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J Biol Chem. 2013;288(13):9058–9065. doi: 10.1074/jbc.M113.452110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pentelute BL, Sharma O, Collier RJ. Chemical dissection of protein translocation through the anthrax toxin pore. Angew Chem Int Ed Engl. 2011;50(10):2294–2296. doi: 10.1002/anie.201006460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pentelute BL, Barker AP, Janowiak BE, Kent SB, Collier RJ. A semisynthesis platform for investigating structure-function relationships in the N-terminal domain of the anthrax Lethal Factor. ACS Chem Biol. 2010;5(4):359–364. doi: 10.1021/cb100003r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yamada S, Kuroda T, Fuchs BC, He X, Supko JG, Schmitt A, et al. Oncolytic herpes simplex virus expressing yeast cytosine deaminase: relationship between viral replication, transgene expression, prodrug bioactivation. Cancer Gene Ther. 2012;19(3):160–170. doi: 10.1038/cgt.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Simpson GR, Horvath A, Annels NE, Pencavel T, Metcalf S, Seth R, et al. Combination of a fusogenic glycoprotein, pro-drug activation and oncolytic HSV as an intravesical therapy for superficial bladder cancer. Br J Cancer. 2012;106(3):496–507. doi: 10.1038/bjc.2011.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boland EL, Van Dyken CM, Duckett RM, McCluskey AJ, Poon GM. Structural complementation of the catalytic domain of Pseudomonas exotoxin A. J Mol Biol. 2014;426(3):645–655. doi: 10.1016/j.jmb.2013.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dong Z, Nor JE. Transcriptional targeting of tumor endothelial cells for gene therapy. Adv Drug Deliv Rev. 2009;61(7–8):542–553. doi: 10.1016/j.addr.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu Y. Transcriptionally regulated, prostate-targeted gene therapy for prostate cancer. Adv Drug Deliv Rev. 2009;61(7–8):572–588. doi: 10.1016/j.addr.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 92.Robson T, Hirst DG. Transcriptional targeting in cancer gene therapy. J Biomed Biotechnol. 2003;2003(2):110–137. doi: 10.1155/S1110724303209074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Galarneau A, Primeau M, Trudeau LE, Michnick SW. Beta-lactamase protein fragment complementation assays as in vivo and in vitro sensors of protein interactions. Nat Biotechnol. 2002;20(6):619–622. doi: 10.1038/nbt0602-619. [DOI] [PubMed] [Google Scholar]
- 94.Ulbrich K, Etrych T, Chytil P, Jelinkova M, Rihova B. HPMA copolymers with pH controlled release of doxorubicin: in vitro cytotoxicity and in vivo antitumor activity. J Control Release. 2003;87(1–3):33–47. doi: 10.1016/S0168-3659(02)00348-6. [DOI] [PubMed] [Google Scholar]
- 95.Chandran SS, Nan A, Rosen DM, Ghandehari H, Denmeade SR. A prostate-specific antigen-activated N-(2-hydroxypropyl) methacrylamide copolymer prodrug as dual-targeted therapy for prostate cancer. Mol Cancer Ther. 2007;6(11):2928–2937. doi: 10.1158/1535-7163.MCT-07-0392. [DOI] [PubMed] [Google Scholar]
- 96.Huang B, Desai A, Tang S, Thomas TP, Baker JR. The synthesis of a c(RGDyK) targeted SN38 prodrug with an indolequinone structure for bioreductive drug release. Org Lett. 2010;12(7):1384–1387. doi: 10.1021/ol1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.de Groot FM, Broxterman HJ, Adams HP, van Vliet A, Tesser GI, Elderkamp YW, et al. Design, synthesis, and biological evaluation of a dual tumor-specific motive containing integrintargeted plasmin-cleavable doxorubicin prodrug. Mol Cancer Ther. 2002;1(11):901–911. [PubMed] [Google Scholar]
- 98.Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF, et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood. 2003;102(4):1458–1465. doi: 10.1182/blood-2003-01-0039. [DOI] [PubMed] [Google Scholar]
- 99.Sanderson RJ, Hering MA, James SF, Sun MMC, Doronina SO, Siadak AW, et al. In vivo drug-linker stability of an anti-CD30 dipeptide-linked auristatin immunoconjugate. Clin Cancer Res. 2005;11(2):843–852. [PubMed] [Google Scholar]
- 100.Omelyanenko V, Gentry C, Kopečková P, Kopeček J. HPMA copolymer-anticancer drug-OV-TL16 antibody conjugates. II. Processing in epithelial ovarian carcinoma cells in vitro. Int J Cancer. 1998;75(4):600–608. doi: 10.1002/(SICI)1097-0215(19980209)75:4<600::AID-IJC18>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]