

Abstract
Activator protein 1 (AP-1) is a pivotal transcription factor that regulates a wide range of cellular processes including proliferation, apoptosis, differentiation, survival, cell migration, and transformation. Accumulating evidence supports that AP-1 plays an important role in several severe disorders including cancer, fibrosis, and organ injury, as well as inflammatory disorders such as asthma, psoriasis, and rheumatoid arthritis. AP-1 has emerged as an actively pursued drug discovery target over the past decade. Excitingly, a selective AP-1 inhibitor T-5224 (51) has been investigated in phase II human clinical trials. Nevertheless, no effective AP-1 inhibitors have yet been approved for clinical use. Despite significant advances achieved in understanding AP-1 biology and function, as well as the identification of small molecules modulating AP-1 associated signaling pathways, medicinal chemistry efforts remain an urgent need to yield selective and efficacious AP-1 inhibitors as a viable therapeutic strategy for human diseases.
1. Introduction
Activator protein 1 (aka activating protein 1, AP-1) is a critical transcription factor that participates in a wide range of cellular processes including proliferation, apoptosis, differentiation, survival, cell migration, and transformation. AP-1 has emerged as an actively pursued drug discovery target and has received particular attention over the past 2 decades with a resurgence of interest in recent years (Figure 1). Accumulating evidence suggests that AP-1 plays an important role in several severe disorders including cancer, fibrosis, and organ injury, as well as inflammatory disorders such as asthma, psoriasis, rheumatoid arthritis, and transplant rejection.1−4 Despite the great therapeutic potential of this target and the tremendous academic and industrial efforts dedicated to it, only one selective AP-1 inhibitor has been advanced into human clinical trials. 3-{5-[4-(Cyclopentyloxy)-2-hydroxybenzoyl]-2-[(3-hydroxy-1,2-benzisoxazol-6-yl)methoxy]phenyl}propionic acid (T-5224, 51),5 a novel AP-1 inhibitor co-developed by Toyama Chemical and Kitasato University, has proven to prevent joint destruction, pannus formation, and osteoclastogenesis in collagen-induced arthritis (CIA) in rats. 51 shows good promise as a new drug for the treatment of arthritis and is currently in phase II human clinical trials in Japan. Additionally, 51 is under investigation for other inflammatory diseases in which AP-1 is involved. This review is structured to provide the readers with a brief summary of AP-1 family proteins, structures, functions, AP-1 associated signaling pathways, and their roles in various human diseases, as well as the development of AP-1 inhibitors and hit-to-lead optimizations over the past 2 decades from a medicinal chemistry perspective.
Figure 1.
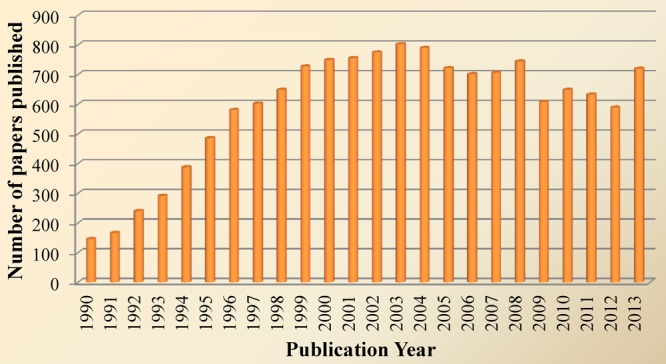
Number of papers published between 1990 and 2013 according to recent PubMed search using “AP-1”.
2. AP-1 Family Proteins, Structures, and Functions
The AP-1 family of transcription factors is composed of homodimers and heterodimers of Jun (v-Jun, c-Jun, JunB, and JunD), Fos (v-Fos, c-Fos, FosB, Fral, and Fra2), ATF (ATF2, ATF3/LRF1, B-ATF, JDP1, and JDP2), and MAF (c-Maf, MafB, MafA, MafG/F/K, and Nrl) protein families,6,7 which are characterized by highly conserved dimeric basic leucine zipper (bZIP) DNA-binding domains. The leucine zipper is a structural motif that forms an extended α-helix in which every seventh amino acid is a leucine.7 The carboxy-terminal regions of α-helixes align to form parallel “coiled coils”, while the amino-terminal regions make base-specific contacts with DNA in the major groove (Figure 2, c-Fos/c-Jun, PDB code 1Fos).8 There are a number of crystal structures of AP-1 protein domains and complexes available from the Protein Data Bank, which have significantly facilitated the understanding of the AP-1 family proteins and their structural diversities.
Figure 2.
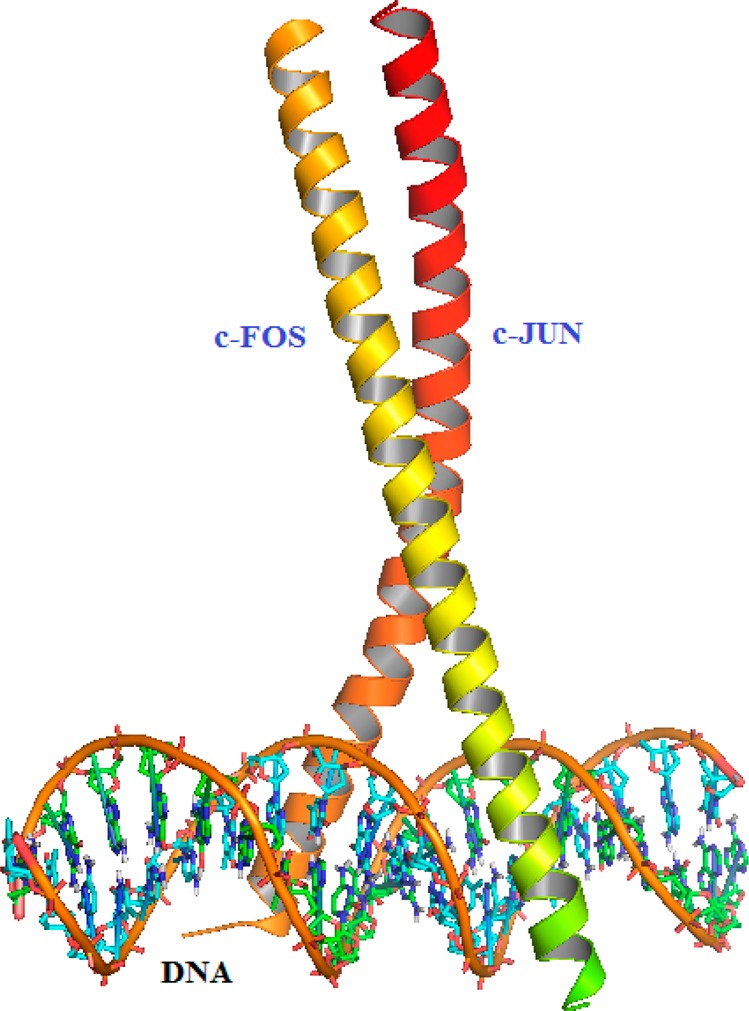
Structures of AP-1 (c-Fos/c-Jun, PDB code 1Fos).
These multiple family members are expressed in a cell- and stage-dependent manner during development and mediate the transcription of specific genes at different levels.7,9 Among them, the Jun and Fos subfamilies are the most studied and the major AP-1 proteins. Although members of the Jun and Fos families share a high degree of structural homology, the individual AP-1 dimers exert significant differences in their DNA binding affinity and their capability of activating or suppressing gene expression, suggesting specific functions in gene regulation for individual AP-1 dimers.2 The Jun proteins can both homo- and heterodimerize with members of Fos and ATF subfamilies, whereas Fos protein can only heterodimerize with Jun proteins rather than homodimerize among themselves. Jun-Jun and Jun-Fos dimers prefer to bind to a heptamer consensus sequence known as the TPA-responsive element (TRE, 5′-TGA(C/G)TCA-3′), whereas Jun-ATF dimers or ATF homodimers preferentially bind to a different consensus sequence known as the cAMP-responsive element (CRE, 5′-TGACGTCA-3′).10 In addition, AP-1 proteins can also interact with non-bZIP proteins, including the p65 subunit of NF-κB, CBP/p300, and Rb, further expanding the combinatorial diversity of AP-1 family proteins and the spectrum of regulated genes.11
A variety of AP-1 associated biological functions in development and disease have been revealed from extensive analyses of mice and cells harboring genetic modifications of distinct Fos and Jun genes. As summarized in Table 1,12−14 many important insights have been provided though gain-of-function and loss-of-function experiments using transgenic and embryonic stem (ES) cell technology.
Table 1. Functions of Jun and Fos proteins.
| phenotype | affected organs/cell types | |
|---|---|---|
| Transgene/Promoters | ||
| c-Fos/H2Kb | Osteosarcoma | Bone/osteoblasts |
| FosB/H2Kb | None | Bone |
| ΔFosB/TCRβ | Impaired T cell differentiation | Thymus/immature thymocytes |
| ΔFosB/NSE | Osteosclerosis | Bone/osteoblasts |
| Fra1/H2Kb | Osteosclerosis | Bone/osteoblasts |
| Fra2/CMV | Occular malformations | Anterior eye structure |
| Fra2a/H2Kb | Increased bone mass/fibrosis | Bone/internal organs/skin |
| c-Jun/H2Kb | None | None |
| JunB/ubiquitin C | Increase bone mass | Not defined |
| JunB/CD4 | Enhanced Th2 maturation | Thymus/CD4 thymocytes |
| JunD/Ubiquitin C | Peripheral T cells and B cells reduced | Lymphocytes |
| Knock-Out | ||
| c-Fos | Osteopetrosis | Bone/osteoclasts |
| FosB | Nurturing defect | Brain/hypothalamus |
| Fra1 | Embryonic lethality (E9.5) | Extra-embryonic tissue/yolk sac; placenta/labyrinth layer |
| Fra2 | Lethal at birth | Bone/osteroclasts |
| c-Jun | Embryonic lethality (E12.5) | Live/hepatoblasts; heart/outflow tract |
| JunB | Embryonic lethality (E8.5-10) | Extra-embryonic tissue/yolk sac, giant trophoblasts; placenta/labyrinth layer |
| JunD | Male sterility | Testis/spermatogenesis |
| Conditional Knock-Out | ||
| c-Jun/Alfp-cre | Impaired liver regeneration | Liver/hapatocytes |
| c-Jun/Col2a1 | Scoliosis | Bones/notochordal cells |
| c-Jun/K5-cre | Eye open at birth reduced tumors | Keratinocytes |
| c-Jun/Nestin-cre | Axonal regeneration defect | Central nervous system/motoneurons |
| JunB/MORE-cre | Osteopenia | Bone/osteoclasts, osteoblasts |
| c-Fos/Nestin-cre | Learning defects | Brain/hippocampal neurons |
| Fra1/MORE-cre | Osteopenia | Bone/osteoblasts |
| JunB + c-Jun/K5-creERT | Psoriasis, psoriatic arthritis | Skin, joint/keratinocytes |
3. AP-1 Signaling Pathways
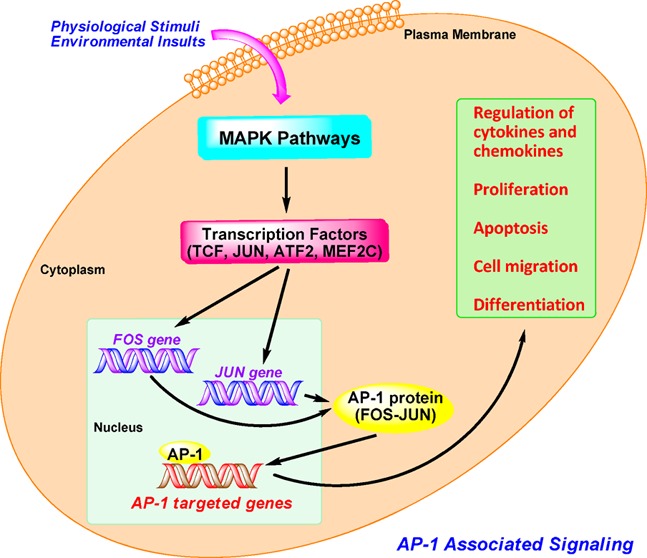
AP-1 activity is induced by a plethora of physiological stimuli and environmental insults, such as the phorbol ester tumor promoter TPA, growth factors, neurotransmitters, polypeptide hormones, cell–matrix interactions, cytokines, UV irradiation, and bacterial and viral infections.2,15,16 AP-1 protein is primarily regulated at the level of both Jun and Fos gene transcription involving mitogen-activated protein kinases (MAPKs) pathways and by post-translational modification via phosphorylation and dephosphorylation.15
As depicted in Figure 3, MAPK cascades consist of three- or four-tiered signaling modules in which the MAPK is activated by a MAPK kinase (MAPKK), which in turn is activated by a MAPK kinase kinase (MAPKKK). The MAPKKK itself is activated by a small G-protein such as Ras, either directly or via another upstream kinase.17 Among these, the JNKs and ERKs, together with the later discovered p38s, constitute three separate groups of MAPKs. The JNKs are activated by the MAPKKs (MKK4 and MKK7). The ERKs are activated by the MEK1 and MEK2, and the p38 is activated by the MKK3 and MKK6.18 After MAPKs are activated, they are able to regulate downstream transcription factors that induce the transcription of Fos and Jun genes, thereby increasing the expression of AP-1 complexes. The expression of Fos is induced by TCFs, which are activated through phosphorylation by the p38, JNKs, and ERKs. The expression of Jun is induced by MEF2C, ATF2, and JUN, which are activated through phosphorylation by the p38 and JNK.18 Once AP-1 and other associated cellular factors are activated, they can participate in regulating altered gene expression, cellular proliferation, apoptosis, differentiation, and migration in response to cytokines and growth factors, noxious stimuli, and oncogenic transformation.
Figure 3.

AP-1 associated signaling pathways.
In addition, various kinases via post-translational phosphorylation also regulate AP-1 activity, including its transactivating potential, DNA-binding capacity, and the stability of AP-1 components; for instance, casein kinase II (CKII), glycogen synthase kinase-3β (GSK-3β), and ribosomal S6 kinase 2 (RSK2). Recently, it has also been shown that the AP-1 protein can be regulated and activated by several other mechanisms, including genetic interaction with other oncoproteins or ancillary proteins, messenger RNA turnover, and protein stability.2
4. AP-1 as a Potential Target for Human Diseases
4.1. AP-1 and Inflammatory Diseases
Inflammation is a part of the complex physiological response of the human body to harmful stimuli, such as pathogen invasion, irritants, and tissue injury. Inflammation is widely recognized as the body’s attempt at self-protection and a beginning of the healing process. In the course of inflammation, immune cells of the innate and/or adaptive immune system are activated, and a variety of different cytokines and chemokines, which are predominantly regulated by AP-1 and other transcription factors including NF-κB, NFATs, and STATs, are recruited to the site of inflammation. Inappropriate activation of the immune system, such as in the case when inflammatory cells and proteins attack and destroy healthy tissue, can result in the overproduction of immune cells, inflammatory cytokines, and tissue-destructive enzymes, thereby giving rise to inflammatory disorders including osteoarthritis, periodontal disease, rheumatoid arthritis, multiple sclerosis, asthma, inflammatory bowel disease, and psoriasis. Interestingly, many cytokine genes are cooperatively regulated by a transcription factor complex consisting of AP-1 and NFAT. AP-1 is a proinflammatory element that is believed to directly control the expression of the cytokines such as tumor necrosis factor α (TNF-α), interleukin 1 (IL-1), interleukin 2 (IL-2), IFNγ, GM-CSF, and matrix-degrading matrix metalloproteases (MMPs) such as collagenase 1 and stromelysin at the level of mRNA synthesis by directly binding their promoter AP-1 binding motifs.7,19−31 Moreover, AP-1 also induces many of the genes that are dependently regulated by NFAT.32
Increasing evidence supports that AP-1 plays a vital role in the initiative and development of inflammatory disorders. It is believed that AP-1 activation is a necessary step in the chain of events that lead to joint erosion. Systematic administration of an AP-1 decoy oligodeoxynucleotide to competitively inhibit AP-1 DNA binding was found to reduce arthritic joint destruction in mice with collagen-induced arthritis.27 Moreover, increased activation and expression of AP-1 have been shown in the airway of asthmatic patients.33,34 The gene for Muc5B, which contains a putative AP-1 consensus site in its promoter, is responsible for the airways mucus production.35 Furthermore, in psoriatic lesions, epidermal keratinocytes have decreased expression of JunB (19p13.2), a gene localized in the psoriasis susceptibility region PSORS6 (19p13).36 Both human and inducible mouse models have demonstrated that down-regulation of JunB in keratinocytes is one initiating event in the etiology of psoriasis that is characterized by increased cell proliferation and deregulated cytokine expression.37−39
Taken together, inhibition of AP-1 may act as a promising therapeutic strategy to provide the benefit of treating the underlying inflammatory process and reducing the production of inflammatory cytokines and chemokines, which play a key role in inflammatory disorders.
4.2. AP-1 and Cancer
In 1982, murine c-Fos proteins were first identified as the viral oncoproteins v-Fos in the Finkel–Biskis–Jinkins osteosarcoma virus and showed increased transforming activity.40−42 Later, mouse c-Jun protein was also first found in its viral counterparts v-Jun in the avian sarcoma virus 17.42 Other Jun (JunB and JunD) and Fos (FosB, Fra1 and Fra2) proteins have also been identified as oncoproteins, all of which are components of AP-1. Several AP-1 proteins such as c-Fos, FosB, and c-Jun have potent transactivation domains,12 which are identified by their ability to induce target–gene transcription and efficiency in transforming cells in culture. Other AP-1 proteins have either a weak transforming activity (Fra1 and Fra243,44) or no transforming activity (JunB and JunD45) due to the lack of potent transactivation domains. Nevertheless, all of these AP-1 proteins are actively involved in tumor development and progression.
Numerous studies have shown that the AP-1 family of transcription factors play a crucial role in proliferation, apoptosis, angiogenesis, oncogene-induced transformation, and invasiveness (Table 2) and are involved in many cancers including breast, ovarian, liver, skin, bone, lung, endometrial, and colorectal tumors.18 When widely overexpressed in mice, c-Fos causes osteosarcoma formation by the transformation of chondroblasts and osteoblasts, which identifies these two cell types as cellular targets of c-Fos-induced tumorigenesis.46,47 It has also been revealed that c-Fos is required for breast cancer cell growth.48 Although c-Jun is variably expressed in human breast tumors with only approximately 20–40%, c-Jun overexpression in MCF-7 breast cancer cells can produce highly invasive and hormone-resistant tumors.49,50 It is found that blockade of c-Jun with the expression of a dominant-negative c-Jun (TAM67) reduces the expression of cyclin Ds, E2Fs, and DP1, resulting in suppressed cell proliferation in vitro and in vivo.51,52 Conditional inactivation of c-Jun by overexpressing TAM67 in mammary epithelial cells, basal keratinocytes, or the liver reduces c-Jun activity and interferes with the development of transgene- or chemical-induced ER-negative mammary tumor, papillomas, and liver tumors, respectively,51,53,54 suggesting that c-Jun is an important target in the development of ER-negative breast cancer, skin and liver tumors. In addition, overexpression of Fra1 and Fra2 in transgenic mice can lead to the development of lung tumors and epithelial tumors, respectively.55 Furthermore, loss of the JunB transcription factor induces a myeloproliferative disease (MPD) arising from the hematopoietic stem cell (HSC) compartment.56JunB inactivation has been observed in a spectrum of human myeloid malignancies including CML,57 and down-regulation of JunB expression has also been found in the HSC compartment of patients with acute myeloid leukemia.58 Therefore, JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal.59
Table 2. AP-1 Target Genes in Tumor Development and Suppression.
| main regulator | gene product | activity |
|---|---|---|
| c-Fos (up-regulates) | DNMT1 | DNA methylation60 |
| FASL | Stimulates apoptosis61 | |
| VEGFD | Angiogenesis62 | |
| MMP1, MMP3, CD44, | Invasiveness63−65 | |
| c-Jun (up-regulates) | EGFR, HB-EGF, GM-CSF, KGF, | Stimulates proliferation66 |
| FASL, BIM | Stimulates apoptosis67 | |
| BCL 3 | Inhibits apoptosis68 | |
| Proliferin | Angiogenesis | |
| CD44 | Invasiveness69 | |
| c-Jun (down-regulates) | WAF-1, p53, INK4A, | Inhibits proliferation52,53,70,71 |
| Cyclin Ds, E2F2, DP1 | ||
| P53, FAS | Stimulates apoptosis72 | |
| JunB (up-regulates) | EGFR | Stimulates proliferation73 |
| INK4A | Stimulates apoptosis74 | |
| Proliferin | Angiogenesis75 | |
| JunB (down-regulates) | GM-CSF, KGF, cyclin D1 | Stimulates proliferation71,76,77 |
| BCL 3, BCL-XL | Inhibits apoptosis56 | |
| JunD (down-regulates) | ARF | Inhibits proliferation78 |
| Fra1 (up-regulates) | uPA, uPAR | Angiogenesis79 |
| MMP3, MMP1 | Invasiveness80 |
In summary, accumulating studies support that increased expression of AP-1 is associated with a variety of human cancers, and AP-1 is actively implicated in tumor progression and development. The available findings suggest that AP-1 may act as a promising target for cancer prevention and therapy and that AP-1 inhibitors may have great potential to be developed as effective drugs for oncogene-induced transformation, premalignant lesions, and tumor cell growth.
4.3. AP-1 and Other Human Diseases
Of the several potential therapeutic indications by modulating AP-1 activity, inflammation and cancer represent the two most promising major areas. Moreover, compelling evidence using genetically modified mice has provided novel insights into the fundamental functions of AP-1 and shown that up-regulation or down-regulation of AP-1 expression also has a significant contribution in the progression or initiation of various other human diseases, including hepatitis, pulmonary fibrosis, atherosclerosis, cardiovascular diseases, and Parkinson’s disease (PD).
4.3.1. Hepatitis and Liver Injury
c-Jun and JunB are strongly expressed in the liver of human and murine with hepatitis. In hepatocytes, the dimeric transcription factor c-Jun is a major mediator of cell survival during hepatitis (inflammation of the liver). Mice lacking c-Jun in hepatocytes display increased liver cell death and mortality upon administration of Con A, which induces liver inflammation and injury. Wagner and colleagues81 found that this phenotype was caused by impaired expression of inducible nitric oxide synthase (NOS2), a direct transcriptional target of c-Jun, and reduced production of hepatoprotective nitric oxide (NO). Moreover, increased hepatotoxicity in mutant mice is likely caused by hypoxia and oxidative stress and can be pharmacologically rescued by liver-specific NO delivery. Thus, c-Jun is hepatoprotective during acute hepatitis by regulating NOS2/NO expression and thus functionally antagonizes the cytokine-induced cell death-promoting functions of JNK during hepatitis.81 Surprisingly, c-Jun does not seem to be critical for immune response in hepatitis. Mice specifically lacking JunB in hepatocytes are also found to display a mild increase in Con A-induced liver damage. Using loss-of-function mouse models for JunB, Wagner and co-workers82 have subsequently demonstrated that JunB promotes cell death during acute hepatitis by regulating IFN-γ production in NK and NKT cells and thus functionally antagonizes the hepatoprotective function of c-Jun in hepatocytes.
4.3.2. Pulmonary Fibrosis
In transgenic mice, ectopic expression of Fra 2 in various organs results in generalized fibrosis with predominant manifestation in the lung, possibly by linking vascular remodeling and fibrogenesis.83 Strong expression of Fra 2 was also observed in human samples of idiopathic and autoimmune-mediated pulmonary fibrosis.83 Fra2 has been considered as a contributing pathogenic factor of pulmonary fibrosis in humans. However, different from Fra2, Fra1 plays a protective role in lung fibrosis84 and modulates early profibrotic cellular responses.85 Fra1 mediates antifibrotic effects through the modulation of expression of proinflammatory, profibrotic, and fibrotic gene both in vitro and in vivo.84 Thus, either Fra1 or Fra2 transcription factors may act as potential targets for pulmonary fibrosis, a progressive disorder with poor prognosis and treatment.
4.3.3. Atherosclerotic Disease
In vitro and in vivo animal studies implicate AP-1 as a critical common inflammatory transcription factor correlated in the initiation and progression of vascular dysfunction and atherogenesis.86−90 Lindeman and colleagues91 have demonstrated that abundant AP-1 activation is associated with all stages of atherosclerosis by performing systematic histological evaluation. Lai and colleagues92 have revealed that irbesartan, an angiotensin II receptor antagonist, may modulate inflammation-based atherosclerotic diseases through a cell-mediated mechanism involving suppression of human T-lymphocytes activation via down-regulation of AP-1 activity. Hence, AP-1 inhibition may represent a promising strategy to prevent progression of atherosclerotic disease.
4.3.4. Other Cardiovascular Diseases
Early immediate up-regulation of AP-1 in response to cardiac hypertrophic stimuli was reported in the 1990s.93−96 Two members of the AP-1 family of transcription factors, JunD and Fra1, have been found to play an important role in regulation of heart growth during hypertrophic response.97,98 JunD, the only Jun protein constitutively and highly expressed in mammalian heart, can attenuate phenylephrine-mediated cardiomyocyte hypertrophy by inhibiting AP-1 transcriptional activity and may effectively function as an endogenous dominant negative regulator, buffering against extensive hypertrophic growth of cardiomyocytes in response to pathophysiological stress.99 In several in vitro studies, c-Jun and c-Fos have been suggested to be required for induction of fetal gene expression and cardiomyocyte hypertrophy in response to different stimuli.100−105 In fact, both Jun and Fos are not essential for postnatal cardiac hypertrophy as well as heart growth in response to mechanical pressure overload from a genetic evidence in vivo.106 Remarkably, however, deletion of Jun but not Fos can result in progressive myocardial fibrosis, cardiomyocyte apoptosis, and changes in sarcomeric organization.106 Additionally, the AP-1 components FosB and JunB regulate the intrinsic matrix metalloproteinase 2 (MMP-2) promoter in vivo following ischemia–reperfusion injury.107 MMP-2, which influences ventricular performance, is a central component of the response to injury in the heart.
4.3.5. Parkinson’s Disease (PD)
AP-1 is involved in the neuropathology of PD patients, whose phenotype can be closely mimicked by MPP+-induced neurotoxicity in vitro. Using an S-type human neuroblastoma cell line (SH-EP1) as a model, Feng and colleagues108 investigated the involvement of NF-κB and AP-1 pathways in MPP+-induced neurotoxicity. Besides NF-κB, JNK and c-Jun are also activated upon MPP+ stimulation. Inhibition of c-Jun activation, by a dominant negative c-Jun or c-Jun inhibitor such as curcumin, can significantly attenuate MPP+-mediated cell death, suggesting that c-Jun activation is proapoptotic.108 Thus, down-regulating the activation of NF-κB, JNK, and c-Jun may represent a new strategy for the treatment of PD.
5. Development of Small Molecules Targeting AP-1
As AP-1 plays important pathophysiological roles in various human conditions, AP-1 inhibitors are currently under intense development preclinically and clinically in many therapeutic areas, especially for inflammatory disease and cancers. Several core molecular scaffolds have been identified to be associated with anti-AP-1 properties and led to development of a number of potent AP-1 inhibitors. In addition, some natural products bearing AP-1 inhibitory activity are also described herein. There appears to be no generally accepted system of classifying such molecules, and this review relies on broad structural similarity and seeks to provide an update on research advances of the past 2 decades in the development of chemical entities as AP-1 inhibitors.
5.1. SP100030 Analogues
SP100030 (1, Figure 4) is one of the first reported potent small molecule inhibitors of AP-1 and NF-κB transcription activation with an IC50 value of 0.05 μM.107 It is about 10-fold more potent than the earlier hit 2 (IC50 = 0.5 μM), which has been identified by using automated high-throughput assays with stably transfected human Jurkat T-cells.109,110 Hit 2 was reported to have an inhibitory effect on the production of IL-2 and IL-8 levels in stimulated cells, and it was found to be efficacious in an animal model of inflammation.109 In order to increase the potency of hit 2, hundreds of compounds have been designed and synthesized through the use of solution-phase parallel chemistry and targeted synthesis and 1 was identified as the most potent analogue. 1 has been demonstrated to selectively inhibit CD8(+) T-cells and mRNA expression of both Th1 and Th2 cytokines in vivo instead of inhibiting allergen-induced airway eosinophilia and bronchial hyperresponsiveness (BHR) in a rat model of asthma.52 In addition, 1 was confirmed active in a dose-dependent manner in several animal models of inflammation and immunosuppression (ip, 10–20 mg/kg).110 Moreover, daily subcutaneous injection of cachectic Yoshida AH-130 ascites hepatoma-bearing rats with 1 at a dose of 1 mg/kg resulted in a clear amelioration of the cachectic effect, especially at the level of skeletal muscle.111 At this dose, 1 acts as an effective inhibitor of AP-1, while the NF-κB transcription factor is not affected, suggesting that the AP-1 signaling cascade plays an important role in the signaling of muscle wasting associated with disease.111 However, the low aqueous solubility and high lipophilicity of 1 likely gave reason for the lack of poor oral activity in the animal models, consistent with its poor permeability in the gastrointestinal cell line, Caco-2 (apparent permeability coefficient Papp = (11 ± 4) × 10–7 cm/s).112
Figure 4.

Chemical structures of 1 and its representative analogues 2–20.
To improve its potential oral bioavailability and Caco-2 permeability, a series of analogues of 1 modified at the 2, 4, 5, and 6 positions of the pyrimidine ring as shown in Figure 4 have been prepared by using a solution-phase parallel synthesis technique. All these compounds meet the criteria of Lipinski’s “rule of five”. The introduction of a fluoro group in the place of 2-chloro of 1 results in compound 3 with a comparable activity (IC50 of 0.01 vs 0.05 μM). However, other substitutions at the 2-position lead to a loss of activity, such as compounds 4 and 5 (Table 3). The trifluoromethyl group at the 4-position can be replaced with a methyl (6), chloro (7), or phenyl (8) with slight changes of activity. The carboxamide group at the 5-position is critical for activity. When it is moved to the 6-position (compounds 9 and 10), the activity is totally lost. Among these, the 2-methyl analogue (6) shows comparable in vitro activity to 1 (IC50 of 0.02 vs 0.05 μM), with improved Caco-2 permeability (Papp of (62 ± 6) × 10–7 vs (11 ± 4) × 10–7 cm/s) and potential oral bioavailability.112
Table 3. Inhibition of SP100030 Analogues on AP-1 and NF-κB Mediated Transcriptional Activation in Luciferase Reporter Assays on Jurkat T-Cells.
| IC50, μM |
||
|---|---|---|
| compd | AP-1 | NF-κB |
| 1 | 0.05 | 0.05 |
| 2 | 0.50 | 0.50 |
| 3 | 0.1 | 0.4 |
| 4 | >10 | >10 |
| 5 | >10 | >10 |
| 6 | 0.02 | 0.05 |
| 7 | 0.2 | 0.6 |
| 8 | 0.4 | 0.4 |
| 9 | >10 | >10 |
| 10 | >10 | >10 |
| 11 | >30 | >30 |
| 12 | 10 | 10 |
| 13 | 1.7 | 1.7 |
| 14 | 4.0 | 4.0 |
| 21 | 10 | 10 |
| 22 | 9.4 | 13 |
| 23 | 9.4 | 24 |
| 24 | 6.1 | 4.2 |
| 25 | 8.5 | 8.0 |
| 26 | 10 | 10 |
| 27 | 1.9 | 1.3 |
| 28 | 0.1 | 0.2 |
Palanki and co-workers112 also investigated the importance of the pyrimidine by replacement with several other ring systems while retaining the N-(3′,5′-bis(trifluoromethyl)phenyl)carboxamide framework. Phenyl analogue 11 is completely inactive, while pyridazines (12 and 13) and pyrazine 14 are less potent than 1.112 Surprisingly, compound 13 with a 5-hydrogen is more potent than the 5-trifluoromethyl compound 12 (IC50 of 1.7 vs 10 μM),112 which is somewhat different from the compounds with a pyrimidine ring system. However, compound 12 is 34-fold less potent than 1 (IC50 of 1.7 vs 0.05 μM) and exhibits a comparable potency to its 1,4-isomer 14 (IC50 of 1.7 vs 4 μM).112 In addition, replacing the 2-chloro-4-trifluorobenzamide in 11 with 2-halogen-4-hydroxylbenzamide results in compounds 15–17, which display good inhibitory effects on AP-1 mediated transcriptional activation in the order of Br > Cl > I (Table 4) and are more potent than their corresponding 3,5-dichlorophenyl analogues 18–20.113
Table 4. Inhibition of AP-1 Mediated Transcriptional Activation in HeLa Cell Line Based in Vitro Assay.
| inhibition, % |
||
|---|---|---|
| compd | 10 μg/mL | 1 μg/mL |
| 15 | 89.1 | 42.4 |
| 16 | 91.2 | 48.4 |
| 17 | 82.4 | 25.4 |
| 18 | 74.8 | 22.7 |
| 19 | 83.8 | 39.3 |
| 20 | 75.4 | NDa |
ND: not determined.
On the other hand, in an effort to increase the druglike properties of highly lipophilic 1, Palanki and co-workers114 have also designed several conformationally restricted analogues in which the pyrimidine ring and the aniline ring are connected through a second “bridge”. In fact, their original plan was only to simply substitute one or two trifluoromethyl groups in 1 with any other groups, but all initial attempts resulted in a loss of activity.115 As shown in Figure 5, compounds 21–23 by restricting one or two dihedral angles are about 200-fold less potent than 1 (Table 3). All three dihedral angles are restricted in compounds 24–28. The direct bridged analogue 24, the carbon-bridged 25, and the nitrogen-bridged 26 display weaker activity and are 120- to 200-fold less potent than 1. However, the oxygen-bridged analogue 27 is 3- to 5-fold more potent than 24–26. Interestingly, the potency of seven-membered sulfur-bridged analogue 28 bearing only one trifluoromethyl group is comparable to that of 1 (IC50 of 0.1 vs 0.05 μM), representing the most potent in this series. The seven-membered sulfur-bridged analogues thus provide a new class of inhibitors for AP-1 and NF-κB mediated transcriptional activation.114
Figure 5.

Chemical structures of 21–28.
5.2. SPC-839 Analogues
By screening their diversified compound library using automated high-throughput assays with stably transfected human Jurkat T-cells, Palanki and colleagues116,117 have also identified another two novel hits, 29 and its isomer 39 (Figures 6 and 7), which display a slightly lower potency than their earlier hit 2 (IC50 of 2.0 and 1.0 vs 0.5 μM, respectively). Both hits 29(117) and 39(116) were found to inhibit AP-1 and NF-κB mediated transcriptional activation without blocking basal transcription driven by the β-actin promoter and to exhibit a similar inhibitory effect on the production of IL-2 and IL-8 levels in stimulated Jurkat T-cells.
Figure 6.

Chemical structures of analogues based on modifications of hit 29.
Figure 7.

Chemical structures of analogues based on modifications of hit 39.
To improve the potency of 29, different substituents around the 2, 4, and 5 positions of the pyrimidine ring were explored.117 As shown in Figure 6, all these compounds display similar IC50 values in both AP-1 and NF-κB assays. The removal of a methyl group or the introduction of a phenyl group in place of methyl group or the introduction of another methyl group on the citraconamido ring of the 2-position of the pyrimidine ring in 29 results in a slight change in activity (IC50 of 1.6–3.9 vs 2.0 μM, Table 5). However, removal of the citraconamido ring leads to a much weaker potency (IC50 = 30 μM). When the hydrogen of NH group at the 2-position is replaced with an alkyl, substituted carbonyl, urea, or carbamate group, all these compounds exhibit 2- to 6-fold improvement in potency (IC50 of 0.3–0.83 vs 2.0 μM). Among them, N-methylated 2-amino group of 29 as the most potent compound displays an improved potency in comparison with 30 (IC50 of 0.3 vs 2.0 μM). At the 4-position of the pyrimidine ring, replacement of the trifluoromethyl with a methyl group results in a compound with a comparable potency, while that with an ethyl (31) or pentafluoroethyl group leads to an improved potency (IC50 of 0.2–0.4 vs 2.0 μM). Moreover, replacement of the trifluoromethyl of 30 with an ethyl group can further improve its potency about another 8-fold (32, IC50 of 0.035 vs 0.3 μM). Substitutions with bulky groups such as phenyl and benzyl are not favorable to improve the potency in general, while 2-(5-methylthienyl) analogue (33) displays a better potency with 40-fold improvement with an IC50 value of 0.045 μM. The introduction of a bulkier alkyl ester group such as tert-butyl ester (34) instead of ethyl ester at the 5-position leads to about a 10-fold increase in potency (IC50 of 0.21 vs 2.0 μM). Other substitution groups such as a carboxylic acid, carboxamide, or N,N-dimethylcarboxamide result in a loss of potency (IC50 = 30 μM). Compounds 35 and 36 with a methyl ketone or a phenyl ketone at the 5-position exhibit an improved potency with an IC50 value of 4.4 and 0.098 μM, respectively. Several bioisosteres of ethyl ester such as oxazoline, isoxazole, oxadiazole, tetrazole, or phenyloxazole moieties were found to display a lower potency than 29 (IC50 of 2.8–10 μM), while analogues 37 and 38 with the methyloxazole exhibit a slightly increased potency.117
Table 5. Inhibition of SPC-839 Analogues on AP-1 and NF-κB Mediated Transcriptional Activation in Luciferase Reporter Assays on Jurkat T-Cells.
| IC50, μM |
||
|---|---|---|
| compd | AP-1 | NF-κB |
| 29 | 2 | 2 |
| 30 | 0.3 | 0.3 |
| 31 | 0.4 | 0.4 |
| 32 | 0.035 | 0.035 |
| 33 | 0.045 | 0.045 |
| 34 | 0.21 | 0.21 |
| 35 | 4.4 | 4.4 |
| 36 | 0.098 | 0.098 |
| 37 | 0.83 | 0.83 |
| 38 | 0.76 | 0.76 |
| 39 | 1 | 1 |
| 40 | 0.1 | 0.1 |
| 41 | 0.02 | 0.02 |
| 42 | 0.008 | 0.008 |
| 43 | 0.003 | 0.003 |
Upon the basis of hit 39 as the chemical lead, different substituents at the 2 and 4 positions of the pyrimidine ring have also been investigated to improve its potency.116 As shown in Figure 7, the substitutions with small alkyl groups such as methyl and ethyl instead of trifluoromethyl moiety at the 2-position generally result in a loss of activity, while bulkier groups such as tert-butyl group can retain the activity with a comparable potency. The substituted or unsubstituted heterocyclic rings at 2-position of the hit generally lead to a reduced activity. However, analogues 40 and 41 with a phenyl or 2-thienyl moiety exhibit a 10- to 50-fold improved activity (IC50 of 0.1–0.02 vs 1.0 μM, Table 4). Other substituents on the phenyl ring of 40 or the thienyl ring of 41 appear to be less favorable. The removal of a methyl group or the introduction of a phenyl group instead of methyl or the introduction of an additional methyl group into the citraconamido ring, as well as methylation or acetylation of the NH group at the 4-position of the pyrimidine ring in 39 results in a decreased activity. Unfortunately, the most potent compound 41 was found to display no oral activity in rat PK studies and poor Caco-2 permeability likely due to the existence of the carboxylate moiety.118 In addition, several analogues of 41 with oxazoline, isoxazole, oxadiazole, or tetrazole as the carboxylate bioisosteres at the 5-position of pyrimidine ring turn out to be less potent.116
To improve the druglikeness of highly lipophilic lead 41, a new class of inhibitors without a carboxylate ester function have been designed and synthesized by introducing a fused phenyl ring on the pyrimidine with a methoxy group at its 5-position (Figure 7).118 By changing the position and number of the methoxy groups on the phenyl of the quinazoline ring, as well as replacement with other electron-withdrawing or -donating groups, Palanki and co-workers118 have identified several improved compounds with a methoxy group at the 5-position (42) or 6-position (43) with 2.5- to 6.7-fold improvement compared to 41 (IC50 of 0.008–0.003 vs 0.02 μM). Compound 42 (SPC-839) displays higher permeability than 43 (Pc of (1.41 ± 0.4) × 10–5 vs (1.62 ± 0.2) × 10–6 cm/s). Moreover, 42 has also been demonstrated to be more efficacious in an adjuvant-induced arthritis rat model by reducing the swelling by 65% in the noninjected foot.118
On the basis of compound 42 as the advanced chemical lead and utilizing the bioisosterism and other medicinal chemistry optimization approaches, Giri and colleagues119 have designed and investigated a series of new compounds with the (2-(2,4-disubstituted-thiazole-5-yl)-3-aryl-3H-quinazolin-4-one scaffold as potential inhibitors of NF-kB and/or AP-1 mediated transcriptional activation for developing anti-inflammatory agents (Figure 7). 44 (IC50 = 5.5 μM) and 45 (IC50 = 5.5 μM) have been identified to be more selective toward inhibiting AP-1 mediated transcriptional activity over NF-κB in luciferase reporter assays on HEK293 cells. Compound 48 turns out to be the most potent dual inhibitors of NF-κB and AP-1 mediated transcriptional activation with an IC50 value of 0.2 and 0.5 μM, respectively.12048 has also been demonstrated to have significant in vivo efficacy in a carrageenan injection-induced inflammation model with 56% inhibition of rat paw edema.120 Additionally, Giri and co-workers121 have also evaluated the potential of the novel scaffold mentioned above for cancer by inhibiting multiple pathways in luciferase reporter assays on HEK293 cells. 46 and 47 (IC50 of 1.2–3.0 μM) are more selective toward inhibiting AP-1 mediated transcriptional activity, while 49 is a potent dual inhibitor of both NF-κB and AP-1 mediated transcriptional activation with IC50 values of 3.3 and 4.3 μM, respectively. By replacement of the trisubstituted thiazole moiety with a thiophenyl ring, a series of 2-(2,3-disubstituted-thiophen-5-yl)-3H-quinazolin-4-one analogues have been designed and explored as potential anti-inflammatory and anticancer agents.121 Compound 50 is the most potent dual inhibitor of NF-κB and AP-1 mediated transcriptional activation from the entire series, with IC50 values of 10 and 5 μM for NF-κB and AP-1, respectively.
5.3. T-5224 Analogues
51 (Figure 8), an inhibitor of the c-Fos/AP-1, has been identified by converting cyclic disulfide decapeptides122 to a series of nonpeptidic benzophenone derivatives123 using a lead-hopping approach based on a 3D pharmacophore model. 51 specifically inhibits the DNA binding activity of c-Fos/c-Jun without affecting those of other transcription factors including C/EBPa and ATF-2 (bZIP domain), MyoD (basic helix–loop–helix domain), Sp-1 (zinc-finger domain), and NF-κB/p65 (Rel homology domain), as well as the levels of c-Fos family protein members themselves.124 Administration of 51 at a dose of 30 mg/kg was found to resolve type II collagen-induced arthritis (CIA) in a preclinical model by reducing the amount of inflammatory cytokines including interleukin 1β and matrix-degrading MMPs in vivo in sera and joints as well as in vitro in synovial cell and chondrocyte cultures.12451 also synergizes with antitumor necrosis factor α (TNFα), a signaling molecule immediately downstream of c-Fos, to inhibit arthritis.124
Figure 8.

Discovery of 51, a selective AP-1 inhibitor in phase II human clinical trial.
Animal studies revealed that the major metabolites of 51 were glucuronides.5 In addition, glucuronides were also found to be major metabolites in human urine. By use of human liver microsomes (HLMs), human intestinal microsomes (HIMs), recombinant human cytochrome P450 (CYP450), and UDP-glucuronosyltransferases (UGTs) isoforms expressed in baculovirus-infected insect cells, it was predicted that 51 was converted to its acyl O-glucuronide by UGT1A1 and UGT1A3 and to its hydroxyl O-glucuronide by several UGTs, but it was not metabolized by the P450.5 Moreover, the glucuronidation of 51 was estimated to predominantly occur in the liver by comparing the intrinsic clearances (CLint) between HLM and HIM.
On the basis of its promising pharmacological effects in arthritis, 51 has been developed by Toyama Chemical as a potential therapeutic agent for rheumatoid arthritis and has advanced into human phase II clinical trials in Japan. Recently, it has also been reported that 51 not only can ameliorate lipopolysaccharide (LPS) induced liver injury through decreasing production of proinflammatory cytokines and chemokines in endotoxemic mice125 but also has a potential inhibitory effect against endotoxin-induced acute kidney injury (AKI) by suppressing the TNF-α inflammatory response and other downstream effectors.126
The cyclic disulfide decapeptides, especially the most potent inhibitor Ac-c[Cys-Gly-Gln-Leu-Asp-Leu-Ala-Asp-Gly-Cys]-NH2 (52), have been designed and synthesized as inhibitors of the c-Fos/AP-1 de novo by three-dimensional pharmacophore modeling122 based on the X-ray crystal structure of the bZIP domain of the AP-1–DNA complex.8 A hypothetical 3D pharmacophore model was then constructed for generating new c-Fos/AP-1 inhibitors based on an alanine scan experiment, molecular dynamics simulation of the bZIP–52 complex, and NMR measurement of the peptide in water.122 The pharmacophore consists of three hydrophobic groups, one hydrogen bond acceptor or donor, and one acidic group. Using a lead-hopping strategy based on the 3D pharmacophore model, Tsuchida and co-workers123 discovered new nonpeptidic small-molecule AP-1 inhibitors based on 1-thia-4-azaspiro[4.5]decane and benzophenone derivatives, which were synthetically accessible and easy to optimize.
As shown in Figure 8, compounds 53 and 54 bearing the scaffold of 1-thia-4-azaspiro[4.5]decane and compounds 55 and 56 bearing the scaffold of benzophenone display lower inhibitory activity than peptide 52 (IC50 = 64 μM) on the binding of AP-1 bZIP and oligonucleotides containing the AP-1 binding site (IC50 of 420–650 μM) using an ELISA-based AP-1 DNA-binding assay. Additionally, these compounds also inhibit the expression of AP-1-luciferase by TPA-stimulated NIH3T3 cell (IC50 of 5.0–13.3 μM). However, 51 can fit the cyclic peptide 52 -derived model well, with an IC50 value of ∼10 μM for most of the in vitro cellular assays including luciferase assays. Moreover, in vivo ED50 of 51 is ∼1–10 mg/kg, and the Cmax is 0.03–0.5 μM (15–240 ng/mL).124 Compound 51 is the only selective AP-1 inhibitor that has been advanced into human clinical trials to date.
5.4. Natural Products and Other Compounds
K1115 A (57, Figure 9), a new anthraquinone derivative, was isolated from the culture broth of Streptomyces griseorubiginosus (Mer-K1115).12757 inhibits the direct binding of AP-1 to AP-1 oligonucleotide (IC50 = 100 μM) and the production of collagenase in IL-1α-stimulated rat synovial cells (IC50 = 60 μM) in vitro. Moreover, 57 can attenuate the inflammatory response mediated by AP-1 through decreasing the ornithine decarboxylase (ODC) activity of phorbol myristate acetate (PMA) induced mice. Given that certain truncated peptides would have the potential to bind the DNA AP-1 consensus sequence under the same conditions as the native AP-1 protein, Patterson and co-workers128 have designed a series of peptide–anthraquinone conjugates as inhibitors of AP-1 transcription factor. Truncated AP-1-like peptides, with five to seven residues, bearing a highly conserved sequence motif lysine–cysteine–arginine (KCR) were attached at the N-terminus to an intercalating anthraquinone moiety of 57 through an amino acid type linker. All these anthraquinone–peptides were found to displace AP-1 protein binding from its DNA consensus sequence much more effectively than their respective free peptides using the electrophoretic mobility shift assay (EMSA). Moreover, the peptide conjugates containing the more basic sequences 2-AKCRNA (58), 2-AKCRKA (59), 2-AKCRNRA (60), and 2-AKCRKRA (61) were demonstrated to be the most effective inhibitors in this series.128
Figure 9.

Chemical structures of K1115A, curcumin, and their analogues. Abbreviations in the peptides are the following: A, Ala; K, Lys; C, Cys; N, Asp; R, Arg.
Curcumin (62, Figure 9), a hydrophobic polyphenol derived from Curcuma longa, a plant of the ginger family, is well-known to have diverse biological functions such as anti-inflammatory, antitumor, antioxidative, cytotoxic, antifungal, antibacterial, and antihepatotoxic activities.129 Curcumin was reported to inhibit TPA-induced expression of c-Fos and c-Jun protooncogene mRNA and to repress c-Jun binding to its cognate motif in NIH3T3 mouse fibroblast cells.130,131 Additionally, suppression of AP-1 binding to DNA was also observed in human leukemia cells132 and transformed keratinocytes.133 Furthermore, curcumin displays an inhibitory effect against the complex formation of the Fos–Jun dimer and the DNA consensus sequence with an IC50 value of 6.9 nM and is about 30-fold more potent than dihydroguaiaretic acid (DHGA, 63), isolated from the aryls of Myristicafragrans (IC50 = 0.21 μM).134 However, the analogue of DHGA, nordihydroguaiaretic acid (NDGA, 64), exhibits a potency comparable to that of curcumin (IC50 = 7.9 nM). Both DHGA and NDGA are capable of suppressing leukemia and lung and colon cancer in the MTT-based bioassay.134 Curcumin has also progressed to human clinical trials, but it is a nonselective AP-1 inhibitor mixed with activities of other targets including various transcription factors, thereby significantly limiting its potential as a targeted therapy.
To improve the potency and selectivity of curcumin-based compounds, Yang and colleagues135 synthesized a series of symmetrical curcumin analogues and evaluated their inhibitory activity on Fos–Jun complex formation though the EMSA experiment. As shown in Figure 9, curcuminoids 65 and 66 exhibit an IC50 value of 8.98 and 5.40 μM, respectively, which are 60- to 100-fold more potent than curcumin (IC50 = 540 μM). In addition, through inhibition of AP-1 transcription, thereby down-regulating the expression of angiogenesis-associated genes (VEGF and MMP-9), curcumin analogues 67 and 68 were found to show the angiogenesis inhibitory effect on the developmental neovascularization of chicken embryonic with 68–88% inhibition of embryos exposed to 10 μg of the compound treatment.136 Their inhibitory effects on angiogenesis have also been confirmed by wound migration, invasion, and tube formation assays.136
Momordin I (69), isolated from Ampelopsis radix, and its disaccharide and carboxylic acid modified derivatives 70 and 71 (Figure 10) have demonstrated a remarkable inhibitory effect on the formation of Fos–Jun DNA complex.137 Momordin I derivatives 70 and 71 display an IC50 value of approximately 4.0 μM in EMSA assay, which is 30-fold more potent than momordin I (IC50 = 0.13 mM) and about 125-fold more effective than curcumin (IC50 = 0.48 mM).
Figure 10.

Momordin I and its analogues.
Microbial transformation is an area of great interest for applying biocatalysis to selectively convert synthetic and natural products to biological compounds that are difficult to obtain by conventional chemical methods.138,139 Special attention has been paid to filamentous fungi because of their capability of catalyzing regio- and stereoselective hydroxylation of a variety of nonfunctionalized hydrocarbon centers of a great variety of substrates.140 Isosteviol (72, Figure 11), with a rigid skeleton comprising four fused rings similar to the steroid skeleton, possesses various biological activities.141 Isosteviol lactone (73), an ent-beyerane tetracyclic diterpenoid prepared by reacting isosteviol (72) with m-chloroperbenzoic acid, was investigated for its activity on mitochondrial metabolism.142 A number of hydroxylated diterpenoids have been generated from the microbial transformation of isosteviol lactone (73) with Mucorrecurvatus MR 36, Aspergillusniger BCRC 31130, or Absidiapseudocylindrospora ATCC 24169. As shown in Table 6, compounds 73–80 exhibit significant inhibitory effects on AP-1 activation in lipopolysaccharide-stimulated RAW 264.7 macrophages by AP-1-mediated luciferase reporter gene assay, while 79 is more potent than the reference compound of dexamethasone.142 Given that the important biological properties of steroids are dependent upon its D-ring, a series of compounds with modified D ring have been produced by microbial transformation of isosteviol oxime (81) with Aspergillus niger BCRC 32720 and Absidia pseudocylindrospora ATCC 24169.143 Among the compounds tested, 82–86 significantly inhibit AP-1 activation in LPS-stimulated RAW 264.7 macrophages, and in particular, 85 displays an inhibitory activity more potent than dexamethasone.143
Figure 11.

Isosteviol and its analogues.
Table 6. Luciferase Activity of Isosteviol Analoguesa.
| compd | luciferase activity | compd | luciferase activity |
|---|---|---|---|
| 73 | 2.38 ± 0.18 | 81 | 4.87 ± 2.15 |
| 74 | 2.69 ± 0.92 | 82 | 2.69 ± 0.45 |
| 75 | 2.39 ± 0.64 | 83 | 2.98 ± 0.35 |
| 76 | 2.73 ± 0.58 | 84 | 2.38 ± 0.18 |
| 77 | 3.02 ± 0.71 | 85 | 2.22 ± 0.35 |
| 78 | 2.58 ± 0.86 | 86 | 2.85 ± 0.48 |
| 79 | 1.88 ± 0.39 | control | 3.95 ± 0.53 |
| 80 | 2.66 ± 0.99 | dexamethasone | 2.33 ± 0.36 |
The concentration of each test compound was 10 μM. All luciferase activities were normalized to Renilla luciferase activity. The data were expressed as multiples of luciferase activity compared to the no-treatment (control) group. Dexamethasone is the reference compound.
Other than diterpenes, norditerpenes and triterpenes can also inhibit the AP-1 activity. As shown in Figure 12, norditerpenes nagilactone (87) and inumakilactone (88),144 isolated from an organic solvent extract of the root bark of Podocarpus latifolius (Thunb.) collected in Tanzania, have also been demonstrated to be capable of inhibiting phorbol ester TPA-induced activation of AP-1 activity at the concentrations tested, with IC50 values estimated from dose–response curves of 1.5 and 4.0 μM, respectively. However, both of them also appear to be toxic, with cell survival of <50% in each case at more than 2.5 μM.144 Quassinoids glaucarubinone (89) and nothospondin (90) also display potent, dose-dependent AP-1 inhibition at noncytotoxic concentrations by a β-lactamase driven reporter assay using fluorescence resonance energy transfer (FRET) technology, with EC50 values of 1.49 and 0.13 μM, respectively.145 The potent AP-1 inhibitory activity of 89 may be ascribed to its ether bridge between C-17 and C-11, which is seen in many other AP-1 active quassinoids, such as 6α-senecionylchaparrin (91) and ailanthinone (92).146 However, 90 is the first quassinoid without an ether linkage that can inhibit AP-1, albeit at a significantly reduced potency.145 In addition, 89 is noncytotoxic at a high concentration of 80 μM by an XTT assay, while 90 shows some cytotoxicity with an IC50 of approximately 10 μM.145
Figure 12.

Chemical structures of quassinoids with AP-1 inhibitory effects.
Citrifolinin A (93, Figure 13), a new unusual iridoid isolated from the leaves of Morindacitrifolia, shows significant inhibition of UVB-induced AP-1 activity in cell cultures, with an IC50 of 69.6 μM.147 Citrifolinoside (94)148 is another iridoid isolated from the leaves of Morindacitrifolia and displays a significant inhibitory effect on UVB-induced AP-1 activity and is 2.4-fold more potent than 93 (IC50 = 29.0 μM).
Figure 13.

Natural products 93–96 and other AP-1 inhibitors 97 and 98.
From an extract of the Palauan cyanobacterium Lyngbya majuscule, grassypeptolides F (95, Figure 13) and G (96), bis-thiazoline-containing cyclic depsipeptides with a rare β-amino acid, extensive N-methylation, and a large number of d-amino acids, were isolated and found to have moderate inhibitory activity against the transcription factor AP-1 (IC50 of 5.2 and 6.0 μM, respectively) in HEK293T cells.149
In addition, compound (−)-97, a cycloadduct synthesized by tandem retro-Diels–Alder/Diels–Alder cycloadditions of o-quinol dimer under microwave irradiation, shows selective inhibition against AP-1 at 4 μM in luciferase reporter assays on HEK293 cells (Figure 13).150 Moreover, (−)-97 does not display inhibition of HIF-2, NF-κB, or SRE dependent transcription at the same concentration. However, during the experiments in the NCI 60-cell screen at 10 μM, (−)-97 did not pass the threshold for further evaluation of cell growth inhibition.
Retinoic acid is effective for chemotherapy and chemoprevention of cancer mediated either by blocking AP-1 activity or by activating retinoic acid response element (RARE).151 Using an AP-1-luciferase transgenic mouse as a carcinogenesis, Dong and colleagues152 provided the first in vivo evidence that 98 (SR11302), an AP-1 inhibition-specific retinoid, markedly inhibited both 12-O-tetradecanoylphorbol-13-acetate-induced papilloma formation and AP-1 activation in 7,12-dimethylbenzanthracene-initiated mouse skin. Interestingly, 98 does not activate transcription from the RARE and displays no activity at retinoic acid receptors (EC50 > 1 μM for RARα, RARβ, RARγ, and RXRα).152 As an AP-1 inhibitor that displays antitumor effects in vivo, 98 serves as an important research tool compound that is commercially available from Tocris. In addition, some other nonspecific AP-1 inhibitors such as 6-shogaol and 6-gingerol have also been reported to inhibit 12-O-tetradecanoylphorbol-13-acetate-induced tumor promotion in mice.153
6. Conclusions and Future Directions
Since the discovery of AP-1 as the pivotal transcription factor in 1987,154 significant progress has been made by many pioneers in understanding AP-1 biology and function and identifying AP-1 as an important and a valid therapeutic target for various human diseases. There is a great potential for the development of AP-1 inhibitors as preventive or therapeutic agents for cancer and inflammation, as well as a variety of other human conditions including hepatitis, pulmonary fibrosis, atherosclerosis, cardiovascular diseases, and Parkinson’s disease. Although currently available drugs are helpful in alleviating many of the symptoms of these diseases, more targeted therapeutic approaches based upon the underlying mechanisms remain an urgent need for better specificity of the treatment with lower side effects. To this end, targeting AP-1 with potent and specific inhibitors may represent an attractive molecular approach for new medications.
The proof of concept by developing AP-1 inhibitors as a novel therapy for inflammation has been confirmed by the investigation of clinical drug candidate 51, although its fate is dependent on the outcomes of phase II human clinical trials. Other inhibitors, such as analogues of 1 and 42, also have the potential to be developed as novel therapeutic agents. Nevertheless, researchers have spent tremendous efforts on hit-to-lead optimizations improving their druglike properties of the hits from high-throughput screening, especially their DMPK properties in vivo. Thus, safety concerns and in vivo pharmacokinetic alerts should be considered early in the drug discovery process. Fragment-based drug design (FBDD), by taking advantage of privileged fragments, may also provide an efficient approach to identify more effective and selective AP-1 inhibitors with better druglikeness.155−157
On the other hand, it is worth noting that AP-1 may act as a double-edged sword in tumor development. By regulating genes involved in cell proliferation, differentiation, apoptosis, angiogenesis, and tumor invasion, AP-1 not only can be oncogenic but also can be antioncogenic.18,158 AP-1 activity in cancer seems to depend upon AP-1 dimer composition and tumor type as well as its differentiation state, tumor stage, and the genetic background of tumor.18 Generally, c-Jun mainly has oncogenic functions, while JunB and JunD have antioncogenic effects. As such, it would be quite challenging for medicinal chemists to design potent and specific AP-1 inhibitors as potential therapy for cancer. Individualized treatment may provide a solution to this problem by selecting appropriate patient populations. Furthermore, if the antioncogenic function of some AP-1 proteins is properly exploited, it may provide a novel, unexpected, and more tractable direction for the discovery of anticancer treatments that can induce the formation of specific dimers for the selective and efficient killing of cancer cells, thereby actively interfering or antagonizing tumor development.
Despite the advances in high-throughput screening, structure, and ligand-based molecular modeling and hit-to-lead optimizations, which have identified numerous compounds capable of inhibiting AP-1 transcription activation or its DNA binding activity, only one selective AP-1 inhibitor 51 has progressed to human clinical trials. Meanwhile, it has to be pointed out that many of the currently available compounds display polypharmacological profiles by targeting various other transcription factors including NF-κB. It is the opinion of the authors that because of the combinatorial diversity and expressed dependence of AP-1 family proteins, the multidisciplinary approaches with combined techniques in molecular pharmacology, chemical biology, and structural biology based on X-ray crystallography of protein–inhibitor complexes, as well as computational chemistry, will provide new insights into rational drug design and facilitate the search of novel AP-1 family selective inhibitors. Although those identified inhibitors of dual or multiple transcription factors may also be of value for treatment or prevention of diseases, discovery and development efforts remain an urgent need for medicinal chemists to yield more potent, efficacious, and specific AP-1 inhibitors as a viable therapeutic strategy for use in the human clinic.
Acknowledgments
This work was supported by Grants P50 CA097007, P30 DA028821, and R21 MH093844 from the National Institutes of Health, R. A. Welch Foundation Chemistry and Biology Collaborative Grant from Gulf Coast Consortia (GCC) for Chemical Genomics, a training fellowship from the Keck Center for Interdisciplinary Bioscience Training of the GCC (NIGMS Grant T32 GM089657), John Sealy Memorial Endowment Fund, and the Center for Addiction Research (CAR) at University of Texas Medical Branch.
Glossary
Abbreviations Used
- CIA
collagen-induced arthritis
- ATF
activating transcription factor
- MAF
musculoaponeurotic fibrosarcoma
- TPA
phorbol 12-O-tetradecanoate-13-acetate
- NF-κB
nuclear factor κ light chain enhancer of activated B cells
- MAPK
mitogen-activated protein kinase
- JNK
JUN amino terminal kinase
- ES
embryonic stem
- ERK
extracellular-signal-regulated kinase
- HLM
human liver microsome
- HIM
human intestinal microsome
- PD
Parkinson’s disease
- TCF
ternary-complex factor
- MEF2C
myocyte-enhancer factor 2C
- LPS
lipopolysaccharide
- AKI
acute kidney injury
- CKII
casein kinase II
- GSK-3β
glycogen synthase kinase 3β
- RSK2
ribosomal S6 kinase 2
- NFAT
nuclear factor of activated T cells
- STAT
signal transducer and activator of transcription
- VEGF
vascular endothelial growth factor
- MMP9
matrix metallopeptidase 9
- MPP+
1-methyl-4-phenylpyridinium ion
- IC50
50% inhibitory concentration
- ED50
50% effective dose
- Cmax
maximum plasma concentration
- SRE
serum response element
- RARE
retinoic acid response element
- RAR
retinoic acid receptor
- CYP450
cytochrome P450
- CLint
intrinsic clearance
- UGT
UDP-glucuronosyltransferase
- FBDD
fragment-based drug design
- DMPK
drug metabolism and pharmacokinetics
Biographies
Na Ye earned her Ph.D. in Medicinal Chemistry from Shanghai Institute of Materia Medica, Chinese Academy of Sciences, under the supervision of Dr. Ao Zhang in 2013. She is currently pursuing her postdoctoral training under the direction of Dr. Jia Zhou at University of Texas Medical Branch. Her research topics focus on the target-based drug design and chemical synthesis of bioactive small molecules including AP-1, EPAC-specific inhibitors as pharmacological probes, and drug candidates for the treatment of CNS disorders, cancer, and other human diseases.
Ye Ding obtained his Ph.D. in Medicinal Chemistry from China Pharmaceutical University under the tutelage of Dr. Yihua Zhang in 2011. During the subsequent 2 years, he worked as a Research Scientist at Research & Development Center, Nanjing Sanhome Pharmaceutical Co., Ltd. Dr. Ding is currently pursuing his postdoctoral training at the Chemical Biology Program, Department of Pharmacology and Toxicology at University of Texas Medical Branch under the supervision of Dr. Jia Zhou. His research interests currently focus on rational design and synthesis of target-based small molecules and natural product analogues as potential therapeutics for CNS disorders, cancer, and other human diseases.
Christopher Wild studied biology and chemistry at the California State University, Northridge, where he received his Bachelor and Master of Science degrees under the direction of Dr. Gagik Melikyan. Subsequently, he worked as a program chemist for ChemicoMays, CA, and a research chemist at Celenese, TX, where he was a member of the acetyl catalyst development team under the tutelage of Dr. Michael Nutt. Currently, Christopher is a member of the chemistry faculty at San Jacinto College, TX, and is pursuing a Ph.D. as a Keck Research Fellow at University of Texas Medical Branch under the supervision of Dr. Jia Zhou. His research focus is on the design and synthesis of mechanism-based small molecule inhibitors and allosteric modulators as potential medications.
Qiang Shen received his medical education in Kunming Medical University in China and a Ph.D. degree in Cell Biology from the University of Texas Medical Branch in 2002. Then he was trained as a Postdoctoral Fellow at Baylor College of Medicine with Dr. Powel Brown in the areas of breast cancer biology and molecular cancer prevention from 2002 to 2006, and promoted to instructor at Baylor from 2006 to 2009. Dr. Shen joined The University of Texas M. D. Anderson Cancer Center in 2009 as a tenure-track Assistant Professor in the Department of Clinical Cancer Prevention. Dr. Shen studies transcription factors including AP-1 and STAT3 in the development of breast and other cancers, has published more than 30 peer-reviewed research articles, and is a co-inventor of two patents.
Jia Zhou received his Ph.D. in Organic Chemistry in 1997 from Nankai University, China. Then he joined the chemistry faculty at the same university and was promoted to Associate Professor there. In 1999, he started his postdoctoral training in organic chemistry with Dr. Sidney M. Hecht at the University of Virginia. After further postdoctoral training in medicinal chemistry with Dr. Alan P. Kozikowski at Georgetown University, Washington, DC, he conducted research at Acenta Discovery and PsychoGenics, Inc. as a Senior Principal Scientist for 7 years. Dr. Zhou is currently a tenured Associate Professor at the Chemical Biology Program, Department of Pharmacology and Toxicology at University of Texas Medical Branch, leading a drug discovery research group. He is an author of more than 70 papers and an inventor of 10 patents.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Palanki M. S. Inhibitors of AP-1 and NF-kappa B mediated transcriptional activation: therapeutic potential in autoimmune diseases and structural diversity. Curr. Med. Chem. 2002, 9, 219–227. [DOI] [PubMed] [Google Scholar]
- Shaulian E.; Karin M. AP-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [DOI] [PubMed] [Google Scholar]
- Hess J.; Angel P.; Schorpp-Kistner M. AP-1 subunits: quarrel and harmony among siblings. J. Cell Sci. 2004, 117, 5965–5973. [DOI] [PubMed] [Google Scholar]
- Wagner E. F.; Eferl R. Fos/AP-1 proteins in bone and the immune system. Immunol. Rev. 2005, 208, 126–140. [DOI] [PubMed] [Google Scholar]
- Uchihashi S.; Fukumoto H.; Onoda M.; Hayakawa H.; Ikushiro S.; Sakaki T. Metabolism of the c-Fos/activator protein-1 inhibitor T-5224 by multiple human UDP-glucuronosyltransferase isoforms. Drug Metab. Dispos. 2011, 39, 803–813. [DOI] [PubMed] [Google Scholar]
- Hai T. W.; Liu F.; Allegretto E. A.; Karin M.; Green M. R. A family of immunologically related transcription factors that includes multiple forms of ATF and AP-1. Genes Dev. 1988, 2, 1216–1226. [DOI] [PubMed] [Google Scholar]
- Angel P.; Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1991, 1072, 129–157. [DOI] [PubMed] [Google Scholar]
- Glover J. N.; Harrison S. C. Crystal structure of the heterodimeric bZIP transcription factor c-Fos-c-Jun bound to DNA. Nature 1995, 373, 257–261. [DOI] [PubMed] [Google Scholar]
- Ryseck R. P.; Bravo R. c-JUN, JUN B, and JUN D differ in their binding affinities to AP-1 and CRE consensus sequences: effect of FOS proteins. Oncogene 1991, 6, 533–542. [PubMed] [Google Scholar]
- Hai T.; Curran T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 3720–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinenov Y.; Kerppola T. K. Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 2001, 20, 2438–2452. [DOI] [PubMed] [Google Scholar]
- Jochum W.; Passegue E.; Wagner E. F. AP-1 in mouse development and tumorigenesis. Oncogene 2001, 20, 2401–2412. [DOI] [PubMed] [Google Scholar]
- Zenz R.; Eferl R.; Scheinecker C.; Redlich K.; Smolen J.; Schonthaler H. B.; Kenner L.; Tschachler E.; Wagner E. F. Activator protein 1 (Fos/Jun) functions in inflammatory bone and skin disease. Arthritis Res. Ther. 2008, 10, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenz R.; Wagner E. F. Jun signalling in the epidermis: from developmental defects to psoriasis and skin tumors. Int. J. Biochem. Cell Biol. 2006, 38, 1043–1049. [DOI] [PubMed] [Google Scholar]
- Suto M. J.; Ransone L. J. Novel approaches for the treatment of inflammatory diseases: inhibitors of NF-kappaB and AP-1. Curr. Pharm. Des 1997, 5, 515–528. [Google Scholar]
- Wang A.; Al-Kuhlani M.; Johnston S. C.; Ojcius D. M.; Chou J.; Dean D. Transcription factor complex AP-1 mediates inflammation initiated by Chlamydia pneumoniae infection. Cell Microbiol. 2013, 15, 779–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger G. R.; Gerwins P.; Widmann C.; Jarpe M. B.; Johnson G. L. MEKKs, GCKs, MLKs, PAKs, TAKs, and tpls: upstream regulators of the c-Jun amino-terminal kinases?. Curr. Opin. Genet. Dev. 1997, 7, 67–74. [DOI] [PubMed] [Google Scholar]
- Eferl R.; Wagner E. F. AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 2003, 3, 859–868. [DOI] [PubMed] [Google Scholar]
- Angel P.; Imagawa M.; Chiu R.; Stein B.; Imbra R. J.; Rahmsdorf H. J.; Jonat C.; Herrlich P.; Karin M. Phorbol ester-inducible genes contain a common cis element recognized by a TPA-modulated trans-acting factor. Cell 1987, 49, 729–739. [DOI] [PubMed] [Google Scholar]
- Schonthal A.; Herrlich P.; Rahmsdorf H. J.; Ponta H. Requirement for fos gene expression in the transcriptional activation of collagenase by other oncogenes and phorbol esters. Cell 1988, 54, 325–334. [DOI] [PubMed] [Google Scholar]
- Gutman A.; Wasylyk B. The collagenase gene promoter contains a TPA and oncogene-responsive unit encompassing the PEA3 and AP-1 binding sites. EMBO J. 1990, 9, 2241–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirum-Connolly K.; Brinckerhoff C. E. Interleukin-1 or phorbol induction of the stromelysin promoter requires an element that cooperates with AP-1. Nucleic Acids Res. 1991, 19, 335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroki Y.; Shiozawa S.; Sugimoto T.; Fujita T. Constitutive expression of c-fos gene inhibits type 1 collagen synthesis in transfected osteoblasts. Biochem. Biophys. Res. Commun. 1992, 182, 1389–1394. [DOI] [PubMed] [Google Scholar]
- Shiozawa S.; Tanaka Y.; Fujita T.; Tokuhisa T. Destructive arthritis without lymphocyte infiltration in H2-c-fos transgenic mice. J. Immunol. 1992, 148, 3100–3104. [PubMed] [Google Scholar]
- Miyauchi A.; Kuroki Y.; Fukase M.; Fujita T.; Chihara K.; Shiozawa S. Persistent expression of proto-oncogene c-fos stimulates osteoclast differentiation. Biochem. Biophys. Res. Commun. 1994, 205, 1547–1555. [DOI] [PubMed] [Google Scholar]
- Karin M.; Liu Z.; Zandi E. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [DOI] [PubMed] [Google Scholar]
- Shiozawa S.; Shimizu K.; Tanaka K.; Hino K. Studies on the contribution of c-fos/AP-1 to arthritic joint destruction. J. Clin. Invest. 1997, 99, 1210–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J.; Porte D.; Munz C.; Angel P. AP-1 and Cbfa/runt physically interact and regulate parathyroid hormone-dependent MMP13 expression in osteoblasts through a new osteoblast-specific element 2/AP-1 composite element. J. Biol. Chem. 2001, 276, 20029–20038. [DOI] [PubMed] [Google Scholar]
- Kawasaki H.; Komai K.; Ouyang Z.; Murata M.; Hikasa M.; Ohgiri M.; Shiozawa S. c-Fos/activator protein-1 transactivates wee1 kinase at G(1)/S to inhibit premature mitosis in antigen-specific Th1 cells. EMBO J. 2001, 20, 4618–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y.; Wenger L.; Brinckerhoff C. E.; Misra R. R.; Cheung H. S. Basic calcium phosphate crystals induce matrix metalloproteinase-1 through the Ras/mitogen-activated protein kinase/c-Fos/AP-1/metalloproteinase 1 pathway. Involvement of transcription factor binding sites AP-1 and PEA-3. J. Biol. Chem. 2002, 277, 1544–1552. [DOI] [PubMed] [Google Scholar]
- Kawasaki H.; Komai K.; Nakamura M.; Yamamoto E.; Ouyang Z.; Nakashima T.; Morisawa T.; Hashiramoto A.; Shiozawa K.; Ishikawa H.; Kurosaka M.; Shiozawa S. Human wee1 kinase is directly transactivated by and increased in association with c-Fos/AP-1: rheumatoid synovial cells overexpressing these genes go into aberrant mitosis. Oncogene 2003, 22, 6839–6844. [DOI] [PubMed] [Google Scholar]
- Macian F.; Lopez-Rodriguez C.; Rao A. Partners in transcription: NFAT and AP-1. Oncogene 2001, 20, 2476–2489. [DOI] [PubMed] [Google Scholar]
- Demoly P.; Basset-Seguin N.; Chanez P.; Campbell A. M.; Gauthier-Rouviere C.; Godard P.; Michel F. B.; Bousquet J. c-fos proto-oncogene expression in bronchial biopsies of asthmatics. Am. J. Respir. Cell Mol. Biol. 1992, 7, 128–133. [DOI] [PubMed] [Google Scholar]
- Teo J.-L.; Kahn M. AP-1 as a potential therapeutic target in allergic airways inflammation. Drugs Future 2004, 29, 693–703. [Google Scholar]
- Van Seuningen I.; Perrais M.; Pigny P.; Porchet N.; Aubert J. P. Sequence of the 5′-flanking region and promoter activity of the human mucin gene MUC5B in different phenotypes of colon cancer cells. Biochem. J. 2000, 348Part 3675–686. [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Chiba Y.; Sakai H.; Misawa M. Possible involvements of nuclear factor-kappa B and activator protein-1 in the tumor necrosis factor-alpha-induced upregulation of matrix metalloproteinase-12 in human alveolar epithelial A549 cell line. J. Pharmacol. Sci. 2010, 112, 83–88. [DOI] [PubMed] [Google Scholar]
- Vandal K.; Rouleau P.; Boivin A.; Ryckman C.; Talbot M.; Tessier P. A. Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J. Immunol. 2003, 171, 2602–2609. [DOI] [PubMed] [Google Scholar]
- Zenz R.; Eferl R.; Kenner L.; Florin L.; Hummerich L.; Mehic D.; Scheuch H.; Angel P.; Tschachler E.; Wagner E. F. Psoriasis-like skin disease and arthritis caused by inducible epidermal deletion of Jun proteins. Nature 2005, 437, 369–375. [DOI] [PubMed] [Google Scholar]
- Haider A. S.; Duculan J.; Whynot J. A.; Krueger J. G. Increased JunB mRNA and protein expression in psoriasis vulgaris lesions. J. Invest. Dermatol. 2006, 126, 912–914. [DOI] [PubMed] [Google Scholar]
- Curran T.; Teich N. M. Candidate product of the FBJ murine osteosarcoma virus oncogene: characterization of a 55,000-dalton phosphoprotein. J. Virol 1982, 42, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran T.; Peters G.; Van Beveren C.; Teich N. M.; Verma I. M. FBJ murine osteosarcoma virus: identification and molecular cloning of biologically active proviral DNA. J. Virol 1982, 44, 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt P. K. Fortuitous convergences: the beginnings of JUN. Nat. Rev. Cancer 2002, 2, 465–469. [DOI] [PubMed] [Google Scholar]
- Foletta V. C.; Sonobe M. H.; Suzuki T.; Endo T.; Iba H.; Cohen D. R. Cloning and characterisation of the mouse fra-2 gene. Oncogene 1994, 9, 3305–3311. [PubMed] [Google Scholar]
- Bergers G.; Graninger P.; Braselmann S.; Wrighton C.; Busslinger M. Transcriptional activation of the fra-1 gene by AP-1 is mediated by regulatory sequences in the first intron. Mol. Cell. Biol. 1995, 15, 3748–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandel L.; Pfarr C. M.; Huguier S.; Loiseau L.; Sergeant A.; Castellazzi M. Increased transforming activity of JunB and JunD by introduction of an heterologous homodimerization domain. Oncogene 1995, 10, 495–507. [PubMed] [Google Scholar]
- Wang Z. Q.; Grigoriadis A. E.; Mohle-Steinlein U.; Wagner E. F. A novel target cell for c-fos-induced oncogenesis: development of chondrogenic tumours in embryonic stem cell chimeras. EMBO J. 1991, 10, 2437–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriadis A. E.; Schellander K.; Wang Z. Q.; Wagner E. F. Osteoblasts are target cells for transformation in c-fos transgenic mice. J. Cell Biol. 1993, 122, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Shen Q.; DuPre E.; Kim H.; Hilsenbeck S.; Brown P. H. cFos is critical for MCF-7 breast cancer cell growth. Oncogene 2005, 24, 6516–6524. [DOI] [PubMed] [Google Scholar]
- Smith L. M.; Wise S. C.; Hendricks D. T.; Sabichi A. L.; Bos T.; Reddy P.; Brown P. H.; Birrer M. J. cJun overexpression in MCF-7 breast cancer cells produces a tumorigenic, invasive and hormone resistant phenotype. Oncogene 1999, 18, 6063–6070. [DOI] [PubMed] [Google Scholar]
- Shen Q.; Uray I. P.; Li Y.; Zhang Y.; Hill J.; Xu X. C.; Young M. R.; Gunther E. J.; Hilsenbeck S. G.; Colburn N. H.; Chodosh L. A.; Brown P. H. Targeting the activator protein 1 transcription factor for the prevention of estrogen receptor-negative mammary tumors. Cancer Prev. Res. 2008, 1, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Lu C.; Shen Q.; Munoz-Medellin D.; Kim H.; Brown P. H. AP-1 blockade in breast cancer cells causes cell cycle arrest by suppressing G1 cyclin expression and reducing cyclin-dependent kinase activity. Oncogene 2004, 23, 8238–8246. [DOI] [PubMed] [Google Scholar]
- Shen Q.; Uray I. P.; Li Y.; Krisko T. I.; Strecker T. E.; Kim H. T.; Brown P. H. The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene 2008, 27, 366–377. [DOI] [PubMed] [Google Scholar]
- Young M. R.; Li J. J.; Rincon M.; Flavell R. A.; Sathyanarayana B. K.; Hunziker R.; Colburn N. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9827–9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R.; Ricci R.; Kenner L.; Zenz R.; David J. P.; Rath M.; Wagner E. F. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell 2003, 112, 181–192. [DOI] [PubMed] [Google Scholar]
- Jochum W.; David J. P.; Elliott C.; Wutz A.; Plenk H. Jr.; Matsuo K.; Wagner E. F. Increased bone formation and osteosclerosis in mice overexpressing the transcription factor Fra-1. Nat. Med. 2000, 6, 980–984. [DOI] [PubMed] [Google Scholar]
- Passegue E.; Jochum W.; Schorpp-Kistner M.; Mohle-Steinlein U.; Wagner E. F. Chronic myeloid leukemia with increased granulocyte progenitors in mice lacking junB expression in the myeloid lineage. Cell 2001, 104, 21–32. [DOI] [PubMed] [Google Scholar]
- Yang M. Y.; Liu T. C.; Chang J. G.; Lin P. M.; Lin S. F. JunB gene expression is inactivated by methylation in chronic myeloid leukemia. Blood 2003, 101, 3205–3211. [DOI] [PubMed] [Google Scholar]
- Steidl U.; Rosenbauer F.; Verhaak R. G.; Gu X.; Ebralidze A.; Otu H. H.; Klippel S.; Steidl C.; Bruns I.; Costa D. B.; Wagner K.; Aivado M.; Kobbe G.; Valk P. J.; Passegue E.; Libermann T. A.; Delwel R.; Tenen D. G. Essential role of Jun family transcription factors in PU.1 knockdown-induced leukemic stem cells. Nat. Genet. 2006, 38, 1269–1277. [DOI] [PubMed] [Google Scholar]
- Santaguida M.; Schepers K.; King B.; Sabnis A. J.; Forsberg E. C.; Attema J. L.; Braun B. S.; Passegue E. JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 2009, 15, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakin A. V.; Curran T. Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science 1999, 283, 387–390. [DOI] [PubMed] [Google Scholar]
- Kasibhatla S.; Brunner T.; Genestier L.; Echeverri F.; Mahboubi A.; Green D. R. DNA damaging agents induce expression of Fas ligand and subsequent apoptosis in T lymphocytes via the activation of NF-kappa B and AP-1. Mol. Cell 1998, 1, 543–551. [DOI] [PubMed] [Google Scholar]
- Marconcini L.; Marchio S.; Morbidelli L.; Cartocci E.; Albini A.; Ziche M.; Bussolino F.; Oliviero S. c-fos-induced growth factor/vascular endothelial growth factor D induces angiogenesis in vivo and in vitro. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9671–9676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennigan R. F.; Hawker K. L.; Ozanne B. W. Fos-transformation activates genes associated with invasion. Oncogene 1994, 9, 3591–3600. [PubMed] [Google Scholar]
- Jooss K. U.; Muller R. Deregulation of genes encoding microfilament-associated proteins during Fos-induced morphological transformation. Oncogene 1995, 10, 603–608. [PubMed] [Google Scholar]
- Spence H. J.; Johnston I.; Ewart K.; Buchanan S. J.; Fitzgerald U.; Ozanne B. W. Krp1, a novel kelch related protein that is involved in pseudopod elongation in transformed cells. Oncogene 2000, 19, 1266–1276. [DOI] [PubMed] [Google Scholar]
- Park J. M.; Adam R. M.; Peters C. A.; Guthrie P. D.; Sun Z.; Klagsbrun M.; Freeman M. R. AP-1 mediates stretch-induced expression of HB-EGF in bladder smooth muscle cells. Am. J. Physiol. 1999, 277, C294–C301. [DOI] [PubMed] [Google Scholar]
- Whitfield J.; Neame S. J.; Paquet L.; Bernard O.; Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron 2001, 29, 629–643. [DOI] [PubMed] [Google Scholar]
- Rebollo A.; Dumoutier L.; Renauld J. C.; Zaballos A.; Ayllon V.; Martinez A. C. Bcl-3 expression promotes cell survival following interleukin-4 deprivation and is controlled by AP1 and AP1-like transcription factors. Mol. Cell. Biol. 2000, 20, 3407–3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb R. F.; Hennigan R. F.; Turnbull K.; Katsanakis K. D.; MacKenzie E. D.; Birnie G. D.; Ozanne B. W. AP-1-mediated invasion requires increased expression of the hyaluronan receptor CD44. Mol. Cell. Biol. 1997, 17, 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber M.; Kolbus A.; Piu F.; Szabowski A.; Mohle-Steinlein U.; Tian J.; Karin M.; Angel P.; Wagner E. F. Control of cell cycle progression by c-Jun is p53 dependent. Genes Dev. 1999, 13, 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E.; Schreiber M.; Piu F.; Beeche M.; Wagner E. F.; Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell 2000, 103, 897–907. [DOI] [PubMed] [Google Scholar]
- Ivanov V. N.; Bhoumik A.; Krasilnikov M.; Raz R.; Owen-Schaub L. B.; Levy D.; Horvath C. M.; Ronai Z. Cooperation between STAT3 and c-jun suppresses Fas transcription. Mol. Cell 2001, 7, 517–528. [DOI] [PubMed] [Google Scholar]
- Zenz R.; Scheuch H.; Martin P.; Frank C.; Eferl R.; Kenner L.; Sibilia M.; Wagner E. F. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev. Cell 2003, 4, 879–889. [DOI] [PubMed] [Google Scholar]
- Passegue E.; Wagner E. F. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000, 19, 2969–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft D. J.; Rosenberg S. B.; Bergers G.; Volpert O.; Linzer D. I. Reactivation of proliferin gene expression is associated with increased angiogenesis in a cell culture model of fibrosarcoma tumor progression. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 13055–13059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakiri L.; Lallemand D.; Bossy-Wetzel E.; Yaniv M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: a role in the control of cyclin D1 expression. EMBO J. 2000, 19, 2056–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabowski A.; Maas-Szabowski N.; Andrecht S.; Kolbus A.; Schorpp-Kistner M.; Fusenig N. E.; Angel P. c-Jun and JunB antagonistically control cytokine-regulated mesenchymal-epidermal interaction in skin. Cell 2000, 103, 745–755. [DOI] [PubMed] [Google Scholar]
- Weitzman J. B.; Fiette L.; Matsuo K.; Yaniv M. JunD protects cells from p53-dependent senescence and apoptosis. Mol. Cell 2000, 6, 1109–1119. [DOI] [PubMed] [Google Scholar]
- Kustikova O.; Kramerov D.; Grigorian M.; Berezin V.; Bock E.; Lukanidin E.; Tulchinsky E. Fra-1 induces morphological transformation and increases in vitro invasiveness and motility of epithelioid adenocarcinoma cells. Mol. Cell. Biol. 1998, 18, 7095–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E.; Mueller E.; Oliviero S.; Papaioannou V. E.; Johnson R.; Spiegelman B. M. Targeted disruption of the c-fos gene demonstrates c-fos-dependent and -independent pathways for gene expression stimulated by growth factors or oncogenes. EMBO J. 1994, 13, 3094–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasselblatt P.; Rath M.; Komnenovic V.; Zatloukal K.; Wagner E. F. Hepatocyte survival in acute hepatitis is due to c-Jun/AP-1-dependent expression of inducible nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 17105–17110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen M. K.; Bakiri L.; Hasenfuss S. C.; Hamacher R.; Martinez L.; Wagner E. F. JUNB/AP-1 controls IFN-gamma during inflammatory liver disease. J. Clin. Invest. 2013, 123, 5258–5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eferl R.; Hasselblatt P.; Rath M.; Popper H.; Zenz R.; Komnenovic V.; Idarraga M. H.; Kenner L.; Wagner E. F. Development of pulmonary fibrosis through a pathway involving the transcription factor Fra-2/AP-1. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 10525–10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran S.; Vaz M.; Reddy S. P. Fra-1/AP-1 transcription factor negatively regulates pulmonary fibrosis in vivo. PLoS One 2012, 7, e41611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekaran S.; Reddy N. M.; Zhang W.; Reddy S. P. Expression profiling of genes regulated by Fra-1/AP-1 transcription factor during bleomycin-induced pulmonary fibrosis. BMC Genomics 2013, 14, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning M. W.; Cassis L. A.; Daugherty A. Differential effects of doxycycline, a broad-spectrum matrix metalloproteinase inhibitor, on angiotensin II-induced atherosclerosis and abdominal aortic aneurysms. Arterioscler., Thromb., Vasc. Biol. 2003, 23, 483–488. [DOI] [PubMed] [Google Scholar]
- Kim T. J.; Yun Y. P. Potent inhibition of serum-stimulated responses in vascular smooth muscle cell proliferation by 2-chloro-3-(4-hexylphenyl)-amino-1,4-naphthoquinone, a newly synthesized 1,4-naphthoquinone derivative. Biol. Pharm. Bull. 2007, 30, 121–127. [DOI] [PubMed] [Google Scholar]
- Muslin A. J. MAPK signalling in cardiovascular health and disease: molecular mechanisms and therapeutic targets. Clin. Sci. 2008, 115, 203–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osto E.; Matter C. M.; Kouroedov A.; Malinski T.; Bachschmid M.; Camici G. G.; Kilic U.; Stallmach T.; Boren J.; Iliceto S.; Luscher T. F.; Cosentino F. c-Jun N-terminal kinase 2 deficiency protects against hypercholesterolemia-induced endothelial dysfunction and oxidative stress. Circulation 2008, 118, 2073–2080. [DOI] [PubMed] [Google Scholar]
- Wang J.; An F. S.; Zhang W.; Gong L.; Wei S. J.; Qin W. D.; Wang X. P.; Zhao Y. X.; Zhang Y.; Zhang C.; Zhang M. X. Inhibition of c-Jun N-terminal kinase attenuates low shear stress-induced atherogenesis in apolipoprotein E-deficient mice. Mol. Med. 2011, 17, 990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer C. A.; Le Haen P. A.; van Dijk R. A.; Hira M.; Hamming J. F.; van Bockel J. H.; Lindeman J. H. Activator protein-1 (AP-1) signalling in human atherosclerosis: results of a systematic evaluation and intervention study. Clin. Sci. 2012, 122, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S. M.; Yang S. P.; Ho L. J.; Tsao T. P.; Chang D. M.; Lai J. H. Irbesartan inhibits human T-lymphocyte activation through downregulation of activator protein-1. Br. J. Pharmacol. 2004, 142, 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komuro I.; Kaida T.; Shibazaki Y.; Kurabayashi M.; Katoh Y.; Hoh E.; Takaku F.; Yazaki Y. Stretching cardiac myocytes stimulates protooncogene expression. J. Biol. Chem. 1990, 265, 3595–3598. [PubMed] [Google Scholar]
- Parker T. G.; Schneider M. D. Growth factors, proto-oncogenes, and plasticity of the cardiac phenotype. Annu. Rev. Physiol. 1991, 53, 179–200. [DOI] [PubMed] [Google Scholar]
- Schneider M. D.; McLellan W. R.; Black F. M.; Parker T. G. Growth factors, growth factor response elements, and the cardiac phenotype. Basic Res. Cardiol. 1992, 87Suppl. 233–48. [DOI] [PubMed] [Google Scholar]
- Sadoshima J.; Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol. 1997, 59, 551–571. [DOI] [PubMed] [Google Scholar]
- Hilfiker-Kleiner D.; Hilfiker A.; Kaminski K.; Schaefer A.; Park J. K.; Michel K.; Quint A.; Yaniv M.; Weitzman J. B.; Drexler H. Lack of JunD promotes pressure overload-induced apoptosis, hypertrophic growth, and angiogenesis in the heart. Circulation 2005, 112, 1470–1477. [DOI] [PubMed] [Google Scholar]
- Ricci R.; Eriksson U.; Oudit G. Y.; Eferl R.; Akhmedov A.; Sumara I.; Sumara G.; Kassiri Z.; David J. P.; Bakiri L.; Sasse B.; Idarraga M. H.; Rath M.; Kurz D.; Theussl H. C.; Perriard J. C.; Backx P.; Penninger J. M.; Wagner E. F. Distinct functions of junD in cardiac hypertrophy and heart failure. Genes Dev. 2005, 19, 208–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker-Kleiner D.; Hilfiker A.; Castellazzi M.; Wollert K. C.; Trautwein C.; Schunkert H.; Drexler H. JunD attenuates phenylephrine-mediated cardiomyocyte hypertrophy by negatively regulating AP-1 transcriptional activity. Cardiovasc. Res. 2006, 71, 108–117. [DOI] [PubMed] [Google Scholar]
- Rosenzweig A.; Halazonetis T. D.; Seidman J. G.; Seidman C. E. Proximal regulatory domains of rat atrial natriuretic factor gene. Circulation 1991, 84, 1256–1265. [DOI] [PubMed] [Google Scholar]
- Bishopric N. H.; Jayasena V.; Webster K. A. Positive regulation of the skeletal alpha-actin gene by Fos and Jun in cardiac myocytes. J. Biol. Chem. 1992, 267, 25535–25540. [PubMed] [Google Scholar]
- Kovacic-Milivojevic B.; Gardner D. G. Divergent regulation of the human atrial natriuretic peptide gene by c-jun and c-fos. Mol. Cell. Biol. 1992, 12, 292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis P.; MacLellan W. R.; Belaguli N. S.; Schwartz R. J.; Schneider M. D. Serum response factor mediates AP-1-dependent induction of the skeletal alpha-actin promoter in ventricular myocytes. J. Biol. Chem. 1996, 271, 10827–10833. [DOI] [PubMed] [Google Scholar]
- Omura T.; Yoshiyama M.; Yoshida K.; Nakamura Y.; Kim S.; Iwao H.; Takeuchi K.; Yoshikawa J. Dominant negative mutant of c-Jun inhibits cardiomyocyte hypertrophy induced by endothelin 1 and phenylephrine. Hypertension 2002, 39, 81–86. [DOI] [PubMed] [Google Scholar]
- Taimor G.; Schluter K. D.; Best P.; Helmig S.; Piper H. M. Transcription activator protein 1 mediates alpha- but not beta-adrenergic hypertrophic growth responses in adult cardiomyocytes. Am. J. Physiol.: Heart Circ. Physiol. 2004, 286, H2369–H2375. [DOI] [PubMed] [Google Scholar]
- Windak R.; Muller J.; Felley A.; Akhmedov A.; Wagner E. F.; Pedrazzini T.; Sumara G.; Ricci R. The AP-1 transcription factor c-Jun prevents stress-imposed maladaptive remodeling of the heart. PLoS One 2013, 8, e73294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfonso-Jaume M. A.; Bergman M. R.; Mahimkar R.; Cheng S.; Jin Z. Q.; Karliner J. S.; Lovett D. H. Cardiac ischemia–reperfusion injury induces matrix metalloproteinase-2 expression through the AP-1 components FosB and JunB. Am. J. Physiol.: Heart Circ. Physiol. 2006, 291, H1838–1846. [DOI] [PubMed] [Google Scholar]
- Yang H. J.; Wang L.; Xia Y. Y.; Chang P. N.; Feng Z. W. NF-kappaB mediates MPP+-induced apoptotic cell death in neuroblastoma cells SH-EP1 through JNK and c-Jun/AP-1. Neurochem. Int. 2010, 56, 128–134. [DOI] [PubMed] [Google Scholar]
- Sullivan R. W.; Bigam C. G.; Erdman P. E.; Palanki M. S.; Anderson D. W.; Goldman M. E.; Ransone L. J.; Suto M. J. 2-Chloro-4-(trifluoromethyl)pyrimidine-5-N-(3′,5′-bis(trifluoromethyl)phenyl)-carboxamide: a potent inhibitor of NF-kappa B- and AP-1-mediated gene expression identified using solution-phase combinatorial chemistry. J. Med. Chem. 1998, 41, 413–419. [DOI] [PubMed] [Google Scholar]
- Goldman M. E.; Ransone L. J.; Anderson D. W.; Gaarde W. A.; Suto M. J.; Sullivan R. W.; Shorthouse R.; Morikawa M.; Morris R. E. SP100030 is a novel T-cell-specific transcription factor inhibitor that possesses immunosuppressive activity in vivo. Transplant. Proc. 1996, 28, 3106–3109. [PubMed] [Google Scholar]
- Moore-Carrasco R.; Busquets S.; Almendro V.; Palanki M.; Lopez-Soriano F. J.; Argiles J. M. The AP-1/NF-kappaB double inhibitor SP100030 can revert muscle wasting during experimental cancer cachexia. Int. J. Oncol. 2007, 30, 1239–1245. [PubMed] [Google Scholar]
- Palanki M. S.; Erdman P. E.; Gayo-Fung L. M.; Shevlin G. I.; Sullivan R. W.; Goldman M. E.; Ransone L. J.; Bennett B. L.; Manning A. M.; Suto M. J. Inhibitors of NF-kappaB and AP-1 gene expression: SAR studies on the pyrimidine portion of 2-chloro-4-trifluoromethylpyrimidine-5-[N-(3′,5′-bis(trifluoromethyl)phenyl)carboxamide]. J. Med. Chem. 2000, 43, 3995–4004. [DOI] [PubMed] [Google Scholar]
- Muto S.; Itai A.. Preparation of phenol or phenyl acetate derivatives as inhibitors against the activation of activator protein-1 (AP-1) and nuclear factor of activated T- cells (NFAT). WO 2003/103647 A1, 2003.
- Palanki M. S.; Erdman P. E.; Goldman M. E.; Suto C.; Suto M. J. Synthesis and structure–activity relationship studies of conformationally restricted analogs of 2-chloro-4-trifluoromethylpyrimidine-5-[N-(3′,5′-bis(trifluoromethyl)phenyl)]carboxamide. Med. Chem. Res. 2000, 10, 19–29. [Google Scholar]
- Lewis A. J.The prospect for improved medicines. In Emerging Drugs, 3rd ed.; Bowman W. C., Fitzgerald J. D., Taylor J. B., Eds.; Ashley Publication: London, 1998; pp 31–47. [Google Scholar]
- Palanki M. S.; Erdman P. E.; Manning A. M.; Ow A.; Ransone L. J.; Spooner C.; Suto C.; Suto M. Novel inhibitors of AP-1 and NF-kappaB mediated gene expression: structure–activity relationship studies of ethyl 4-[(3-methyl-2,5-dioxo(3-pyrrolinyl))amino]-2-(trifluoromethyl)pyrimidine-5-carboxylate. Bioorg. Med. Chem. Lett. 2000, 10, 1645–1648. [DOI] [PubMed] [Google Scholar]
- Palanki M. S.; Gayo-Fung L. M.; Shevlin G. I.; Erdman P.; Sato M.; Goldman M.; Ransone L. J.; Spooner C. Structure–activity relationship studies of ethyl 2-[(3-methyl-2,5-dioxo(3-pyrrolinyl))amino]-4-(trifluoromethyl)pyrimidine-5-carboxylate: an inhibitor of AP-1 and NF-kappaB mediated gene expression. Bioorg. Med. Chem. Lett. 2002, 12, 2573–2577. [DOI] [PubMed] [Google Scholar]
- Palanki M. S.; Erdman P. E.; Ren M.; Suto M.; Bennett B. L.; Manning A.; Ransone L.; Spooner C.; Desai S.; Ow A.; Totsuka R.; Tsao P.; Toriumi W. The design and synthesis of novel orally active inhibitors of AP-1 and NF-kappaB mediated transcriptional activation. SAR of in vitro and in vivo studies. Bioorg. Med. Chem. Lett. 2003, 13, 4077–4080. [DOI] [PubMed] [Google Scholar]
- Giri R. S.; Thaker H. M.; Giordano T.; Williams J.; Rogers D.; Sudersanam V.; Vasu K. K. Design, synthesis and characterization of novel 2-(2,4-disubstituted-thiazole-5-yl)-3-aryl-3H-quinazoline-4-one derivatives as inhibitors of NF-kappaB and AP-1 mediated transcription activation and as potential anti-inflammatory agents. Eur. J. Med. Chem. 2009, 44, 2184–2189. [DOI] [PubMed] [Google Scholar]
- Giordano A.; Giri R. S.; Inamdar G. S.; Sudarsanam V.; Thakar H. M.; Vasu Kamala K.; Yerande S. G.. Thiazole and thiophene analogues, and their use in treating autoimmune diseases and cancers. WO 2007/118149 A3, 2007.
- Giri R. S.; Thaker H. M.; Giordano T.; Chen B.; Nuthalapaty S.; Vasu K. K.; Sudarsanam V. Synthesis and evaluation of quinazolinone derivatives as inhibitors of NF-kappaB, AP-1 mediated transcription and eIF-4E mediated translational activation: inhibitors of multi-pathways involve in cancer. Eur. J. Med. Chem. 2010, 45, 3558–3563. [DOI] [PubMed] [Google Scholar]
- Tsuchida K.; Chaki H.; Takakura T.; Yokotani J.; Aikawa Y.; Shiozawa S.; Gouda H.; Hirono S. Design, synthesis, and biological evaluation of new cyclic disulfide decapeptides that inhibit the binding of AP-1 to DNA. J. Med. Chem. 2004, 47, 4239–4246. [DOI] [PubMed] [Google Scholar]
- Tsuchida K.; Chaki H.; Takakura T.; Kotsubo H.; Tanaka T.; Aikawa Y.; Shiozawa S.; Hirono S. Discovery of nonpeptidic small-molecule AP-1 inhibitors: lead hopping based on a three-dimensional pharmacophore model. J. Med. Chem. 2006, 49, 80–91. [DOI] [PubMed] [Google Scholar]
- Aikawa Y.; Morimoto K.; Yamamoto T.; Chaki H.; Hashiramoto A.; Narita H.; Hirono S.; Shiozawa S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nat. Biotechnol. 2008, 26, 817–823. [DOI] [PubMed] [Google Scholar]
- Izuta S.; Ueki M.; Ueno M.; Nishina K.; Shiozawa S.; Maekawa N. T-5224, a selective inhibitor of c-Fos/activator protein-1, attenuates lipopolysaccharide-induced liver injury in mice. Biotechnol. Lett. 2012, 34, 2175–2182. [DOI] [PubMed] [Google Scholar]
- Miyazaki H.; Morishita J.; Ueki M.; Nishina K.; Shiozawa S.; Maekawa N. The effects of a selective inhibitor of c-Fos/activator protein-1 on endotoxin-induced acute kidney injury in mice. BMC Nephrol. 2012, 13, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto M.; Masegi M.; Yamauchi T.; Chiba K.; Kuboi Y.; Harada K.; Naruse N. K1115 A, a new anthraquinone derivative that inhibits the binding of activator protein-1 (AP-1) to its recognition sites. I. Biological activities. J. Antibiot. 1998, 51, 539–544. [DOI] [PubMed] [Google Scholar]
- Ijaz T.; Tran P.; Ruparelia K. C.; Teesdale-Spittle P. H.; Orr S.; Patterson L. H. Anthraquinone-peptides as inhibitors of AP-1 transcription factor. Bioorg. Med. Chem. Lett. 2001, 11, 351–353. [DOI] [PubMed] [Google Scholar]
- Ammon H. P.; Wahl M. A. Pharmacology of Curcuma longa. Planta Med. 1991, 57, 1–7. [DOI] [PubMed] [Google Scholar]
- Huang T. S.; Lee S. C.; Lin J. K. Suppression of c-Jun/AP-1 activation by an inhibitor of tumor promotion in mouse fibroblast cells. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 5292–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakar S. S.; Roy D. Curcumin inhibits TPA induced expression of c-fos, c-jun and c-myc proto-oncogenes messenger RNAs in mouse skin. Cancer Lett. 1994, 87, 85–89. [DOI] [PubMed] [Google Scholar]
- Han S. S.; Keum Y. S.; Seo H. J.; Surh Y. J. Curcumin suppresses activation of NF-kappaB and AP-1 induced by phorbol ester in cultured human promyelocytic leukemia cells. J. Biochem. Mol. Biol. 2002, 35, 337–342. [DOI] [PubMed] [Google Scholar]
- Balasubramanian S.; Eckert R. L. Curcumin suppresses AP1 transcription factor-dependent differentiation and activates apoptosis in human epidermal keratinocytes. J. Biol. Chem. 2007, 282, 6707–6715. [DOI] [PubMed] [Google Scholar]
- Park S.; Lee D. K.; Yang C. H. Inhibition of fos-jun-DNA complex formation by dihydroguaiaretic acid and in vitro cytotoxic effects on cancer cells. Cancer Lett. 1998, 127, 23–28. [DOI] [PubMed] [Google Scholar]
- Kim H. K.; Yang C. H. Synthetic curcumin derivatives inhibit Jun-Fos-DNA complex formation. Bull. Korean Chem. Soc. 2004, 25, 1769–1774. [Google Scholar]
- Hahm E. R.; Gho Y. S.; Park S.; Park C.; Kim K. W.; Yang C. H. Synthetic curcumin analogs inhibit activator protein-1 transcription and tumor-induced angiogenesis. Biochem. Biophys. Res. Commun. 2004, 321, 337–344. [DOI] [PubMed] [Google Scholar]
- Lee J.; Park C.; Kim W.; Hwan Y. H.; Jeong K.; Yang C. Inhibitory effects of momordin I derivatives on the formation of Fos-Jun-AP-1 DNA complex. Bull. Korean Chem. Soc. 2006, 27, 535–538. [Google Scholar]
- Venisetty R. K.; Ciddi V. Application of microbial biotransformation for the new drug discovery using natural drugs as substrates. Curr. Pharm. Biotechnol. 2003, 4, 153–167. [DOI] [PubMed] [Google Scholar]
- Arantes S. F.; Hanson J. R. The biotransformation of sesquiterpenoids by mucor plumbeus. Curr. Org. Chem. 2007, 11, 657–663. [Google Scholar]
- Lehman L. R.; Stewart J. D. Filamentous fungi potentially useful catalysts for the biohydroxylations of non-activated carbon centers. Curr. Org. Chem. 2001, 5, 439–470. [Google Scholar]
- Chang S. F.; Yang L. M.; Lo C. H.; Liaw J. H.; Wang L. H.; Lin S. J. Microbial transformation of isosteviol and bioactivities against the glucocorticoid/androgen response elements. J. Nat. Prod. 2008, 71, 87–92. [DOI] [PubMed] [Google Scholar]
- Chou B. H.; Yang L. M.; Chang S. F.; Hsu F. L.; Lo C. H.; Lin W. K.; Wang L. H.; Liu P. C.; Lin S. J. Fungal transformation of isosteviol lactone and its biological evaluation for inhibiting the AP-1 transcription factor. Phytochemistry 2009, 70, 759–764. [DOI] [PubMed] [Google Scholar]
- Chang S. F.; Chou B. H.; Yang L. M.; Hsu F. L.; Lin W. K.; Ho Y.; Lin S. J. Microbial transformation of isosteviol oxime and the inhibitory effects on NF-kappaB and AP-1 activation in LPS-stimulated macrophages. Bioorg. Med. Chem. 2009, 17, 6348–6353. [DOI] [PubMed] [Google Scholar]
- Devkota K. P.; Ratnayake R.; Colburn N. H.; Wilson J. A.; Henrich C. J.; McMahon J. B.; Beutler J. A. Inhibitors of the oncogenic transcription factor AP-1 from Podocarpus latifolius. J. Nat. Prod. 2011, 74, 374–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diyabalanage T.; Ratnayake R.; Wilson J. A.; Henrich C. J.; Beutler J. A.; Colburn N. H.; McMahon J. B.; Gustafson K. R. Nothospondin, a new AP-1 inhibitory quassinoid from the Cameroonian plant Nothospondias staudtii. Bioorg. Med. Chem. Lett. 2011, 21, 4397–4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beutler J. A.; Kang M. I.; Robert F.; Clement J. A.; Pelletier J.; Colburn N. H.; McKee T. C.; Goncharova E.; McMahon J. B.; Henrich C. J. Quassinoid inhibition of AP-1 function does not correlate with cytotoxicity or protein synthesis inhibition. J. Nat. Prod. 2009, 72, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- S S.; HE K.; Liu G.; Zhu N.; Wang M.; Jhoo J.; Zheng Q.; Dong Z.; Ghai G.; Rosen R. T.; Ho C. T. Citrifolinin A, a new unusual iridoid with inhibition of activator protein-1 (AP-1) from the leaves of noni (Morinda citrifolia L.). Tetrahedron Lett. 2001, 42, 1823–1825. [DOI] [PubMed] [Google Scholar]
- Sang S.; He K.; Liu G.; Zhu N.; Cheng X.; Wang M.; Zheng Q.; Dong Z.; Ghai G.; Rosen R. T.; Ho C. T. A new unusual iridoid with inhibition of activator protein-1 (AP-1) from the leaves of Morinda citrifolia L. Org. Lett. 2001, 3, 1307–1309. [DOI] [PubMed] [Google Scholar]
- Popplewell W. L.; Ratnayake R.; Wilson J. A.; Beutler J. A.; Colburn N. H.; Henrich C. J.; McMahon J. B.; McKee T. C. Grassypeptolides F and G, cyanobacterial peptides from Lyngbya majuscula. J. Nat. Prod. 2011, 74, 1686–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong S.; Cahill K. J.; Kang M. I.; Colburn N. H.; Henrich C. J.; Wilson J. A.; Beutler J. A.; Johnson R. P.; Porco J. A. Microwave-based reaction screening: tandem retro-Diels–Alder/Diels–Alder cycloadditions of o-quinol dimers. J. Org. Chem. 2011, 76, 8944–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanjul A.; Dawson M. I.; Hobbs P. D.; Jong L.; Cameron J. F.; Harlev E.; Graupner G.; Lu X. P.; Pfahl M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 1994, 372, 107–111. [DOI] [PubMed] [Google Scholar]
- Huang C.; Ma W. Y.; Dawson M. I.; Rincon M.; Flavell R. A.; Dong Z. Blocking activator protein-1 activity, but not activating retinoic acid response element, is required for the antitumor promotion effect of retinoic acid. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 5826–5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H.; Hsieh M. C.; Lo C. Y.; Liu C. B.; Sang S.; Ho C. T.; Pan M. H. 6-Shogaol is more effective than 6-gingerol and curcumin in inhibiting 12-O-tetradecanoylphorbol 13-acetate-induced tumor promotion in mice. Mol. Nutr. Food. Res. 2010, 54, 1296–1306. [DOI] [PubMed] [Google Scholar]
- Lee W.; Haslinger A.; Karin M.; Tjian R. Activation of transcription by two factors that bind promoter and enhancer sequences of the human metallothionein gene and SV40. Nature 1987, 325, 368–372. [DOI] [PubMed] [Google Scholar]
- Rees D. C.; Congreve M.; Murray C. W.; Carr R. Fragment-based lead discovery. Nat. Rev. Drug Discovery 2004, 3, 660–672. [DOI] [PubMed] [Google Scholar]
- Chen H.; Yang Z.; Ding C.; Chu L.; Zhang Y.; Terry K.; Liu H.; Shen Q.; Zhou J. Fragment-based drug design and identification of HJC0123, a novel orally bioavailable STAT3 inhibitor for cancer therapy. Eur. J. Med. Chem. 2013, 62, 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Wang C. Z.; Ding C.; Wild C.; Copits B.; Swanson G. T.; Johnson K. M.; Zhou J. A combined bioinformatics and chemoinformatics approach for developing asymmetric bivalent AMPA receptor positive allosteric modulators as neuroprotective agents. ChemMedChem 2013, 8, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E. AP-1—the Jun proteins: oncogenes or tumor suppressors in disguise?. Cell. Signalling 2010, 22, 894–899. [DOI] [PubMed] [Google Scholar]