

Abstract
Increasing evidence of a role of chronic inflammation in type 2 diabetes progression has led to the development of therapies targeting the immune system. We develop a model of interleukin-1β dynamics in order to explain principles of disease onset. The parameters in the model are derived from in vitro experiments and patient data. In the framework of this model, an IL-1β switch is sufficient and necessary to account for type 2 diabetes onset. The model suggests that treatments targeting glucose bear the potential of stopping progression from pre-diabetes to overt type 2 diabetes. However, once in overt type 2 diabetes, these treatments have to be complemented by adjuvant anti-inflammatory therapies in order to stop or decelerate disease progression. Moreover, the model suggests that while glucose-lowering therapy needs to be continued all the way, dose and duration of the anti-inflammatory therapy needs to be specifically controlled. The model proposes a framework for the discussion of clinical trial outcomes.
Author Summary
Insulin resistance and relative insulin deficiency are two hallmarks of type 2 diabetes. While insulin resistance is always present in the early phase, it is β-cell failure that determines the pace of the disease onset. Increasing evidence that the immune system is activated and plays an important role in type 2 diabetes has stimulated efforts of developing drugs targeting inflammatory cytokines. We built a model to describe the principles of type 2 diabetes onset under the influence of interleukin-1β. The disease onset is understood in terms of bifurcation. It is found that inflammatory cytokines are required to be suppressed for a limited time only, while glucose has to be controlled over the long term. These structural insights may serve as a guideline for future clinical trials.
Introduction
Despite more than 350 million patients worldwide and the concomitant expensive socioeconomic burden, the pathogenesis of type 2 diabetes (T2D) is not yet completely understood. T2D is a progressive disease. The most important physiological components of T2D are insulin resistance, which is characterized by impaired response to insulin in insulin-sensitive tissues, and β-cell failure, which is characterized by β-cell dysfunction and reduced β-cell mass. The progression of T2D is clearly divided into at least two phases, pre-diabetes and overt diabetes [1]–[5]. In the pre-diabetes phase, insulin resistance is compensated by increased single β-cell secretion capacity and/or β-cell number. If insulin resistance is not completely compensated, the blood glucose level would grow slowly, manifested as higher fasting glucose (impaired fasting glucose, IFG) and/or higher post-load glucose (impaired glucose tolerance, IGT) [6]. Overt T2D is characterized by compensation failure and continuous loss of functional β-cells [7], [8], hence accompanied by continuously aggravated hyperglycaemia.
Although insulin resistance is usually present in the early phase of pre-diabetes, it is the pace of β-cell failure that determines the onset of overt T2D [9]. The mechanisms leading to the transition from pre-diabetes to overt T2D are unclear [10]. However, there is evidence that the transition from β-cell compensation to β-cell failure happens in a comparably short time span [2], [4], typically within 3 years [2]. This is further supported by a recent longitudinal study in a large population [5]. The trajectory of glycaemia before diagnosis of T2D was shown to be composed of a slow and stable adaptation, which lasts 12 years, followed by a rapid rise of glucose to overt T2D within 2 years [5]. Once overt T2D is started, hyperglycaemia continues to worsen regardless of treatments based on oral anti-diabetic agents [11]–[13].
The evidence of a role of inflammatory responses in the pathogenesis of T2D was increasing in recent years. Interleukin-1β (IL-1β) has been reported to contribute to β-cell failure [14]–[18]. β-cells themselves secrete IL-1β upon glucose stimulation [14]. Furthermore, IL-1β stimulates its own production in β-cells [17] and attracts macrophages [18] which can act as an extra source of IL-1β and other cytokines. Although it is currently unclear whether inflammatory responses are a primary cause or a secondary effect in T2D progression, therapies targeting IL-1β have shown encouraging progress albeit diverse results in different clinical trials [19]–[26].
These results motivated our working hypothesis that the pre-diabetic and overt T2D might be characterised by two qualitatively different states and that IL-1β is a potential candidate for promoting the transition between these two states. The hypothesis is consistent with results from clinic trials in which IL-1β blockade by interleukin-1-receptor antagonist (IL-1Ra), a naturally occurring competitive inhibitor of IL-1β, induced sustained improvements of β-cell function and the systemic inflammation state in patients with a mean disease duration of 11 years, even 39 weeks after cessation of treatment [19], [20]. IL-1Ra competes with IL-1β for IL-1 receptors but does not trigger any signalling event. It is an important regulator of the effect of IL-1β in many cell types, including human pancreatic β-cells which secrete IL-1Ra themselves [27], [28]. In several liver, autoimmune, and infectious diseases IL-1Ra is a better indicator of disease severity than IL-1β [29]. Results from a recent longitudinal study show that an accelerated increase in circulating IL-1Ra starts about 5 years before diagnosis of T2D [30], coinciding with the accelerated deterioration of insulin-sensitivity and the compensation of β-cell function [5]. In addition, long-term effects of temporary intensive insulin therapy on diabetes remission (or at least a prolonged normoglycaemia phase) in newly diagnosed diabetic patients may be partially attributed to the anti-inflammatory activity of insulin [31]. All these results highlight the role of inflammatory responses in the pre-diabetes phase and the possibility that the long-term effects induced by temporary anti-inflammatory therapy are mediated by a switch, i.e. a sudden transition, between the qualitatively different compensation and overt T2D states. Interestingly, in vitro experiments show that the effect of IL-1β on β-cell mass and insulin secretion is twofold [32]: low concentration of IL-1β stimulates β-cell proliferation, inhibits β-cell apoptosis and enhances glucose-stimulated insulin secretion while high concentration of IL-1β has opposite effects. In other words, IL-1β may contribute to both the β-cell compensation phase and the β-cell failure phase.
Here, we present a mathematical model showing the possibility that T2D onset is induced by a sudden transition of IL-1β to a high level. The IL-1β switch results from the coexistence of two different states, which is often termed as bi-stability. Bi-stable switch models have been widely used in modeling developmental processes, such as cell cycle progression, cellular differentiation and apoptosis. The characteristics of bi-stable switches include sudden transition, and hysteresis. The latter means that the state of a system depends not only on the current environment but also on its past state. We combine the IL-1β bi-stable switch model with a previously published T2D progression model [33], by assuming extrapolated β-cell turnover rates caused by exogenous IL-1β [32]. Similar concepts describing diabetes progression by different steady states have been already developed for both type 1 and 2 diabetes [33]–[35]. Our model falls into the same class as those previously published by Topp et al. [33] and De Gaetano et al. [35], which considered the evolution of β-cell mass, insulinemia and glycaemia over a time-scale of years and rely on glucose toxicity. β-cell mass is controlled by an empirical parabolic function of glucose in [33]. Subsequently a physiologically amenable concept of pancreatic reserve was introduced, which controls the direction of β-cell mass change [35]. Both β-cell mass and the pancreatic reserve were modeled phenomenologically as functions of glucose, where some parameters bear the potential to act as bifurcation parameters. The model presented here is built on these results. The phenomenological implementation of glucose toxicity is replaced by a model of IL-1β, which is explicitly defined and relatively easy to measure. We focus on an IL-1β bi-stable switch giving rise to hysteresis which is not discussed in previous studies. IL-1β hysteresis turns out to be of utmost importance because it widely shapes the strategy of anti-inflammatory therapies. While the complexity of the disease will never be captured by a mathematical model, the structural insights suggested by the model will help exploiting treatment approaches more efficiently and elaborating the effect of different therapies on β-cell mass and disease progression.
The IL-1β bi-stable switch model implies that T2D onset and later irreversible progression of β-cell failure arise from altered stability properties of the β-cell compensation state. Therefore, the strategy of glucose control would be more effective in the compensation phase than after the onset of overt T2D. This model result is consistent with recent data from the Diabetes Prevention Program Outcome Study (DPPOS) showing that IGT people who once regressed to normoglycaemia have a 56% lower relative risk of developing diabetes [36]. While glucose-lowering therapy bears the potential of stopping disease progression in the compensation phase, the IL-1β bi-stable switch model suggests that a combined glucose-lowering and anti-inflammatory therapy is necessary to stop the loss of functional β-cell mass as well as hyperglycaemia progression in the overt T2D phase, and eventually reestablish the compensation phase.
Materials and Methods
A full description of the model (Fig. 1), the assumptions, and the derivation of all model parameters from experimental data (summarized in Table 1) are provided in this section. Scripts of all calculations are provided in the supplement, including the model file and the parameter estimation procedures.
Figure 1. Model scheme: IL-1Ra (A) and IL-1β (L) compete for IL-1 receptors giving rise to a fraction of IL-1β bound receptors (F) which determines subsequent signalling in β-cells (B).

These control insulin (I) release and, via the influence of insulin resistance, blood glucose level (G). Arrows: activation effect, line with bar end: inhibition effect. Kinetic terms corresponding to each interaction are labelled. Variables evolving on different time scales are marked by different colours.
Table 1. Summary of parameters used in the model.
| Description | Symbol | Value | Unit | Ref. |
| glucose | G | |||
| initial value | 5.47(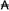 ) ) |
mM | [5] | |
5.25( ) ) |
mM | |||
| glucose production | r 0 | 48 | mM d−1 | [33] |
| insulin-independent Glucose uptake | d G | 1.44 | d−1 | [33] |
| insulin | I | - | ||
| initial value | 70.73(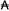 ) ) |
pM | DBSS | |
74.28( ) ) |
pM | |||
| maximal rate of insulin secretion by β-cells | h | 300.02 | pM d−1 | [33] |
| insulin clearance rate | dI | 432 | d−1 | [33] |
| glucose stimulated insulin secretion | s(G) | - | ||
| K G | 7.86 | mM2 | [33] | |
| β-cell mass | B | - | ||
| initial value | 312.1(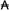 ) ) |
mgmg | DBSS | |
346.7( ) ) |
DBSS | |||
| ratio of proliferation to apoptosis | λ | 2.33 | DBSS | |
| β-cell proliferation | p(F) | - | ||
| a p | −0.048 | by fit | ||
| b p | −0.46 | by fit | ||
| c p | 0.62 | by fit | ||
| β-cell apoptosis rate | a(F) | - | ||
| a a | 0.047 | by fit | ||
| b a | 0.57 | by fit | ||
| c a | 2.36 | by fit | ||
| β-cell turnover time constant | τ | 0.00052 | d−1 | FBNH |
| IL-1Ra | A | |||
| initial value | 6000(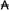 ) ) |
pg ml−1 | * | |
5982( ) ) |
pg ml−1 | DBSS | ||
| IL-1β stimulated IL-1Ra production | - | |||
| k 1 | 9.47e5 | pg ml−1 d−1 | DBSS | |
| k 2 | 3.12e7 | pg ml−1 d−1 | DBSS | |
| k 3 | 2.65e4 | d−1 | DBSS | |
| degradation rate of IL-1Ra | d A | 166.36 | d−1 | [29] |
| dissociation constant of IL-1Ra | K A | 1300 | pg ml−1 | [29] |
| IL-1β | L | |||
| initial value | 12(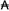 ) ) |
pg ml−1 | * | |
11.25( ) ) |
pg ml−1 | DBSS | ||
| inhibitory effects of basal IL-1β mRNA | b(L) | - | ||
| u | 5.8 | by fit | ||
| r | 2 | by fit | ||
| coefficient relating IL-1β mRNA level to its concentration | k 4 | 0.047 | pg−1 ml | DBSS |
| glucose stimulated IL-1β production | g(G) | - | ||
| s | 2.77 | by fit | ||
| v | 3.52 | mM | by fit | |
| k 5 | 3.745e3 | pgml−1 d−1 | DBSS | |
| IL-1β auto-stimulation | l(F) | |||
| t | 3 | by fit | ||
| K F | 0.1575 | by fit | ||
| k 6 | 1.048e9 | pgml−1 d−1 | DBSS | |
| degradation rate of IL-1β | d L | 55.45 | d−1 | [63] |
| dissociation constant of IL-1β | K L | 1300 | pg ml−1 | [29] |
| insulin resistance | R | |||
| R 0 | 9.643 | pM d | [33] | |
| m | 1.688 | pM d | FBNH |
Parameter values of the mathematical model and references.  : initial value of incident diabetes cases;
: initial value of incident diabetes cases;  : initial value of non-diabetic control; FBNH: fitted by glucose history; DBSS: determined by steady state conditions;
: initial value of non-diabetic control; FBNH: fitted by glucose history; DBSS: determined by steady state conditions;
*: parameter discussed in the section “steady state conditions”.
Overview
The mathematical model (Fig. 1) is constructed in three steps. First, we build the core of the model which describes the competition between IL-1β and IL-1Ra, where glucose stimulates IL-1β production. Since the effect of IL-1β on β-cells is mediated by the binding to its receptor, which is also the target of IL-1Ra antagonism, the fraction of IL-1β bound receptor F is used as a measure for IL-1β stimulation.
Second, the interaction between glucose (G) and insulin (I) is modeled with β-cell mass being a parameter. This subsystem is mainly adopted from [33], which is a single-compartment model justified by the slow dynamics of glucose and insulin over a time-scale of days to years. G and I are associated with long-term fasting glucose and insulin levels. As short-term (daily) fluctuations are averaged out, the model cannot describe disease onset derived from details of glucose and insulin dynamics.
Third, the two subsystems which describe the inflammatory signals and glucose-dependent insulin secretion are connected. For that purpose the IL-1β-dependent β-cell turnover rates are adopted from human islets which were cultured with exogenous IL-1β. The data suggest a bimodal effect of IL-1β on the β-cell mass [32]: IL-1β stimulates β-cell proliferation and inhibits β-cell apoptosis when presented in low concentrations. Conversely, at high concentrations IL-1β enhances β-cell apoptosis and reduces β-cell proliferation. The unknown model parameters are determined from steady state conditions of the IL-1β/IL-1Ra subsystem and from the natural history of fasting glucose in the pre-diabetic phase [5].
There are three different time scales in the model (Fig. 1). The IL-1β/IL-1Ra subsystem evolves on the fastest scale, since ligands and receptors bind/unbind on a time scale of seconds and proteins are synthesised/secreted on a time scale half an hour. The glucose/insulin subsystem evolves on an intermediate time scale of days [33]. β-cell evolves on the slowest scale. The turnover rate of β-cell is rather slow: typically, one or two cells divide per month. Since our main interest lies on long-term evolution of fasting glucose, the IL-1β/IL-1Ra subsystem is assumed in steady-state equilibrium and β-cell mass is assumed to be constant on this relatively fast time scale of fasting glucose. The different time-scales in the model make it possible to separate the model into different subsystems and justify our three-step modelling approach.
The antagonist effect of IL-1Ra (A)
All effects of IL-1β (L) are mediated by the IL-1 receptor and are blocked by IL-1Ra (A). IL-1Ra competes with IL-1β for the receptor but does not trigger any signalling event. Therefore, ligand-receptor binding and unbinding are considered to describe the antagonist effect of IL-1Ra. The ligand-receptor binding/unbinding happens on a time scale of seconds and is fast compared to other time-scales in the model, such as IL-1β and IL-1Ra production and secretion which happens on a time-scale of tens of minutes. Therefore, it is reasonable to assume that ligand and receptor are always in equilibrium, which enables us to describe the fraction of IL-1β bound receptors (F) by
| (1) |
where K L and K A are the dissociation constants of IL-1β and IL-1Ra, respectively. We assume a constant number of IL-1 receptors on the β-cell membrane. Consequently, F can be considered as a measure of IL-1β stimulation.
Estimate of endogenous IL-1β (L) and IL-1Ra (A) in cultured islets
F is used in the following to fit certain functions in the model to corresponding experimental data. In these experiments, different amounts of exogenous IL-1β were added to cultured human islets and the corresponding effects were measured, while endogenous IL-1Ra and IL-1β were not measured. It is safe to neglect endogenous IL-1β and IL-1Ra when exogenous IL-1β is dominant, which is the case for most measured data points. However, it is necessary to estimate the endogenous IL-1β and IL-1Ra concentrations for data points with zero exogenous IL-1β.
Secretion of IL-1Ra from cultured human islets was measured after 4 days which led to the endogenous IL-1Ra concentration of 114 pg/ml [32]. Endogenous IL-1β is estimated to be 0.228 pg/ml using the ratio of IL-1Ra/IL-1β of 500 [27].
IL-1Ra (A) dynamics
IL-1Ra (A) stimulation by IL-1β (L) is bimodal [32]. IL-1Ra is induced by IL-1β at exogenous concentrations below 20 pg/ml. At higher concentrations, IL-1β restores IL-1Ra to nearly the basal level. This nonlinear effect is phenomenologically captured by the nonlinear equation
| (2) |
where d A is the natural degradation rate of IL-1Ra. The parameters k 1 to k 3 are determined in the Section “steady states conditions” (see below).
IL-1β (L) dynamics
IL-1β dynamics are described by
| (3) |
Glucose induces β-cells to secrete IL-1β [17] (k 5 g(G)). IL-1β is also secreted from islets via auto-stimulation [17] (k 6 l(F)). High levels of IL-1β mRNA inhibit the stimulating effect of glucose and of auto-stimulation [17] (b(L)). IL-1β degrades with a rate d L. In the following, the functions g(G), l(F), and b(L) are derived from experimental data. The parameters k 5 and k 6 are determined in the Section “steady state conditions”.
1. IL-1β induction by glucose (g(G))
We use a Hill function g(G)
| (4) |
to fit data from cultured human islet describing the increase of IL-1β mRNA (Fig. 4A in [17]) as a function of glucose (Fig. 2 A). This implicitly assumes that IL-1β mRNA level is proportional to IL-1β concentration. Since the available data do not include glucose levels below 5.5 mM, we assume that half of the control rate is achieved at 3 mM (marked in Fig. 2A by the black square), which is the usual limit of hypoglycaemia. The choice of this value does not alter the model results. Other values, such as 2 or 4 mM, only affect the values of the model parameters (k 1–6) determined by steady state conditions (see Section below). We ignore the data point at 40 mM glucose, since such a high glucose level is not physiological. ag = 1 is used in the model, since the constant is merged with k5.
Figure 2. Parameter fit from experimental data.

A: Data are reproduced from Fig. 4A in [17]. The function g(G) in Eq. (4) is fitted to these data. Note a = 1 is used in the model, since the constant could be merged with k 5. B: Data were reproduced from Fig. 4B in [17]. The function l(F) in Eq. (5) is fitted to these data. Note a = 1 is used in the model. C: Data are reproduced from Fig. 1E in [17]. Eq. (6) is fitted to these data. Note the interchanged axes and a = 1 is used in the model. D: Data are reproduced from Fig. 1C in [32]. The function a(F) in Eq. (13) is fitted to these data. E: Data are reproduced from Fig. 1B in [32]. The function p(F) in Eq. (14) is fitted to these data.
2. IL-1β auto-stimulation (l(F))
IL-1β auto-stimulation is described by
| (5) |
and is fitted to data (Fig. 4B in [17]) which determine the increase of IL-1β mRNA of cultured human islets in dependence on exogenous IL-1β concentrations (Fig. 2 B). al = 1 is used in the model, since the constant is merged with k6.
3. Inhibition of the effects of glucose and IL-1β by IL-1β mRNA (b(L))
The stimulatory effect of glucose is inhibited by basal IL-1β mRNA [17]. IL-1β auto-stimulation is linearly related to glucose stimulated IL-1β in human islet preparations [17]. This linear relationship suggests a common regulatory mechanism of glucose-stimulated and auto-stimulated IL-1β production. This is represented in Eq. (3) by basal IL-1β mRNA-dependent inhibition of both glucose stimulation and IL-1β auto-stimulation using the sigmoidal function
| (6) |
where the scaling parameter k 4 relates IL-1β mRNA level to IL-1β protein concentration. Eq. (6) is fitted to the data (Fig. 1E in [17]) as a function of basal IL-1β (Fig. 2 C) and k 4 is determined in the Section “steady state conditions”. ab = 1 is used in the model, since the constant is merged with k5 and k6.
Glucose (G) and insulin (I) dynamics
G and I are quantities associated with long-term changes (days to years) of fasting levels of glucose and insulin, respectively. Therefore, a single-compartment model for glucose and insulin is used. The ordinary differential equations describing glucose and insulin are adapted from Topp et al. [33] with the modification that the dynamics of insulin resistance (R) is included as input to the model.
1. Glucose dynamics (G)
Glucose is produced with a constant rate (r 0) and consumed by an insulin-dependent (I/R) and an insulin-independent rate (d G)
| (7) |
where R denotes insulin resistance.
2. Insulin dynamics (I)
Insulin dynamics is modeled by a mass balance equation
| (8) |
with a secretion rate s(G) and a clearance rate (d I). Secretion is stimulated by glucose according to the sigmoidal function
| (9) |
and is proportional to the β-cell mass (B). The maximal rate of insulin secretion h, the insulin clearance rate d I, and the half concentration KG of glucose stimulated insulin secretion are taken from [33].
3. Insulin resistance (R)
Insulin sensitivity provided in [5] is homeostatic model assessment (HOMA) index. The one compartment model of glucose and insulin dynamics is a simplification of the minimal model [37]. Insulin resistance derived from the minimal model exhibits a linear relationship to the HOMA-IR index on logarithmic scale [38]. Hence,
| (11) |
is used to convert the HOMA index value into R. Here SH is the insulin sensitivity index (HOMA2 %S) reported in [5]. The parameter m is determined by fitting the model to the natural history of fasting glucose in the pre-diabetic phase [5].
β-cell mass dynamics (B)
The dynamics of the β-cell mass
| (12) |
is controlled by IL-1β induced β-cell apoptosis and proliferation. The apoptosis rate a(F)
| (13) |
and the proliferation rate p(F)
| (14) |
are fitted to data from human islets [32], which were cultured for 4 days with different IL-1β concentrations applied at day 0 (Fig. 2 D and E, respectively).
We assume that the initiation of apoptosis and proliferation is faster than IL-1β-induced IL-1β and IL-1Ra secretion. Thus, the IL-1β and IL-1Ra dynamics are de-coupled from β-cell turnover and the initial F (at day 0 of the experiment) can be used as an approximation in fitting the data. Since the apoptosis and the proliferation rates depend on log(F), eventual modifications of F caused by newly secreted molecules would anyway remain small on the logarithmic scale.
β-cell apoptosis and proliferation were measured as percentage of control (i.e. no IL-1β added) [32] such that the absolute rates remained undetermined. Scaling of the rates is represented by an extra parameter τ (Eq. (12)), which is fitted to the natural history of fasting glucose in the pre-diabetic phase [5]. The relative importance of apoptosis and proliferation is controlled by the parameter λ in Eq. (12), which is determined in the Section “steady state conditions”.
Steady state conditions
While a large number of model parameters could be determined by experimental constraints, the parameters k 1–6 and λ remained undetermined and are derived from steady state conditions for Eqs. (2,3,12). Eq. (12) is used to determine λ. Eq. (2) and (3) each exhibit two stable steady states and a bifurcation point, such that these 6 additional conditions can be used to determine k 1–6.
1. Steady state of the compensation phase
Reports from the Whitehall II longitudinal study [5] show that 13 years before diagnosis of T2D, the fasting glucose level of the incident diabetic participants is already higher than that of control (5.47 vs. 5.25 mM, Table 2). The corresponding serum IL-1Ra concentration of the incident diabetic participants is 300 pg/ml [30]. Using the reported in vitro ratio of IL-1Ra/IL-1β, which is 500 [27], the corresponding serum IL-1β concentration is estimated to be 0.6 pg/ml. This value is consistent with the average serum IL-1β level (0.57±0.93 pg/ml) reported in [39]. Since IL-1β/IL-1Ra are locally produced in the pancreas islets and their half-lives are short (5∼10 minutes), we assume 20 fold more IL-1β/IL-1Ra in the pancreas than in the blood under healthy conditions (note that the blood-tissue ratio might change after T2D onset), inferring 12 and 6000 pg/ml (Table 2) of IL-1β and IL-1Ra, respectively, in the islets. As this choice is arbitrary, other choices of the tissue-blood ratio, such as 200 or 2000, with fixed ratio of IL-1Ra/IL-1β, were tested. The ratio does not change the model behaviour since the fraction of IL-1β bound receptors F rather than the level of IL-1β determines the inflammatory stimulus. However, the choice of the tissue-blood ratio affects the values of the model parameters (λ and k 1–6) derived from steady state conditions.
Table 2. Steady state conditions for the IL-1β/IL-1 Ra subsystem.
| Glucose(mM) | IL-1β (pg/ml) | IL-1 Ra (pg/ml) | |
| (G) | (L) | (A) | |
| compensation | 5.47 | 12 | 6000 |
| bifurcation | 5.84 | Lb (unknown) | Ab (unknown) |
| overt diabetes | 13.4 | 12*55.8 | 6000*1.8 |
2. Steady state of β-cell mass determines λ
β-cell proliferation and apoptosis are assumed to be exactly balanced at a healthy glucose level (4.5 mM), which determines the parameter λ assuming Eq. (12) in steady state using F 0(G0 = 4.5 mM). If the glucose level is less than 4.5 mM, the apoptosis rate would surpass the proliferation rate which would decrease the β-cell mass. When the glucose level increases, more IL-1β is produced which stimulates β-cell proliferation and increases the β-cell mass. Increasing insulin resistance detunes this equilibrium.
3. Steady state at the critical (bifurcation) point
A sudden rapid increase in fasting glucose happens about two years before diagnosis of T2D. The average fasting glucose level at the inflection point of glucose history is 5.84 mM [5] (Table 2).
4. Steady state in the overt diabetes phase
IL-1β mRNA expression was measured in β-cells from T2D and non-diabetic cadaver donors [17]. Real-time quantitative PCR revealed in average 55.8 fold more IL-1β mRNA in T2D patients compared to healthy controls. As discussed above, we assume a linear relation of mRNA and protein concentration. The average fasting glucose of those diabetic donors was 13.4 mM (Table 2) [17]. Because of the bimodal dependency of IL-1Ra on IL-1β (see Fig. 2E in [32]), two IL-1Ra levels may be consistent with the 55.8 fold increase of IL-1β: Either the basal level of IL-1Ra or a 1.8 fold increased level. We tested both solutions by parameter scanning (see below) and only the latter solution yielded consistent results and is, therefore, taken in the model (Table 2).
5. Steady state equations
The steady state equations for Eq. (2) and (3) read
where i = {c,b,d} correspond to the compensation, bifurcation and diabetes state, respectively. The values are summarized in Table 2. Note that IL-1β (L b) and IL-1Ra (A b) are not determined by experimental data.
6. Determine the range of IL-1β and IL-1Ra at bifurcation by parameter scan
The L b-A b parameter plane is scanned in order to identify possible IL-1β and IL-1Ra concentrations at the bifurcation point. The values are restricted by the side condition that a switch of IL-1β and IL-1Ra exists (Fig. 3 A and B). This is done by numerically checking the determinant of the Jacobian and then checking the number of solutions on both sides of the bifurcation point Gb (the Matlab script is included in Dataset S1). While a switch of IL-1β exists in a large area of the L b-A b plane (red region, Fig. 3 A), the required switch to a 55.8 fold IL-1β level is only achieved for a small subset of values (Fig. 3 B). Most parameter combinations led to a smaller switch of IL-1β. This determined the choice of the values for L b and A b. Note that the scan for different basal IL-1β/IL-1Ra levels (120/6×104 or 1200/6×105 with IL-1Ra/IL-1β ratio fixed at the reported value [27]) yielded similar results (Fig. 3).
Figure 3. Parameter determined by scanning.

A: Random points are selected in the L b-A b plane (normalised with L c and A c) and used in the steady state equations (Eq. (2,3)) to determine k 1–6. If k 1–6 are non-negative, points either induce an IL-1β switch (red) or do not (blue). B: The IL-1β level L d/L c after the switch is shown as a function of IL-1Ra concentration at the bifurcation point.
Fitting the natural history of fasting glucose
The last two undetermined parameters, m and τ defined in Eq. 11 and 12, are determined by fitting the model to the fasting glucose history of both incident diabetes cases and the non-diabetics controls [5]. The best fit is shown in Fig. 4A. Interpolated insulin sensitivity [5] (Fig. 4B) which is an input to the model during fitting, the behaviour of the β-cell mass (Fig. 4C) and of IL-1β (Fig. 4D) are also shown. The fitting is implemented in the SBTOOLBOX2 [40] using a differential evolution algorithm, which reports multiple possible parameter sets (Fig. 5A). A strong correlation between m and τ was identified (Fig. 5B), which gives rise to large variations of glucose and β-cell mass in the simulations of overt diabetes (Fig. S1). In other words, the pre-diabetes glucose trajectory does not allow for a precise determination of m and τ. The values from the best fit are reported in Table 1 and used throughout the paper.
Figure 4. Fitting the model to the fasting glucose history to determine m and τ.

Shown are the best fit results of incident diabetes cases before T2D diagnosis and the data (black line) (red line cross) and non-diabetic controls (red line) (A), interpolated insulin resistance as model input (B), corresponding behaviour of β-cell mass (C) and IL-1β (D).
Figure 5. There exist multiple reasonable fits (shadow) of incident diabetes cases before T2D diagnosis and the data (black line) and non-diabetic controls (red line) (A).

Fitted parameters exhibit correlations (B).
A complementary approach
The above approach demonstrates that the bifurcation of the IL-1β/IL-1Ra subsystem is sufficient to account for the fitted data set. To investigate whether the bifurcation of the IL-1β/IL-1Ra subsystem is necessary, a complementary approach is employed: the model is fitted to the data without bifurcation as a presupposed condition. The steady state equations for Eq. (2) and (3), in the compensation phase and in the overt diabetes phase, allow reducing the degree of freedom from 8 (k1∼6, m and τ) to 4: k1, k2, k5, and k6 are explicitly expressed as functions of k3 and k4, and only k3, k4, m and τ are fitted. Strong correlations are found to exist between m and τ, which cause large variations of glucose and β-cell mass in the simulations of the overt diabetes (Fig. S2 A and B). Strong correlations are also found between m and k4, which cause large variations of the time point of the IL-1β/IL-1Ra switch (Fig. S2 C and D). Please note that the complementary approach found, although with low probability, cases allowing an extra transition (see Fig. S2 C and D).
Although some parameters cannot be determined precisely, the key qualitative property, i.e. the switch of the IL-1β/IL-1Ra subsystem before T2D diagnosis, remains robust. Taking together the results of both complementary approaches, it is suggested that the bifurcation of the IL-1β/IL-1Ra subsystem is both sufficient and necessary to account for the data set, in the framework of the current model. Please note that despite the model being validated by quantitative experimental data, the results presented here remain of qualitative nature. In particular, in the overt T2D state we expect that additional physiological process as well as medication will alter the results on a quantitative level.
Results
The IL-1β/IL-1Ra subsystem has multiple steady states
An IL-1β switch is observed in a large range of parameter values including the physiological relevant range. Below the critical glucose level of 5.84 mM the model gives rise to five steady states, three stable and two unstable ones (Fig. 6A). The stable steady state with lowest IL-1β (green in Fig. 6 A) is stable only if glucose is below the critical level. It is interpreted as corresponding to the compensation phase. The hyperglycaemia parts of the two other stable steady states may be associated with overt T2D.
Figure 6. A: The bifurcation diagram of the IL-1β-IL-1Ra subsystem as glucose (G) varies.

This subsystem exhibits three stable (full lines) and two unstable (dotted blue lines) steady states. Above a critical glucose level (5.84 mM) the system loses stability of the pre-diabetic state (green line) and progresses to a high IL-1β state (red line), which is stable across the whole physiological glucose range. A third stable state (black line) with even higher IL-1β exists, which, in the hyperglycaemia range, may be associated with advanced T2D. B: A transition from five to three steady states is found that corresponds to the transition from pre-diabetes to overt T2D. Steady states appear as crossing points of the nullclines. The nullclines of IL-1β (red) and IL-1Ra (black) cross in five points at normal glucose level (4.5 mM). When glucose increases, the IL-1β nullcline raises its minimum (inset, transition from red to blue to green curve) such that the low IL-1β and low IL-1Ra steady state vanishes. The system is forced to switch to a high IL-1β state which may be associated with overt T2D. The corresponding glucose level is 4.5, 5.84 and 6.84 mM for the red, blue and green IL-1β nullcline in the inset, respectively.
Nullcline analysis facilitates understanding the transition between the different inflammatory levels. Steady states appear as the crossing points of the IL-1Ra versus IL-1β nullclines where both quantities become static (Fig. 6B). The IL-1β nullcline (Fig. 6B red line) exhibits a nadir followed by a peak, which is the typical feature of an auto-stimulation species. The IL-1Ra nullcline (Fig. 6B black line) exhibits one peak. This implies that at large IL-1β its further increase is accompanied by decreased IL-1Ra. The measured non-linear dependence of IL-1Ra on IL-1β [32] is at the origin of the third stable steady state with highest IL-1β. If IL-1Ra is stimulated by IL-1β in a linear manner, only two stable steady states would have been found. Such a dysregulation of IL-1Ra may also happen in other diseases involving IL-1β, such as chronic myelogenous leukaemia and hairy cell leukaemia [29]. Hence, dysregulation of IL-1Ra at high IL-1β concentrations might be associated with a boost of disease progression.
The compensation steady state is lost upon progression to overt T2D
The stable steady state with lowest IL-1β corresponds to the pre-diabetic state. As glucose influences IL-1β production [14], [17] the nadir of the IL-1β nullcline moves up in response to increased glucose (Fig. 6B inset) while the IL-1Ra nullcline remains unchanged (Fig. 6B, black line). The two involved steady states approach each other and ultimately merge into one steady state (Fig. 6B inset, blue line). At even higher glucose this steady state disappears and the system makes a transition (Fig. 6B, black arrow) to the next available stable steady state, which is characterised by substantially higher IL-1β and moderately increased IL-1Ra. This transition is interpreted to correspond to the sudden transition from the compensation phase to disease progression.
At even higher levels of IL-1β, the effect of glucose on IL-1β production is inhibited by IL-1β itself (represented in Eq. 3 by b(l)), such that the IL-1β nullcline remains unaffected by glucose at high IL-1β concentrations. Therefore, the current model only reflects the transition from the compensation phase to the mild disease phase and not to the strong disease phase. An extended model including detailed IL-1β mRNA dynamics and other factors like free fatty acids, leptin or tissue resident macrophages might generate two transitions.
Glucose-lowering therapy cannot cure or stop progression of T2D
Glucose-lowering therapy, simulated in silico by fixing the level of glucose at normal level (5.47 mM), is represented by the IL-1β nullcline which now, as in the pre-diabetic state, intersects with the IL-1Ra nullcline (Fig. 7A) around its nadir at low IL-1β. The therapy reconstitutes the existence of the pre-diabetic stable steady state (Fig. 7A, red circle). However, the original pathological steady state (Fig. 7A, black square) is neither deleted nor does it lose stability. The disease state remains stable irrespective of the glucose levels reached by glucose therapy. Therefore, the glucose-lowering therapy does not induce a transition from overt T2D to pre-diabetes and the functional β-cell mass is further decreasing (Fig. 7D, black squares).
Figure 7. T2D therapy strategies and their effect on β-cell mass.

A: Glucose-lowering therapy. The pre-diabetic steady state is restored at the IL-1β (red line) and IL-1Ra (black line) nullcline intersection (red circle). But the system stays in the stable T2D steady state (black square). B: Permanent IL-1Ra therapy. The IL-1β (red line) and IL-1Ra (black dotted line) nullclines do not intersect at low IL-1β and the system starts in the stable overt T2D steady state (black square). Clamping IL-1Ra to levels below the peak of the IL-1β nullcline (lower blue line) reduces IL-1β (red cross). Clamping IL-1Ra to levels above the peak (upper blue line) makes IL-1β jump to low levels (green triangle). C: Combined IL-1Ra and glucose-lowering therapy. Above threshold clamp of IL-1Ra (dashed dotted line) induces a substantial IL-1β reduction (red dot). Simultaneous control of glucose restores a stable steady state at low IL-1β (red and black nullclines intersect at the blue circle). A release of IL-1Ra clamp induces a transition to this steady state. D: Effect of the different treatments on β-cell mass. Therapy as in A (black squares), B (red crosses for below threshold IL-1Ra clamp; green triangles for above threshold IL-1Ra clamp), C (red dots for combined IL-1Ra and glucose-lowering therapy; blue circles after release of IL-1Ra clamp). No treatment (black line).
The pre-diabetic state cannot be restored by IL-1Ra therapy of overt T2D
IL-1Ra is a naturally occurring inhibitor of IL-1β, and it is upregulated in obesity [28] and reduced in poorly controlled T2D patients [27]. By injecting exogenous IL-1Ra, IL-1Ra therapy aims at inhibiting IL-1β signalling. This is simulated in silico by fixing the level of IL-1Ra at a higher concentration (Fig. 7B, red cross). The simulated therapy slows down the loss of β-cell mass (Fig. 7D, red crosses) because increased IL-1Ra drives IL-1β into a less pathological regime. The higher IL-1Ra is set, the more IL-1β is reduced. At a threshold level of IL-1Ra, which is determined by the peak of the IL-1β nullcline (Fig. 6B, red line), the response of IL-1β becomes qualitatively different and it is reset below its physiological normal level (Fig. 7B, green triangle). Though the speed of β-cell loss is significantly reduced, it is not stopped (Fig. 7D, green triangles). Releasing IL-1Ra fixation leads to a restoration of the T2D state associated with accelerated loss of β-cell mass (Fig. 7B, black square), because the stable pre-diabetic steady state associated with low IL-1β is not restored by this therapy (Fig. 7B).
The pre-diabetic state can be restored by combined glucose-lowering and IL-1Ra therapy
A combined IL-1Ra and glucose-lowering therapy (Fig. 7C) is required to induce functional β-cell mass reconstitution. Strong IL-1Ra therapy induces a transition from the overt T2D steady state to a transient state at low IL-1β (Fig. 7C, from black square to red dot) associated with reduced speed of β-cell loss (Fig. 7D, red dots). A concurrent glucose-lowering therapy restores a stable steady state at low IL-1β (Fig. 7C, blue circle). Upon withdrawal of the IL-1Ra fixation, the in silico patient makes a transition to the restored stable steady state (Fig. 7C, from red dot to blue circle) which is associated with β-cell growth (Fig. 7D, blue circles), thus, inducing a long-standing improvement of the disease state.
The IL-1Ra therapy must be released to achieve a disease state improvement
Long-term combined IL-1Ra and glucose-lowering therapy prohibits β-cell mass regrowth in silico (Fig. 7D, red dots). The model predicts that IL-1Ra therapy, simulated in the model as the fixation of IL-1Ra, must be released in order to allow for functional β-cell mass reconstitution. β-cell mass regrowth is suppressed during IL-1Ra therapy as it reduces IL-1β to levels below the pre-diabetic state.
Discussion
An IL-1β switch model of T2D onset after long-term compensation was presented and a strategy for an anti-inflammatory therapy of T2D was proposed. The model is characterised by a pre-diabetic state during which increasing insulin resistance drives hyperglycaemia. This ultimately leads to a changed steady state configuration and progression into the overt T2D disease state. The model parameters are derived from in vitro islet measurements, from steady state conditions, and from glucose history of T2D patients, such that the model parameters are fully determined. In the following we first discuss its relevance to the empirical theory of five-stage β-cell dysfunction [3], then describe the predictions of the model, its relevance and potential contributions to clinical trials [23], [26], and its relation to previous modelling works [33], [35]. Last but not least, we discuss its limitations.
Interestingly, the model shows three steady-states of IL-1β. This result is beyond our expectations when designing the model but is consistent with the five-stage diabetes development theory [3]. This theory is mainly based on clinical observations, where stage 2, “adaptation” (termed as compensation in this paper) corresponds to the model state of lowest IL-1β (green in Fig. 6A). Stage 3 “transient unstable early de-compensation” corresponds to the sudden switch of IL-1β/IL-1Ra in the model, where a 55 fold increase in IL-1β level is accompanied with only 2 fold more IL-1Ra, and the transient adaptation time thereafter. Stage 4 “stable de-compensation” corresponds to the state with medium IL-1β level (red in Fig. 6A) and stage 5 “severe de-compensation” corresponds to the state with highest IL-1β level (black in Fig. 6A). Stage 1 “normal” is not represented in the model because glucose level is already higher even 14 years before T2D diagnosis (see Fig. 4A and [5]). Thus, the IL-1β switch model qualitatively reflects all stages of T2D development, starting from the compensation phase and progressing to the mild disease and ultimately to the strong disease state.
Stage 3 is observed to be transient, persisting a few weeks in different animal models that were subjected to partial pancreatectomies [41] or islets transplantation [42], [43]. Interestingly, islets transplantation [43] and pancreatectomies [41] in rodents gave rise to glucose levels in two discrete groups: either normal glucose or severe diabetes, with almost no glucose levels in between, a pattern consistent with the idea of two distinct states in the presented model. Stage 3, transient and unstable, lies between the clinically non-pathologic and pathologic states, and thus, may be of great interest in terms of providing a time window during which therapies are more efficient in reversing the pathological processes right after diagnosis. Results from newly diagnosed diabetic patients who received intensive insulin therapy supports this notion [31]. However, in the current model, the switch of IL-1β/IL-1Ra, which is relevant to stage 3, is instantaneous (see Fig. 6 and Fig. S2) because the model does not describe the dynamics of the IL-1β/IL-1Ra subsystem during the transition. The equations of the IL-1β/IL-1Ra are mainly based on assumed steady state relations, which were derived from in vitro data. In addition, the model does not describe the dynamics of insulin resistance, the improvement of which has been assumed to be responsible of preventing/postponing the transition during stage 3 [3]. Rather, insulin resistance is implemented as an input to the glucose/insulin subsystem. These limitations of the current modeling approach (see also later discussions about the meaning of G and I in the model) make information about the duration of stage 3 unavailable from the model.
The IL-1β bi-stable switch model suggests four requirements for a successful anti-inflammatory treatment of T2D:
Glucose-lowering therapy is combined with IL-1Ra therapy;
IL-1Ra is kept at an above threshold level;
IL-1Ra fixation is released when IL-1β levels are down;
Glucose-lowering therapy is continued on long-term.
Then, the functional β-cell mass is predicted to increase. But even if it reaches its healthy level, the in silico T2D patient would remain in the pre-diabetic state, since the release of glucose control would restart T2D progression due to the pathological state of insulin resistance. The power of this therapy is not to cure T2D but to keep the in silico patient in the pre-diabetic state.
Dose and duration of anti-IL-1β therapy are critical. On the one hand, an above threshold IL-1Ra is required for inhibition of IL-1β auto-stimulation and the reverse transition from β-cell failure to compensation. On the other hand, IL-1β has to be kept above its healthy level for β-cell reconstitution. This infers a U-shaped dose-effect relationship of anti-IL-1β therapy in silico, which has been confirmed in a phase 1 clinical trial using a monoclonal antibody of human IL-1β [23].
Large scale clinical trials with anti-IL-1β agents revealed that glycaemia was less efficiently reduced in patient groups with short (6 years) compared to groups with long (11 years) disease duration [26]. We have performed simulations of IL-1Ra therapy on patients with short and long T2D duration, where short and long T2D are associated with mild and strong T2D steady states, respectively (Fig. 8). The strong T2D state is initiated by manually increasing IL-1β and decreasing IL-1Ra to the levels shown in Fig. 6B at 11 years. Then, anti-IL-1β therapy (ignoring other adjuvant medication) is applied by an IL-1Ra fixation for 90 days to both in silico patient groups. The IL-1Ra dose-effect relationship for the two groups, expressed by the glycaemia improvement versus control, is compared (Fig. 8). In agreement with the clinical-trials results, optimal IL-1Ra dosage induced more efficient glucose improvement for strong than for mild T2D. This happens because in strong T2D the aggravation of blood glucose levels is faster in the control group. The optimal dose of IL-1Ra is different in both groups in silico (Fig. 8). This calls for an individualised treatment according to the endogenous inflammatory state. These simulations do not aim at explaining the diverse outcomes from clinical trials because other adjuvant medications are neglected during the simulations. Furthermore, the capability of the current model is limited to the low to medium hyperglycaemia/IL-1β region (see later discussion). However, the model still provides a framework in which different outcomes can be discussed in respect to patients' internal inflammatory levels and strategies may be suggested for patient stratification with respect to their inflammatory levels at the beginning of a clinical trial.
Figure 8. The dose-effect relationship of IL-1Ra therapy for mild (red circle) and strong T2D (black dot) in silico are shown.

Following the protocol of the clinical trial, glycaemia improvement after 90 days of therapy versus control, is used as a measure. The IL-1Ra threshold predicted by the model is reflected in the jump of the resulting glucose level for both T2D groups. The optimal IL1-Ra dose for strong T2D is substantially larger because of the lower endogenous level of IL-1Ra. The best achieved glucose improvement in response to the therapy is three-fold higher in the case of strong than in mild T2D. The starting glucose level is 11.96 and 18.02 mM for mild and strong T2D, respectively. The final glucose level of the both control groups is 12.21 and 18.82 mM, respectively.
The model proposed by De Gaetano et al [35] is able to account for different arms of the Diabetes Prevention Program (DPP) results [44], by modeling the effects of different drugs on insulin resistance development. Some differences and similarities are worth noting here, besides those already discussed in the introduction. In both models glucose toxicity introduces a positive feedback in the disease progression. Therefore, both models emphasize the importance of an early treatment of hyperglycaemia. The unique property of the current model is that the inflammatory signal, once switched on by glucose, cannot be switched off by glucose-lowering therapies. It has to be treated separately. This point seems to be consistent with recently discovered anti-inflammatory effects of some long-standing anti-diabetic agents. In fact, insulin has long been known for its anti-inflammatory effect, which, early presumed to be due to its glucose-lowering ability, recently was established to be a complement to its metabolic role [for a recent review see 45]. Additionally, there is emerging evidence suggesting that metformin, the first-line diabetic drug, have anti-inflammatory effects that are independent of its hypoglycaemia effect [46], even though its anti-inflammatory effect in T2D being not as robust as in other diseases, such as hypertension [47]–[49]. The impact of currently employed T2D drugs onto the inflammatory state of the patients prohibits falsification or verification of the model prediction that the T2D disease state cannot be alleviated by controlling glycaemia alone.
The presence of the IL-1β switch suggests that the system is operating in two distinct regimes, which defines a clear threshold for an anti-inflammatory therapy. A striking prediction of the model is that the anti-inflammatory therapy has to be released after a certain time. These inferences are derived from the IL-1β/IL-1Ra hysteresis and are consistent with some long-standing findings [11], [12], as well as recent progresses in the field, such as multistage development of the disease [3], [5], long-term effect of temporary IL-1Ra in overt-diabetes [20] and the efficiency of glucose control in pre-diabetes but not overt-diabetes [31], [36].
The present IL-1β switch model is built on in vitro data and several assumptions, both of which cast limitations on its applicability. The in vitro data show regulation of IL-1β and IL-1Ra by glucose and exogenously added IL-1β [14], [17], [27], [32]. Other factors involved in in vivo IL-1β control, such as free fatty acids [50], infiltrated macrophages and islet amyloid polypeptides [51] are not considered. These factors are associated with serious hyperglycaemia and overt T2D. However, they do not or only weakly affect the results of the core IL-1β model, especially concerning the onset bifurcation at glucose levels below 6 mM.
IL-1β induced β-cell proliferation and apoptosis were extrapolated from data of cultured islets [32]. It is unclear to what degree the in vitro data can reflect relevant in vivo processes, but several weak points of this assumption could be delineated. There is evidence that β-cells can re-enter cell cycle in vivo and that β-cell mass can adapt to metabolic needs [52], both supporting adaptive β-cell proliferation in vivo. However, it is not the only possible source of functional β-cells. Dedifferentiation of other cell types with a common precursor [53], as well as reactivation of dysfunctional β-cells [8], contributes to β-cell homeostasis. Furthermore, data from autopsy indicate that the proliferation rate of existing β-cells is very low. Consequently, increased β-cell mass seen in the compensation phase may be mainly attributed to new islet formation from other cell types [54]. A more realistic description of β-cell mass dynamics may further improve the model and extend its validity to later stages of the disease.
By representing the inflammatory stimulus via F instead of IL-1β, it is implicitly assumed that the amount of membrane IL-1β receptor per β-cell is constant. In support of this, there is evidence that the amount of IL-1β receptor in the β-cell membrane is much higher than in other cell types [55], suggesting that it is saturated. However, it is unlikely that the receptor is not down-regulated at high concentrations of IL-1β. Consequently, the model may overestimate the inflammatory signal at high IL-1β levels. This limitation has to be kept in mind when interpreting model results at high IL-1β.
Another important assumption concerns the IL-1β mRNA quantity used in the model. The IL-1β mRNA quantity is assumed to be proportional to the IL-1β protein level, in order to establish the quantitative relationships in IL-1β production. However, IL-1β production is characterized by the dissociation of transcription and translation. As a result, high levels of IL-1β mRNA are always associated with low levels of protein [29]. Alternatively, a sigmoidal relationship may be considered instead of the linear one used here. While it is difficult to estimate the quantitative error induced by the assumed linear relationship, it is straightforward to predict that the alternative sigmoidal relationship would increase the nonlinearity of IL-1β auto-stimulation, and of the interaction between glucose and IL-1β. Therefore, the essential elements for the bi-stable switch remain unchanged (or even would be increased), such that the qualitative results of the IL-1β switch model are robust.
It is difficult to define the exact physiological meaning of G and I in the model [35]. Since G and I are described by one-compartment dynamics in the model, only changes in long-term fasting levels are accessible to the model. Hence, we propose to interpret G as an abstract parameter associated with long-term fasting glucose on a time scale of days to weeks. Fasting levels represent more the internal property of the body's glucose control system than the history of how it was perturbed by meal or exercise [56]. However, the driving stimulus of IL-1β production may depend on peaks of glycaemia rather than on fasting levels. It is possible that dysregulations of fasting glucose levels and postprandial peaks are independent, as evidenced by patients with impaired glucose tolerance but normal fasting glucose. Eventually, the modifications of the dynamics of insulin release (first-phase insulin) are more important for glycaemia control than the long-term fasting levels of glucose. We would like to emphasize that these details of glucose and insulin dynamics are beyond the resolution of the model presented here and have to be addressed with a model focusing on the short-term dynamics of these quantities.
T2D is a very complex disease. The current model is simplified on purpose in order to generate structural insight. Other players of the glucose control system, for example glucagon, also play very important roles in glucose homeostasis and T2D development. Augmented glucagon secretion, together with impaired β-cell function, happens in the very early phase of pre-diabetes [57], [58]. The underlying islet defects were identified as reduced maximal insulin response and reduced glucose-sensitivity of β-/α-cell [58]. According to the physiological integral rein control theory [56], [59], glucose-sensitivity of β-/α-cell are important factors in setting steady state glucose level. In fact, recent studies support an independent role of glucose-sensitivity in hyperglycaemia development [60], [61]. Future improvement of the model should include the glucose-sensitivity of β-/α-cell to account for the rising of fasting glucose in the compensation phase. In particular, this might improve the fitting to the data in Fig. 4A during the compensation phase.
In addition, post-load glucose of incident diabetes cases showed a rapid increase 5 years before diagnosis [5], which is followed by a rapid decrease in insulin sensitivity. This important change is not reflected in the history of fasting glucose while it is represented in the model output (Fig. 4A). On the one hand, the absence of a corresponding change in fasting glucose at ∼5 years before diagnosis suggests that fasting glucose level is dominated by factors other than insulin resistance. Again, this might point to a role of glucagon. On the other hand, the coincident post-load glucose increase and insulin sensitivity decrease at ∼5 years before diagnosis might suggest a cause-effect relationship between β-cell function and insulin resistance [62]. The complexity of the defects during the pre-diabetes phase highlights the necessity of the mathematical modeling approach in the field.
Although results from both in vitro experiments and animal models were promising, the actual effect of anti-IL-1β agents in clinical trials of T2D was a matter of debate because the observed effect was very modest so far. Besides the possible role of polymorphisms of the IL1RN gene which encodes IL-1Ra [20], more research is needed to clarify for example, drug interaction with other IL-1 family members, such as soluble IL-1 receptors and soluble IL-1 receptor accessory proteins, in the pancreatic local environment as well as in blood and liver.
On the structural level, the present analysis proposes that the transition from pre-diabetes to overt T2D is associated with changed stability properties of the endocrine-immune system. The transition is initiated at a threshold value of glucose and associated with qualitatively different inflammatory states. The notion of an IL-1β hysteresis is supported by data from different studies, which showed the effectiveness of glucose control in pre-diabetes [36] or newly diagnosed patients [31] and the ineffectiveness of glucose control in long-standing T2D patients [11]–[13]. It is also supported by the clinical trial based on a combined IL-1Ra and glucose-lowering therapy: the improvement of the disease state remained for a year after release of the IL-1Ra therapy [20]. The existence of the IL-1β hysteresis requires a combined glucose-lowering and anti-inflammatory therapy for overt T2D patients, where the anti-inflammatory therapy should be kept for only a limited time.
Supporting Information
This zip files contains the matlab scripts for reproducing the figures in the manuscript. It contains a readme.txt where more details are present.
(ZIP)
Simulation of glucose (A) and β-cell mass (B) evolution to overt-diabetes based on multiple fitted parameters. Bifurcation is used as a side condition.
(EPS)
Simulation of glucose (A), β-cell mass (B), IL-1β (C) and IL-1Ra (D) evolution to overt-diabetes based on multiple fitted parameters. Bifurcation is not used as a side condition.
(EPS)
Acknowledgments
We thank Michele Solimena and his group for many fruitful discussions.
Funding Statement
This work was supported by the Helmholtz Association cross-program activity “Metabolic Dysfunction and Human Disease”. MMH was supported by the BMBF GerontoSys initiative GerontoShield, by the HFSP, and by the BMBF eMED SYSIMIT and SysStomach project. KM is supported by Competence network diabetes mellitus funded by the Federal Ministry of Education and Research (BMBF) and the European Research Council (ERC 260336). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Bagust A, Beale S (2003) Deteriorating beta-cell function in type 2 diabetes: a long-term model. QJM 96: 281–288. [DOI] [PubMed] [Google Scholar]
- 2. Ferrannini E, Nannipieri M, Williams K, Gonzales C, Haffner SM, et al. (2004) Mode of Onset of Type 2 Diabetes from Normal or Impaired Glucose Tolerance. Diabetes 53: 160–165 10.2337/diabetes.53.1.160 [DOI] [PubMed] [Google Scholar]
- 3. Weir GC, Bonner-Weir S (2004) Five Stages of Evolving Beta-Cell Dysfunction During Progression to Diabetes. Diabetes 53: S16–S21 10.2337/diabetes.53.suppl3.S16 [DOI] [PubMed] [Google Scholar]
- 4. Mason CC, Hanson RL, Knowler WC (2007) Progression to Type 2 Diabetes Characterized by Moderate Then Rapid Glucose Increases. Diabetes 56: 2054–2061 10.2337/db07-0053 [DOI] [PubMed] [Google Scholar]
- 5. Tabák AG, Jokela M, Akbaraly TN, Brunner EJ, Kivimäki M, et al. (2009) Trajectories of glycaemia, insulin sensitivity, and insulin secretion before diagnosis of type 2 diabetes: an analysis from the Whitehall II study. The Lancet 373: 2215–2221 10.1016/S0140-6736(09)60619-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. DeFronzo RA (2009) From the Triumvirate to the Ominous Octet: A New Paradigm for the Treatment of Type 2 Diabetes Mellitus. Diabetes 58: 773–795 10.2337/db09-9028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC (2008) Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab 10 Suppl 4: 32–42 10.1111/j.1463-1326.2008.00969.x [DOI] [PubMed] [Google Scholar]
- 8. Ferrannini E (2010) The stunned beta cell: a brief history. Cell Metab 11: 349–352 10.1016/j.cmet.2010.04.009 [DOI] [PubMed] [Google Scholar]
- 9. Levy J, Atkinson AB, Bell PM, McCance DR, Hadden DR (1998) Beta-cell deterioration determines the onset and rate of progression of secondary dietary failure in Type 2 diabetes mellitus: the 10-year follow-up of the Belfast Diet Study. Diabetic Medicine 15: 290–296 doi:;10.1002/(SICI)1096-9136(199804)15:4<290::AID-DIA570>3.0.CO;2-M [DOI] [PubMed] [Google Scholar]
- 10. Muoio DM, Newgard CB (2008) Molecular and metabolic mechanisms of insulin resistance and [beta]-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9: 193–205 10.1038/nrm2327 [DOI] [PubMed] [Google Scholar]
- 11. U.K. prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group (1995) Diabetes 44: 1249–1258. [PubMed] [Google Scholar]
- 12. Turner RCCC (1999) Glycemic control with diet, sulfonylurea, metformin, or insulin in patients with type 2 diabetes mellitus: Progressive requirement for multiple therapies (ukpds 49). JAMA 281: 2005–2012 10.1001/jama.281.21.2005 [DOI] [PubMed] [Google Scholar]
- 13. Lencioni C, Lupi R, Del Prato S (2008) Beta-cell failure in type 2 diabetes mellitus. Curr Diab Rep 8: 179–184. [DOI] [PubMed] [Google Scholar]
- 14. Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, et al. (2002) Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110: 851–860 10.1172/JCI15318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Donath MY, Størling J, Berchtold LA, Billestrup N, Mandrup-Poulsen T (2008) Cytokines and beta-cell biology: from concept to clinical translation. Endocr Rev 29: 334–350 10.1210/er.2007-0033 [DOI] [PubMed] [Google Scholar]
- 16. Donath MY, Shoelson SE (2011) Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11: 98–107 10.1038/nri2925 [DOI] [PubMed] [Google Scholar]
- 17. Böni-Schnetzler M, Thorne J, Parnaud G, Marselli L, Ehses JA, et al. (2008) Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta -cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J Clin Endocrinol Metab 93: 4065–4074 10.1210/jc.2008-0396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, et al. (2007) Increased Number of Islet-Associated Macrophages in Type 2 Diabetes. Diabetes 56: 2356–2370 10.2337/db06-1650 [DOI] [PubMed] [Google Scholar]
- 19. Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, et al. (2007) Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N Engl J Med 356: 1517–1526 10.1056/NEJMoa065213 [DOI] [PubMed] [Google Scholar]
- 20. Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, et al. (2009) Sustained Effects of Interleukin-1 Receptor Antagonist Treatment in Type 2 Diabetes. Dia Care 32: 1663–1668 10.2337/dc09-0533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ramos-Zavala MG, González-Ortiz M, Martínez-Abundis E, Robles-Cervantes JA, González-López R, et al. (2011) Effect of Diacerein on Insulin Secretion and Metabolic Control in Drug-Naïve Patients With Type 2 Diabetes A randomized clinical trial. Dia Care 34: 1591–1594 10.2337/dc11-0357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Van Asseldonk EJP, Stienstra R, Koenen TB, Joosten LAB, Netea MG, et al. (2011) Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. J Clin Endocrinol Metab 96: 2119–2126 10.1210/jc.2010-2992 [DOI] [PubMed] [Google Scholar]
- 23. Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, et al. (2012) Effects of Gevokizumab on Glycemia and Inflammatory Markers in Type 2 Diabetes. Dia Care 35: 1654–1662 10.2337/dc11-2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rissanen A, Howard CP, Botha J, Thuren T (2012) Investigators for the G (2012) Effect of anti-IL-1β antibody (canakinumab) on insulin secretion rates in impaired glucose tolerance or type 2 diabetes: results of a randomized, placebo-controlled trial. Diabetes, Obesity and Metabolism 14: 1088–1096 10.1111/j.1463-1326.2012.01637.x [DOI] [PubMed] [Google Scholar]
- 25. Sloan-Lancaster J, Abu-Raddad E, Polzer J, Miller JW, Scherer JC, et al. (2013) Double-Blind, Randomized Study Evaluating the Glycemic and Anti-inflammatory Effects of Subcutaneous LY2189102, a Neutralizing IL-1β Antibody, in Patients With Type 2 Diabetes. Diabetes Care 36: 2239–46 10.2337/dc12-1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mandrup-Poulsen T (2012) Perspective: Testing failures. Nature 485: S17–S17 10.1038/485S17a [DOI] [PubMed] [Google Scholar]
- 27. Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, et al. (2004) Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci USA 101: 8138–8143 10.1073/pnas.0305683101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Glas R, Sauter N, Schulthess F, Shu L, Oberholzer J, et al. (2009) Purinergic P2X7 receptors regulate secretion of interleukin-1 receptor antagonist and beta cell function and survival. Diabetologia 52: 1579–1588 10.1007/s00125-009-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dinarello C (1996) Biologic basis for interleukin-1 in disease. Blood 87: 2095–2147. [PubMed] [Google Scholar]
- 30. Carstensen M, Herder C, Kivimäki M, Jokela M, Roden M, et al. (2010) Accelerated Increase in Serum Interleukin-1 Receptor Antagonist Starts 6 Years Before Diagnosis of Type 2 Diabetes. Diabetes 59: 1222–1227 10.2337/db09-1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Retnakaran R, Drucker DJ (2008) Intensive insulin therapy in newly diagnosed type 2 diabetes. The Lancet 371: 1725–1726 10.1016/S0140-6736(08)60736-9 [DOI] [PubMed] [Google Scholar]
- 32. Maedler K, Schumann DM, Sauter N, Ellingsgaard H, Bosco D, et al. (2006) Low concentration of interleukin-1beta induces FLICE-inhibitory protein-mediated beta-cell proliferation in human pancreatic islets. Diabetes 55: 2713–2722 10.2337/db05-1430 [DOI] [PubMed] [Google Scholar]
- 33. Topp B, Promislow K, deVries G, Miura RM, Finegood DT (2000) A model of beta-cell mass, insulin, and glucose kinetics: pathways to diabetes. J Theor Biol 206: 605–619 10.1006/jtbi.2000.2150 [DOI] [PubMed] [Google Scholar]
- 34. Blasio BFD, Bak P, Pociot F, Karlsen AE, Nerup J (1999) Onset of type 1 diabetes: a dynamical instability. Diabetes 48: 1677–1685 10.2337/diabetes.48.9.1677 [DOI] [PubMed] [Google Scholar]
- 35. Gaetano AD, Hardy T, Beck B, Abu-Raddad E, Palumbo P, et al. (2008) Mathematical models of diabetes progression. Am J Physiol Endocrinol Metab 295: E1462–E1479 10.1152/ajpendo.90444.2008 [DOI] [PubMed] [Google Scholar]
- 36. Perreault L, Pan Q, Mather KJ, Watson KE, Hamman RF, et al. (2012) Effect of regression from prediabetes to normal glucose regulation on long-term reduction in diabetes risk: results from the Diabetes Prevention Program Outcomes Study. Lancet 379: 2243–2251 10.1016/S0140-6736(12)60525-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bergman RN, Lovejoy JC (1997) The Minimal Model Approach and Determinants of Glucose Tolerance. Louisiana State University Press. 406 p. [Google Scholar]
- 38. García-Estévez DA, Araújo-Vilar D, Fiestras-Janeiro G, Saavedra-González A, Cabezas-Cerrato J (2003) Comparison of several insulin sensitivity indices derived from basal plasma insulin and glucose levels with minimal model indices. Horm Metab Res 35: 13–17 10.1055/s-2003-38385 [DOI] [PubMed] [Google Scholar]
- 39. Spranger J, Kroke A, Möhlig M, Hoffmann K, Bergmann MM, et al. (2003) Inflammatory Cytokines and the Risk to Develop Type 2 Diabetes Results of the Prospective Population-Based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 52: 812–817 10.2337/diabetes.52.3.812 [DOI] [PubMed] [Google Scholar]
- 40. Schmidt H, Jirstrand M (2006) Systems Biology Toolbox for MATLAB: a computational platform for research in systems biology. Bioinformatics 22: 514–515 10.1093/bioinformatics/bti799 [DOI] [PubMed] [Google Scholar]
- 41. Laybutt DR, Glandt M, Xu G, Ahn YB, Trivedi N, et al. (2003) Critical reduction in beta-cell mass results in two distinct outcomes over time. Adaptation with impaired glucose tolerance or decompensated diabetes. J Biol Chem 278: 2997–3005 10.1074/jbc.M210581200 [DOI] [PubMed] [Google Scholar]
- 42. Alejandro R, Cutfield RG, Shienvold FL, Polonsky KS, Noel J, et al. (1986) Natural history of intrahepatic canine islet cell autografts. J Clin Invest 78: 1339–1348 10.1172/JCI112720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Suzuki K, Bonner-Weir S, Trivedi N, Yoon KH, Hollister-Lock J, et al. (1998) Function and survival of macroencapsulated syngeneic islets transplanted into streptozocin-diabetic mice. Transplantation 66: 21–28. [DOI] [PubMed] [Google Scholar]
- 44. Hardy T, Abu-Raddad E, Porksen N, Gaetano AD (2012) Evaluation of a mathematical model of diabetes progression against observations in the Diabetes Prevention Program. Am J Physiol Endocrinol Metab 303: E200–E212 10.1152/ajpendo.00421.2011 [DOI] [PubMed] [Google Scholar]
- 45. Hyun E, Ramachandran R, Hollenberg MD, Vergnolle N (2011) Mechanisms behind the anti-inflammatory actions of insulin. Crit Rev Immunol 31: 307–340. [DOI] [PubMed] [Google Scholar]
- 46. Pruski M, Krysiak R, Okopien B (2009) Pleiotropic action of short-term metformin and fenofibrate treatment, combined with lifestyle intervention, in type 2 diabetic patients with mixed dyslipidemia. Diabetes Care 32: 1421–1424 10.2337/dc08-2335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gómez-García A, Torres GM, Ortega-Pierres LE, Rodríguez-Ayala E, Álvarez-Aguilar C, et al. (2007) Rosuvastatin and Metformin Decrease Inflammation and Oxidative Stress in Patients With Hypertension and Dyslipidemia. Rev Esp Cardiol 60: 1242–1249. [DOI] [PubMed] [Google Scholar]
- 48. Stumvoll M, Goldstein BJ, van Haeften TW (2005) Type 2 diabetes: principles of pathogenesis and therapy. The Lancet 365: 1333–1346 10.1016/S0140-6736(05)61032-X [DOI] [PubMed] [Google Scholar]
- 49. Kita Y, Takamura T, Misu H, Ota T, Kurita S, et al. (2012) Metformin Prevents and Reverses Inflammation in a Non-Diabetic Mouse Model of Nonalcoholic Steatohepatitis. PLoS ONE 7: e43056 10.1371/journal.pone.0043056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dinarello CA, Donath MY, Mandrup-Poulsen T (2010) Role of IL-1β in type 2 diabetes. Current Opinion in Endocrinology, Diabetes and Obesity 17: 1 10.1097/MED.0b013e32833bf6dc [DOI] [PubMed] [Google Scholar]
- 51. Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, et al. (2010) Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1β in type 2 diabetes. Nature Immunology 11: 897–904 10.1038/ni.1935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Butler PC, Meier JJ, Butler AE, Bhushan A (2007) The replication of beta cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab 3: 758–768 10.1038/ncpendmet0647 [DOI] [PubMed] [Google Scholar]
- 53. Talchai C, Xuan S, Lin HV, Sussel L, Accili D (2012) Pancreatic β Cell Dedifferentiation as a Mechanism of Diabetic β Cell Failure. Cell 150: 1223–1234 10.1016/j.cell.2012.07.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, et al. (2003) Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 52: 102–110. [DOI] [PubMed] [Google Scholar]
- 55. Böni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, et al. (2009) Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150: 5218–5229 10.1210/en.2009-0543 [DOI] [PubMed] [Google Scholar]
- 56. Larsson H, Ahrén B (2000) Glucose intolerance is predicted by low insulin secretion and high glucagon secretion: outcome of a prospective study in postmenopausal Caucasian women. Diabetologia 43: 194–202 10.1007/s001250050029 [DOI] [PubMed] [Google Scholar]
- 57. Ahrén B (2009) β- and α-Cell Dysfunction in Subjects Developing Impaired Glucose Tolerance Outcome of a 12-Year Prospective Study in Postmenopausal Caucasian Women. Diabetes 58: 726–731 10.2337/db08-1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Saunders PT, Koeslag JH, Wessels JA (1998) Integral Rein Control in Physiology. Journal of Theoretical Biology 194: 163–173 10.1006/jtbi.1998.0746 [DOI] [PubMed] [Google Scholar]
- 59. Saunders PT, Koeslag JH, Wessels JA (2000) Integral Rein Control in Physiology II: a General Model. Journal of Theoretical Biology 206: 211–220 10.1006/jtbi.2000.2118 [DOI] [PubMed] [Google Scholar]
- 60. Mari A, Tura A, Natali A, Laville M, Laakso M, et al. (2010) Impaired beta cell glucose sensitivity rather than inadequate compensation for insulin resistance is the dominant defect in glucose intolerance. Diabetologia 53: 749–756 10.1007/s00125-009-1647-6 [DOI] [PubMed] [Google Scholar]
- 61. Ferrannini E, Natali A, Muscelli E, Nilsson PM, Golay A, et al. (2011) Natural history and physiological determinants of changes in glucose tolerance in a non-diabetic population: the RISC Study. Diabetologia 54: 1507–1516 10.1007/s00125-011-2112-x [DOI] [PubMed] [Google Scholar]
- 62. Matveyenko AV, Liuwantara D, Gurlo T, Kirakossian D, Dalla Man C, et al. (2012) Pulsatile portal vein insulin delivery enhances hepatic insulin action and signaling. Diabetes 61: 2269–2279 10.2337/db11-1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kudo S, Mizuno K, Hirai Y, Shimizu T (1990) Clearance and Tissue Distribution of Recombinant Human Interleukin 1β in Rats. Cancer Res 50: 5751–5755. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This zip files contains the matlab scripts for reproducing the figures in the manuscript. It contains a readme.txt where more details are present.
(ZIP)
Simulation of glucose (A) and β-cell mass (B) evolution to overt-diabetes based on multiple fitted parameters. Bifurcation is used as a side condition.
(EPS)
Simulation of glucose (A), β-cell mass (B), IL-1β (C) and IL-1Ra (D) evolution to overt-diabetes based on multiple fitted parameters. Bifurcation is not used as a side condition.
(EPS)