

Abstract
The critically endangered New Zealand parrot, the kakapo, is subject to an intensive management regime aiming to maintain bird health and boost population size. Newly hatched kakapo chicks are subjected to human intervention and are frequently placed in captivity throughout their formative months. Hand rearing greatly reduces mortality among juveniles, but the potential long-term impact on the kakapo gut microbiota is uncertain. To track development of the kakapo gut microbiota, fecal samples from healthy, prefledged juvenile kakapos, as well as from unrelated adults, were analyzed by using 16S rRNA gene amplicon pyrosequencing. Following the original sampling, juvenile kakapos underwent a period of captivity, so further sampling during and after captivity aimed to elucidate the impact of captivity on the juvenile gut microbiota. Variation in the fecal microbiota over a year was also investigated, with resampling of the original juvenile population. Amplicon pyrosequencing revealed a juvenile fecal microbiota enriched with particular lactic acid bacteria compared to the microbiota of adults, although the overall community structure did not differ significantly among kakapos of different ages. The abundance of key operational taxonomic units (OTUs) was correlated with antibiotic treatment and captivity, although the importance of these factors could not be proven unequivocally within the bounds of this study. Finally, the microbial community structure of juvenile and adult kakapos changed over time, reinforcing the need for continual monitoring of the microbiota as part of regular health screening.
INTRODUCTION
The kakapo (Strigops habroptilus) is a flightless, nocturnal parrot native to New Zealand. It possesses few defenses against introduced mammalian predators (1), has a low reproductive rate, and usually lays only a single egg. Although kakapos were once common throughout New Zealand (2), the population has declined to 125 individuals at the time of publication, confined to three predator-free islands off the coast of New Zealand. Since the 1980s, kakapos have been subjected to an intensive management program focused on preserving the species and eventually restoring the population to self-sustaining levels (3–5). In an effort to optimize management practices, the New Zealand Department of Conservation has collaborated with researchers from a wide range of biological disciplines, including behavioral ecology, physiology, genetics, nutrition, and, recently, microbiology (6–12).
The kakapo has an array of biological characteristics that make it an unusual animal to study. Apart from being the world's heaviest parrot, the only flightless parrot, and the only parrot to carry out lek breeding (6), the kakapo has been identified as a potential foregut fermenter due to its herbivorous diet and lack of ceca (13). Microbially mediated foregut fermentation is a common trait in mammals but is rare among avians, with only the South American hoatzin being known to perform this process (14). There are notable differences in feeding strategies between the kakapo and hoatzin, however, with kakapos rarely ingesting fibrous plant material, instead extracting the juices from shoots and leaves and discarding the undigested “chews” (15, 16). There is currently no empirical evidence to support or dispute the occurrence of foregut fermentation in kakapos. Intriguingly, the reproductive cycle of the kakapo is linked to the fruiting of particular native New Zealand trees. The rimu tree undergoes a mast season every 3 or 4 years, during which time kakapos mate and rear young. While the link between kakapo and rimu is well established, the causal link between these two phenomena is unclear and is not simply a matter of additional dietary energy enabling reproduction (17).
The role of microbial symbionts in the gastrointestinal (GI) tract of vertebrates is well documented, and a range of mechanisms through which microbes contribute to nutrition (18–21) and development of the host gut (22–24) has been identified. Our previous research into the GI tract-associated bacteria of the kakapo revealed a community dominated by only a few operational taxonomic units (OTUs), mainly from the phyla Proteobacteria and Firmicutes and apparently lacking in archaea (12, 25). The kakapo GI tract appears to have a low phylum-level diversity of bacteria compared to other birds (25, 26), and this has led to speculation of a population bottleneck for the gut microbiota. The kakapo microbiota is not well understood, but it is likely that current management practices have an impact on the microbial community, as kakapos are removed from the wild and given veterinary care at the first sign of sickness (27). This captivity results in a change in diet and often includes antibiotic treatment, both of which are frequently linked to shifts in microbial community structure (23, 28–31).
Of major relevance to kakapo microbiology is the fact that the developmental pattern of the kakapo gut microbiota is completely unknown, thereby making it difficult to address questions regarding the effect of diet or captivity on the gut microbiota. Previous research into the development of the gut microbiota in other host species has shown that the microbiota of juveniles differs significantly from that of adults in both avians and mammals (32–36), although it is not clear whether this pattern is reflected in the kakapo (12). In many avian species, the juvenile microbiota is a dynamically changing community (37–39) that gradually develops toward the adult community structure (33, 36), but the changes in microbiota as the subject ages vary by host. For example, chickens are enriched in Lactobacillaceae in the first week of life (38), while juvenile turkeys appear to harbor a large proportion of Clostridiales until around 10 weeks of age (39). Moreover, even genetically related individuals undergo different developmental patterns when geographically isolated (40). In order to better understand the temporal dynamics of the kakapo microbiota, and potentially gain insights into the impact of human intervention, samples were collected from juvenile kakapos born during the 2011 breeding season, spanning four time points throughout the first year and a half of life. The aims of this study were to compare differences in the juvenile and adult fecal microbiota, to understand the time required for juvenile kakapos to develop a “normal” adult gut microbiota, and to investigate the potential effects of captivity on the juvenile microbiota.
MATERIALS AND METHODS
Sample collection.
In the 2011 breeding season, 11 kakapo chicks were hatched on Codfish Island, off the coast of New Zealand (46°47′S, 167°38′E). Samples were collected from these juveniles at four time points, which are summarized in Table 1. During the nesting period, each juvenile required a period of captivity, due to either low weight gain or sickness. While undergoing the period of captivity, the juveniles were fed a diet of fruit, lactated Ringer's solution, and the proprietary parrot hand rearing formula Exact (Kaytee Products Inc., Chilton, WI). Eight of the captive juveniles were also treated with the commercially available antibiotics Augmentin and Clavulox, which combine a β-lactam (amoxicillin) and a β-lactamase inhibitor (clavulanic acid) (Tables 1 and 2). Samples collected during this period were taken following 5 days of captivity. Following release from captivity, juveniles were given a “recovery” period of 2 weeks, after which additional fecal samples were collected. A final sample was collected approximately 1 year later, at which point the “juveniles” had fledged from the nest and were now independent adults.
TABLE 1.
Sample sizes, mean ages of individuals, and additional notes taken at the time of samplinga
| Sample time point | No. of individuals | Mean age (days) | Sample yr | Description |
|---|---|---|---|---|
| Juvenile_First | 8 | 16 | 2011 | Fecal sample from wild juvenile kakapo |
| Juvenile_Second | 10 | 51 | 2011 | Fecal sample from captive juvenile kakapo fed an artificial diet of lactated Ringer's solution and fresh fruit; 8 juveniles were also treated with antibiotics during captivity (amoxicillin-clavulanic acid formulas) |
| Juvenile_Third | 5 | 69 | 2011 | Fecal sample from juvenile kakapo ∼2.5 wk following release |
| Juvenile_Fourth | 8 | 569 | 2012 | Fecal sample from juvenile kakapo during the next round of health screening; at this point, individuals were mature, independent birds |
| Adult_First | 10 | 2011 | Fecal sample from wild adult kakapo collected at the same time as Juvenile_First | |
| Adult_Second | 7 | 2012 | Fecal sample from wild adult kakapo collected at the same time as Juvenile_Fourth |
For “adult” kakapos, age data are incomplete, but the youngest adults were ≥6 years old in 2011.
TABLE 2.
List of individuals sampled at each point in the juvenile surveya
| Time point | Individuals sampled |
|---|---|
| Juvenile_First | Hakatere |
| Ian | |
| Ihi | |
| Stella | |
| Taonga | |
| Tia | |
| Tutoko | |
| Waa | |
| Juvenile_Second | Atareta |
| Hakatere* | |
| Ian* | |
| Ihi* | |
| Stella* | |
| Taonga* | |
| Tia* | |
| Tutoko* | |
| Waa* | |
| Waikawa | |
| Juvenile_Third | Atareta |
| Ian | |
| Taonga | |
| Tutoko | |
| Waikawa | |
| Juvenile_Fourth | Hakatere |
| Ian | |
| Ihi | |
| Stella | |
| Tia | |
| Tutoko | |
| Waa | |
| Waikawa |
Individuals marked with an asterisk received antibiotic treatment with the commercially available antibiotic amoxicillin-clavulanic acid formulas.
Fresh fecal samples were also collected from adult kakapos in the same manner. Samples were taken from adults with no history of sickness and no history of captivity other than the original translocation to Codfish Island, at time points coinciding with the Juvenile_First and Juvenile_Fourth samplings. The adult age data are incomplete, as some birds were born wild and introduced to Codfish Island only later in life. However, the youngest adults sampled were born on Codfish Island in 2005, making the adult kakapo representative of a substantially older subpopulation than individuals representing the juvenile data set, even though at the Juvenile_Fourth time point, the original juvenile kakapos were mature kakapos.
Fresh fecal samples were collected aseptically during routine health inspections of the juveniles and stored in sterile polypropylene tubes on ice until they were able to be frozen, a period of <1 h following collection. Sample sizes varied throughout the study due to difficulty with the capture and recapture of birds.
DNA extraction, PCR amplification, and amplicon pyrosequencing.
DNA extraction was performed on samples by using a previously described bead-beating method (12). PCR was performed by using the following 16S rRNA gene-specific primers targeting bacteria: 533f (5′-GTGCCAGCAGCYGCGGTMA-3′) and 907R (5′-CCGTCAATTMMYTTGAGTTT-3′) (41). Each primer was synthesized with an FLX Titanium adaptor sequence (CCA TCT CAT CCC TGC GTG TCT CCG AC for 533f and CCT ATC CCC TGT GTG CCT TGG CAG TC for 907R), and a 10-bp multiplex identifier (MID) barcode was attached to the forward primer by using commercially available barcode sequences (Roche Diagnostics Corporation, Branford, CT). For each DNA extraction, three PCRs with 25-μl mixtures were performed with a specific barcoded primer. Reaction mixtures contained 20 mM Tris-HCl, 50 mM KCl, 1.5 mM MgCl2, 100 μM deoxynucleoside triphosphate (dNTP) mixture, 2.5 μM forward and reverse primers, 2% bovine serum albumin, 0.5 units Taq polymerase, and 10 ng of template DNA. Cycling conditions were as follows: an initial denaturation step at 94°C for 5 min; 20 cycles of touchdown PCR (94°C for 30 s, 60°C for 30 s, and 72°C for 45 s, with a 0.5°C decrease in annealing temperature per cycle); 10 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 45 s; and a final elongation step at 72°C for 10 min, with one negative-control run for each primer pair. Following amplification, PCR products from each sample were pooled and purified by using the Agencourt AMPure XP bead system (Agencourt, Beckman Coulter, MA, USA), according to the manufacturer's instructions. Product size was measured by using the Agilent DNA 1000 kit (Agilent Technologies, Waldbronn, Germany) with the Agilent 2100 Bioanalyzer platform. The purified product was quantified by using the Qubit Quant-iT DNA high-sensitivity assay, and DNA concentration and amplicon size were used to equalize the number of molecules pooled per sequencing run. Samples were randomized across sequencing runs, and pyrosequencing was performed on a Roche GS-FLX Titanium platform (Roche, NJ, USA) by Macrogen Inc. (Seoul, South Korea).
Bacterial community analysis.
Sequence reads were processed by using mothur version 1.31.2 (42). Briefly, pyrosequencing flowgrams were denoised by using the mothur implementation of AmpliconNoise (43). Sequences with a length of <200 bp or homopolymers of >8 bp were removed, as were sequences with more than one MID barcode mismatch or two primer mismatches. Sequences were then aligned against a reference database (http://www.mothur.org/wiki/Silva_reference_alignment), and sequences that could not be aligned were removed. Chimeras identified by using the UCHIME algorithm (44) were removed from the data set, and the remaining sequences were classified by using a previously reported method (41, 45). Sequences classified as chloroplast were removed from the data set, as were sequences that could not be classified even to the domain level. From a starting total of 216,759 sequences, the above-mentioned procedures led to a final total of 208,121 high-quality sequence reads for further analysis.
Following these initial quality control steps, sequence data were binned into genus- and species-approximating OTUs using definitions of ≥95% and ≥97% sequence similarity (OTU0.95 and OTU0.97), respectively. Yue-Clayton theta, Jaccard index (46), and phylogeny-based UniFrac distances (47) were calculated between each group by using each OTU definition. Average distances between communities were calculated by randomly subsampling 1,400 sequences per sample and calculating distance measures 10,000 times. Apparent changes in community structure were tested by using analysis of molecular variance (AMOVA) (48, 49). Changes in OTU0.97 abundance between sample groups were tested by using metastats (50) but are reported only if the OTU of interest had a relative abundance of ≥1% of the obtained sequences across any sample, as rarer OTUs were frequently observed in only a single kakapo individual and therefore were not informative when surveying the overall microbiota. Correlations between OTUs that differed significantly between captive and wild individuals were measured by using the Point-Biserial correlation coefficient (rpb) in the R software environment (version 2.15.2 [http://www.r-project.org/]).
Coverage of sequencing was estimated by calculating Good's coverage index after subsampling the OTU0.97 table to a depth of 1,400 OTUs per sample. Richness and diversity estimators were also calculated on the subsampled data by using Shannon diversity and evenness indices, Simpson diversity index, and Chao1 and ACE (abundance coverage-based estimator) estimators. In order to test how well previously reported clone library data represented the full fecal microbiota, representative OTU0.97 sequences were mapped against full-length clone sequences by using usearch (51) with a minimum similarity value of 0.97. The results of the usearch mapping were recorded both in terms of the total proportion of reads that were mapped against the clone library data and as a proportion of the total number of OTU0.97 generated during analysis.
The core microbiota was calculated by scoring the presence or absence of each OTU in each sample and then calculating presence as a proportion of all samples. An overall core was calculated by using all available wild samples (excluding Juvenile_Second) as well as separate cores consisting of exclusively adult kakapos (Adult_First, Adult_Second, and Juvenile_Fourth) and exclusively healthy juvenile kakapos (Juvenile_First and Juvenile_Third). A final core was calculated from the captive, antibiotic-treated kakapos (n = 8) but not from the untreated pair of birds, due to the small sample size. The core microbiota was categorized as “core” microbes, present in >90% of individuals surveyed, and “variable” microbes, present in >60% of individuals surveyed. All other OTUs were considered either transient or individual specific.
Nucleotide sequence accession numbers.
Sequence data were submitted to the NCBI Sequence Read Archive under accession numbers SAMN02369276 to SAMN02369329 and SAMN02420182 to SAMN02420188.
RESULTS
The kakapo microbiota is of low diversity and is dominated by Firmicutes and Proteobacteria.
Amplicon pyrosequencing yielded an average of 4,648 sequences per sample, compared to ∼77 obtained via clone libraries in our previous study (12). Good's coverage values show that amplicon pyrosequencing was able to describe essentially the entire microbiota of each sample group, while ecological diversity measures indicate an uneven bacterial community of low diversity (Table 3). This unevenness was reflected in the low Shannon evenness index calculated for each sample. The Shannon evenness index is the ratio of the Shannon diversity index of a sample to the maximum possible Shannon index for the sample, with a value of 1 being perfectly even. The evenness of all samples was low according to this metric (Table 3), and this was apparent visually (Fig. 1). Community richness (Chao1 and ACE) was lowest during the juvenile captivity period, although this was not reflected clearly in the diversity estimators. Similarly, when amplicon sequences were mapped to previous kakapo gut microbiota clone library data, the diversity within the clone library accounted for a comparatively large proportion of the total reads sequenced but a much smaller proportion of the total OTUs (Table 3). Similar to previous findings, the phylum-level membership consisted predominantly of Proteobacteria and Firmicutes. In contrast to the clone library data, Bacteroidetes and Actinobacteria were also frequently, though not universally, detected (in ∼70% and ∼33% of samples, respectively).
TABLE 3.
Common diversity and richness estimators calculated by using OTUs of ≥97% sequence similaritya
| Sample group | Good's coverage | Shannon diversity index | Shannon evenness index | Simpson diversity index | Chao1 estimator | ACE estimator | Reads mapped (%) | OTUs mapped (%) |
|---|---|---|---|---|---|---|---|---|
| Adult_First | 0.995 | 0.73 | 0.10 | 0.63 | 25.2 | 38 | 99.4 | 46.4 |
| Adult_Second | 0.979 | 1.10 | 0.15 | 0.52 | 100.1 | 228.8 | 24.8 | 8.9 |
| Juvenile_First | 0.991 | 0.90 | 0.12 | 0.49 | 33.3 | 110.3 | 95.4 | 39.0 |
| Juvenile_Second | 0.998 | 0.88 | 0.12 | 0.61 | 9.8 | 17.4 | 88.4 | 53.9 |
| Juvenile_Third | 0.984 | 1.17 | 0.16 | 0.42 | 42.2 | 224.1 | 99.0 | 27.7 |
| Juvenile_Fourth | 0.973 | 0.91 | 0.12 | 0.71 | 152.5 | 348.7 | 10.9 | 7.1 |
The median value for each sample group is reported. Reads mapped refers to the proportion of amplicon sequences that could be mapped to preexisting clone library data. OTUs mapped refers to the number of representative OTUs that could be mapped to preexisting clone library data.
FIG 1.

Phylogenetic distribution of bacterial OTUs in the kakapo fecal microbiota. High-level taxonomic information is provided as per classification. OTUs are defined as groups of 16S rRNA gene sequences that share >97% similarity and are ordered by phylum and then subordered by class. For clarity, only the 50 most abundant OTUs are plotted, representing 98.0% of the total reads, with 477 OTUs comprising the remainder of the reads following removal of singletons. OTU abundances are scaled as a proportion of all sequences in the respective sample. OTU02 (mentioned in the text) is noted with an asterisk.
In order to create a baseline for future kakapo microbiology research, we defined core and variable communities of OTUs that were observed in the kakapo fecal microbiota (Table 4). The core microbiota consisted of OTUs present in >90% of individuals sampled, and the variable microbiota consisted of OTUs present in >60% of individuals. These groupings were further stratified by age (adult core and juvenile core), and treatment core was calculated for juvenile kakapos under the influence of antibiotics (treated core). In all core calculations of wild birds, two OTUs were classified as core, taxonomically assigned as Escherichia and Streptococcus. The variable microbiota differed between juvenile and adult birds, with more OTUs recruiting to the variable microbiota of juveniles, consistent with the higher diversity and richness indices reported for the juvenile population (Table 3). The treated core microbiota differed from that of the juvenile core and accounted for more of the microbiota than in wild samples. It is important to note that the classification scheme used was based only on presence or absence of OTUs and did not account for their relative abundance in the sample, although abundance carries biological significance in the interpretation of these data. With a single exception, core OTUs accounted for >5% of the amplicon reads in their respective groupings (Table 4). Some variable OTUs, including OTU02 in the antibiotic-treated microbiota, OTU04 in the adult microbiota and OTU06 in the wild juvenile microbiota, accounted for a large proportion of the microbiota in some individuals but were not commonly observed among different individuals.
TABLE 4.
Differences in core kakapo microbiota based on different partitioning of samplesa
| OTU | OTU taxonomy | Mean relative abundance (%) |
|||
|---|---|---|---|---|---|
| Overall | Adult | Juvenile (wild) | Juvenile (antibiotics) | ||
| OTU01 | Escherichia | 34.45 | 38.17 | 29.03 | 0.84 |
| OTU02 | Unclassified Proteobacteria OTU | 42.46 | |||
| OTU03 | Streptococcus | 16.33 | 5.81 | 31.69 | 0.89 |
| OTU04 | Clostridium | 5.73 | 8.74 | 1.33 | |
| OTU05 | Enterococcus | 1.04 | 0.05 | 37.97 | |
| OTU06 | Lactobacillus | 12.24 | 9.75 | ||
| OTU07 | Clostridium | 7.62 | |||
| OTU10 | Pseudomonas | 1.29 | |||
| OTU11 | Lactobacillus | 3.20 | |||
| Total | 57.55 | 52.72 | 83.25 | 95.11 | |
OTU labels are provided to allow consistency for comparisons with other tables that report OTU abundances and changes. Values report the mean relative abundance (percent) of an OTU in the overall microbiota for its grouping. Values in boldface type denote core OTUs in their group, while values in lightface type indicate that an OTU was variable. The final row reports the total proportion of the microbiota that is accounted for by these OTUs (percent).
The microbial community structure does not vary between juvenile and adult individuals but changes with time.
Adult and juvenile community structures were not significantly different from each other under any distance-OTU combination when juvenile and adult samples from matching time points were compared. Comparison of the juvenile samples sequentially revealed no differences in community structure (AMOVA, all distances, and all OTUs). The community structures of the first and last juvenile samples were significantly different (P < 0.001 for all distances and all OTUs), and this difference was also observed for the adult samples (P < 0.001 for all distances and all OTUs). The progression of the community structure over time is shown in Fig. 2.
FIG 2.
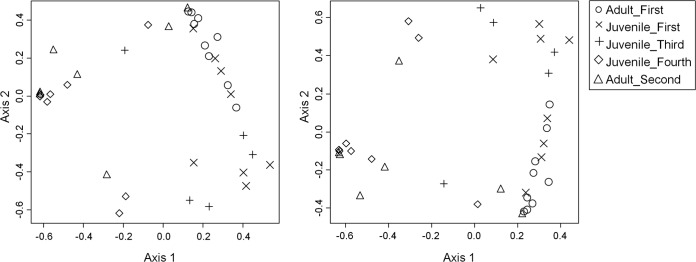
Changes in community structure in wild kakapo samples. Shown is nonmetric multidimensional scaling of the weighted UniFrac distances between individual samples obtained from wild kakapos. (Left) Distances calculated based on OTU0.97 (stress = 0.15; r2 = 0.90). (Right) Distances calculated based on OTU0.95 (stress = 0.15; r2 = 0.91).
Relative OTU abundance changes within fecal microbiota.
Although the community structure overall did not vary significantly between early juvenile and adult sample groups, statistically significant changes in the relative abundances of OTUs were detected. Six OTUs, generally classified as lactic acid bacteria, were present at significantly higher levels in the Juvenile_First microbiota than in the time-equivalent Adult_First microbiota (Table 5). These OTUs were reduced in abundance in the juvenile microbiota, and there were no significantly different OTU abundances between the Adult_First and Juvenile_Third communities. We conclude that the apparent increase in abundance of these OTUs is associated with the young age of the Juvenile_First cohort, with the exception of two OTUs (Table 5, asterisks) that were observed in only two of the individuals comprising the Juvenile_First cohort and that may represent individual-specific variation in the microbiota.
TABLE 5.
Statistically significant changes in OTU abundance between sample groups of interesta
| OTU | OTU taxonomy | Mean relative abundance (%) |
q value | |
|---|---|---|---|---|
| Juvenile_First | Adult_First | |||
| OTU06 | Lactobacillus | 11.16 | 0.01 | <0.001 |
| OTU07 | Clostridium | 4.49 | 0.00 | <0.001 |
| OTU10 | Pseudomonas | 1.41 | 0.01 | <0.001 |
| OTU16 | Lactobacillus* | 1.91 | 0.00 | <0.001 |
| OTU19 | Lactobacillus | 2.58 | 0.01 | <0.001 |
| OTU50 | Clostridium* | 1.78 | 0.00 | <0.001 |
Statistical testing and q value corrections were performed across the entire OTU table, but only OTUs that accounted for ≥1% of the bacterial community are reported. OTUs marked with an asterisk were observed in ≤2 individuals within the Juvenile_First cohort and thus may reflect random interindividual variation rather than a cohort-related difference. OTU labels are provided to allow consistency for comparisons with other tables that report OTU abundances and changes.
The potentially confounding effects of captivity on the microbiota of Juvenile_Second and its subsequent development were a cause for concern within this study, although given the nature of kakapo conservation, this was unavoidable. Due to the differential treatment of juveniles in captivity and the rich wild-type data from the Adult_First, Juvenile_First, and Juvenile_Third groups, some inference can be made from the data, although pending additional testing with an adequate control group, these findings remain speculative. OTUs that fluctuated significantly throughout the captivity period are reported in Fig. 3 and are summarized as follows: OTU03 (Streptococcus) showed a strong, negative correlation with the β-lactam antibiotic treatment kakapo group compared to any nontreated group (rpb = −0.45; P = 0.009) and a weaker, statistically insignificant correlation with captivity overall (rpb = −0.16; P = 0.37). Conversely, OTU05 (Enterococcus) was enriched in the antibiotic-treated (rpb = 0.7; P < 0.0001) and captive (rpb = 0.59; P = 0.0003) kakapos compared to the nontreated groups. Samples obtained from kakapos in captivity without antibiotic treatment showed no differences that could not be explained by the observed differences between Juvenile_First and Juvenile_Third or the lack of differences between Juvenile_Third and Adult_First (i.e., some OTUs were enriched in the samples from the youngest juveniles but not in the later samples or adult samples, implying that individual age was driving the decline in abundance).
FIG 3.
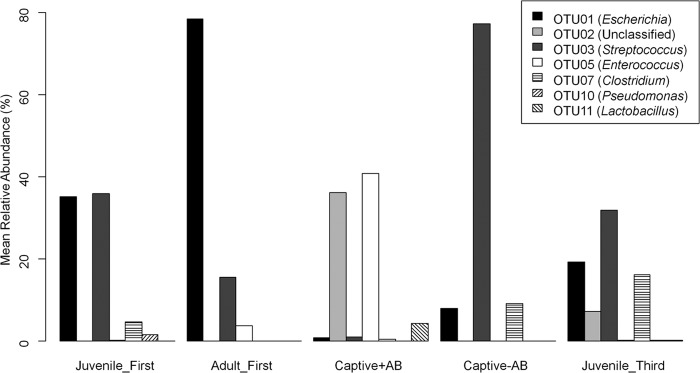
Statistically significant changes in relative OTU abundance during captivity. The Captive+AB and Captive−AB groups refer to the cohort Juvenile_Second split by whether or not antibiotics were administered. Excluding changes in OTU abundance that could be attributed to bird age, only OTU03 and OTU05 were significantly different in antibiotic-treated samples compared to all other kakapo samples (including captive kakapos without antibiotic treatment).
DISCUSSION
The gut microbiota of the kakapo is likely to contribute greatly to the health and well-being of the bird; however, until recently (12, 25), it remained virtually unstudied. Here we document, for the first time, temporal changes in the bacterial communities within the kakapo GI tract but do not find consistent differences between juvenile and adult birds at the community-wide level. Amplicon pyrosequencing confirmed our previous conclusion that the kakapo microbiota is an uneven, low-diversity community. Although differences in experimental factors and sampling depth mean that between-study comparisons must be treated with caution, the diversity and richness estimators calculated in this study are nonetheless low compared to those obtained for other avians. A recent analysis of the emu hindgut reported a mean Shannon diversity index of 3.4 (kakapo = 0.95) and a mean Chao1 richness index of 624 (kakapo = 60.5) (52), while a clone library-based analysis of chicken ceca reported a Chao1 value of 121 (53). A molecular analysis of the hoatzin cecum (54) reported a median inverse Simpson diversity index of >400 (kakapo = 1.78). Kakapo and hoatzin are frequently compared, with the kakapo occasionally being mentioned as a potential candidate for avian foregut fermentation. Despite the marked difference in microbial diversity between the kakapo and hoatzin hindguts, the taxonomic differences between these microbiota are not great. Hierarchical clustering of phylotyped data obtained from hoatzin hindgut analyses (54) revealed no separation between the kakapo juvenile and adult fecal microbiota and the hoatzin cecal microbiota (see Fig. S1 in the supplemental material). While the microbiota of the adult kakapo crop is unknown, it is interesting to observe a degree of convergence in the hindgut microbiota of these two geographically isolated birds.
The greater sequencing depth afforded by amplicon pyrosequencing has allowed us to examine the microbiota in greater detail than our previous efforts (12). Indeed, while the core microbiota in the current analysis consisted of only Gammaproteobacteria and Firmicutes, members of additional phyla were also frequently detected in the birds tested. Representatives of the Bacteroidetes were detected in approximately 70% of samples although often at levels lower than our previous methodology was capable of detecting (12). This finding is somewhat at odds with our previous conclusions and emphasizes the value of increased sequencing depth, even in apparently low-diversity environments. Bacteroidetes-associated OTUs occurred in an individual-specific manner, so although Bacteroidetes were observed in many samples, no Bacteroidetes OTUs were classified in the kakapo core microbiota. In other avian systems, the most abundant bacterial phyla detected with molecular methods are the Firmicutes and Bacteroidetes, followed by Actinobacteria and Proteobacteria (26), which is notably different from the bacterial community profile of the kakapo. The functional implications of this differing community composition compared to those of other herbivorous birds are a matter for future investigation, and knowledge of the core bacteria that comprise the kakapo microbiota will be of benefit for potential future kakapo bacteriotherapy and probiotic development (25).
The lack of differentiation between juvenile and adult community structures is an interesting finding, as strong differences have been reported for other avian systems in which the juveniles were of an age similar to (32, 36) or older than (33) the juveniles studied here. Despite the lack of difference in community structure, some OTUs varied significantly in abundance between younger and older birds, likely reflecting at least some role of bird age in shaping the microbiota. Due to concerns regarding the handling of nesting adults, adults sampled in this study were not the parents of the studied juveniles, and to our knowledge, there was no contact between these juvenile and adult birds. We speculate that this apparently homogenous bacterial community may be due to the small size of Codfish Island, with all adult kakapos utilizing an almost identical diet. It has been observed that kakapos on Codfish Island share overlapping home ranges, with individuals often sharing feeding stations when supplemental feed is provided. Further study into the development of the juvenile microbiota may better resolve this pattern by using a finer time scale to investigate the community structure much sooner after hatching. Although age did not appear to influence community structure, the relative abundance of particular OTUs (including those within the core microbiota) varied with age and time, consistent with the broader avian literature (32, 38, 39). In general, microbial OTUs that were classified as part of the overall core microbiota were present at high levels within the fecal samples, and in each sample grouping, the core and variable OTUs represented over half of the microbiota (Table 4), indicating that these microbes are likely of biological significance in the kakapo. It was interesting that the core microbiota of the juvenile samples accounted for more of the total microbiota and comprised more OTUs than that of the adults. The core microbiota was even more conserved in the antibiotic-treated kakapos, which may be a consequence of a reduction in available niches brought on by a controlled diet and antibiotic pressure.
The need to maintain the health of juvenile kakapos is clearly paramount from a conservation standpoint, and accordingly, it was not possible to maintain a wild juvenile control group when individuals needed to be taken into captivity. With the entire juvenile kakapo population in captivity, there was no adequate control group for comparison other than the adult group, but we nevertheless attempted to identify significant changes in the microbiota that correlated with this period. Studying kakapos that were captive but not treated with antibiotics allowed us to tease apart the influence of diet and antibiotic treatment albeit with a smaller sample size than is ideal. The reported changes in the microbiota were more strongly correlated with antibiotic treatment (OTU03 rpb = −0.45; OTU05 rpb = 0.7) than with captivity in general (OTU03 rpb = −0.16; OTU05 rpb = 0.59), implying that antibiotic treatment was the primary factor driving the observed differences during captivity. We also observed changes in the microbiota of captive, antibiotic-treated individuals, which adds further evidence that captivity alters the kakapo microbiota. The lack of a juvenile control group makes these findings tentative, but there appears to be no long-term impact of these changes, as the bacterial community structures of treated juveniles and untreated adults converged in later samples, and no differences in relative OTU abundance were detected between adults and juveniles following release from captivity. In future breeding seasons, it would be desirable to attempt to repeat this experiment with finer time scales and more rigid control groups to validate these data (bird health permitting).
A previous meta-analysis (55) reported that microbial communities vary over time across a range of natural ecosystems. Consistent with this observation, the kakapo fecal microbiota varied over time, with juvenile and adult samples differing after approximately 1 year between samples. It is possible that this change is related to the change in diet, as reproduction in the kakapo correlates with the fruiting of specific trees native to New Zealand. In addition, supplementary feeding is a common practice during breeding seasons, and these changes in kakapo ecology provide a mechanism that may account for the apparent changes in gut microbiota over time. When the early and later samples were compared, a Proteobacteria-associated OTU (OTU02) (Fig. 3) was observed only sporadically in the early samples but was one of the most abundant OTUs at the final time point (Fig. 1). This OTU could not be classified below the phylum level, with results generally alternating between the classes Betaproteobacteria and Gammaproteobacteria. Additional chimera testing was performed by using both UCHIME and the chimera.slayer command in mothur against the SILVA gold database (http://www.mothur.org/wiki/Silva_reference_files), but this OTU was not determined to be chimeric in either test. The presence and identity of this OTU will be important for future analyses of the kakapo microbiota, as its identity may be better resolved with more sequence data or a different amplicon region. Changes in the microbiota over time carry significance for the wider field of disease ecology, as they emphasize the need for up-to-date knowledge of animal-associated microbiota in order to better enable pathogen detection (56).
Cutoff values of 95% and 97% sequence similarity are commonly reported in the literature, although there is concern regarding which value is appropriate (57–59). In order to account for potential biases introduced by the level of OTU similarity, sequence data were binned into two different OTU definitions (OTU0.95 and OTU0.97), and statistical testing was performed between each sample group using each OTU cutoff. The overall robustness of a finding was based on not only statistical significance but also whether a finding was consistent across different OTU definitions and distance calculations. Statistically significant findings were preserved across OTU definitions, giving confidence that the conclusions regarding community structure were not merely artifacts of the bioinformatic approach.
Overall, our findings suggest that the fecal community structure of kakapos is not influenced by host age per se but does change over time. The finding that age is not a significant factor in shaping the community structure of the kakapo microbiota contrasts with patterns seen in other avian hosts, although age-related differences in OTU abundance indicate that age still plays a role in shaping at least a subset of the kakapo microbiota although possibly on a shorter time scale than normally observed. By comparing our juvenile data to data obtained from adult kakapos, we have teased apart age- and time-based differences and established a working core set of bacteria for further studies of kakapo microbiology. Finally, in confirming that the microbiota changes over time, we have reaffirmed the need for continuous sampling of the microbiota of this endangered bird in order to ensure that knowledge of the healthy microbiota is accurate. The described data, in combination with ongoing ecological and genetic studies, should contribute to the management and, ultimately, the survival of this ancient parrot.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by funding from The University of Auckland Faculty Research Development Fund (grant 9841 3626187). D.W.W. was supported by a University of Auckland doctoral scholarship.
We gratefully thank Ron Moorhouse, Deidre Vercoe, and Jo Ledington (Department of Conservation) for their support with this project. We also thank Peter Deines and Siân Morgan-Waite for helpful comments on the manuscript.
Footnotes
Published ahead of print 16 May 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00975-14.
REFERENCES
- 1.Lloyd BD, Powlesland RG. 1994. The decline of kakapo Strigops habroptilus and attempts at conservation by translocation. Biol. Conserv. 69:75–85. 10.1016/0006-3207(94)90330-1 [DOI] [Google Scholar]
- 2.Wood JR. 2006. Subfossil kakapo (Strigops habroptilus) remains from near Gibraltar Rock, Cromwell Gorge, Central Otago, New Zealand. Notornis 53:191–193 http://notornis.osnz.org.nz/system/files/Notornis_53_1_191.pdf [Google Scholar]
- 3.Clout MN, Merton DV. 1998. Saving the kakapo: the conservation of the world's most peculiar parrot. Bird Conserv. Int. 8:281–296 [Google Scholar]
- 4.Powlesland RG, Merton DV, Cockrem JF. 2006. A parrot apart: the natural history of the kakapo (Strigops habroptilus), and the context of its conservation management. Notornis 53:3–26 http://notornis.osnz.org.nz/system/files/Notornis_53_1_3.pdf [Google Scholar]
- 5.Eason DK, Elliott GP, Merton DV, Jansen PW, Harper GA, Moorhouse RJ. 2006. Breeding biology of kakapo (Strigops habroptilus) on offshore island sanctuaries, 1990-2002. Notornis 53:27–36 http://notornis.osnz.org.nz/system/files/Notornis_53_1_27.pdf [Google Scholar]
- 6.Merton DV, Morris RB, Atkinson IAE. 1984. Lek behaviour in a parrot: the kakapo Strigops habroptilus of New Zealand. Ibis 126:277–283 [Google Scholar]
- 7.Moorhouse RJ, Powlesland RG. 1991. Aspects of the ecology of kakapo Strigops habroptilus liberated on Little Barrier Island (Hauturu), New Zealand. Biol. Conserv. 56:349–365. 10.1016/0006-3207(91)90066-I [DOI] [Google Scholar]
- 8.McNab BK, Salisbury CA. 1995. Energetics of New Zealand's temperate parrots. N. Z. J. Zool. 22:339–349. 10.1080/03014223.1995.9518050 [DOI] [Google Scholar]
- 9.Atkinson IAE, Merton DV. 2006. Habitat and diet of kakapo (Strigops habroptilis) in the Esperance Valley, Fiordland, New Zealand. Notornis 53:37–54 http://notornis.osnz.org.nz/system/files/Notornis_53_1_37.pdf [Google Scholar]
- 10.Cockrem JF. 2006. The timing of breeding in the kakapo (Strigops habroptilus). Notornis 53:153–159 http://notornis.osnz.org.nz/system/files/Notornis_53_1_153.pdf [Google Scholar]
- 11.Cottam Y, Merton DV, Hendriks W. 2006. Nutrient composition of the diet of parent-raised kakapo nestlings. Notornis 53:90–99 http://notornis.osnz.org.nz/system/files/Notornis_53_1_90.pdf [Google Scholar]
- 12.Waite DW, Deines P, Taylor MW. 2012. Gut microbiome of the critically endangered New Zealand parrot, the kakapo (Strigops habroptilus). PLoS One 7:e35803. 10.1371/journal.pone.0035803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clench MH, Mathias JR. 1995. The avian cecum: a review. Wilson Bull. 107:93–121 [Google Scholar]
- 14.Grajal A, Strahl SD, Pabra R, Dominguez MG, Neher A. 1989. Foregut fermentation in the hoatzin, a neotropical leaf-eating bird. Science 245:1236–1238. 10.1126/science.245.4923.1236 [DOI] [PubMed] [Google Scholar]
- 15.Oliver WRB. 1955. New Zealand birds, 2nd ed. AH & AW Reed, Wellington, New Zealand [Google Scholar]
- 16.Horrocks M, Salter J, Braggins J, Nichol S, Moorhouse R, Elliott G. 2008. Plant microfossil analysis of coprolites of the critically endangered kakapo (Strigops habroptilus) parrot from New Zealand. Rev. Palaeobot. Palynol. 149:229–245. 10.1016/j.revpalbo.2007.12.009 [DOI] [Google Scholar]
- 17.Houston D, McInnes K, Elliott G, Eason D, Moorhouse R, Cockrem J. 2007. The use of a nutritional supplement to improve egg production in the endangered kakapo. Biol. Conserv. 138:248–255. 10.1016/j.biocon.2007.04.023 [DOI] [Google Scholar]
- 18.Hill MJ. 1997. Intestinal flora and endogenous vitamin synthesis. Eur. J. Cancer Prev. 6:S43–S45. 10.1097/00008469-199703001-00009 [DOI] [PubMed] [Google Scholar]
- 19.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. 2006. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444:1027–1031. 10.1038/nature05414 [DOI] [PubMed] [Google Scholar]
- 20.Torok VA, Hughes RJ, Mikkelsen LL, Perez-Maldonado R, Balding K, MacAlpine R, Percy NJ, Ophel-Keller K. 2011. Identification and characterization of potential performance-related gut microbiotas in broiler chickens across various feeding trials. Appl. Environ. Microbiol. 77:5868–5878. 10.1128/AEM.00165-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanley D, Denman SE, Hughes RJ, Geier MS, Crowley TM, Chen H, Haring VR, Moore RJ. 2012. Intestinal microbiota associated with differential feed conversion efficiency in chickens. Appl. Microbiol. Biotechnol. 96:1361–1369. 10.1007/s00253-011-3847-5 [DOI] [PubMed] [Google Scholar]
- 22.Stappenbeck TS, Hooper LV, Gordon JI. 2002. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proc. Natl. Acad. Sci. U. S. A. 99:15451–15455. 10.1073/pnas.202604299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahimi S, Grimes JL, Fletcher O, Oviedo E, Sheldon BW. 2009. Effect of a direct-fed microbial (Primalac) on structure and ultrastructure of small intestine in turkey poults. Poult. Sci. 88:491–503. 10.3382/ps.2008-00272 [DOI] [PubMed] [Google Scholar]
- 24.Cao GT, Xiao YP, Yang CM, Chen AG, Liu TT, Zhou L, Zhang L, Ferket PR. 2012. Effects of Clostridium butyricum on growth performance, nitrogen metabolism, intestinal morphology and cecal microflora in broiler chickens. J. Anim. Vet. Adv. 11:2665–2671. 10.3923/javaa.2012.2665.2671 [DOI] [Google Scholar]
- 25.Waite DW, Deines P, Taylor MW. 2013. Quantifying the impact of storage procedures for faecal bacteriotherapy in the critically endangered New Zealand parrot, the kakapo (Strigops habroptilus). Zoo Biol. 32:620–625. 10.1002/zoo.21098 [DOI] [PubMed] [Google Scholar]
- 26.Kohl KD. 2012. Diversity and function of the avian gut microbiota. J. Comp. Physiol. B 182:591–602. 10.1007/s00360-012-0645-z [DOI] [PubMed] [Google Scholar]
- 27.Eason DK, Moorhouse RJ. 2006. Hand-rearing kakapo (Strigops habroptilus), 1997-2005. Notornis 53:116–125 http://notornis.osnz.org.nz/system/files/Notornis_53_1_116.pdf [Google Scholar]
- 28.Tannock GW. 1997. Gastrointestinal microbiology. Chapman and Hall, New York, NY [Google Scholar]
- 29.Dethlefsen L, Huse S, Sogin ML, Relman DA. 2008. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6:e280. 10.1371/journal.pbio.0060280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammons S, Oh PL, Martínez I, Clark K, Schlegel VL, Sitorius E, Scheideler SE, Walter J. 2010. A small variation in diet influences the Lactobacillus strain composition in the crop of broiler chickens. Syst. Appl. Microbiol. 33:275–281. 10.1016/j.syapm.2010.04.003 [DOI] [PubMed] [Google Scholar]
- 31.Hill DA, Hoffmann C, Abt MC, Du Y, Kobuley D, Kirn TJ, Bushman FD, Artis D. 2010. Metagenomic analyses reveal antibiotic-induced temporal and spatial changes in intestinal microbiota with associated alterations in immune cell homeostasis. Mucosal Immunol. 3:148–158. 10.1038/mi.2009.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gong J, Yu H, Liu T, Gill JJ, Chambers JR, Wheatcroft R, Sabour PM. 2008. Effects of zinc bacitracin, bird age and access to range on bacterial microbiota in the ileum and caeca of broiler chickens. J. Appl. Microbiol. 104:1372–1382. 10.1111/j.1365-2672.2007.03699.x [DOI] [PubMed] [Google Scholar]
- 33.Godoy-Vitorino F, Goldfarb KC, Brodie EL, Garcia-Amado MA, Michelangeli F, Domínguez-Bello MG. 2010. Developmental microbial ecology of the crop of the folivorous hoatzin. ISME J. 4:611–620. 10.1038/ismej.2009.147 [DOI] [PubMed] [Google Scholar]
- 34.Lozupone C, Stombaugh JU, Gordon JI, Jansson JK, Knight R. 2012. Diversity, stability and resilience of the human gut microbiota. Nature 489:220–230. 10.1038/nature11550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI. 2012. Human gut microbiome viewed across age and geography. Nature 486:222–227. 10.1038/nature11053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Dongen WFD, White J, Brandl HB, Moodley Y, Merkling T, Leclaire S, Blanchard P, Danchin É, Hatch SA, Wagner RH. 2013. Age-related differences in the cloacal microbiota of a wild bird species. BMC Ecol. 13:11. 10.1186/1472-6785-13-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Der Wielen PWJJ, Keuzenkamp DA, Lipman LJA, Van Knapen F, Biesterveld S. 2002. Spatial and temporal variation of the intestinal bacterial community in commercially raised broiler chickens during growth. Microb. Ecol. 44:286–293. 10.1007/s00248-002-2015-y [DOI] [PubMed] [Google Scholar]
- 38.Lu J, Idris U, Harmon B, Hofacre C, Maurer JJ, Lee MD. 2003. Diversity and succession of the intestinal bacterial community of the maturing broiler chicken. Appl. Environ. Microbiol. 69:6816–6824. 10.1128/AEM.69.11.6816-6824.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scupham AJ. 2007. Succession in the intestinal microbiota of preadolescent turkeys. FEMS Microbiol. Ecol. 60:136–147. 10.1111/j.1574-6941.2006.00245.x [DOI] [PubMed] [Google Scholar]
- 40.Stanley D, Geier MS, Hughes RJ, Denman SE, Moore RJ. 2013. Highly variable microbiota development in the chicken gastrointestinal tract. PLoS One 8:e84290. 10.1371/journal.pone.0084290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simister R, Taylor MW, Tsai P, Fan L, Bruxner TJ, Crowe ML, Webster N. 2012. Thermal stress responses in the bacterial biosphere of the Great Barrier Reef sponge, Rhopaloeides odorabile. Environ. Microbiol. 14:3232–3246. 10.1111/1462-2920.12010 [DOI] [PubMed] [Google Scholar]
- 42.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75:7537–7541. 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. 2011. Removing noise from pyrosequenced amplicons. BMC Bioinformatics 12:38. 10.1186/1471-2105-12-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N, Perez T, Rodrigo A, Schupp PJ, Vacelet J, Webster N, Hentschel U, Taylor MW. 2012. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. ISME J. 6:564–576. 10.1038/ismej.2011.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yue JC, Clayton MK. 2005. A similarity measure based on species proportions. Commun. Stat. Theory Methods 34:2123–2131. 10.1080/STA-200066418 [DOI] [Google Scholar]
- 47.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235. 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Excoffier L, Smouse PE, Quattro JM. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26:32–46. 10.1111/j.1442-9993.2001.01070.pp.x [DOI] [Google Scholar]
- 50.White JR, Magarajan N, Pop M. 2009. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 5:e1000352. 10.1371/journal.pcbi.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 52.Bennett DC, Tun HM, Kim JE, Leung FC, Cheng KM. 2013. Characterization of cecal microbiota of the emu (Dromaius novaehollandiae). Vet. Microbiol. 166:304–310. 10.1016/j.vetmic.2013.05.018 [DOI] [PubMed] [Google Scholar]
- 53.Gong J, Weiduo S, Forster RJ, Huang R, Yu H, Yin Y, Tang C, Han Y. 2007. 16S rRNA gene-based analysis of mucosa-associated bacterial community and phylogeny in the chicken gastrointestinal tracts: from crops to ceca. FEMS Microbiol. Ecol. 59:147–157. 10.1111/j.1574-6941.2006.00193.x [DOI] [PubMed] [Google Scholar]
- 54.Godoy-Vitorino F, Goldfarb KC, Karaoz U, Leal S, Garcia-Amado MA, Hugenholtz P, Tringe SG, Brodie EL, Dominguez-Bello MG. 2012. Comparative analyses of foregut and hindgut bacterial communities in hoatzins and cows. ISME J. 6:531–541. 10.1038/ismej.2011.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shade A, Caporaso JG, Handelsman J, Knight R, Fierer N. 2013. A meta-analysis of changes in bacterial and archaeal communities with time. ISME J. 7:1493–1506. 10.1038/ismej.2013.54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Belden LK, Harris RN. 2007. Infectious diseases in wildlife: the community ecology context. Front. Ecol. Environ. 5:533–539. 10.1890/060122 [DOI] [Google Scholar]
- 57.Keswani J, Whitman WB. 2001. Relationship of 16S rRNA sequence similarity to DNA hybridization in prokaryotes. Int. J. Syst. Evol. Microbiol. 51:667–678. http://ijs.sgmjournals.org/content/51/2/667.long [DOI] [PubMed] [Google Scholar]
- 58.Stackebrandt E, Ebers J. 2006. Taxonomic parameters revisited: tarnished gold standards. Microbiol. Today 33:152–155 [Google Scholar]
- 59.Koeppel AF, Wu M. 2013. Surprisingly extensive mixed phylogenetic and ecological signals among bacterial operational taxonomic units. Nucleic Acids Res. 41:5175–5188. 10.1093/nar/gkt241 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.