

Abstract
Brain inflammation is a common occurrence following responses to varied insults such as bacterial infections, stroke, traumatic brain injury and neurodegenerative disorders. A common mediator for these varied inflammatory responses is prostaglandin E2 (PGE2), produced by the enzymatic activity of cyclooxygenases (COX) 1 and 2. Previous attempts to reduce neuronal inflammation through COX inhibition, by use of nonsteroidal anti-inflammatory drugs (NSAIDs), have met with limited success. We are proposing the two-hit model for neuronal injury—an initial localized inflammation mediated by PGE2 (first hit) and the simultaneous release of adenosine triphosphate (ATP) by injured cells (second hit), which significantly enhances the inflammatory response through increased synthesis of PGE2. Several evidences on the role of exogenous ATP in inflammation have been reported, including contrary instances where extracellular ATP reduces inflammatory events. In this review, we will examine the current literature on the role of P2 receptors, to which ATP binds, in modulating inflammatory reactions during neurodegeneration. Targeting the P2 receptors, therefore, provides a therapeutic alternative to reduce inflammation in the brain. P2 receptor-based anti-inflammatory drugs (PBAIDs) will retain the activities of essential COX enzymes, yet will significantly reduce neuroinflammation by decreasing the enhanced production of PGE2 by extracellular ATP.
Keywords: ATP, microglia, neuroinflammation, NSAIDs, P2 receptors, PBAIDs, prostaglandin E2
Inflammation within the brain
Various environmental factors can lead to inflammation within the brain. These range from bacterial infections that cause acute inflammation to neurodegenerative disorders that mediate chronic inflammation. The inflammation may be restricted to a local region in focal ischemia or occur in a wider zone during traumatic brain injury. Inflammation could also result from an autoimmune response such as multiple sclerosis or in response to toxins and nerve agents (for general reviews, see Lucas et al., 2006; Aguzzi et al., 2013). Recent reports implicate inflammation contributing to the pathology of psychiatric disorders such as stress, depression and schizophrenia (Najjar et al., 2013), in metabolic syndromes such as obesity and type 2 diabetes (Purkayastha and Cai, 2013), and even as a response to increased neuronal activity (Xanthos and Sandkuhler, 2014). Irrespective of the type of inflammation, the molecular mediators are oftentimes the same –prostaglandin E2 (PGE2) or cytokines such as interleukin-1β(IL-1β), produced by the activity of resident microglial cells. Despite brain inflammation playing such a major role in various CNS disorders, successful therapeutic strategies to overcome it are still lacking.
PGE2 is produced by the action of cyclooxygenases (COX) which mediate the first committing step in its synthesis from arachidonic acid (Akundi et al., 2005). The constitutively active COX-1 isoform is believed to be responsible for the majority of PGE2 formed in the body. However, it is the growth factor-, cytokine- or mitogen-inducible COX-2 that emerged as the isoform responsible for the massive release of PGE2 during inflammation of all types—systemic, central, acute or chronic. The COX enzymes have been a therapeutic target in a multitude of disorders, ranging from fever and pain to cancer, rheumatoid arthritis, and Alzheimer’s disease (AD; Yedgar et al., 2007). Their importance can be judged from the widespread use of aspirin as an analgesic and antipyretic; and the promise of nonsteroidal anti-inflammatory drugs (NSAIDs) against spreading neurodegeneration in AD (Szekely and Zandi, 2010). However clinical trials failed to not only halt the progression of dementia in AD patients but also showed increased risks of myocardial infarction and stroke (Jüni et al., 2004). An essential lesson learnt from the debacle of NSAIDs was that the two isoforms of COX do not functionally substitute one another but each remains indispensable in certain functions. COX-2 stands on a delicate balance—the neuronal isoform plays an important role in synaptic plasticity, memory consolidation and cortical development while the microglial isoform mediates neuroinflammation. Targeting COX enzymes, therefore, requires a careful consideration of the benefit-to-risk ratio.
Neuroinflammation: a two-hit model
An interesting observation in the past decade and half showed that the inflammatory response of microglia—the release of mature IL-1β from lipopolysaccharide (LPS)-primed cells—could be significantly modulated with the addition of exogenous adenosine triphosphate (ATP; Ferrari et al., 1997). More diversified studies showed that ATP is able to mediate the release of PGE2 in IL-1β-treated astrocytes (Xu et al., 2003) or in LPS-activated macrophages (Barberà-Cremades et al., 2012). We found a similar synergistic effect of ATP on LPS-mediated PGE2 release in primary rat microglial cells (unpublished observation). These studies conclusively showed that exogenous ATP significantly modulates inflammation. In this review, we are proposing the two-hit model for neuroinflammation. The first hit is the injury itself—nerve injury, bacterial infections, hypoxia-ischemia, autoimmune reactions or proteopathies associated with neurodegeneration—leading to the activation of glial cells (Figure 1). The second hit is the release of large pools of cytosolic ATP from damaged neurons into the extracellular milieu, in response to direct injury or following glial cell activation. This excess ATP, despite mechanisms regulating their concentration outside the cell, activates a wide variety of purinergic receptors present on cells in the vicinity, thus modulating glial activity and neuronal response to inflammation. Such a model was earlier proposed for the release of mature IL-1β following bacterial infections (Ferrari et al., 2006). Identifying the pro-inflammatory receptors of ATP, and targeting them pharmacologically, will significantly diminish the dramatic release of prostanoids and cytokines to clinically manageable levels; thus, balancing their functional roles in active defence and tissue repair.
Figure 1.
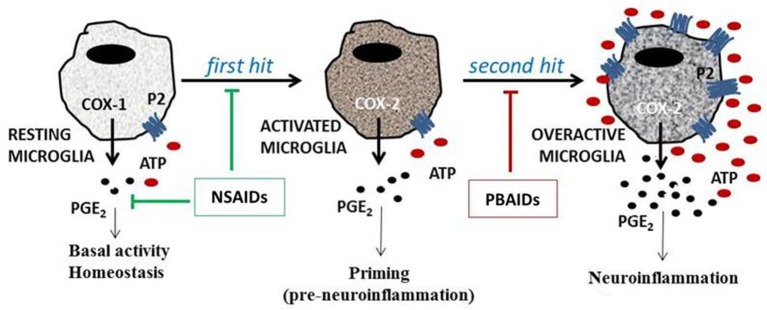
The two-hit model of neuroinflammation. The ATP-mediated enhancement of neuroinflammation can be explained through the two-hit model. A variety of insults, such as bacterial LPS, various cytokines, or amyloid peptides, can act as the first hit, resulting in microglial activation, COX-2 induction and PGE2 release. The second hit, following neuronal injury, death or persistent glial cell activation, results in the release of ATP, which acts on both neuronal and glial P2 receptors, leading to enhanced microglial PGE2 release. NSAIDs target COX enzymes affecting the housekeeping roles of PGE2. ATP potentiates the effects of first hit multi-fold, and thus, would be the most relevant target for therapeutic intervention. By acting on P2 receptors, PBAIDs are believed to reduce PGE2 to pre-inflammatory levels without affecting the activity of COX enzymes.
ATP: the common denominator for varied inflammatory insults
Both healthy neurons and glial cells carry ATP, in millimolar concentrations, within presynaptic vesicles and granules, respectively (Abbracchio et al., 2009). Neuronal ATP serves as a neurotransmitter while astrocytic ATP allows distant astrocytes to communicate with each other and modulate neuronal response. However, the release of ATP by neurons or astrocytes is usually very low, in the nanomolar range. Furthermore, the extracellular concentration of ATP is dependent on the regional distribution and local activity of synaptic ectonucleotidases CD28/CD39 which convert ATP to ADP and AMP, CD73 which converts AMP to adenosine, and nucleoside diphosphate kinase whose transphosphorylating activity maintains the exogenous levels of various nucleotides in steady state (Lazarowski et al., 2003). This steady state balance is, however, disrupted during pathological conditions when damaged neurons and chronically activated glial cells release dramatic levels of ATP, uridine triphosphate (UTP) and other intracellular nucleotides.
Not only neuropathological conditions even systemic inflammation leads to an increase in exogenous ATP within the CNS (Gourine et al., 2007). In fact the release of ATP in response to tissue injury is a universal phenomenon also seen in plants at sites of physical wounding (Choi et al., 2014). Efflux of ATP into the extracellular space is a common universal “stress signal”, leading to the evolution of receptors for ATP to recognize this “danger” and initiate a stress response. Mammals evolved purinergic receptors with varying specificities for ligands such as ATP, ADP, UTP, UDP, UDP-sugars or adenosine, and diverse range of intracellular signaling mechanisms downstream to receptor activation. Nucleotides act on P2 receptors, with seven ionotropic P2X receptors gating Na+, K+, and especially Ca2+ ions, and eight G protein-coupled metabotropic P2Y receptors (Abbracchio et al., 2009). Adenosine, on the other hand, acts on adenosine receptors, of which A1 and A3 adenosine receptors inhibit, while A2A and A2B adenosine receptors stimulate adenylyl cyclase (Fredholm, 2010). Recently ATP receptors have also been identified in plants which are activated in response to tissue wound (Choi et al., 2014). Called DORN1 (Does not Respond to Nucleotides 1), these receptors, much like their mammalian counterparts, show high affinity to ATP and alter Ca2+ flow.
Exogenous ATP has both positive and negative roles in inflammation
The presence of functionally active purinergic receptors on microglia indicates the likelihood of astrocyte-microglia crosstalk (Verderio and Matteoli, 2001). Such a communication enhances microglial surveillance system and their response to inflammation within the CNS. Indeed, neuronal and astrocytic release of ATP during traumatic brain injury causes rapid microglial chemotactic response (Davalos et al., 2005). At the site of injury, exogenous ATP mediates the release of pro-inflammatory cytokines and PGE2 (Xu et al., 2003; Ferrari et al., 2006; Xia and Zhu, 2011). The end effect of this synergism is the production of pathological levels of inflammatory cytokines and prostanoids.
While the above reports suggest extracellular ATP as proinflammatory, others have reported to the contrary. In LPS-primed cells, ATP inhibited the release of cytokines from spinal cord microglia (Ogata et al., 2003), nitric oxide (NO) release in BV-2 microglia (Brautigam et al., 2005), and pro-inflammatory markers such as tumor necrosis factor α (TNF-α), interleukin 6 (IL-6) and NO in primary microglia (Boucsein et al., 2003). Since TNF-α and IL-6 also have neuroprotective roles (Suzuki et al., 2004; Noguchi et al., 2013), the above inhibitory action of ATP may actually be detrimental to the organism. Increases in exogenous ATP need not always be hazardous—extracellular levels of adenosine increases 6- to 31-fold within the hippocampus of patients with epilepsy, but acts as a natural anticonvulsant terminating seizure (During and Spencer, 1992). Interestingly, cancer cells evade surveillance by up-regulating a subpopulation of regulatory T cells expressing ectonucleotidases CD39 and CD73 to exploit the immunosuppressive nature of adenosine (Whiteside and Jackson, 2013). Early development of nervous system is dependent on purinergic signals, which in concert with growth factors, regulate the number of proliferating and differentiating neural stem cells (Ulrich et al., 2012).
P2 receptors determine the eventual effect of extracellular ATP
P2X7 receptors
The specific roles of P2 receptors in neuroinflammation are still being uncovered. The subject is still in its teens, starting with the discovery of ATP enhancing IL-1β release in activated immune cells (Ferrari et al., 1997). LPS-mediated activation of toll-like receptor 4 leads to the formation of the inflammasome complex wherein IL-1β processing occurs (Martinon et al., 2002). However, release of IL-1β requires loading of the inflammasome complex into the secretory lysosome, or the formation of membrane blebs—either mechanism triggered through P2X7 receptor-mediated K+ efflux (Ferrari et al., 2006; di Virgilio, 2007). As a result, the effect of ATP is dependent on cells primed with LPS, and conversely, LPS does not release IL-1β in the absence of P2X7 receptors (Solle et al., 2001). Pannexin 1, a gap junction-related protein, has been shown to be responsible for the release of ATP from dying cells, leading to the activation of the inflammasome and recruitment of phagocytes (Dahl and Keane, 2012). Intraperitoneal injection of LPS results in two-four-fold higher detection of ATP in the mouse peritoneum (Barberà-Cremades et al., 2012). Systemic administration of LPS markedly increases the expression of P2X7 receptors in the brain (Choi et al., 2007). LPS- or IL-1β-mediated febrile response is greatly reduced in mice with genetic or pharmacological loss of P2X7 receptors (Barberà-Cremades et al., 2012). ATP and the preferential P2X7 agonist, 2′(3′)-O-(4-benzoylbenzoyl) ATP (BzATP) induce the secretion of cytokines IL-6 and TNF-α in wildtype microglia but not in cells derived from P2X7−/− mice (Shieh et al., 2014). These reports underline the importance of P2X7 receptors in mediating inflammation, especially in the release of IL-1β (Ferrari et al., 2006). The low affinity of P2X7 receptors for extracellular ATP ensures their activation occurs only under pathological conditions where excess ATP is found, further supporting the notion of exogenous ATP as an “alarm” signal.
Immunohistochemical analysis of AD brains reveal significant levels of P2X7 receptors colocalized with activated microglia, an observation that was also found in the hippocampus of rats injected with Aβ1-42 (McLarnon et al., 2006). Aβ triggers ATP release, membrane permeabilization and IL-1β secretion in wild-type but not in P2X7−/− mouse (Sanz et al., 2009). In fact, overexpression of P2X7 receptor itself, in the absence of any pro-inflammatory stimuli, can drive the activation and proliferation of microglial cells (Monif et al., 2009). Similarly, exposure to high levels of extracellular ATP can also tilt the signaling mechanism from a P2X7-phosphatidylinositol 3-kinase/Akt-mediated growth pathway to a novel P2X7-AMPK-mammalian target of rapamycin (mTOR)-mediated autophagic pathway, as observed in tumor cells (Bian et al., 2013). Rapamycin reduces neuroinflammation and brain lesions in a mouse model of Leigh syndrome (Johnson et al., 2013). In astrocytes, inhibition of mTOR significantly reduces the stability of inducible nitric oxide synthase (iNOS) mRNA (Lisi et al., 2011). However, in microglia, blocking of mTOR pathway in activated cells leads to enhanced PGE2 synthesis (de Oliveira et al., 2012). Activated microglia downregulate microRNA, miRNA-200b, which leads to increased c-Jun N-terminal kinase (JNK) activity leading to increased iNOS expression (Jadhav et al., 2014). These observations suggest that the role of mTOR in neuroinflammation is cell-type specific and depends on both epigenetic factors and the presence of inflammatory stimuli.
Gene expression studies in Aβ-treated microglia derived from human post-mortem brains, in fact, suggest that the expression of pro-inflammatory genes are largely up-regulated at the expense of genes involved in Aβ phagocytosis and removal (Walker et al., 2006). Activated P2X7 receptors impair lysosomal function and instead stimulate the release of autolysosomal contents into the extracellular space, possibly leading to the increased secretion of IL-1β or amyloidogenic proteins (Takenouchi et al., 2009). In corollary, silencing of P2X7 receptors in Aβ-stimulated cells leads to a decreased release of pro-inflammatory cytokines and a marked increase in the phagocytosis of Aβ1-42 peptide (Ni et al., 2013). P2X7 receptors, therefore, turn phagocytic (neuroprotective) microglia into inflammatory (neurodegenerative) phenotype.
Expression of P2X7 receptors is also up-regulated in Huntington’s disease and amyotrophic lateral sclerosis (ALS; Díaz-Hernández et al., 2009). In microglia isolated from superoxide dismutase SOD1-G93A mutant mouse model of ALS, activation of P2X7 receptors enhances oxidative stress (Apolloni et al., 2013). Oxidative stress drives the nitration of 90 kDa heat-shock protein (Hsp90), which mediates cell death through P2X7 receptors (Franco et al., 2013). Nitrated Hsp90 is found in the motor neurons of patients with ALS; and as expected, deletion of P2X7 receptors prevents the neurotoxic effects of nitrated Hsp90.
Imbalances in energy homeostasis are associated with neurodegenerative disorders (Akundi et al., 2013). Over-activation of poly (ADP-ribose) polymerase 1 (PARP1) contribute towards dopaminergic degeneration in Parkinson’s disease (PD), which is completely absent in PARP1−/− mice (Kim et al., 2013). PARP1 activation leads to depletion of cytosolic NAD+. Replenishment of NAD+ prevents PARP1-mediated neuronal death (Alano et al., 2010). Exogenous NAD+, surprisingly, enters neurons through the dilated P2X7 receptor-gated channels, marking a neuroprotective role for the otherwise proinflammatory P2X7 receptors.
Among other neuroprotective roles, various in vitro models show that activation of P2X7 receptors stimulates α-secretase activity leading to the shedding of non-amyloidogenic soluble amyloid precursor protein (APP; Darmellah et al., 2012). On the contrary, inhibition of P2X7 receptors in a transgenic mouse for mutant human APP show a significant decrease in the number of amyloid plaques through increased activity of α-secretase (Diaz-Hernandez et al., 2012). Such opposing roles could be best explained with the discovery of a shorter, natural, splice variant of P2X7 receptor that exhibits neurotrophic properties (Adinolfi et al., 2010). Though it remains to be investigated, it is probable that the truncated P2X7 receptors induce α-secretase activity while the longer isoforms are inhibitory. The factors that mediate the retention or deletion of the C-terminal part of P2X7 receptors are not yet known. Such contrasting roles of P2X7 receptors have also been identified in other cellular systems such as cancer (Feng et al., 2006). The distribution of short and long isoforms of P2X7 within the receptor heterotrimer most likely determines its overall trophic or toxic nature.
P2X4 receptors
An interesting use of neuronal P2 receptors as “flags” for microglial recognition has been reported. In the mutant superoxide dismutase SOD1 mouse model of ALS, degenerating motor neurons typically express P2X4 receptors for the recruitment and eventual engulfment by activated microglia (Casanovas et al., 2008). Unlike a typical cell undergoing apoptosis, P2X4-positive neurons neither show chromatin condensation nor caspase 3 activity; rather exhibit loss of neuronal NeuN marker and recruitment of microglial cells. It is not just restricted to motor neurons but to other degenerating neurons affected with ALS—serotonergic neurons of raphe nucleus, noradrenergic neurons of locus coeruleus, and Purkinje cells in the cerebellum. In Aβ1-42-treated neurons that do undergo caspase 3-mediated apoptosis, increased surface expression of P2X4 receptors occurs due to the unique presence of a putative caspase 3 cleavage site within the C terminus region (Varma et al., 2009). Hence, overexpression of P2X4 receptors enhances Aβ-induced neuronal death, while receptor inhibition subdues cell death. These reports form the basis for our hypothesis that surface expression of P2X4 receptors may serve as markers for degenerating neurons, attracting microglial cells for eventual engulfment.
On the other hand microglial P2X4 receptor expression is associated with increased neurophagic activity (Cavaliere et al., 2003). Knocking out P2X4 receptors results in poorer microglial activation and loss of PGE2-mediated inflammatory pathway (Ulmann et al., 2010). P2X4 receptor forms a large conductance pore on the cell surface affecting ionic balance, thus mediating the release of proinflammatory substances. Constitutively, P2X4 receptors are trafficked into late endosomes and remain resistant to lysosomal degradation (Robinson and Murrell-Lagnado, 2013). Such a mechanism prevents the “flagging” of healthy neurons or the “activation” of microglia under normal physiology.
Other P2X receptors
Slow neurodegeneration, following axotomy, shows an upregulation of P2X1 and P2X2 receptors, synchronous with upregulation of neuronal nitric oxide synthase (nNOS; Viscomi et al., 2004). P2X receptors further mediate translocation of nNOS to the plasma membrane (Ohnishi et al., 2009). In an animal model of PD, dopamine denervation upregulates P2X1, P2X3, P2X4 and P2X6 receptors on nigral GABAergic neurons to compensate the loss of dopamine (Amadio et al., 2007). Coincidentally, within the substantia nigra, two out of five groups of GABAergic neurons, but none of the five groups of dopaminergic neurons, express nNOS (González-Hernández and Rodríguez, 2000). Whether P2X and nNOS are upregulated within the same cell during neurodegeneration is not known; however, coordinated activation of purinergic and nitrergic mediators seems a likely event during neuroinflammation.
P2Y receptors
The metabotropic P2Y receptors play a major role in neuron-glia communication. Neuronal injuries activate astrocytic P2Y1 receptors leading to the release of PGE2, causing reactive gliosis (Xia and Zhu, 2011), or glutamate, mediating synaptic modulation (Domercq et al., 2006). Blocking of P2Y1 receptors therefore reduces glial activity (Davalos et al., 2005) and improves cognitive outcome following traumatic brain injury (Choo et al., 2013). In the AD brain, P2Y1 receptors are localized in the neurofibrillary tangles and neuritic plaques (Moore et al., 2000). In contrast, there is a selective loss of P2Y2 receptors correlating with worsening neuropathological scores (Lai et al., 2008). This is not surprising since P2Y2 receptors stimulate α-secretase activity (Camden et al., 2005). In addition, P2Y2 receptors mediate microglial phagocytosis of fibrillar forms of Aβ in a mouse model of AD (Ajit et al., 2014). The soluble Aβ peptides are instead cleared through ATP-dependent P2Y4 receptor-mediated pinocytosis (Li et al., 2013). In fact soluble Aβ1-42 itself induces ATP release, auto-stimulating P2Y4 receptors in microglia, thus mediating its own clearance. Degradation of extracellular amyloid peptides is also performed by metallopeptidases such as matrix metalloproteinase 9 (MMP-9), whose secretion is upregulated following inhibition of the tonically active P2Y14 receptors (Kinoshita et al., 2013). These reports suggest that the loss of P2Y2 and P2Y4 receptors or an overactivation of P2Y1 and P2Y14 receptors alter the steady state levels of amyloid peptides leading to AD.
Both P2Y2 and P2Y4 receptors are preferentially expressed in perivascular astrocytes, and in response to exogenous ATP, mediate increased levels of cytosolic calcium within their end-feet processes (da Silva et al., 2009). As a result, P2Y receptors influence the permeability of blood-brain barrier through induction of endothelial nitric oxide synthase (eNOS). Activated glial cells also induce the expression of chemokines such as monocyte chemotactic protein 1 (MCP1) leading to the CNS recruitment of monocytes (Kim et al., 2011). Interestingly, MCP1 deficiency decreases microglial phagocytosis of Aβ oligomers, thus contributing to progressive amyloidosis (Kiyota et al., 2013).
UDP is the ligand of choice for P2Y6 receptors. Activated P2Y6 receptors trigger a change in microglial phenotype—from active motile/surveillance cells to active neuron devouring/phagocytic cells (Koizumi et al., 2007). The neurophagic activity of microglial cells is potentiated by TNF-α, LPS or Aβ peptides, and delayed through P2Y6 receptor antagonists (Neher et al., 2014). Furthermore, activated P2Y6 receptors block dilation of P2X4 receptor-mediated channels, shifting microglial phenotype from inflammatory to phagocytic cells (Bernier et al., 2013). Blocking of P2Y6 receptors increases neuronal survival suggesting that phagocytosis is not limited to degenerating neurons alone but non-specifically targets “stressed-but-otherwise-viable” neurons as well (Emmrich et al., 2013). Irrespective of the type of insult, the release of UDP signals microglia to initiate indiscriminate phagocytosis found in neurodegenerative disorders.
The migration of microglial cells to the site of injury is mediated by P2Y12 receptors (Haynes et al., 2006). In mice lacking P2Y12 receptors, microglia fail to polarize and migrate towards the lesion site while overactivation of P2Y12 receptors enhances neuroinflammation. As a result P2Y12+/− mice show lesser severity of neuronal injury following cerebral ischemia compared to P2Y12+/+ littermates (Webster et al., 2013). Interestingly, loss of the transcriptional factor interferon regulatory factor 8 (IRF8) suppresses microglial chemotaxis (Masuda et al., 2014). Irf8−/− microglia show reduced expression of P2Y12, P2X4 and adenosine A3 receptors—all involved in microglial activation and migration to the site of injury. As a result, Irf8−/− mice are resistant to experimental autoimmune encephalitis (EAE)—a mouse model of multiple sclerosis (Yoshida et al., 2014).
ADP, formed by the activity of ectonucleotidases on extracellular ATP, is the preferred ligand for P2Y13 receptors. Ubiquitination at its C-terminal end leads to proteasomal degradation and poor surface expression (Pons et al., 2014). However its surface expression increases in response to oxidative stress and genotoxins such as cisplatin or UV irradiation (Morente et al., 2014). In the red blood cells, ADP-activated P2Y13 receptors show a negative feedback loop by inhibiting ATP release (Wang et al., 2005). Such a mechanism ensures additional regulation of extracellular ATP during neuronal injuries by restricting the lesion area such that undamaged and far away neurons which are exposed to ADP are not activated.
Adenosine receptors
Enhanced neuroinflammation and microglial activity is a feature of A1 adenosine receptor knockout mice (A1AR−/−), suggesting that activation of A1ARs is neuroprotective under pathological conditions (Luongo et al., 2014). On the other hand, A2A receptors facilitate glutamate release, and their association with ectonucleotidase, CD73, implies A2A receptors are activated under pathological conditions when excess extracellular ATP is found (Augusto et al., 2013). Hence A2A receptor antagonists such as caffeine limit the pathology of neurodegenerative disorders such as AD and PD. Interestingly, in mouse models of senescence, A1ARs significantly decrease with age while A2A receptors increase with age (Castillo et al., 2009). A higher density of A2A receptors in the putamen of PD patients also correlates with increasing motor symptoms (Varani et al., 2010). A2A receptors induce microglial COX-2 expression (Fiebich et al., 1996) and inhibit astrocyte glutamate uptake by interacting with the α2 subunits of Na+/K+-ATPase (Matos et al., 2013). Though activation of A1ARs and inhibition of A2A receptors provide neuroprotection in the adult brain, the opposite is true for the embryonic brain. Chronic hypoxia mediates accelerated maturation of oligodendrocyte progenitor cells leading to hypomyelination and ventriculomegaly in mice (Akundi and Rivkees, 2009). Deletion of A1ARs or early intervention with caffeine rescues embryos against hypoxia-mediated white matter injury (Back et al., 2006). Similarly, hypoxic ischemia-mediated brain damage was more profound in newborn A2A−/− mice compared with their wildtype littermates (Adén et al., 2003). Similarly, it has been observed that A2A receptor agonists show synergy with agonists of certain toll-like receptors, such as TLR2, 4, 7 and 9, in selectively upregulating the expression of vascular endothelial growth factor and downregulating the release of TNF-α (Pinhal-Enfield et al., 2003). Such a synergistic mechanism provides an angiogenic role for macrophages making it relevant in the aftermath of cerebral ischemia.
Adenosine A3 receptors mediate microglial process extension (Ohsawa et al., 2012). Agonists of A3 receptors thereby provide neuroprotection against ischemia (Choi et al., 2011). However, during chronic neuroinflammation microglia undergo process retraction through upregulation of adenosine A2A receptors (Orr et al., 2009). A2A receptor-dependent process retraction is also seen in the substantia nigra of mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) for 5 days (Gyoneva et al., 2014). A2A receptor-mediated loss of process extension (due to A3 receptors) and chemotaxis (due to P2Y12 receptors) thereby dampens microglial response to injury. Microglial cells also express α7 nicotinic acetylcholine receptor. Activation of these receptors attenuates neuroinflammation in a mouse model of MPTP (Liu et al., 2012). Coincidentally, nicotinic acetylcholine receptor stimulation mediates dopamine release in the rat striatum which is negatively regulated by agonists of adenosine A2A receptors (Garção et al., 2013). These observations suggest that adenosine A2A receptors regulate various neurotransmitter systems, and use of specific antagonists of A2A receptors, therefore, show potential as therapeutic alternatives for PD (Threlfell et al., 2012).
Targeting P2 receptors may be an attractive therapeutic approach over COX2 inhibition
Neuroinflammation in AD was promisingly approached through the use of NSAIDs (Szekely and Zandi, 2010). Following failures in clinical trials, the AD Anti-inflammatory Prevention Trial (ADAPT) group found that neither celecoxib nor naproxen prevented AD in adults with a family history of dementia (Breitner et al., 2013). The drawback of using COX inhibitors lies in the ubiquitous presence and role of its product, PGE2. PGE2 plays an important role in gastro-intestinal (GI secretion, bowel motility), cardiovascular (regulates blood pressure), renal (hemodynamics in the glomeruli), and reproductive (embryo implantation, uterine contraction) systems; and within the CNS, regulates body temperature, sleep-wake cycle, memory consolidation and synaptic plasticity. The relative contribution of either COX isoforms in mediating the above functions is unclear. Selective COX-2 antagonists were designed to target the excess PGE2 formed during neuroinflammatory episodes with the consideration that the more ubiquitous COX-1 would suffice for the production of physiological levels of PGE2. However, despite better gastrointestinal safety ratio, selective COX-2 inhibitors showed increased risks of myocardial infarction, stroke, systemic and pulmonary hypertension, and sudden cardiac death (Jüni et al., 2004). The multiple deaths led to the eventual withdrawal of COX-2 inhibitors such as rofecoxib leading to sweeping lawsuits and wider criticism of the drug licensing procedures.
In this scenario targeting P2 receptors, which modulate PGE2 synthesis, comes as a promising therapeutic possibility (Figure 1). The rat COX-2 promoter carries consensus sequences for transcription factors such as nuclear factor κB (NF-κB), NF-IL6, AP-1 and cAMP-responsive element (Tanabe and Tohnai, 2002). In addition, COX-2 can also be epigenetically regulated with hypermethylation responsible for its silencing in various types of cancer (Lodygin et al., 2005; Castells et al., 2006). Epigenetic contribution in the development of multiple sclerosis and neurodegenerative disorders is slowly being recognized, although evidences for such changes on P2 receptor genes is not yet known (Noh et al., 2012; Koch et al., 2013; Qureshi and Mehler, 2013). Furthermore, COX-2 is posttranscriptionally regulated as well. The human COX-2 mRNA contains at least 23 AU-rich elements (AREs) in the 3′-untranslated region (UTR) conferring to its instability (Shaw and Kamen, 1986). The interactions of ARE-binding protein with the 5′-methylguanosine cap-binding protein and polyadenosine tail-binding protein can either further stabilize the mRNA or lead to its degradation through recruitment of deadenylases (Dean et al., 2004). The p38 mitogen-activated protein kinase plays a critical role in the post-transcriptional regulation of several proinflammatory genes through controlling the phosphorylation status of these binding proteins (Clark et al., 2003). Other proteins that bind to COX-2 ARE and lead to its mRNA stabilization include the heat shock protein hsp70 (Kishor et al., 2013), and the RNA-binding protein HuR which inhibits the destabilization of COX-2 mRNA mediated by microRNA miR-16 (Young et al., 2012). Another regulator is the heterogeneous nuclear riboprotein A1 (hnRNP-A1) whose declining levels have been correlated with the severity of symptoms in various neurodegenerative diseases including AD (Bekenstein and Soreq, 2013). It was recently reported to regulate IL-6 transcription, with overexpression of hnRNP-A1 increasing IL-6 expression and knockdown leading to reduced IL-6 synthesis (Zheng et al., 2013). Post-transcriptional and post-translation regulation of various P2 receptors is not yet known, although the various alternate splicing mechanisms as shown in P2X7 (Adinolfi et al., 2010) and adenosine A1 receptor (Ren and Stiles, 1994) suggest their involvement. Transcription factors downstream of P2 receptor activation bind to most of the COX-2 promoter consensus sequences (Brautigam et al., 2005; Ferrari et al., 2006; Lenertz et al., 2011). P2X7 receptor antagonists present themselves as appropriate therapeutic alternatives to specific COX-2 inhibitors based on several evidences implicating them in neuroinflammation. Currently a few P2 receptor antagonists have advanced to clinical trials (Arulkumaran et al., 2011; North and Jarvis, 2013). It sets the stage for the potential role of P2 receptor-based anti-inflammatory drugs (PBAIDs) in targeting neuroinflammation.
A considerable roadblock in the design of PBAIDs is inherent in the diversity of P2 receptors. The conflicting reports on the neuroprotective and pro-inflammatory roles of various P2 receptors stems from the limited understanding of the actual types of receptors expressed, which may differ between cell types, species, age, and physiological status (Crain et al., 2009; Serrano et al., 2012). Nerve injury indiscriminately releases nucleotides of all kinds, including functionally opposing ones. The predominant ligand concentration depends on the distribution and activity of ectonucleotidases. Most studies utilize pharmacological agents which exhibit receptor promiscuity and have been characterized on purified homomers. However, not only surface P2 receptor density changes under pathological conditions, there is also the possibility of formation of heteromultimers (such as P2X2/6, or P2X4/6, and even P2X2/4/6) with altered ligand affinity and functions (Robinson and Murrell-Lagnado, 2013). The long and short splice variants of P2X7 receptor have opposing roles on cell growth and death (Adinolfi et al., 2010). Finally, an ideal PBAID has to overcome potential possibilities of receptor compensation. This was particularly evident in the failure of neuroprotection in P2X7−/− mice where higher numbers of functionally compensating P2X4 receptors were found instead (Hracskó et al., 2011). In another example, P2Y12−/− mice show delayed microglial response to injury; however, other signaling mechanisms did ensure that microglia reached the site of injury despite the delay (Haynes et al., 2006). The P2X2-P2X5 heterotrimer is functionally analogous to P2X7 receptors, including pore dilation, membrane blebbing and phosphatidylserine exposure (Compan et al., 2012). Such rich receptor diversity allows P2X receptors to functionally compensate the loss of other family members. Therefore, more studies are required to correctly identify the aggravating P2 receptor contributing to neuroinflammation. Because of the functional diversity of P2 receptors, PBAIDs should be carefully chosen to target the disease at the appropriate stage where benefit outweighs risk.
Summary
Conventionally COX-2 has been a target in various inflammatory disorders. However, the failure of NSAIDs and selective enzyme inhibitors reveal the importance of COX-2 not only in various physiological activities but also in tissue repair following neuronal injury. The COX enzymes maintain a delicate balance of tissue scavenging and tissue repair during neuroinflammation. An imbalance could lead to excessive PGE2 activity leading to increased tissue damage or chronic inflammation. All cells within the vertebrate system upon damage (hit one) release large amounts of ATP (hit two) into the extracellular space. The effect of released ATP depends on the nature of two downstream factors—(1) the type of receptors present on cells within the vicinity of the injury; and (2) the distribution and activity of hydrolyzing ectonucleotidases. In large quantities, ATP potentiates the inflammatory reaction while other nucleotides have various modulatory roles in shaping the outcome of inflammation (summarized in Figure 2). PBAIDs aim to reduce the effect of second hit by targeting P2 receptors responsible for inflammation-enhancement rather than the COX enzymes mediating PGE2 synthesis. By not interfering with the COX system PBAIDs, unlike NSAIDs, retain the housekeeping functions of PGE2, but vastly reduce the pathology through P2 receptor inhibition. Identifying the target P2 receptor, and designing a selective PBAID, remains a challenge for future therapeutic successes in neuroinflammation. Surface expression of P2 receptors under certain pathological conditions may depend on epigenetic stimuli. Silenced P2 receptors which were once active during neural development could be reprogrammed in the event of tissue injury. A global study of P2 receptor density and mutations that affect their binding to specific nucleotides, may identify newer insights into the susceptibility of neurodegenerative disorders to specific populations. Furthermore, it is essential to understand the activity of various ectonucleotidases since the steady-state levels of various nucleotides have contrasting outcome in neuroinflammation. Therapeutically increasing the activity of specific ectonucleotidases following excessive ATP release is another approach to counter neuroinflammation. Finally, the two-hit hypothesis can also be extended to various other inflammatory disorders such as arthritis, toxin exposures including nerve gas poisoning, in the inflammatory model of cancer, and in psychological stress and depression. More studies in these areas will provide new roles for PBAIDs as effective anti-inflammatory drugs.
Figure 2.

P2 receptors modulate neuroinflammation. A simplified model based on the literature mentioned in this review summarizes the interactions between neurons and glial cells. Pro-inflammatory signals modulate P2X7-mediated release of IL-1β and surface expression of P2X4 receptors in the presence of ATP released by degenerating neurons and reactive astrocytes. On the surface of neurons, P2X7 receptors mediate apoptosis with caspase 3-dependent expression of P2X4 receptors as “flags” for microglial engulfment. Microglial migration to sites of insult is mediated by P2Y12 and adenosine A3 receptors and its neurophagic activity through P2Y6 receptors. While A1 adenosine receptors inhibit general inflammatory pathways, A2a receptors activate COX-2 as well as retract microglial processes. In healthy neurons, truncated P2X7 and P2Y2 receptors enhance α-secretase activity, preventing the formation of amyloid deposits. Amyloid formation is also immediately cleared through microglial phagocytosis, mediated by P2Y2 receptors; pinocytosis, through P2Y4 receptors; and activity of MMP-9, inhibited by the tonic activity of P2Y14 receptors.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Glossary
Abbreviations
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- ATP
adenosine triphosphate
- COX
cyclooxygenase
- IL-1β
interleukin-1β
- IRF8
interferon regulatory factor 8
- LPS
lipopolysaccharide
- MCP1
monocyte chemotactic protein 1
- MMP-9
matrix metalloproteinase 9
- NSAIDs
non-steroidal anti-inflammatory drugs
- PARP
poly (ADP-ribose) polymerase
- PBAIDs
P2 receptor-based anti-inflammatory drugs
- PGE2
prostaglandin E2
- PD
Parkinson’s disease
- TNF-α
tumour necrosis factor α
- UTP
uridine triphosphate
References
- Abbracchio M. P., Burnstock G., Verkhratsky A., Zimmermann H. (2009). Purinergic signalling in the nervous system: an overview. Trends Neurosci. 32, 19–29 10.1016/j.tins.2008.10.001 [DOI] [PubMed] [Google Scholar]
- Adén U., Halldner L., Lagercrantz I., Dalmau I., Ledent C., Fredholm B. B. (2003). Aggravated brain damage after hypoxic ischemia in immature adenosine A2A knockout mice. Stroke 34, 739–744 10.1161/01.str.0000060204.67672.8b [DOI] [PubMed] [Google Scholar]
- Adinolfi E., Cirillo M., Woltersdorf R., Falzoni S., Chiozzi P., Pellegatti P., et al. (2010). Trophic activity of a naturally occurring truncated isoform of the P2X7 receptor. FASEB J. 24, 3393–3404 10.1096/fj.09-153601 [DOI] [PubMed] [Google Scholar]
- Aguzzi A., Barres B. A., Bennett M. L. (2013). Microglia: scapegoat, saboteur, or something else? Science 339, 156–161 10.1126/science.1227901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajit D., Woods L. T., Camden J. M., Thebeau C. N., El-Sayed F. G., Greeson G. W., et al. (2014). Loss of P2Y2 nucleotide receptors enhances early pathology in the TgCRND8 mouse model of Alzheimer’s disease. Mol. Neurobiol. 49, 1031–1042 10.1007/s12035-013-8577-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akundi R. S., Candelario-Jalil E., Hess S., Hull M., Lieb K., Gebicke-Haerter P. J., et al. (2005). Signal transduction pathways regulating cyclooxygenase-2 in lipopolysaccharide-activated primary rat microglia. Glia 51, 199–208 10.1002/glia.20198 [DOI] [PubMed] [Google Scholar]
- Akundi R. S., Rivkees S. A. (2009). Hypoxia alters cell cycle regulatory protein expression and induces premature maturation of oligodendrocyte precursor cells. PLoS One 4:e4739 10.1371/journal.pone.0004739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akundi R. S., Zhi L., Sullivan P. G., Bueler H. (2013). Shared and cell type-specific mitochondrial defects and metabolic adaptations in primary cells from Pink1-deficient mice. Neurodegener. Dis. 12, 136–149 10.1159/000345689 [DOI] [PubMed] [Google Scholar]
- Alano C. C., Garnier P., Ying W., Higashi Y., Kauppinen T. M., Swanson R. A. (2010). NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 30, 2967–2978 10.1523/JNEUROSCI.5552-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amadio S., Montilli C., Picconi B., Calabresi P., Volonte C. (2007). Mapping P2X and P2Y receptor proteins in striatum and substantia nigra: an immunohistological study. Purinergic Signal. 3, 389–398 10.1007/s11302-007-9069-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni S., Parisi C., Pesaresi M. G., Rossi S., Carri M. T., Cozzolino M., et al. (2013). The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1–G93A microglia model of amyotrophic lateral sclerosis. J. Immunol. 190, 5187–5195 10.4049/jimmunol.1203262 [DOI] [PubMed] [Google Scholar]
- Arulkumaran N., Unwin R. J., Tam F. W. (2011). A potential therapeutic role for P2X7 receptor (P2X7R) antagonists in the treatment of inflammatory diseases. Expert Opin. Investig. Drugs 20, 897–915 10.1517/13543784.2011.578068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augusto E., Matos M., Sevigny J., El-Tayeb A., Bynoe M. S., Muller C. E., et al. (2013). Ecto-5′-nucleotidase (CD73)-mediated formation of adenosine is critical for the striatal adenosine A2A receptor functions. J. Neurosci. 33, 11390–11399 10.1523/JNEUROSCI.5817-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back S. A., Craig A., Luo N. L., Ren J., Akundi R. S., Ribeiro I., et al. (2006). Protective effects of caffeine on chronic hypoxia-induced perinatal white matter injury. Ann. Neurol. 60, 696–705 10.1002/ana.21008 [DOI] [PubMed] [Google Scholar]
- Barberà-Cremades M., Baroja-Mazo A., Gomez A. I., Machado F., Di Virgilio F., Pelegrín P. (2012). P2X7 receptor-stimulation causes fever via PGE2 and IL-1β release. FASEB J. 26, 2951–2962 10.1096/fj.12-205765 [DOI] [PubMed] [Google Scholar]
- Bekenstein U., Soreq H. (2013). Heterogeneous nuclear ribonucleoprotein A1 in health and neurodegenerative disease: from structural insights to post-transcriptional regulatory roles. Mol. Cell. Neurosci. 56, 436–446 10.1016/j.mcn.2012.12.002 [DOI] [PubMed] [Google Scholar]
- Bernier L.-P., Ase A. R., Boue-Grabot E., Seguela P. (2013). Inhibition of P2X4 function by P2Y6 UDP receptors in microglia. Glia 61, 2038–2049 10.1002/glia.22574 [DOI] [PubMed] [Google Scholar]
- Bian S., Sun X., Bai A., Zhang C., Li L., Enjyoji K., et al. (2013). P2X7 integrates PI3K/AKT and AMPK-PRAS40-mTOR signalling pathways to mediate tumour cell death. PLoS One 8:e60184 10.1371/journal.pone.0060184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucsein C., Zacharias R., Farber K., Pavlovic S., Hanisch U.-K., Kettenmann H. (2003). Purinergic receptors on microglial cells: functional expression in acute brain slices and modulation of microglial activation in vitro. Eur. J. Neurosci. 17, 2267–2276 10.1046/j.1460-9568.2003.02663.x [DOI] [PubMed] [Google Scholar]
- Brautigam V. M., Frasier C., Nikodemova M., Watters J. J. (2005). Purinergic receptor modulation of BV-2 microglial cell activity: potential involvement of p38 MAP kinase and CREB. J. Neuroimmunol. 166, 113–125 10.1016/j.jneuroim.2005.05.012 [DOI] [PubMed] [Google Scholar]
- Breitner J., Baker L., Drye L., Evans D., Lyketsos C., Ryan L., et al. (2013). Results of a follow-up study to the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT). Alzheimers Dement. 9, 714–723 10.1016/j.jalz.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camden J. M., Schrader A. M., Camden R. E., Gonzalez F. A., Erb L., Seye C. I., et al. (2005). P2Y2 nucleotide receptors enhance alpha-secretase-dependent amyloid precursor protein processing. J. Biol. Chem. 280, 18696–18702 10.1074/jbc.m500219200 [DOI] [PubMed] [Google Scholar]
- Casanovas A., Hernandez S., Tarabal O., Rossello J., Esquerda J. E. (2008). Strong P2X4 purinergic receptor-like immunoreactivity is selectively associated with degenerating neurons in transgenic rodent models of amyotrophic lateral sclerosis. J. Comp. Neurol. 506, 75–92 10.1002/cne.21527 [DOI] [PubMed] [Google Scholar]
- Castells A., Payá A., Alenda C., Rodríguez-Moranta F., Agrelo R., Andreu M., et al. (2006). Cyclooxygenase 2 expression in colorectal cancer with DNA mismatch repair deficiency. Clin. Cancer Res. 12, 1686–1692 10.1158/1078-0432.ccr-05-1581 [DOI] [PubMed] [Google Scholar]
- Castillo C. A., Albasanz J. L., Leon D., Jordan J., Pallas M., Camins A., et al. (2009). Age-related expression of adenosine receptors in brain from the senescence-accelerated mouse. Exp. Gerontol. 44, 453–461 10.1016/j.exger.2009.04.006 [DOI] [PubMed] [Google Scholar]
- Cavaliere F., Florenzano F., Amadio S., Fusco F. R., Viscomi M. T., D’Ambrosi N., et al. (2003). Up-regulation of P2X2, P2X4 receptor and ischemic cell death: prevention by P2 antagonists. Neuroscience 120, 85–98 10.1016/s0306-4522(03)00228-8 [DOI] [PubMed] [Google Scholar]
- Choi I. Y., Lee J. C., Ju C., Hwang S., Cho G. S., Lee H. W., et al. (2011). A3 adenosine receptor agonist reduces brain ischemic injury and inhibits inflammatory cell migration in rats. Am. J. Pathol. 179, 2042–2052 10.1016/j.ajpath.2011.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H. B., Ryu J. K., Kim S. U., McLarnon J. G. (2007). Modulation of the purinergic P2X7 receptor attenuates lipopolysaccharide-mediated microglial activation and neuronal damage in inflamed brain. J. Neurosci. 27, 4957–4968 10.1523/jneurosci.5417-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J., Tanaka K., Cao Y., Qi Y., Qiu J., Liang Y., et al. (2014). Identification of a plant receptor for extracellular ATP. Science 343, 290–294 10.1126/science.343.6168.290 [DOI] [PubMed] [Google Scholar]
- Choo A. M., Miller W. J., Chen Y.-C., Nibley P., Patel T. P., Goletiani C., et al. (2013). Antagonism of purinergic signalling improves recovery from traumatic brain injury. Brain 136, 65–80 10.1093/brain/aws286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A. R., Dean J. L., Saklatvala J. (2003). Post-transcriptional regulation of gene expression by mitogen-activated protein kinase p38. FEBS Lett. 546, 37–44 10.1016/s0014-5793(03)00439-3 [DOI] [PubMed] [Google Scholar]
- Compan V., Ulmann L., Stelmashenko O., Chemin J., Chaumont S., Rassendren F. (2012). P2X2 and P2X5 subunits define a new heteromeric receptor with P2X7-like properties. J. Neurosci. 32, 4284–4296 10.1523/JNEUROSCI.6332-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crain J. M., Nikodemova M., Watters J. J. (2009). Expression of P2 nucleotide receptors varies with age and sex in murine brain microglia. J. Neuroinflammation 6:24 10.1186/1742-2094-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl G., Keane R. W. (2012). Pannexin: from discovery to bedside in 11 ± 4 years? Brain Res. 1487, 150–159 10.1016/j.brainres.2012.04.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmellah A., Rayah A., Auger R., Cuif M.-H., Prigent M., Arpin M., et al. (2012). Ezrin/radixin/moesin are required for the purinergic P2X7 receptor (P2X7R)-dependent processing of the amyloid precursor protein. J. Biol. Chem. 287, 34583–34595 10.1074/jbc.M112.400010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva C. G., Specht A., Wegiel B., Ferran C., Kaczmarek E. (2009). Mechanism of purinergic activation of endothelial nitric oxide synthase in endothelial cells. Circulation 119, 871–879 10.1161/CIRCULATIONAHA.108.764571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos D., Grutzendler J., Yang G., Kim J. V., Zuo Y., Jung S., et al. (2005). ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 10.1038/nn1472 [DOI] [PubMed] [Google Scholar]
- Dean J. L., Sully G., Clark A. R., Saklatvala J. (2004). The involvement of AU-rich element-binding proteins in p38 mitogen-activated protein kinase pathway-mediated mRNA stabilization. Cell. Signal. 16, 1113–1121 10.1016/j.cellsig.2004.04.006 [DOI] [PubMed] [Google Scholar]
- de Oliveira A. C., Candelario-Jalil E., Langbein J., Wendeburg L., Bhatia H. S., Schlachetzki J. C., et al. (2012). Pharmacological inhibition of Akt and downstream pathways modulates the expression of COX-2 and mPGES-1 in activated microglia. J. Neuroinflammation 9:2 10.1186/1742-2094-9-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz-Hernández M., Díez-Zaera M., Sánchez-Nogueiro J., Gómez-Villafuertes R., Canals J. M., Alberch J., et al. (2009). Altered P2X7 receptor level and function in mouse models of Huntington’s disease and therapeutic efficacy of antagonist administration. FASEB J. 23, 1893–1906 10.1096/fj.08-122275 [DOI] [PubMed] [Google Scholar]
- Diaz-Hernandez J. I., Gomez-Villafuertes R., Leon-Otegui M., Hontecillas-Prieto L., Del Puerto A., Trejo J. L., et al. (2012). In vivo P2X7 inhibition reduces amyloid plaques in Alzheimer’s disease through GSK3β and secretases. Neurobiol. Aging 33, 1816–1828 10.1016/j.neurobiolaging.2011.09.040 [DOI] [PubMed] [Google Scholar]
- di Virgilio F. (2007). Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol. Sci. 28, 465–472 10.1016/j.tips.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Domercq M., Brambilla L., Pilati E., Marchaland J., Volterra A., Bezzi P. (2006). P2Y1 receptor-evoked glutamate exocytosis from astrocytes: control by tumour necrosis factor-α and prostaglandins. J. Biol. Chem. 281, 30684–30696 10.1074/jbc.m606429200 [DOI] [PubMed] [Google Scholar]
- During M. J., Spencer D. D. (1992). Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann. Neurol. 32, 618–624 10.1002/ana.410320504 [DOI] [PubMed] [Google Scholar]
- Emmrich J. V., Hornik T. C., Neher J. J., Brown G. C. (2013). Rotenone induces neuronal death by microglial phagocytosis of neurons. FEBS J. 280, 5030–5038 10.1111/febs.12401 [DOI] [PubMed] [Google Scholar]
- Feng Y. H., Li X., Wang L., Zhou L., Gorodeski G. I. (2006). A truncated P2X7 receptor variant (P2X7-j) endogenously expressed in cervical cancer cells antagonizes the full-length P2X7 receptor through hetero-oligomerization. J. Biol. Chem. 281, 17228–17237 10.1074/jbc.m602999200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D., Chiozzi P., Falzoni S., Hanau S., di Virgilio F. (1997). Purinergic modulation of IL-1β release from microglial cells stimulated with bacterial endotoxin. J. Exp. Med. 185, 579–582 10.1084/jem.185.3.579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari D., Pizzirani C., Adinolfi E., Lemoli R. M., Curti A., Idzko M., et al. (2006). The P2X7 receptor: a key player in IL-1 processing and release. J. Immunol. 176, 3877–3883 10.4049/jimmunol.176.7.3877 [DOI] [PubMed] [Google Scholar]
- Fiebich B. L., Biber K., Lieb K., van Calker D., Berger M., Bauer J., et al. (1996). Cyclooxygenase-2 expression in rat microglia is induced by adenosine A2a-receptors. Glia 18, 152–160 [DOI] [PubMed] [Google Scholar]
- Franco M. C., Ye Y., Refakis C. A., Feldman J. L., Stokes A. L., Basso M., et al. (2013). Nitration of Hsp90 induces cell death. Proc. Natl. Acad. Sci. U S A 110, E1102–E1111 10.1073/pnas.1215177110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm B. B. (2010). Adenosine receptors as drug targets. Exp. Cell Res. 316, 1284–1288 10.1016/j.yexcr.2010.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garção P., Szabó E. C., Wopereis S., Castro A. A., Tomé A. R., Prediger R. D., et al. (2013). Functional interaction between presynaptic α6β2-containing nicotinic and adenosine A2A receptors in the control of dopamine release in the rat striatum. Br. J. Pharmacol. 169, 1600–1611 10.1111/bph.12234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Hernández T., Rodríguez M. (2000). Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J. Comp. Neurol. 421, 107–135 [DOI] [PubMed] [Google Scholar]
- Gourine A. V., Dale N., Llaudet E., Poputnikov D. M., Spyer K. M., Gourine V. N. (2007). Release of ATP in the central nervous system during systemic inflammation: real-time measurement in the hypothalamus of conscious rabbits. J. Physiol. 585, 305–316 10.1113/jphysiol.2007.143933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyoneva S., Shapiro L., Lazo C., Garnier-Amblard E., Smith Y., Miller G. W., et al. (2014). Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson’s disease. Neurobiol. Dis. 67, 191–202 10.1016/j.nbd.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes S. E., Hollopeter G. E., Yang G., Kurpius D., Dailey M. E., Gan W. B., et al. (2006). The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 9, 1512–1519 10.1038/nn1805 [DOI] [PubMed] [Google Scholar]
- Hracskó Z., Baranyi M., Csölle C., Gölöncsér F., Madarász E., Kittel A., et al. (2011). Lack of neuroprotection in the absence of P2X7 receptors in toxin-induced animal models of Parkinson’s disease. Mol. Neurodegener. 6:28 10.1186/1750-1326-6-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jadhav S. P., Kamath S. P., Choolani M., Lu J., Dheen S. T. (2014). MicroRNA-200b modulates microglia-mediated neuroinflammation via the cJun/MAPK pathway. J. Neurochem. 130, 388–401 10.1111/jnc.12731 [DOI] [PubMed] [Google Scholar]
- Johnson S. C., Yanos M. E., Kayser E. B., Quintana A., Sangesland M., Castanza A., et al. (2013). mTOR inhibition alleviates mitochondrial disease in a mouse model of Leigh syndrome. Science 342, 1524–1528 10.1126/science.1244360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüni P., Nartey L., Reichenbach S., Sterchi R., Dieppe P. A., Egger M. (2004). Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet 364, 2021–2029 10.1016/s0140-6736(04)17514-4 [DOI] [PubMed] [Google Scholar]
- Kim T. W., Cho H. M., Choi S. Y., Suguira Y., Hayasaka T., Setou M., et al. (2013). (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis. 4:e919 10.1038/cddis.2013.447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B., Jeong H. K., Kim J. H., Lee S. Y., Jou I., Joe E. H. (2011). Uridine 5′-diphosphate induces chemokine expression in microglia and astrocytes through activation of the P2Y6 receptor. J. Immunol. 186, 3701–3709 10.4049/jimmunol.1000212 [DOI] [PubMed] [Google Scholar]
- Kinoshita M., Nasu-Tada K., Fujishita K., Sato K., Koizumi S. (2013). Secretion of matrix metalloproteinase-9 from astrocytes by inhibition of tonic P2Y14 receptor-mediated signal(s). Cell. Mol. Neurobiol. 33, 47–58 10.1007/s10571-012-9869-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishor A., Tandukar B., Ly Y. V., Toth E. A., Suarez Y., Brewer G., et al. (2013). Hsp70 is a novel posttranscriptional regulator of gene expression that binds and stabilizes selected mRNAs containing AU-rich elements. Mol. Cell. Biol. 33, 71–84 10.1128/MCB.01275-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiyota T., Gendelman H. E., Weir R. A., Higgins E. E., Zhang G., Jain M. (2013). CCL2 affects β-amyloidosis and progressive neurocognitive dysfunction in a mouse model of Alzheimer’s disease. Neurobiol. Aging 34, 1060–1068 10.1016/j.neurobiolaging.2012.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M. W., Metz L. M., Kovalchuk O. (2013). Epigenetic changes in patients with multiple sclerosis. Nat. Rev. Neurol. 9, 35–43 10.1038/nrneurol.2012.226 [DOI] [PubMed] [Google Scholar]
- Koizumi S., Shigemoto-Mogami Y., Nasu-Tada K., Shinozaki Y., Ohsawa K., Tsuda M., et al. (2007). UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 446, 1091–1095 10.1038/nature05704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai M. K., Tan M. G., Kirvell S., Hobbs C., Lee J., Esiri M. M., et al. (2008). Selective loss of P2Y2 nucleotide receptor immunoreactivity is associated with Alzheimer disease neuropathology. J. Neural Transm. 115, 1165–1172 10.1007/s00702-008-0067-y [DOI] [PubMed] [Google Scholar]
- Lazarowski E. R., Boucher R. C., Harden T. K. (2003). Mechanisms of release of nucleotides and integration of their action as P2X- and P2Y-receptor activating molecules. Mol. Pharmacol. 64, 785–795 10.1124/mol.64.4.785 [DOI] [PubMed] [Google Scholar]
- Lenertz L. Y., Gavala M. L., Zhu Y., Bertics P. J. (2011). Transcriptional control mechanisms associated with the nucleotide receptor P2X7, a critical regulator of immunologic, osteogenic and neurologic functions. Immunol. Res. 50, 22–38 10.1007/s12026-011-8203-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.-Q., Chen C., Dou Y., Wu H.-J., Liu Y.-J., Lou H.-F., et al. (2013). P2Y4 receptor-mediated pinocytosis contributes to amyloid beta-induced self-uptake by microglia. Mol. Cell. Biol. 33, 4282–4293 10.1128/MCB.00544-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisi L., Navarra P., Feinstein D. L., Dello Russo C. (2011). The mTOR kinase inhibitor rapamycin decreases iNOS mRNA stability in astrocytes. J. Neuroinflammation 8:1 10.1186/1742-2094-8-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Hu J., Wu J., Zhu C., Hui Y., Han Y., et al. (2012). α7 nicotinic acetylcholine receptor-mediated neuroprotection against dopaminergic neuron loss in an MPTP mouse model via inhibition of astrocyte activation. J. Neuroinflammation 9:98 10.1186/1742-2094-9-98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodygin D., Epanchintsev A., Menssen A., Diebold J., Hermeking H. (2005). Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 65, 4218–4227 10.1158/0008-5472.can-04-4407 [DOI] [PubMed] [Google Scholar]
- Lucas S.-M., Rothwell N. J., Gibson R. M. (2006). The role of inflammation in CNS injury and disease. Br. J. Pharmacol. 147, S232–S240 10.1038/sj.bjp.0706400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo L., Guida F., Imperatore R., Napolitano F., Gatta L., Cristino L., et al. (2014). The A1 adenosine receptor as a new player in microglial physiology. Glia 62, 122–132 10.1002/glia.22592 [DOI] [PubMed] [Google Scholar]
- Martinon F., Burns K., Tschopp J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of pro-IL1β. Mol. Cell 10, 417–426 10.1016/S1097-2765(02)00599-3 [DOI] [PubMed] [Google Scholar]
- Masuda T., Nishimoto N., Tomiyama D., Matsuda T., Tozaki-Saitoh H., Tamura T., et al. (2014). IRF8 is a transcriptional determinant for microglial motility. Purinergic Signal. [Epub ahead of print]. 10.1007/s11302-014-9413-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matos M., Augusto E., Agostinho P., Cunha R. A., Chen J. F. (2013). Antagonistic interactions between adenosine A2a receptors and Na+/K+-ATPase-α2 controlling glutamate uptake in astrocytes. J. Neurosci. 33, 18492–18502 10.1523/JNEUROSCI.1828-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarnon J. G., Ryu J. K., Walker D. G., Choi H. B. (2006). Upregulated expression of purinergic P2X7 receptor in Alzheimer disease and amyloid-β peptide-treated microglia and in peptide-injected rat hippocampus. J. Neuropathol. Exp. Neurol. 65, 1090–1097 10.1097/01.jnen.0000240470.97295.d3 [DOI] [PubMed] [Google Scholar]
- Monif M., Reid C. A., Powell K. L., Smart M. L., Williams D. A. (2009). The P2X7 receptor drives microglial activation and proliferation: a trophic role for P2X7R pore. J. Neurosci. 29, 3781–3791 10.1523/JNEUROSCI.5512-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore D., Iritani S., Chambers J., Emson P. (2000). Immunohistochemical localization of the P2Y1 purinergic receptor in Alzheimer’s disease. Neuroreport 11, 3799–3803 10.1097/00001756-200011270-00041 [DOI] [PubMed] [Google Scholar]
- Morente V., Perez-Sen R., Ortega F., Huerta-Cepas J., Delicado E. G., Miras-Portugal M. T. (2014). Neuroprotection elicited by P2Y13 receptors against genotoxic stress by inducing DUSP2 expression and MAPK signalling recovery. Biochim. Biophys. Acta 1843, 1886–1898 10.1016/j.bbamcr.2014.05.004 [DOI] [PubMed] [Google Scholar]
- Najjar S., Pearlman D. M., Alper K., Najjar A., Devinsky O. (2013). Neuroinflammation and psychiatric illness. J. Neuroinflammation 10:43 10.1186/1742-2094-10-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher J. J., Neniskyte U., Hornik T., Brown G. C. (2014). Inhibition of UDP/P2Y6 purinergic signalling prevents phagocytosis of viable neurons by activated microglia in vitro and in vivo. Glia 62, 1463–1475 10.1002/glia.22693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J., Wang P., Zhang J., Chen W., Gu L. (2013). Silencing of the P2X7 receptor enhances amyloid-β phagocytosis by microglia. Biochem. Biophys. Res. Comm. 434, 363–369 10.1016/j.bbrc.2013.03.079 [DOI] [PubMed] [Google Scholar]
- Noguchi Y., Shinozaki Y., Fujishita K., Shibata K., Imura Y., Morizawa Y., et al. (2013). Astrocytes protect neurons against methylmercury via ATP/P2Y(1) receptor-mediated pathways in astrocytes. PLoS One 8:e57898 10.1371/journal.pone.0057898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh K. M., Hwang J. Y., Follenzi A., Athanasiadou R., Miyawaki T., Greally J. M., et al. (2012). Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodelling is critical to ischemia-induced neuronal death. Proc. Natl. Acad. Sci. U S A 109, E962–E971 10.1073/pnas.1121568109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- North R. A., Jarvis M. F. (2013). P2X receptors as drug targets. Mol. Pharmacol. 83, 759–769 10.1124/mol.112.083758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogata T., Chuai M., Morino T., Yamamoto H., Nakamura Y., Schubert P. (2003). Adenosine triphosphate inhibits cytokine release from lipopolysaccharide-activated microglia via P2y receptors. Brain Res. 981, 174–183 10.1016/s0006-8993(03)03028-2 [DOI] [PubMed] [Google Scholar]
- Ohnishi T., Matsumura S., Ito S. (2009). Translocation of neuronal nitric oxide synthase to the plasma membrane by ATP is mediated by P2X and P2Y receptors. Mol. Pain 5:40 10.1186/1744-8069-5-40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsawa K., Sanagi T., Nakamura Y., Suzuki E., Inoue K., Kohsaka S. (2012). Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. J. Neurochem. 121, 217–227 10.1111/j.1471-4159.2012.07693.x [DOI] [PubMed] [Google Scholar]
- Orr A. G., Orr A. L., Li X. J., Gross R. E., Traynelis S. F. (2009). Adenosine A(2A) receptor mediates microglial process retraction. Nat. Neurosci. 12, 872–878 10.1038/nn.2341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinhal-Enfield G., Ramanathan M., Hasko G., Vogel S. N., Salzman A. L., Boons G. J., et al. (2003). An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7 and 9 and adenosine A(2A) receptors. Am. J. Pathol. 163, 711–721 10.1016/s0002-9440(10)63698-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons V., Serhan N., Gayral S., Malaval C., Nauze M., Malet N., et al. (2014). Role of the ubiquitin-proteasome system in the regulation of P2Y13 receptor expression: impact on hepatic HDL uptake. Cell. Mol. Life Sci. 71, 1775–1788 10.1007/s00018-013-1471-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purkayastha S., Cai D. (2013). Neuroinflammatory basis of metabolic syndrome. Mol. Metab. 2, 356–363 10.1016/j.molmet.2013.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi I. A., Mehler M. F. (2013). Epigenetic mechanisms governing the process of neurodegeneration. Mol. Aspects Med. 34, 875–882 10.1016/j.mam.2012.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren H., Stiles G. H. (1994). Posttranscriptional mRNA processing as a mechanism for regulation of human A1 adenosine receptor expression. Proc. Natl. Acad. Sci. U S A 91, 4864–4866 10.1073/pnas.91.11.4864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson L. E., Murrell-Lagnado R. D. (2013). The trafficking and targeting of P2X receptors. Front. Cell. Neurosci. 7:233 10.3389/fncel.2013.00233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz J. M., Chiozzi P., Ferrari D., Colaianna M., Idzko M., Falzoni S., et al. (2009). Activation of microglia by amyloid beta requires P2X7 receptor expression. J. Immunol. 182, 4378–4385 10.4049/jimmunol.0803612 [DOI] [PubMed] [Google Scholar]
- Serrano A., Mo G., Grant R., Pare M., O’Donnell D., Yu X. H., et al. (2012). Differential expression and pharmacology of native P2X receptors in rat and primate sensory neurons. J. Neurosci. 32, 11890–11896 10.1523/JNEUROSCI.0698-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw G., Kamen R. (1986). A conserved AU sequence from the 3′ untranslated region of GM-CSF mRNA mediates selective mRNA degradation. Cell 46, 659–667 10.1016/0092-8674(86)90341-7 [DOI] [PubMed] [Google Scholar]
- Shieh C.-H., Heinrich A., Serchov T., van Calker D., Biber K. (2014). P2X7-dependent, but differentially regulated release of IL-6, CCL-2 and TNF-α in cultured mouse microglia. Glia 62, 592–607 10.1002/glia.22628 [DOI] [PubMed] [Google Scholar]
- Solle M., Labasi J., Perregaux D. G., Stam E., Petrushova N., Koller B. H., et al. (2001). Altered cytokine production in mice lacking P2X7 receptors. J. Biol. Chem. 276, 125–132 10.1074/jbc.m006781200 [DOI] [PubMed] [Google Scholar]
- Suzuki T., Hide I., Ido K., Kohsaka S., Inoue K., Nakata Y. (2004). Production and release of neuroprotective tumour necrosis factor by P2X7 receptor-activated microglia. J. Neurosci. 24, 1–7 10.1523/jneurosci.3792-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely C. A., Zandi P. P. (2010). Non-steroidal anti-inflammatory drugs and Alzheimer’s disease: the epidemiological evidence. CNS Neurol. Disord. Drug Targets 9, 132–139 10.2174/187152710791012026 [DOI] [PubMed] [Google Scholar]
- Takenouchi T., Nakai M., Iwamaru Y., Sugama S., Tsukimoto M., Fujita M., et al. (2009). The activation of P2X7 receptor impairs lysosomal functions and stimulates the release of autophagolysosomes in microglial cells. J. Immunol. 182, 2051–2062 10.4049/jimmunol.0802577 [DOI] [PubMed] [Google Scholar]
- Tanabe T., Tohnai N. (2002). Cyclooxygenase isoenzymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 68–69, 95–114 10.1016/s0090-6980(02)00024-2 [DOI] [PubMed] [Google Scholar]
- Threlfell S., Lalic T., Platt N. J., Jennings K. A., Diesseroth K., Cragg S. J. (2012). Striatal dopamine release is triggered by synchronized activity in cholinergic interneurons. Neuron 75, 58–64 10.1016/j.neuron.2012.04.038 [DOI] [PubMed] [Google Scholar]
- Ulmann L., Hirbec C., Rassendren F. (2010). P2X4 receptors mediate PGE2 release by tissue-resident macrophages and initiate inflammatory pain. EMBO J. 29, 2290–2300 10.1038/emboj.2010.126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich H., Abbracchio M. P., Burnstock G. (2012). Extrinsic purinergic regulation of neural stem/progenitor cells: implications for CNS development and repair. Stem Cell Rev. 8, 755–767 10.1007/s12015-012-9372-9 [DOI] [PubMed] [Google Scholar]
- Varani K., Vincenzi F., Tosi A., Gessi S., Casetta I., Granieri G., et al. (2010). A2A adenosine receptor overexpression and functionality, as well as TNF-α levels, correlate with motor symptoms in Parkinson’s disease. FASEB J. 24, 587–598 10.1096/fj.09-141044 [DOI] [PubMed] [Google Scholar]
- Varma R., Chai Y., Troncoso J., Gu J., Xing H., Stojilkovic S., et al. (2009). Amyloid-β induces a caspase-mediated cleavage of P2X4 to promote purinotoxicity. Neuromol. Med. 11, 63–75 10.1007/s12017-009-8073-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C., Matteoli M. (2001). ATP mediates calcium signalling between astrocytes and microglial cells: modulation by IFN-γ. J. Immunol. 166, 6383–6391 10.4049/jimmunol.166.10.6383 [DOI] [PubMed] [Google Scholar]
- Viscomi M. T., Florenzano F., Conversi D., Bernardi G., Molinari M. (2004). Axotomy dependent purinergic and nitrergenic co-expression. Neuroscience 123, 393–404 10.1016/j.neuroscience.2003.09.030 [DOI] [PubMed] [Google Scholar]
- Walker D. G., Link J., Lue L.-F., Dalsing-Hernandez J. E., Boyes B. E. (2006). Gene expression changes by amyloid beta peptide-stimulated human post-mortem brain microglia identify activation of multiple inflammatory processes. J. Leukoc. Biol. 79, 596–610 10.1189/jlb.0705377 [DOI] [PubMed] [Google Scholar]
- Wang L., Olivecrona G., Gotberg M., Olsson M. L., Winzell M. S., Erlinge D. (2005). ADP acting on P2Y13 receptors is a negative feedback pathway for ATP release from human red blood cells. Circ. Res. 96, 189–196 10.1161/01.res.0000153670.07559.e4 [DOI] [PubMed] [Google Scholar]
- Webster C. M., Hokari M., McManus A., Tang X. N., Ma H., Kacimi R., et al. (2013). Microglial P2Y12 deficiency/inhibition protects against brain ischemia. PLoS One 8:e70927 10.1371/journal.pone.0070927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteside T. L., Jackson E. K. (2013). Adenosine and prostaglandin E2 production by human inducible regulatory T cells in health and disease. Front. Immunol. 4:212 10.3389/fimmu.2013.00212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xanthos D. N., Sandkuhler J. (2014). Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 15, 43–53 10.1038/nrn3617 [DOI] [PubMed] [Google Scholar]
- Xia M., Zhu Y. (2011). Signaling pathways of ATP-induced PGE2 release in spinal cord astrocytes are EGFR transactivation-dependent. Glia 59, 664–674 10.1002/glia.21138 [DOI] [PubMed] [Google Scholar]
- Xu J., Chalimoniuk M., Shu Y., Simonyi A., Sun A. Y., Gonzalez F. A., et al. (2003). Prostaglandin E2 production in astrocytes: regulation by cytokines, extracellular ATP, oxidative agents. Prostaglandins Leukot. Essent. Fatty Acids 69, 437–448 10.1016/j.plefa.2003.08.016 [DOI] [PubMed] [Google Scholar]
- Yedgar S., Krimsky M., Cohen Y., Flower R. J. (2007). Treatment of inflammatory diseases by selective eicosanoid inhibition: a double-edged sword? Trends Pharmacol. Sci. 28, 459–464 10.1016/j.tips.2007.07.005 [DOI] [PubMed] [Google Scholar]
- Yoshida Y., Yoshimi R., Yoshii Y., Kim D., Dey A., Xiong H., et al. (2014). The transcription factor IRF8 activates integrin-mediated TGF-β signalling and promotes neuroinflammation. Immunity 40, 187–198 10.1016/j.immuni.2013.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young L. E., Moore A. E., Sokol L., Meisner-Kober N., Dixon D. A. (2012). The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 10, 167–180 10.1158/1541-7786.MCR-11-0337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D., Worthington J., Timms J. F., Woo P. (2013). HNRNPA1 interacts with a 5’-flanking distal element of interleukin-6 and upregulates its basal transcription. Genes Immun. 14, 479–486 10.1038/gene.2013.41 [DOI] [PMC free article] [PubMed] [Google Scholar]