

Abstract
Upon encountering antigens, mature IgM-positive B lymphocytes undergo class-switch recombination (CSR) wherein exons encoding the default Cμ constant coding gene segment of the immunoglobulin (Ig) heavy-chain (Igh) locus are excised and replaced with a new constant gene segment (referred to as “Ch genes”, e.g., Cγ, Cε, or Cα). The B cell thereby changes from expressing IgM to one producing IgG, IgE, or IgA, with each antibody isotype having a different effector function during an immune reaction. CSR is a DNA deletional-recombination reaction that proceeds through the generation of DNA double-strand breaks (DSBs) in repetitive switch (S) sequences preceding each Ch gene and is completed by end-joining between donor Sμ and acceptor S regions. CSR is a multistep reaction requiring transcription through S regions, the DNA cytidine deaminase AID, and the participation of several general DNA repair pathways including base excision repair, mismatch repair, and classical nonhomologous end-joining. In this review, we discuss our current understanding of how transcription through S regions generates substrates for AID-mediated deamination and how AID participates not only in the initiation of CSR but also in the conversion of deaminated residues into DSBs. Additionally, we review the multiple processes that regulate AID expression and facilitate its recruitment specifically to the Ig loci, and how deregulation of AID specificity leads to oncogenic translocations. Finally, we summarize recent data on the potential role of AID in the maintenance of the pluripotent stem cell state during epigenetic reprogramming.
1. OVERVIEW OF GENOMIC ALTERATIONS IN B CELLS
The aptitude of the vertebrate immune system to recognize a highly diverse set of antigens is achieved via adaptive immune lymphocytes called B and T cells. B cells are integral to the humoral immune response due to the expression of immunoglobulin (Ig) surface receptors that can be secreted as antibodies. Ig molecules are comprised of two heavy and two light chain polypeptides that are held together through disulphide bridges. The heavy and light chains are comprised of a diverse amino-terminal variable region, responsible for antigen recognition and binding, and a carboxy-terminal constant region that determines effector function subsequent to antigen engagement. In humans and mice, three distinct genomic alterations create the enormous diversity of B cells and tailor them for the most efficacious immune response: V(D)J recombination, class-switch recombination (CSR), and somatic hypermutation (SHM). Some species such as sheep and chicken undergo a secondary diversification reaction termed gene conversion; this will not be discussed further here.
1.1. V(D)J recombination
In the fetal liver and adult bone marrow, developing B cells assemble the genes encoding the amino-terminal variable regions of the Ig heavy and light chains from component variable (V), diversity (D), and joining (J) “coding” gene segments through a somatic gene rearrangement process called V(D)J recombination (Schatz & Baltimore, 2004; Tonegawa, 1983). This reaction is initiated by the lymphocyte-specific endonuclease recombination activating genes 1 and 2 (RAG1/2) that generates DNA double-strand breaks (DSBs) at recombination signal sequences (RSSs) flanking the V, D, and J gene segments (Gellert, 2002; Oettinger, Schatz, Gorka, & Baltimore, 1990; Schatz, Oettinger, & Baltimore, et al., 1989). Components of the general classical nonhomologous end-joining (C-NHEJ) pathway joins DSBs between the gene segments to complete V(D)J recombination (Alt, Zhang, Meng, Guo, & Schwer, 2013; Boboila, Alt & Schwer, 2012; Schatz & Ji, 2011). In the Igh locus, the productive assembly of the recombined VDJ segment directly upstream of Cμ generates the Igμ polypeptide, which pairs with a similarly recombined κ or λ Ig light chain to generate an IgM molecule expressed on the surface of a mature naïve B cell. Alternative splicing of the VDJ exon to the Cδ constant region segment allows for expression of IgD. The combinatorial usage of different V, D, and J segments combined with sequence modifications during V(D)J recombination allows the generation of a naïve B cell population expressing a highly varied repertoire of low-affinity surface IgM (or IgD) molecules (Alt et al., 2013; Boboila, Alt & Schwer, 2012; Schatz & Ji, 2011). These mature naïve B cells circulate through peripheral lymphoid organs, where upon encountering their cognate antigens in germinal centers (GC), they undergo two additional genomic alterations in the forms of CSR and SHM (Fig. 1.1). Both CSR and SHM require the activity activation-induced cytidine deaminase (AID) (Muramatsu et al., 2000; Revy et al., 2000).
Figure 1.1.

Secondary immunoglobulin gene diversification. Mature B cells undergo class-switch recombination (CSR) and somatic hypermutation (SHM). During SHM, mutations are introduced into the rearranged variable region genes. CSR is initiated by transcription (grey arrows) through switch (S) regions (ovals) and requires AID and components of several DNA repair pathways. During CSR, the intervening DNA sequence between participating S regions is excised as a switch circle and a new constant region gene is juxtaposed downstream of the variable region exons.
1.2. Somatic hypermutation
During SHM, point mutations, and sometimes deletions and insertions, are introduced at a very high rate (10−2 to 10−3 per base pair per generation) into the recombined, expressed variable region of both the Igh and Igκ/λ genes (McKean et al., 1984; Papavasiliou & Schatz, 2002; Peled et al., 2008; Rajewsky, 1996; Wagner & Neuberger, 1996). While mutations are found throughout the variable region exons, they are more frequently observed at RGYW/WRCY (R = A/G, Y = C/T, W = A/T) “hot spot” motifs (Rogozin & Diaz, 2004). Typically, the mutations accumulate within the complementarity determining region (CDR) loops, which generally reside in the Ig regions that contact antigen. SHM allows for the selection of a B cell with increased affinity for its cognate antigen. It should be noted that characterization of broadly neutralizing antibodies to HIV-1 in humans revealed, for the first time, that the framework regions that scaffold the CDR loops, and are normally thought to be resistant and less tolerant of mutations, also accumulate SHM (Klein et al., 2013). As B cells accrue mutations, they are selected for their antigen-binding affinity, with those harboring mutations that enhance affinity receiving necessary survival signals to predominate the immune response.
SHM requires transcription through the expressed variable region exons, with mutations beginning from 100–200 bp downstream of the promoter and extending up to 1.5–2 kb further downstream, sparing the constant regions (Gearhart & Bogenhagen, 1983; Lebecque & Gearhart, 1990; Pech, Hochtl, Schnell, & Zachau, 1981; Peters & Storb, 1996; Rada, Gonzalez-Fernandez, Jarvis, & Milstein, 1994; Rothenfluh, Blanden, & Steele, et al., 1995; Winter, Sattar, Mai, & Gearhart, 1997). In transgenic SHM substrates, the variable regions could be replaced with non-Ig sequences without loss of mutability, indicating that the primary nucleotide sequence does not contribute to the targeting of the mutational machinery (Michael et al., 2002; Peters & Storb, 1996; Tumas-Brundage & Manser, 1997; Yelamos et al., 1995). In a simplified model for SHM (Fig. 1.2), transcription through the variable region exons promotes the recruitment of AID, which deaminates deoxycytidines (dCs) to deoxyuridines (dUs) (Petersen-Mahrt, Harris, & Neuberger, et al., 2002). Replication across the dU:dG mismatch results in the introduction of transition mutations. On the other hand, removal of dU by the base excision repair (BER) enzyme uracil DNA glycosylase (UNG) prior to replication generates an abasic site, which when replicated across by error-prone DNA polymerases leads to the introduction of both transition and transversion mutations (Petersen-Mahrt et al., 2002). In addition, the dU:dG mismatch could be recognized by mismatch repair (MMR) proteins, which coupled with gap-filling by errorprone DNA polymerases can result in point mutations at A:T residues and/or short insertions and deletions throughout the variable region (Peled et al., 2008; Petersen-Mahrt et al., 2002).
Figure 1.2.
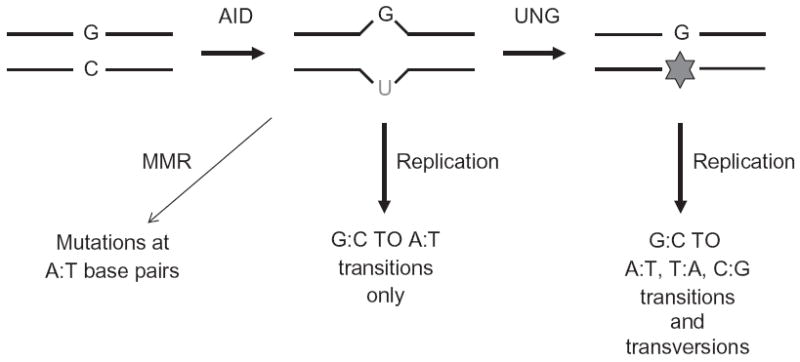
Model for SHM. AID deaminates dCs to dUs within RGYW hotspots in variable region genes. Activities of mismatch repair (MMR) and base excision repair (BER) proteins (e.g., UNG of the BER pathway) coupled with DNA replication generate transition and transversion mutations.
Several excellent recent reviews have delved into the mechanism of SHM and should be consulted for a more in-depth treatise (Di Noia & Neuberger, 2007; Pavri & Nussenzweig, 2011; Saribasak & Gearhart, 2012; Teng & Papavasiliou, 2007). One aspect of SHM that merits brief discussion here is the mechanism by which AID, an exclusively single-stranded (ss) DNA-specific cytidine deaminase, accesses the variable region exons. One possibility is that transcription promotes negative supercoiling of DNA upstream of the elongating RNA polymerase, thus creating DNA topology conducive for AID binding (Kodgire, Mukkawar, Ratnam, Martin, &Storb, 2013; Shen &Storb, 2004). Another, not necessarily mutually exclusive, proposal is that the ssDNA binding protein replication protein A (RPA), which interacts with AID, stabilizes ssDNA bubbles within transcribed variable region exons, allowing AID-mediated deamination to facilitate SHM (Chaudhuri, Khuong, & Alt, et al., 2004). The differential requirement of RPA in CSR and SHM and its phosphorylation-dependent binding to AID will be addressed later.
1.3. Class-switch recombination
Immunoglobulin heavy-chain CSR exchanges the default Cμ exons for an alternative set of downstream Igh C-region (Ch) exons, that is, Cγ, Cε, or Cα (Fig. 1.1). Thereby, the B cell is altered from expressing IgM to one producing a secondary antibody isotype such as IgG, IgE, or IgA, respectively, with each antibody class having distinct effector functions during an immune response (Honjo, Kinoshita, & Muramatsu, et al., 2002; Stavnezer, Guikema, & Schrader, et al., 2008). CSR is a deletional-recombination reaction that occurs between repetitive DNA elements termed “switch” or S regions that precede each Ch gene. According to the prevailing model for CSR, transcription through S regions allows generation of RNA:DNA hybrid structures, such as R-loops, revealing stretches of ssDNA that serve as substrates for AID (Chaudhuri & Alt, 2004; Chaudhuri et al., 2007; Yu & Lieber, 2003). Subsequent processing by components of the BER and MMR pathways convert the deaminated residues into DSBs. End-joining of DSBs between two S regions excises the intervening DNA sequence and completes CSR by placing a new constant gene directly downstream of the rearranged variable region exons. Thus, CSR allows for the generation of Ig molecules with the same affinity for the antigen, but with new effector functions (Boboila, Alt & Schwer, 2012; Stavnezer, 2011).
2. INITIATION OF CSR: S REGIONS AND GERMLINE TRANSCRIPTION
2.1. Requirement of S regions in CSR
The mouse Igh locus is comprised of eight Ch genes, each of which, except Cδ, is preceded by a 1–12 kb long S region DNA element (Fig. 1.1). The human Igh locus is similarly organized, a notable exception being the presence of an uncharacterized σδ repetitive sequence located between Cμ and Cδ that allows for the CSR-like exchange of IgM to IgD (Chen et al., 2009; Kluin et al., 1995). The primary sequences of S regions are not identical, but they do share similarities in that they are unusually G-rich on the template strand and consist of recurring motifs of varying lengths. Sμ, for example, is approximately 3.2 kb long and is comprised of GAGCT pentameric motifs, with the AGCT palindromic sequence representing a canonical RGYW/WRCY sequence. Sγ1 is approximately 10 kb long and has multiple RGYW/WRCY sequences embedded within 49 bp repeat units. Other repeat motifs found in S regions include GGGGA/T in Sμ, Sγ1, Sγ2b, and Sγ3 and GGGCT in Sε and Sα. Most of the characterized switch junctions from cells that have undergone CSR fall within the S regions, thus it was logically postulated that S regions serve as recombination targets to drive CSR (Davis, Kim, & Hood, et al., 1980; Dunnick, Hertz, Scappino, & Gritzmacher, 1993; Dunnick, Rabbitts, & Milstein, et al., 1980; Kataoka, Yamawaki-Kataoka, Yamagishi, & Honjo, 1979; Nikaido, Nakai, & Honjo, et al., 1981; Obata et al., 1980; Sakano, Maki, Kurosawa, Roeder, & Tonegawa, 1980; Stavnezer, 1996; Takahashi et al., 1982; Zarrin, Goff, Senger, & Alt, 2008).
Unequivocal evidence for the requirement of S regions in CSR came from gene targeting studies. Deletion of Sγ1 completely abrogated CSR to IgG1 without affecting CSR to the other isotypes (Shinkura et al., 2003). Deletion of Sμ also led to a severe defect in CSR to IgG1, as would be expected upon loss of the donor S region (Khamlichi et al., 2004; Luby, Schrader, Stavnezer, & Selsing, 2001). A more recent study, however, revealed that Sμ deletion still allowed significant levels of CSR to IgG1, leading to the provocative proposal that in the absence of Sμ, Sγ1 could serve as a donor S region (Zhang et al., 2010). A direct correlation between S region length and CSR frequency was shown, at least for Sγ1, when it was replaced with 49 bp Sγ1 repeats of varying lengths (Zarrin, Tian, Wang, Borjeson, & Alt, 2005). This correlation is consistent with the difference in CSR frequency between IgG1 (12 kb S region) versus IgE (1 kb S region) observed in ex vivo stimulated splenic B cells. While IgG1 was the predominant antibody isotype following switching (~30% of total B cells), a remarkable increase in CSR to IgE (3% to >40% IgE) was observed after deletion of Sγ1 (Misaghi et al., 2010). The unique structural and/or sequence features of S regions are essential for CSR, as substitution of Sγ1 with non-S region sequence was unable to support CSR to IgG1, while replacing it with wildtype or synthetic Sγ2b sequences restored CSR to a level observed for a Sγ1 sequence of similar length (2 kb) (Zarrin et al., 2008). Taken together, these experiments provided strong evidence that S regions serve as targets for the recombination reaction.
2.2. Requirement of germline transcription
The molecular basis for the role of S regions is intricately linked to the essential requirement of “germline” transcription in CSR (Jung, Rajewsky, & Radbruch, et al., 1993; Zhang, Bottaro, Li, Stewart, &Alt, 1993). Individual Ch genes (except Cδ) are organized as transcription units comprised of a cytokine/activation-inducible promoter upstream of an intervening exon (I-exon), an intronic S region, and Ch exons (Lennon & Perry, 1985) (Fig. 1.3). The primary transcript, initiating from the promoter upstream of the I-exon, proceeds through the S region and terminates downstream of the Ch exons. Splicing of the primary transcript removes the intronic S region and joins the I-exon to the Ch exons. The mature transcripts are polyadenylated, but do not code for any proteins, and are referred to as germline or sterile transcripts (Alt, Rosenberg, Casanova, Thomas, & Baltimore, 1982; Alt, Rosenberg, Enea, Siden, & Baltimore, 1982; Stavnezer-Nordgren & Sirlin, 1986; Yancopoulos et al., 1986). Transcription through individual Ch genes induces CSR to that particular isotype (Snapper, Finkelman, & Paul, et al., 1988). For example, ex vivo activation of mouse splenic B cells with bacterial lipopolysaccharides (LPS) activates transcription through Iγ2b-Sγ2b-Cγ2b and Iγ3-Sγ3-Cγ3 transcription units and promotes CSR to IgG2b and IgG3, respectively. Conversely, addition of IL-4 inhibits transcription through γ2b and γ3 units and instead activates transcription through Iγ1-Sγ1-Cγ1 and Iε-Sε-Cε transcription units and CSR to IgG1 and IgE, respectively. Likewise, activation with TGF-β activates the Iα promoter and allows for CSR to IgA (Lutzker, Rothman, Pollock, Coffman, & Alt, 1988; Rothman, Lutzker, Cook, Coffman, & Alt, 1988; Stavnezer et al., 1988). The μ promoter, on the other hand, drives constitutive transcription through Sμ even in unstimulated B cells (Li et al., 1994). It is to be noted that ex vivo activation of mouse splenic B cells in culture with distinct combinations of cytokines/activators has been a convenient and well-established tool to elucidate CSR.
Figure 1.3.
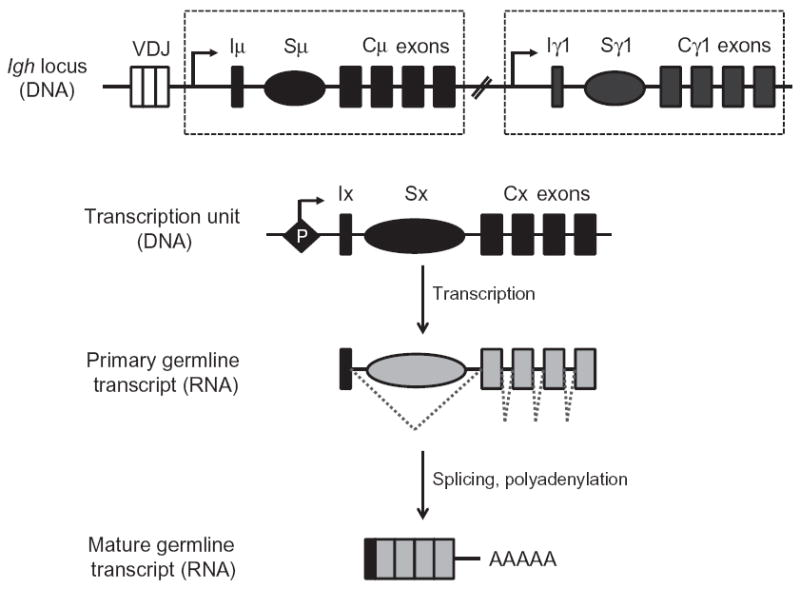
Germline transcription through S regions. Each constant region gene is comprised of a transcription unit with a cytokine-inducible promoter (P), an intervening (I)-exon, S region, and Ch exons. The primary transcript is spliced and polyadenylated to generate a noncoding mature transcript.
The strict correlation between induction of germline transcription and activation of CSR to particular isotypes provided a strong mechanistic link between the two processes. This notion was further validated through mutational analyses. Deletion of I-exon promoters or regulatory regions (3′-RR) at the 3′-end of Igh abolished or severely reduced CSR, while replacing the inducible I-exon promoters with heterologous constitutive promoters drove CSR in a cytokine-independent fashion (Bottaro et al., 1994; Cogne et al., 1994; Jung et al., 1993; Kuzin et al., 2000; Lorenz, Jung, & Radbruch, et al., 1995; Manis, van der Stoep, et al., 1998; Michaelson, Giannini, & Birshtein, et al., 1995; Pinaud et al., 2001; Qiu, Harriman, & Stavnezer, et al., 1999; Seidl et al., 1998; Vincent-Fabert et al., 2010; Zhang et al., 1993). These studies thus provided experimental evidence for the notion that germline transcription of particular Ch genes renders them “accessible” for CSR (Stavnezer-Nordgren & Sirlin, 1986; Yancopoulos et al., 1986). We now know that important components of this accessibility includeRNApolymerase II (Pol II)-dependent recruitment of AID to S regions (Pavri & Nussenzweig, 2011), transcription-coupled modifications of histones at S regions (Jeevan-Raj et al., 2011; Wang, Wuerffel, Feldman, Khamlichi, & Kenter, 2009), and generation of RNA:DNA hybrid structures that provide ssDNA substrates of AID (Chaudhuri et al., 2007).
Several intriguing reports have also suggested that germline switch transcripts might have mechanistic roles in CSR independent of transcription alone. Deletion of the Iγ1-exon splice donor site to inhibit splicing of the primary switch transcripts specifically abrogated CSR to IgG1, even though transcription through Sγ1 was unaffected (Lorenz et al., 1995). Additionally, expression of Sα transcripts from a plasmid enhanced CSR to IgA in a B lymphoma cell line (Muller, Giese, Henry, Mushinski, & Marcu, 1998). Collectively, the findings suggest that germline switch transcripts might have roles in the regulation of CSR, even though their mechanism of action is yet to be elucidated. We will thus focus on the well-characterized role of germline transcription in the generation of substrates compatible for AID-mediated deamination.
It has now been well documented that transcription through S regions in vitro and in vivo generates R-loops in which the template DNA strand is stably hybridized to the RNA while the nontemplate strand is looped out as ssDNA (Daniels & Lieber, 1995; Kao et al., 2013; Mizuta et al., 2003; Reaban & Griffin, 1990; Shinkura et al., 2003; Tian & Alt, 2000; Yu, Chedin, Hsieh, Wilson, & Lieber, 2003) (Fig. 1.4). The looped out nontemplate strand has also been proposed to assume additional structures such as stem-loops (Tashiro, Kinoshita, & Honjo, et al., 2001), or fourstranded G-quartets that are stabilized by Hoogsteen pairing between G-residues (Dempsey, Sun, Hanakahi, & Maizels, 1999; Sen & Gilbert, 1988). However, while R-loop formation (where the displaced template strand is single stranded) at transcribed S regions has been experimentally demonstrated in B cells undergoing CSR (Kao et al., 2013; Mizuta et al., 2003; Yu et al., 2003), there is no direct evidence that the ssDNA assumes additional secondary structures in switching B cells.
Figure 1.4.
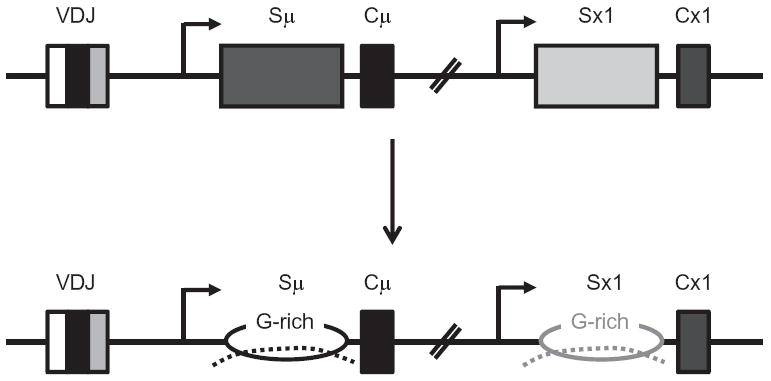
Transcription-driven R-loop formation at S regions. Transcription occurs through each participating S region (indicated by arrows) and the nascent transcripts (dotted lines) remain bound to template DNA due to high GC content of the sequence. This displaces the nontemplate strands as ssDNA substrates, forming structures called R-loops.
Formation of RNA:DNA hybrids at transcribed S regions is predicted from the primary sequence of S regions, given that the thermodynamic stability of G-rich RNA:C-rich DNA is much stronger than that of G-rich DNA:C-rich DNA (Ratmeyer, Vinayak, Zhong, Zon, & Wilson, 1994). This prediction is consistent with in vitro experiments where an S region transcribed in the physiological orientation forms stable R-loops, a structure not observed when the S region is transcribed in a nonphysiological orientation (G-rich template strand, C-rich RNA) (Daniels & Lieber, 1995; Mizuta et al., 2003; Reaban & Griffin, 1990; Shinkura et al., 2003; Tian & Alt, 2000). The physiological relevance of R-loops in CSR came from an elegant genetic study where a 1 kb synthetic S region with a G-rich nontemplate strand and the ability to form R-loops in vitro supported CSR in vivo; however, the same sequence transcribed in the non-R-loop forming reverse orientation was severely impaired at mediating CSR (Shinkura et al., 2003). The generally accepted role of R-loops in CSR is that they provide ssDNA substrates for the key enzyme AID (Chaudhuri et al., 2007).
3. INDUCTION OF DNA LESIONS IN CSR: ESSENTIAL REQUIREMENT OF AID
3.1. Discovery of AID
The discovery of AID was a notable triumph that came at a time when the field was struggling to come to consensus on a cohesive model for the mechanisms of SHM and CSR. The discovery was facilitated by characterization of a B lymphoma line CH12F3 that can be induced to undergo CSR from IgM to IgA with high frequency (Nakamura et al., 1996). Using a PCRbased cDNA subtraction screen, AID was identified as a gene that was strongly induced when CH12F3 cells were stimulated to undergo CSR (Muramatsu et al., 1999). AID, encoded from the Aicda locus, is a protein of 198 amino acids (24 kDa) that is barely detectable in unstimulated CH12F3 cells, but increases in expression a remarkable 10-fold in 6 h following stimulation (Muramatsu et al., 1999). The essential role of AID in SHM and CSR came from two co-published studies. Generation of AID-deficient mice showed a striking and complete defect in both CSR and SHM (Muramatsu et al., 2000), while AID mutations in patients with an autosomal recessive form of hyper-IgM syndrome (HIGM2) resulted in a lack of SHM and CSR (Revy et al., 2000).
3.2. AID is a single-strand DNA deaminase
AID is one of 12 members of the APOBEC family of DNA/RNA cytidine deaminases. APOBEC1, the first to be discovered, is unique in its ability to selectively deaminate a specific C on a defined mRNA (Davidson & Shelness, 2000), while other members deaminate cytidines at multiple residues on ssDNA. In vitro biochemical studies demonstrated that AID is a ssDNA-specific cytidine deaminase, converting dCs to dUs, with no observable activity on double-strand DNA, RNA, or RNA:DNA hybrids (Chaudhuri et al., 2003; Dickerson, Market, Besmer, & Papavasiliou, 2003; Nabel, Lee, Wang, & Kohli, 2013; Pham, Bransteitter, Petruska, & Goodman, 2003; Ramiro, Stavropoulos, Jankovic, & Nussenzweig, 2003; Sohail, Klapacz, Samaranayake, Ullah, & Bhagwat, 2003; Yu, Roy, Bayramyan, Haworth, & Lieber, 2005). Based on bacterial cytidine deaminases that are homologous to APOBEC1 and AID, the deamination reaction proceeds via a direct nucleophilic attack at position 4 of the pyrimidine ring of cytosine by Zn2+ coordinated to AID. While X-ray crystallographic andNMRstudies have revealed the structures of APOBEC family members (Vasudevan et al., 2013), the structure of AID/APOBEC proteins bound to nucleic acids have not been solved, leading to a dearth in our understanding of how AID engages its substrates. Still, in vitro assays identified an 11 amino acid hotspot recognition motif (residues 113–123), distinct from the active site residues (56–58 and 87–90) that confer specificity for the RGYW/WRCY hotspot motif. Replacing this recognition loop with one from another APOBEC family member(APOBEC3G or APOBEC3F)changed the mutation signature of AID toward that of the donor protein (Carpenter, Rajagurubandara, Wijesinghe, & Bhagwat, 2010; Pham, Calabrese, Park, & Goodman, 2011; Wang, Rada, & Neuberger, et al., 2010).
AID was shown to be a processive enzyme in vitro, catalyzing numerous cytidine deaminations on the same ssDNA fragment (Pham et al., 2003, 2011). However, AID is an inefficient enzyme, deaminating only about 3% of the cytidines even at preferred hotspot motifs. Mathematical formulation based on in vitro deamination data led to a “random walk” model wherein AID mainly traverses ssDNA by sliding in a bidirectional fashion, with a deamination probability of 1–7% per hotspot motif encounter (Mak, Pham, Afif, & Goodman, 2013; Pham et al., 2011). It has been suggested that the haphazard and inefficient activity of AID ensures that a wide variety of variable region mutations are tested for antigen affinity during SHM (Mak et al., 2013; Pham et al., 2011). How this inefficiency aids in CSR, where a high density of breaks is favored, is not clear at present. It is to be noted that the majority of in vitro studies have been carried out with bulky tags on AID, whose effect on the efficiency of the reaction has not been determined. Likewise, how the rate of deamination is altered by modifications in AID or its interactions with other proteins remains to be elucidated. Despite these unresolved issues that limit precise examination of its enzymatic activity, it is now unequivocally established that AID mediates CSR (and SHM) by acting as a ssDNA cytidine deaminase.
The ssDNA specificity of AID naturally leads to the question of how such substrates are generated during CSR. As discussed earlier, S regions, when transcribed in vitro in the physiological orientation, form R-loops in which cytidines in the displaced G-rich nontemplate strand can be efficiently deaminated by AID (Chaudhuri et al., 2003) (Fig. 1.4). A more direct correlation between R-loop formation and AID activity came from the observations that a G-rich DNA that supported CSR in vivo was efficiently deaminated by AID in vitro when the reaction was coupled to transcription, while a C-rich DNA that did not target CSR in vivo was a poor AID substrate in vitro (Chaudhuri et al., 2003). These observations led to a plausible and prevailing model wherein transcription through mammalian S regions generates ssDNA R-loop substrates to allow for the cytidine deaminase activity of AID. The deaminated DNA is subsequently processed into DSBs through the activities of BER and MMR proteins (Fig. 1.5). This R-loop-based model for AID access to S region DNA provides a convincing explanation for the essential requirements of S regions and S region transcription during CSR.
Figure 1.5.
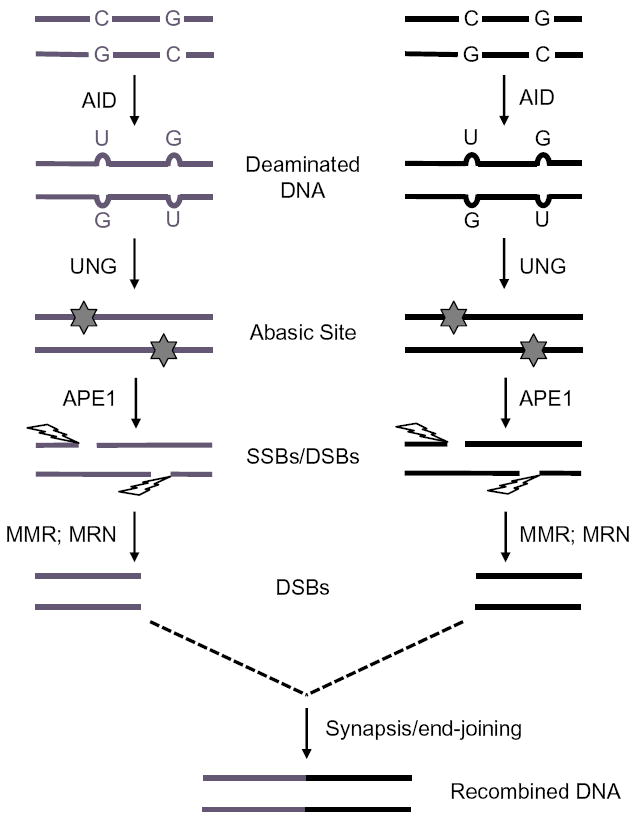
Processing of deaminated DNA during CSR. S regions deaminated by AID are processed by UNG, APE1, and MMR proteins to generate blunt DSBs that are synapsed and ligated to complete CSR.
3.3. RNA editing by AID?
The sequence homology of AID with a bona fide RNA-editing enzyme, APOBEC1, led to the proposal that AID deaminates cytidines in specific mRNAs and that the “edited” mRNAs can then encode proteins required for CSR and SHM (Honjo, 2008). The requirement for de novo protein synthesis for DSB formation during CSR was argued to support this model (Begum et al., 2007, 2009; Begum, Kinoshita, Muramatsu, et al., 2004; Doi, Kinoshita, Ikegawa, Muramatsu, & Honjo, 2003), although an equally plausible explanation is that CSR requires labile proteins that function downstream of DNA deamination. In a variation of this mRNA-editing model, it was proposed that AID modifies microRNAs (miRNAs) that regulate expression of Topoisomerase I (Top1) (Kobayashi et al., 2009). According to this notion, AID modifies miRNAs that regulate Top1 mRNA, leading to a decrease in the amount of Top1 in cells undergoing CSR, which in turn alters DNA structure and induces cleavage in S regions (Kobayashi et al., 2009). However, the mechanism by which Top1 levels can specifically induce DSBs in S regions remains unclear, and the identity of the RNA substrates of AID remain elusive. Furthermore, recombinant AID failed to deaminate cytidines in RNA (Nabel et al., 2013), even though one could argue that AID can only act on a very specific RNA moiety. Overall, at present, RNA-editing by AID remains a provocative idea. On the other hand, the observations that (a) uracils accumulate within S regions in an AID-dependent fashion (Maul et al., 2011), (b) deficiency in processing uridines in DNA severely impairs CSR (discussed below), and (c) AID associates with S regions in vivo (Nambu et al., 2003), provide compelling and overwhelming support for the idea that AID functions in CSR (and SHM)through directDNAdeamination, converting dCs to dUs at S regions.
4. PROCESSING OF DEAMINATED DNA: REQUIREMENTS FOR BER AND MMR PROTEINS
4.1. Removal of uracil residues from deaminated DNA
The detection of episomal circles comprised of the DNA excised between two S regions suggested that CSR proceeds through DSB intermediates (Iwasato, Shimizu, Honjo, & Yamagishi, 1990; Matsuoka, Yoshida, Maeda, Usuda, & Sakano, 1990). Additional evidence came from ligationmediated PCR experiments that detected AID-dependent DSBs at S regions, and from the presence of phosphorylated H2AX (γH2AX) foci, a well-characterized marker of DSBs, at the Igh locus (Petersen et al., 2001; Schrader, Guikema, Linehan, Selsing, & Stavnezer, 2007; Schrader, Linehan, Mochegova, Woodland, & Stavnezer, 2005; Wuerffel, Du, Thompson, & Kenter, 1997). Finally, the requirement for several DSB sensors and repair factors, including 53BP1, ATM, and components of the C-NHEJ pathway, strongly indicates that DSBs are generated during CSR (Alt et al., 2013). It is now generally believed that the BER and MMR pathways contribute to the conversion of deaminated cytidines into DSBs.
Components of the BER pathway play a major role in CSR. Through BER, the AID-introduced dU in S region DNA is removed by UNG to generate an abasic site (Fig. 1.5). The abasic site is recognized by the apurinic/apyrimidinic endonuclease APE1 generating a nick (Petersen-Mahrt et al., 2002). A closely spaced, similarly created nick on the opposite strand generates a staggered DSB. As predicted, mutations in UNG in humans and mice lead to impaired DSB formation at S regions and a severe block in CSR (Di Noia & Neuberger, 2002; Imai et al., 2003; Rada, Williams, et al., 2002; Schrader et al., 2005). Additionally, both mice heterozygous for APE1 deletion and CH12F3 cells with a homozygous deletion of APE1 have significantly impaired CSR (Guikema, Stavnezer, & Schrader, et al., 2010; Masani, Han, & Yu, et al., 2013; Schrader, Guikema, Wu, & Stavnezer, 2009). Overall, there is strong genetic data to support the proposed AID/UNG/APE1 pathway in CSR. In contrast to the wellestablished activities of UNG, it has been suggested that UNG might play a noncanonical role, since a UNG mutant deficient in uracil-removal activity can still promote CSR (Begum et al., 2007, 2009; Begum, Kinoshita, Kakazu, et al., 2004). A simpler, alternative explanation for this observation is that the residual activity in the mutant UNG protein is sufficient to remove uracils during CSR (Stivers, 2004).
During CSR, the dU:dG mismatch could also be processed via MMR (Rada, DiNoia,&Neuberger, et al., 2004). During this process, the Msh2–Msh6 heterodimer recognizes dU:dG mismatches, triggering the recruitment of adaptors and other effectors, such as the endonuclease heterodimer Pms2/Mlh1 and exonuclease 1 (Exo1), to generate nicks distal to the mismatch and subsequent conversion of the nicks into ssDNAgaps. Such activities occurring on opposite DNA strands could generate DSBs (Chaudhuri & Alt, 2004; Stavnezer et al., 2008). In mice, mutations in genes encoding MMR proteins (Msh2, Msh6, Mlh1, Pms2, or Exo1) significantly impair CSR, providing further support for this model (Bardwell et al., 2004; Ehrenstein & Neuberger, 1999; Ehrenstein, Rada, Jones, Milstein, & Neuberger, 2001; Roa et al., 2010; Schrader et al., 2004; Schrader, Vardo & Stavnezer, 2002, 2003).
4.2. Deamination of the template strand
Conversion of deaminated residues into DSBs during CSR requires AID activity on both DNA strands; however, purified AID, in vitro, could efficiently deaminate only the displaced single-stranded nontemplate strand, leaving the template strand largely untouched (Chaudhuri et al., 2003). Several proposals have been put forward to explain how template-strand deamination could be achieved. One possibility is that antisense transcription through S regions might allow AID to access both strands of the DNA (Perlot, Li, & Alt, et al., 2008). Additionally, AID has been shown to interact with components of the RNA exosome complex (Basu et al., 2011) and this interaction, perhaps in combination with RNaseH activity (Yu & Lieber, 2003), might remove the nascent transcript leading to R-loop collapse. In this model, the complementary DNA strands misalign due to the repetitive nature of S regions, resulting in exposed stretches of ssDNA on both DNA strands, allowing for AID access (Yu & Lieber, 2003). Finally, it has been proposed that negative supercoiling upstream of the elongating Pol II can allow access of AID to both strands of DNA (Kodgire et al., 2013; Shen & Storb, 2004). Thus, multiple processes could potentially allow for deamination of the template strand, but the precise mechanism is yet to be fully elucidated.
4.3. Conversion of ssDNA breaks into DSBs
Both BER and MMR have the potential to process deaminated DNA. However, it appears that CSR is more reliant on BER, given that UNG deficiency leads to a profound defect in CSR frequency, reducing it to less than 10% of wild-type levels (Di Noia & Neuberger, 2002; Imai et al., 2003; Rada et al., 2004). Residual CSR was completely abolished in B cells deficient for both UNG and Msh2 (or Msh6), suggesting that MMR plays a minor or back-up role in the initial processing of deaminated DNA (Rada et al., 2004; Shen, Tanaka, Bozek, Nicolae, & Storb, 2006). However, mutations in MMR genes (e.g., Msh2, EXO1) result in a nearly 30% defect in CSR, an effect greater than would be expected if MMR is acting as a mere back-up to the BER pathway in processing deaminated cytidines. It has been argued that MMR plays a role in the conversion of single-strand breaks (SSBs) on opposite DNA strands into DSBs (Stavnezer et al., 2008). If SSBs generated by AID–UNG–APE1 on opposite DNA strands are proximal to each other, they could be spontaneously converted into DSBs; however, if they are distal to each other, Msh2–Msh6 may bind to dU:dG mismatches that have not been processed by UNG and recruit EXO1 to excise DNA from the nearest 5′-SSB to the mismatch, creating a DSB with a 5′-overhang, which can subsequently be filled in by a DNA polymerase (Stavnezer et al., 2008). Indeed, in B cells in which the tandem Sμ repeats have been deleted, CSR is only reduced twofold, but is largely dependent on Msh2 and Mlh1, likely due to the requirement of the MMR proteins in conversion of distal SSBs that occur as a result of fewer AID targets (Min et al., 2003). Thus, while the MMR proteins could provide a complementary route to the major UNG-mediated processing of deaminated DNA, it is likely that the primary requirement of the MMR pathway in CSR lies in its ability to convert nicks and SSBs on opposite DNA strands into DSBs.
The majority of DSBs at S regions are staggered with 5′ or 3′ single-strand overhangs and must be processed into blunt or nearly blunt ends prior to ligation with another S region (Rush, Fugmann,& Schatz et al., 2004). Mre11, a component of the Mre11/RAD50/Nbs1 (MRN) complex, participates in CSR likely through its ability to process single-strand overhangs (Buis et al., 2008; Dinkelmann et al., 2009). Additionally, the structure-specific ERCC1–XPF endonuclease, which excises ssDNA tails at junctions with duplex DNA could participate in this process (Schrader et al., 2004; Tian & Alt, 2000). In contrast to excision or “chewing back” to form DSBs, S region breaks could be filled in by error-prone DNA polymerases, as these regions have been shown to accumulate mutations and deletions after B cell activation for CSR, much like mutations seen in variable region genes during SHM (Dunnick, Wilson, & Stavnezer, et al., 1989). This activity has been associated with polymerase eta, but could also be the result of additional error-prone DNA polymerases filling-in the 5′-overhangs left by DNA excision or partial end processing (Delbos et al., 2005; Dunnick et al., 1989; Faili et al., 2004; Schrader, Vardo & Stavnezer, 2003; Wilson et al., 2005; Zhou, Lottenbach, Barenkamp, & Reason, 2004).
Overall, the conversion of deaminated DNA into DSBs requires a large number of protein factors derived from multiple DNA repair pathways that have evolved to respond to general DNA damage. The mechanism through which this myriad of proteins comes together to effectively process the initial DNA lesion (deamination) without faithfully repairing the damage is an intriguing, unanswered question.
5. COMPLETION OF CSR: SYNAPSIS AND END-JOINING
The completion phase of CSR requires “synapsis” or close juxtaposition of the activated S regions followed by end-joining of DSBs between the participating S regions. The observation that the yeast I-SceI endonuclease can induce Igh class switching in B cells in which the S regions have been replaced with I-SceI sites strongly implicates the general cellular DNA-damage response and DSB-repair pathways in the synapsis and long-range end-joining of S region DSBs (Zarrin et al., 2007).
5.1. S region synapsis
Synapsis between S regions, which could be over a 100 kb apart, is critical for CSR. Unlike V(D)J recombination, where the RAG1/2 endonuclease efficiently cleaves RSSs only in the context of a synaptic complex, AID activity does not rely on prior interaction between two S regions, as constitutively transcribed Sμ or Sγ2b regions integrated randomly into a pro-B cell line undergo a high rate of AID-induced internal deletions (Dudley et al., 2002). It has been suggested that interactions between promoters that drive germline transcription and regulatory elements at the Igh locus (Eμ located 5′ of the Iμ-Sμ-Cμ unit and Eα located downstream of Cα) promote the formation of a three-dimensional genomic conformation that brings the S regions together in a S/S “synaptome” required for CSR (Ju et al., 2007; Kenter et al., 2012; Sellars, Reina-San-Martin, Kastner, & Chan, 2009; Wuerffel et al., 2007). In B cells activated to undergo CSR in culture, internal deletions within Sμ are frequently observed even in cells that are still IgM-positive (Alt, Rosenberg, Casanova, et al., 1982; Gu, Zou, & Rajewsky, et al., 1993). These represent cells in which DSBs at Sμ have not interacted, or synapsed, with DSBs at a distal S region, resulting in resolution through internal deletions in an intra-switch recombination reaction. Mutations in 53BP1 and H2AX lead to severe defects in CSR, but undergo internal Sμ deletions at significantly higher frequency than wild-type B cells, suggesting that these proteins participate in the synapsis of S regions (Manis et al., 2004; Reina-San-Martin, Chen, Nussenzweig, & Nussenzweig, 2007; Ward et al., 2004). Recent studies have shown that 53BP1 acquires the ability to bind the newly identified protein Rif1 upon phosphorylation by ATM (Chapman et al., 2013; Di Virgilio et al., 2013; Escribano-Diaz et al., 2013; Feng, Fong, Wang, Wang,&Chen, 2013; Shi et al., 2013; Zimmermann, Lottersberger, Buonomo, Sfeir, & de Lange, 2013). It has been proposed that the interaction between 53BP1 and Rif1 is required for the protection of DSBs from 5′–3′ end resection. In keeping with this notion, Rif1-deficient B cells are significantly impaired in undergoing CSR (Di Virgilio et al., 2013).
5.2. DNA end-joining
Mammalian cells harbor two major DNA repair pathways: homologous recombination (HR) and C-NHEJ. HR is the major pathway in postreplication repair and requires large stretches of homology, while C-NHEJ requires little or no homology, and operates throughout the cell cycle, but is the primary repair pathway in the G1 phase where HR is not active (Alt et al., 2013). CSR-associated DSBs are primarily observed in G1 phase cells and the recombining S regions do not have the extended homology required for HR; therefore, C-NHEJ is thought to be the major pathway in the joining of DSBs during CSR (Petersen et al., 2001; Schrader et al., 2007; Stavnezer et al., 2008). Indeed, mutations in components of the NHEJ pathway, including the DNA end-binding Ku70/Ku80 proteins, DNA ligase IV, or its cofactor XRCC4, lead to significant defects (20–40% of normal levels) in CSR (Boboila, Jankovic, et al., 2010; Boboila, Yan, et al., 2010; Casellas et al., 1998; Han & Yu, 2008; Manis, Gu, et al., 1998; Soulas-Sprauel et al., 2007; Yan et al., 2007).
Residual CSR in C-NHEJ-deficient B cells is mediated by microhomology-biased alternative end-joining (A-EJ), a poorly defined process that operates in the absence of C-NHEJ using a medley of factors, including XRCC1, Ligase III, Mre11, Parp1, and CtIP, from other DNA repair pathways (Dinkelmann et al., 2009; Lee-Theilen, Matthews, Kelly, Zheng, & Chaudhuri, 2011; Robert, Dantzer, & Reina-San-Martin, et al., 2009; Xie, Kwok, & Scully, et al., 2009). While there is a bias toward microhomology at S region junctions in C-NHEJ-deficient B cells, there is a substantial number of direct joins suggesting that microhomology is neither a signature nor a strict requirement for A-EJ (Alt et al., 2013; Boboila, Alt & Schwer, 2012; Boboila, Jankovic, et al., 2010; Boboila, Oksenych, et al., 2012; Deriano & Roth, 2013; Lieber, 2010). While C-NHEJ (with A-EJ) plays a major role in joining Igh DSBs during CSR, it is believed that HR repairs DSBs at non-Ig regions. This is evident from the observation that deficiency of the Rad51 paralog XRCC2, a key component of HR-mediated repair, leads to the generation of high levels of genome-wide AID-dependent DSBs (Hasham et al., 2010, 2012).
End-joining of S region DSBs in the absence of C-NHEJ to mediate CSR unmasks a significant mechanistic difference between CSR and V(D)J recombination, since the latter is entirely reliant on C-NHEJ. This likely reflects the ability of RAG1/2 to stably bind RSSs and actively shepherd DSBs to the NHEJ pathway (Lee, Neiditch, Salus, & Roth, 2004). Even though AID likely recruits proteins that participate downstream of DNA deamination (discussed below), it has not been shown to enforce engagement of C-NHEJ in the repair of DSBs at S regions.
6. AID PHOSPHORYLATION AT SERINE-38 AND A ROLE BEYOND DNA DEAMINATION
Mutations at the C-terminus of AID or deletion of the last 10 amino acids lead to a severe block in CSR without affecting AID deamination activity or binding to the Sμ region (Barreto, Reina-San-Martin, Ramiro, McBride, & Nussenzweig, 2003; Ta et al., 2003). These observations suggested that the C-terminus of AID might participate in recruiting factors specific to CSR, such as those required for S region synapsis of endjoining downstream ofDNAdeamination. In keeping with this notion, AID lacking the C-terminus, unlike the wild-type protein, does not interact with DNA-PKcs (Wu, Geraldes, Platt, & Cascalho, 2005). However, DNAPKcs-deficient cells show significant levels of CSR (Franco et al., 2008). Additionally, B cells expressing the mutant AID protein are inefficient in recruiting UNG and Msh2–Msh6 to S regions during CSR (Ranjit et al., 2011). Conflictingly, mutations at Sμ, which are dependent on UNG and Msh2–Msh6, occur at normal levels in cells expressing the mutant protein (Barreto et al., 2003). Overall, it is still not clear how the C-terminus of AID participates in CSR. The role of AID in steps downstream of deamination, however, has been extensively studied in the context of AID phosphorylation and is discussed below.
6.1. Role of AID phosphorylated at Serine-38 in DSB formation
Purification of AID from B cells undergoing CSR followed by mass spectrometric analyses demonstrated that AID is phosphorylated at serine residue-38 (S38) (Basu et al., 2005; McBride et al., 2006). The S38 residue lies within a protein kinase A (PKA) consensus phosphorylation site and can be phosphorylated in vitro and in vivo by PKA (Basu et al., 2005; McBride et al., 2006; Pasqualucci, Kitaura, Gu, & Dalla-Favera, 2006; Vuong et al., 2009). The association of PKA with transcribed S regions undergoing CSR strongly suggested that PKA-mediated AID phosphorylation occurs at S regions (Vuong et al., 2009).
B cells from mice with an S38A knock-in mutation (AIDS38A) are substantially impaired in mediating CSR, indicating that AID phosphorylated at S38 (pS38-AID) plays a critical role in CSR (Cheng et al., 2009; McBride et al., 2008). AIDS38A is as active as wild-type AID in its ability to deaminate ssDNA and to bind S region DNA, indicating that neither the S38 residue nor its phosphorylation is required for DNA enzymatic activity on ssDNA or association with S regions (Basu et al., 2005; Vuong et al., 2009). However, AIDS38A B cells are impaired in their ability to generate DSBs at S regions, leading to the suggestion that this specific phosphorylation event might be required at a step downstream of DNA deamination (Vuong et al., 2013). Indeed, immunoprecipitation experiments demonstrated that pS38- AID interacts with APE1, the enzyme critical for the generation of DNA breaks downstream of AID and UNG activities during CSR (Vuong et al., 2013) (Fig. 1.6). Taken together, these observations provide strong evidence that phosphorylation of AID at S38 promotes its ability to interact with APE1 and facilitate formation of DSBs during CSR. The interaction between AID and APE1 is not direct and proteins that mediate this interaction have not yet been described.
Figure 1.6.
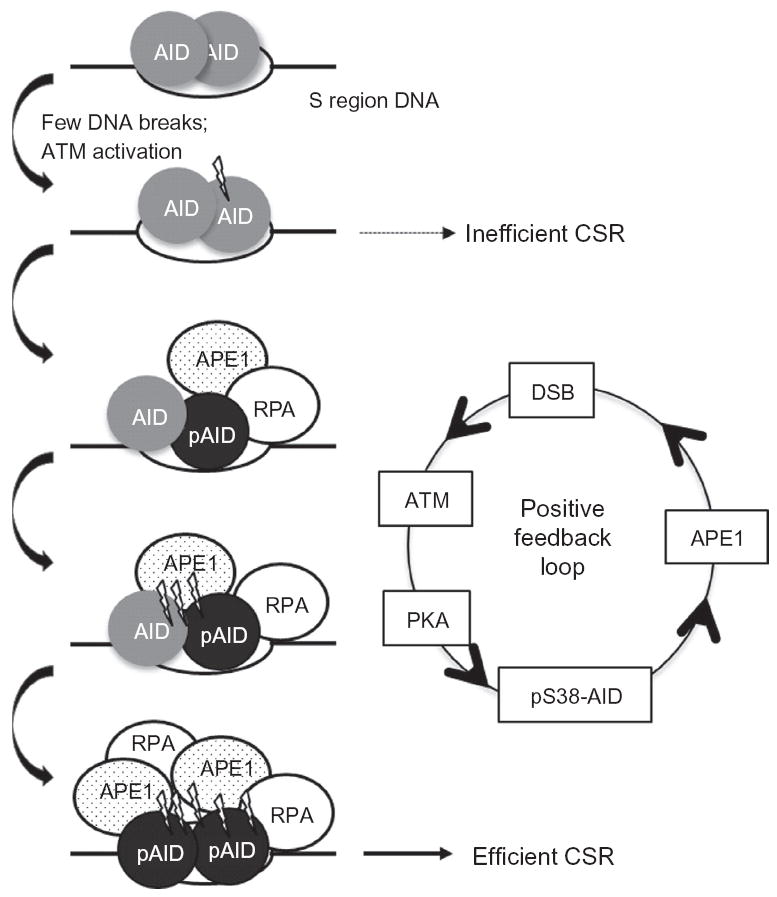
Phosphorylation and DSB-dependent positive feedback loop amplifies DSB formation. Multiple AID molecules are assembled at S region DNA. An initial DNA break due to AID-mediated DNA deamination of cytidines is generated and can result in inefficient CSR. ATM-dependent S38 phosphorylation of AID induces the recruitment of APE1, which increases the number of DNA breaks generated. This induces additional phosphorylation of AID at S38 and APE1 recruitment to generate the high density of DSBs required for efficient CSR. AID phosphorylation at S38 also allows for interaction with RPA, which promotes efficient CSR potentially by recruiting downstream repair factors.
Attempts to understand the mechanism by which AID phosphorylation is initially induced led to the striking observation that a catalytically inactive mutant of AID bound to S regions was not efficiently phosphorylated and failed to interact with APE1, suggesting thatDNAlesions activate AID phosphorylation (Vuong et al., 2013). Consistent with this idea, AID phosphorylation and APE1 interaction are significantly subdued in B cells deficient for both UNG and Msh2 where DSBs are not induced. Treating UNG–Msh2 mutant B cells or cells expressing catalytically inactive AID with ionizing radiation to induce genome-wide DSBs restored both AID phosphorylation and APE1 binding. Thus, DSBs induce AID phosphorylation at S38.
The above findings elicit the question of what senses DSBs to induce AID phosphorylation. A primary participant in the general cellular response to DSBs is ATM, a kinase that phosphorylates a vast number of substrates required for responding to and repairing DSBs (Bensimon, Aebersold, & Shiloh, et al., 2011). B cells lacking ATM are severely impaired in their ability to undergo CSR, and this defect was attributed primarily to a failure to react to and repair DSBs (Lumsden et al., 2004; Reina-San-Martin, Chen, Nussenzweig, & Nussenzweig, 2004). Assessing AID phosphorylation in ATM-deficient cells demonstrated that the phosphorylation-dependent interaction between AID and APE1 was significantly subdued, thereby unmasking a novel role of ATM in inducing DSBs (Vuong et al., 2013). Thus, a single protein, ATM, regulates both the generation and repair of DSBs during CSR, thereby coordinating the DNA-damage response with DNA repair. It is to be noted that while ATM is required for efficient AID phosphorylation, AID is not thought to be a direct target of ATM kinase; rather, PKA is likely the direct AID kinase (Vuong et al., 2009). The mechanism through which ATM activation is transduced into PKA activation remains unclear.
6.2. Positive feedback loop in amplifying DSBs
The observation that DSBs were both dependent on and required for AID phosphorylation led to the proposal that a positive feedback loop amplifies DNA breaks at S regions through AID phosphorylation (Fig. 1.6). Envisioning a likely scenario where a high density of DSBs promotes end-joining between DSBs generated in two distal S regions necessitates a process of DNA break amplification for efficient CSR. A positive feedback loop provides a model whereby even after assembly of AID and PKA at S regions (Vuong et al., 2009), AID will not be efficiently phosphorylated until a DNA break is generated. Once a DNA break is formed, the rapid activation of AID phosphorylation and DSB formation will result in the synchronous activation of many molecules of AID bound to an S region. The high density of DSBs in S regions thus generates many broken DNA ends that promote the ligation of distal DSBs, thereby subverting normal DNA repair. When AID phosphorylation is blocked, as in AIDS38A B cells or reduced, as in PKA hypomorphic mutant B cells, the low density of DSBs induced at individual S regions could be resolved as inefficient CSR (Vuong et al., 2009). Thus, while APE1 can passively access the abasic sites generated through basal AID activity, its active recruitment to the sites of AID-induced deamination through its interaction with pS38-AID would facilitate the rapid and efficient conversion of abasic sites into DNA breaks.
A DSB-inducing positive feedback loop requires coordinate recruitment of both AID and PKA to recombining S regions, which may be a regulatory mechanism to limit AID activity at non-Ig genes. While AID can bind and deaminate several non-Ig genes, without concomitant PKA binding very few of these lesions would be converted into DSBs in the absence of AID phosphorylation. Thus, the two-tiered mode of AID activation (recruitment to S regions and subsequent phosphorylation by PKA) provides a mechanism to generate a high density of DSBs specifically at Igh S regions during CSR while restricting DSB formation at non-Ig sites (Fig. 1.6).
If ATM is critical for the generation of a high density of DSBs in S regions, how does one then explain the increased DSBs and chromosomal translocations in ATM-deficient B cells (Franco et al., 2006)? One possibility is that ATM forces CSR to proceed through the canonical BER pathway, the major effector of CSR (Vuong et al., 2013). If true, ATM-deficient cells would not actively engage BER proteins, especially APE1, to process the dU:dG mismatches; rather, the MMR proteins would convert the deaminated residues into DNA breaks. ATM-deficient B cells are impaired in inter-, but not intra-S region recombination, indicating reduced synapsis of DSBs at S regions (Reina-San-Martin et al., 2004). This defect, when combined with a failure to enforce cell-cycle checkpoints at the G1–S transition, could explain the persistence of DSBs that can partake in aberrant chromosomal translocations (Franco et al., 2006). Therefore, the AID– APE1–DSB–ATM loop represents a positive feedback mechanism to generate a threshold number of DNA breaks precisely within transcribed S regions that not only promotes the joining of distal S regions to drive successful CSR, but also suppresses oncogenic translocations.
6.3. Role of AID in recruiting RPA to S regions
In addition to interacting with APE1, pS38-AID also gains the ability to interact with the ssDNA binding protein RPA. The phosphorylationdependent interaction between AID and RPA was discovered in biochemical assays to identify factors that allowed AID to deaminate variable region genes, which do not form R-loops upon transcription (Basu et al., 2005; Basu, Wang, & Alt, et al., 2008; Chaudhuri et al., 2004). Thus, it was reasonable to propose that the pS38-AID_RPA complex, by virtue of the ssDNA binding ability of RPA, stabilizes ssDNA within transcription bubbles at variable region genes, thereby generating AID substrates during SHM (Chaudhuri et al., 2004). Consistent with this notion, B cells from AIDS38A mice were substantially impaired in mediating SHM (Cheng et al., 2009; McBride et al., 2008). The defect was particularly pronounced in mice haploinsufficient for AIDS38A where the frequency of SHM was reduced to near background levels. Additionally, Xenopus laevis S regions, which are A:T rich and do not form R-loops, could still mediate CSR in mouse B cells by deaminating AGCT sequences in a pS38-AID_RPA complexdependent SHM-like reaction (Zarrin et al., 2004). Taken together, these observations have led to the model that pS38-AID, through its ability to bind RPA, is critical for the recruitment of AID to variable region genes during SHM (Chaudhuri et al., 2004).
While identified as an AID interactor to facilitate SHM, RPA was also shown to bind S regions in a pS38-AID-dependent fashion (Chaudhuri et al., 2004; Vuong et al., 2009; Yamane et al., 2011). The CSR defect in AIDS38A B cells could thus be a combination of a failure of AID to recruit both APE1 and RPA to S region DNA. The observation that second-site mutations in AIDS38A (AIDS38AT40D) restores RPA binding to AID in the absence of phosphorylation, and partially rescues the CSR defect of AIDS38A protein, suggests that RPA plays a direct role in the recombination reaction (Basu et al., 2008). The role of RPA in CSR has not been fully elucidated. RPA bound to S regions may function downstream of deamination to recruit UNG or MMR proteins, which convert deaminated cytidines to DSBs. Additionally, RPA might recruit proteins such as 53BP1 and H2AX to DSBs to promote synapsis of DSBs between distal S regions. The known requirements of UNG,MMRproteins, 53BP1, and H2AX in CSR, and the reported interactions of these proteins with RPA, support a role for RPA downstream of DNA deamination (Vuong & Chaudhuri, 2012). It is also possible that RPA does not participate directly in the recombination reaction, but rather is required in the salvage of unrepaired AID-instigated DSBs at Ig and non-Ig genes by homology-based pathways (Yamane et al., 2013). A formal demonstration of the requirement of RPA in recruiting AID to variable region genes during SHM and defining the role of RPA in CSR remain outstanding issues.
7. MULTIFACETED REGULATION OF AID EXPRESSION AND ACTIVITY
AID is a general mutator and it comes as no surprise that it is regulated at multiple levels, including almost exclusive activated B cell-specific expression, miRNA-mediated regulation of Aicda mRNA stability, subcellular localization, phosphorylation, and specific recruitment to the Ig locus (Fig. 1.7). Below we outline our current understanding of the regulatory mechanisms that restrain AID activity.
Figure 1.7.

Regulation of AID. AID expression, localization and activity are regulated through multiple processes.
7.1. Transcriptional regulation
AID is expressed primarily, though not exclusively, in activated B cells in GCs and can be induced in mature B cells activated ex vivo with cytokines or bacterial LPS through a combination of transcriptional activators and repressors (Lee-Theilen & Chaudhuri, 2010). The Aicda locus, which encodes AID (Muramatsu et al., 2000), contains four distinct regions of transcription factor-binding sites, with two regions lying upstream of the transcription start site (TSS), one within the first intron and the final residing approximately 17 kb downstream of the promoter (Crouch et al., 2007; Tran et al., 2010; Yadav et al., 2006) (Fig. 1.8).
Figure 1.8.
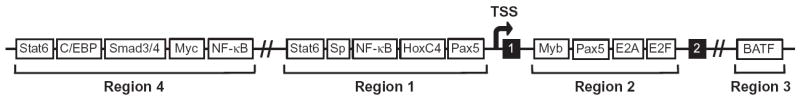
Transcription factor-binding sites in Aicda locus. The Aicda locus is regulated by four regions of transcription factor-binding sites. Region 1 is just 5’ of the transcription start site (TSS), region 2 is located between exons 1 and 2 (shown in black), region 3 is ~17 kb downstream of the TSS, and region 4 is ~8 kb upstream of the TSS. Adapted from Lee-Theilen and Chaudhuri (2010).
Region 4 (RIV), lying ~8 kb upstream of the promoter, contains binding motifs for Stat6, Smad3/4, and NF-κB, which are expressed in response to IL-4, TGF-β, and CD40 ligation, respectively (Huong le et al., 2013; Tran et al., 2010). Two C/EBP binding sites located within this region were shown to be indispensible for the enhancer activity of RIV on AID expression in activated B cells (Huong le et al., 2013; Tran et al., 2010). In addition, c-Myc, has been shown to be required for normal CSR due, in part, to its transcriptional induction of AID gene expression via binding to the RIV regulatory region (Dominguez-Sola et al., 2012; Fernandez et al., 2013).
Region 1 (RI), located immediately upstream of the TSS, contains binding sites for NF-κB (p50), Stat6, HoxC4, and Pax5 (Park et al., 2009; Yadav et al., 2006). Signaling via IL-4 and CD40 ligation were shown to synergize and induce Stat6 and NF-κB binding, respectively, to RI, while mice deficient in Stat6 or p50 exhibited impaired AID expression in response to these stimuli (Dedeoglu, Horwitz, Chaudhuri, Alt, & Geha, 2004). HoxC4, expressed in GC B cells upon CD40 engagement and cytokine stimulation, induces AID expression through binding a highly conserved site at the Aicda promoter (Park et al., 2009, 2013).
The intronic regulatory region 2 (RII) contains sites for both activating (Pax5 and E-proteins) and inhibitory (c-Myb and E2F) proteins (Gonda et al., 2003; Huong le et al., 2013; Sayegh, Quong, Agata, & Murre, 2003; Tran et al., 2010). It has been proposed that B-cell-specific induction of the Aicda gene is achieved by the expression of Pax5 and E-proteins upon B cell activation (Gonda et al., 2003; Sayegh et al., 2003; Tran et al., 2010). Both c-Myb and E2F are factors expressed in naïve and non-B cells, and it is thought that the inhibition of Aicda promoter activation by the binding of these factors may either be blocked, perhaps by Pax5 and E-protein binding, or overcome by cytokine-induced stimulating transcription factors (Huong le et al., 2013; Tran et al., 2010). Ectopic expression of Pax5 was shown to be sufficient to induce Aicda expression in a plasmacytoma cell line in the absence of additional stimuli that activate NF-κB expression (Dege & Hagman, 2013). In addition to the binding of c-Myb and E2F to RII, it is believed that active inhibition of Aicda gene transcription is achieved via the antagonistic activity of inhibitor of differentiation (Id) proteins (Gonda et al., 2003; Sayegh et al., 2003). Id proteins, which lack a DNA-binding domain, inhibit transcription of target genes by forming heterodimers with activating transcription factors such as E2A. Both Id2 and Id3 have been implicated in forming heterodimers with Pax5 and E-proteins, respectively, and impair their binding to RII of the Aicda locus (Gonda et al., 2003; Sayegh et al., 2003).
Furthest from the TSS is region 3 (RIII), with a BATF binding site that is believed to act as an enhancer element, as Batf deletion leads to abrogation of AID expression and CSR (Betz et al., 2010; Crouch et al., 2007; Ise et al., 2011). Interestingly, a recent study employing an Aicda locus transgenic mouse system has shown that deletion of both RII and RIV abrogates normal Aicda expression in activated B cells; thus the presence of RIII cannot compensate for the loss of these two regions in inducing AID expression (Huong le et al., 2013).
The sex hormones estrogen and progesterone have also been associated with the modulation of AID expression in murine splenic B cells activated to undergo CSR (Mai et al., 2010; Pauklin & Petersen-Mahrt, 2009; Pauklin, Sernandez, Bachmann, Ramiro, & Petersen-Mahrt, 2009). Estrogen-bound estrogen receptor induced AID transcription either by directly binding and activating the AID promoter, or indirectly by binding and activating the HoxC4 gene (Mai et al., 2010; Pauklin & Petersen-Mahrt, 2009; Pauklin et al., 2009). In contrast, progesterone-bound progesterone receptor reportedly inhibits transcription of the Aicda locus by binding around the RI region upstream of the promoter (Pauklin & Petersen-Mahrt, 2009). The regulation of AID by hormonal factors could have implications in hormone-based oncogenesis and autoimmunity; however, their importance in the normal CSR mechanism remains to be elucidated (Incorvaia, Sicouri, Petersen-Mahrt, & Schmitz, 2013; Maul & Gearhart, 2009; Pauklin & Petersen-Mahrt, 2009; Pauklin et al., 2009).
7.2. Posttranscriptional control of Aicda mRNA
The binding of small RNA species or miRNAs to a target mRNA sequence results in mRNA silencing by translational repression and mRNA degradation. Several miRNAs, including miR-155, miR-181b, and miR-361, have been implicated in regulating the vigor of AID mRNA by binding to its 3′-untranslated region (3′-UTR) (Borchert, Holton, & Larson, et al., 2011; de Yebenes et al., 2008; Dorsett et al., 2008; Teng et al., 2008). In addition to playing a role in general GC structure and function, miR-155 was found to be upregulated in murine B cells undergoing CSR (Teng et al., 2008; Thai et al., 2007). Disruption of the miR-155 binding site within the 3′-UTR of AID mRNA led to increased AID expression, CSR, and c-Myc:Igh translocations (Dorsett et al., 2008; Teng et al., 2008). Interestingly, the transcriptional repressor Bcl6, whose expression is integral for GC formation and development, regulates miR-155 expression. Bcl6 binds to the promoter of miR-155, as well as to the intronic region upstream of the miR-155 sequence and represses miR-155 expression (Basso & Dalla-Favera, 2012). A second miRNA sequence shown to be a target for Bcl6 binding and repression, miR-361, was also identified as an AID 3′-UTR binding miRNA (Basso & Dalla-Favera, 2012). The role, if any, of miR- 361 in the regulation of AID mRNAstability during theGCreaction and/or CSR remains to be determined. Finally, the expression of miR-181b in activated murine B cells impairs CSR efficiency and leads to decreased AID mRNA and protein levels (de Yebenes et al., 2008).
7.3. Compartmentalization of AID activity
Regulating subcellular localization of proteins is regularly employed by cells to sequester enzymes away from their substrates. Not surprisingly, this strategy is employed with AID,which is predominately cytoplasmic in theBcell and thus limited in its access to its DNA substrate in the nucleus (Ito et al., 2004; Pasqualucci et al., 2004; Rada, Jarvis & Milstein, 2002). This is achieved, in part, by active export via its C-terminal nuclear export signal (NES), which mediates exportin1-dependent nuclear export (Brar, Watson, & Diaz, et al., 2004; Ito et al., 2004; McBride, Barreto, Ramiro, Stavropoulos, & Nussenzweig, 2004). In addition, nuclear exclusion by a “cytoplasmic retention determinant” also present in the C-terminus of AID, but distinct fromthe NES, has been implicated in the cytoplasmic sequestering of AID (Patenaude et al., 2009). It is of note that the NES region overlaps with the C-terminal amino acid residues shown to be indispensible for CSR, but that are not required for SHM (Barreto et al., 2003; Ta et al., 2003).
In addition to a C-terminal NES, AID contains a bipartite nuclear localization signal (NLS) at its N-terminal domain. While, there are conflicting reports regarding the requirement of the NLS in the import of AID into the nucleus (Brar et al., 2004; Hu et al., 2013; Patenaude et al., 2009), it has been demonstrated that positively charged residues throughout the protein sequence, in both the N and C-terminal regions, come together to mediate nuclear transport (Hu et al., 2013). Thus, AID can be shuttled between the nucleus and cytoplasm in an unbalanced, cytoplasmic-dominant manner. AID was recently found to accumulate in subnuclear nucleolar structures, which are hubs for RNA metabolism (Hu et al., 2013). Mutations that abrogated AID localization to these structures resulted in reduced levels of CSR (Hu et al., 2013). Whether subnuclear compartmentalization of AID is important for its interaction with potential targeting factors or for the regulation of AID activity by further sequestration remains an open question.
AID that does make its way into the nucleus has been shown to have a short half-life due to destabilization via polyubiquitination (Aoufouchi et al., 2008). The importance of degrading nuclear AID was underscored when proteasome inhibition resulted in nuclear-restricted AID unleashing enhanced mutagenicity within the Ig loci and throughout the genome (Aoufouchi et al., 2008). A ubiquitin-independent mechanism to degrade nuclear AID involving REGγ, a proteasome activating protein, has also been reported (Uchimura, Barton, Rada, & Neuberger, 2011). Splenic B cells from REGγ-deficient mice underwent higher levels of CSR, as compared to littermate controls (Uchimura et al., 2011). Therefore, regulation of the ability of AID to enter and remain in the nucleus modulates its mutational potential. However, sequences in AID that regulate its residence in the nucleus overlap with residues required for its CSR or SHM activities, thereby complicating interpretation of mutational analysis.
7.4. AID phosphorylation
In addition to S38, AID is phosphorylated at several other residues—serine 3 (S3), threonine 27 (T27), T140, and tyrosine-184 (Y184) (Basu et al., 2005; Chaudhuri et al., 2004; Gazumyan et al., 2011; McBride et al., 2006, 2008; Pasqualucci et al., 2006) (Fig. 1.7). The S3 residue was identified as a site on recombinant AID that could be phosphorylated in vitro by protein kinase C (PKC) (Gazumyan et al., 2011). Mutation of S3 to alanine (AIDS3A) does not affect the DNA deaminase activity of AID (Gazumyan et al., 2011); however, expression of AIDS3A in AID-deficient B cells and fibroblasts increases CSR and SHM, suggesting that phosphorylation of S3 is an inactivating event. Consistent with this notion, expression of the mutant protein significantly increased c-Myc:Igh translocation frequency over that induced by the wild-type AID protein (Gazumyan et al., 2011). The precise mechanism through which phosphorylation at S3 regulates AID function is yet unknown.
Mass spectrometric analysis of AID purified from B cells identified T140 and Y184 as phosphorylated residues (Basu et al., 2005; McBride et al., 2006, 2008). Mutation of T140 to alanine in AID (AIDT140A) does not affectDNA deaminase activity (McBride et al., 2008). While B cells from mice expressing AIDT140A displayed only a mild defect in CSR, a more profound defect in SHM was observed (McBride et al., 2008). The mechanism by which T140 phosphorylation modulates AID activity during SHM is not known, but it is likely that this phosphorylation event modulates the ability of AID to interact with an unidentified factor(s) that is required for SHM but not CSR. Mutation of Y184 to alanine does not affect the ability of AID to mediate CSR (Basu et al., 2005). T27 was identified as a residue phosphorylated by PKA, and mutation of T27 to alanine severely impairs CSR without affecting the ssDNA deaminase activity of AID (Basu et al., 2005; Demorest, Li, & Harris, 2011; Pasqualucci et al., 2006). However, it is not clear if the T27 residue is phosphorylated in vivo and how it influences CSR. Additional phosphorylation sites at S41 and S43 were detected in Sf9 cells expressing AID, but whether these residues are functionally relevant during CSR or SHM is not known (Pham et al., 2008). In general, none of the currently identified AID phosphorylation events regulates its DNA deaminase activity; rather, they appear to mediate the interaction of AID with other proteins to modulate CSR and/or SHM.
8. TARGETING OF AID TO THE IG LOCI
The specific recruitment of AID to the appropriate DNA substrates (variable region genes for SHM and S regions for CSR) is not only essential for its function but also critical to protect the rest of the genome from AID-induced DNA damage. Indeed, mistargeting of AID activity to non-Ig genes has been implicated in chromosomal translocations and pathogenesis of B-cell lymphomas (Chiarle et al., 2011; Klein et al., 2011; Pasqualucci et al., 2008; Ramiro et al., 2004, 2006; Robbiani et al., 2008, 2009). AID expressed even at physiological levels in GC B cells or in ex vivo stimulated B cells associates with and deaminates a large number of non-Ig genes (Liu et al., 2008; Pavri et al., 2010; Yamane et al., 2011). It is generally believed that differential DNA repair protects non-Ig loci from sustained AID-induced damage, leaving only the Ig loci affected by AID activity (Liu et al., 2008). This hypothesis arose from the observation that AID activity could be detected outside the Ig loci on more than half of the transcribed genome (Liu et al., 2008). Nonetheless, while AID might appear to indiscriminately affect large parts of the genome, mistargeting of AID to non-Ig genes occurs at a much lower frequency. Indeed, the rate of mutation at the Ig loci is 20- to 100-fold greater than that found in any other loci (Liu et al., 2008), indicating the existence of an active Ig loci-specific AID-targeting mechanism in B cells.
8.1. Transcription-dependent AID recruitment
The observations that CSR, SHM, and mutations of non-Ig genes are tightly linked to transcription (Betz et al., 1994; Fukita, Jacobs, & Rajewsky. et al., 1998; Peters & Storb, 1996; Stavnezer-Nordgren & Sirlin, 1986; Winter et al., 1997; Yancopoulos et al., 1986) and that AID interacts with Pol II (Nambu et al., 2003; Pavri et al., 2010; Sun et al., 2013), suggested that transcription and Pol II-associated proteins might facilitate the binding of AID to target DNA sequences. In keeping with this notion, a short-hairpin RNA screen for effectors of CSR revealed that the Pol II-associated factor Spt5, which interacts with AID and colocalizes with paused Pol II, is required for CSR (Pavri et al., 2010). Depletion of Spt5 markedly reduces recruitment of AID to Ig and non-Ig sequences. Remarkably, occupancy of Spt5 on stalled Pol II sites is predictive of AID-dependent mutations at the corresponding DNA sequence (Pavri et al., 2010). It is reasonable to conjecture that pausing of Pol II could be facilitated by R-loops impeding transcription elongation.
Histone modifications, like trimethyl histone H3 lysine 4 (H3K4me3) and hyperacetylated H3K9 (Ac-H3K9), that serve as markers of active chromatin conformation at promoter-proximal sites are observed throughout transcribed S regions (Wang, Whang, Wuerffel, & Kenter, 2006; Wang et al., 2009). A direct link between histone acetylation and CSR frequency was shown when inhibiting histone deacetylases through tricostatin A treatment led to increased Ac-H3K9 at S regions and CSR frequency (Wang et al., 2009). Additionally, deletion of PTIP, a component of several histone methyl transferase complexes, reduced H3K4me3 modifications at S regions and concomitantly impaired CSR (Daniel et al., 2010). Taken together, these data support a role for transcription-linked histone modifications in promoting the chromatin accessibility required for CSR.
8.2. GANP and 14-3-3 adaptors
The GC associated nuclear protein GANP (Kuwahara et al., 2004), which is induced in GC B cells and binds variable region transcripts has been reported to recruit AID to variable region genes during SHM. GANP deficiency impairs SHM without affecting CSR; however, the mechanism through which GANP mediates AID targeting to DNA during SHM is unclear (Kuwahara et al., 2004; Maeda et al., 2010).
The 14-3-3 adaptors represent a family of proteins with the ability to bind DNA with altered conformations (e.g., cruciform DNA) and have been implicated in numerous functions including DNA replication. In B cells undergoing CSR, expression of 14-3-3 proteins was rapidly induced through NF-κB signaling and sustained by E2A (Mai et al., 2013). The 14-3-3 proteins bind to repeating RGYW motifs and transcribed S regions, and were shown to directly interact with AID, leading to the proposal that this family of scaffold proteins recruits AID to S regions. Surprisingly, the 14-3-3:AID interaction requires the C-terminus of AID (Xu et al., 2010), yet AID with C-terminal mutations is still able to target Sμ (Barreto et al., 2003). Thus, the relevance of this interaction is not clear at present.
8.3. PTBP2: An AID interactor that promotes binding to S regions
Polypyrimidine-tract binding protein-2 (PTBP2), an RNA-binding protein originally thought to be expressed exclusively in the brain (thus referred to as brPTB or nPTB) and to regulate alternative mRNA splicing (Black, 2003), was subsequently detected in B cells in a proteomic screen to identify AIDinteracting proteins (Nowak, Matthews, Zheng, & Chaudhuri, 2011). Knock-down of PTBP2 in B cells significantly reduced the binding of AID to S regions and consequently impaired CSR. The mechanism by which PTBP2 targets AID to the Igh locus during CSR is not clear. PTBP2 could be recruited to S region DNA, possibly through interactions with other chromatin-associated factors to inhibit splicing, thus enhancing the stability of R-loops and facilitating CSR. Alternatively, the role of PTBP2 in CSR could be independent of its role in splicing. In vitro studies show that PTBP2 binds to sense and antisense S region RNA suggesting that PTBP2 may recruit AID to S regions through its interaction with S region transcripts (Nowak et al., 2011). Additional work is required to elucidate the precise function of PTBP2 in CSR and its requirement, if any, in SHM.
In summary, several scenarios to explain the specific recruitment of AID to S regions have been proposed; yet the exact molecular mechanism(s) involved is still unclear. Our knowledge of the processes that determine AID targeting to variable region exons during SHM is even more limited, in part because there is no convenient assay or cell line to elucidate SHM. The use of the CH12F3 cell line and splenic B cells activated in culture have been valuable tools to identify not only AID but also other effectors of CSR; however, cell lines that undergo SHM (Ramos, BL2), which require weeks of growth in culture, have not similarly contributed to our understanding of SHM effectors. Additionally, the CH12F3 cell line and splenic B cells stimulated ex vivo do not undergo SHM, thus SHM has to be studied in the context of GC cells that are relatively difficult to acquire. Therefore, identification of SHM-specific AID-targeting factors awaits the identification of cell lines that undergo rapid, easily measurable SHM.
9. AID ACTIVITY BEYOND THE IG LOCI
While the variable region exons and S region DNA serve as natural targets of AID, it is becoming increasingly clear that AID can act at several non-Ig genes. “Off-target” AID activity could have pathological consequences such as in the development of B-cell lymphomas or could play a physiological role in the poorly understood process of epigenetic reprogramming.
9.1. AID in B-cell lymphomagenesis
GC B cells undergo robust proliferation and simultaneously undergo processes that induce mutations and DSBs, thereby predisposing these cells to oncogenesis (Kuppers, 2005). DNA-damage checkpoints and DNA repair proteins help to eliminate cells harboring unrepaired DSBs or oncogenic translocations (Franco et al., 2008). Yet, despite these safeguards, AIDinduced aberrations do lead to oncogenesis. In fact, among human lymphomas it is estimated that about 95% are of B cell origin, and of these, a majority are classified as being derived from GC or post-GC B cells (Kuppers, 2005).
A hallmark of many B-cell lymphomas is chromosomal translocation between the Ig loci and a proto-oncogene, such as Bcl2, Bcl6, or Myc (Kuppers, 2005; Nussenzweig & Nussenzweig, 2010). These translocation events can place the oncogene under the control of the active Ig locus and result in constitutive oncogene expression. In this context, enhancers such as the 3′-RR can activate c-Myc over long distances and drive oncogene overexpression (Gostissa et al., 2009). In mouse models, AID was shown to be required for c-Myc:Igh translocations, and such translocations were readily observed in ex vivo stimulated splenic B cells (Ramiro et al., 2004, 2006). Significantly, while S regions serve as physiological substrates for AID, an elegant study demonstrated that DSBs at c-Myc were also AID dependent (Robbiani et al., 2008). Additionally, studies mapping DSBs or highthroughput genome-wide translocation sequencing demonstrated that AID generates off-target DSBs at many genes in activated B cells, including many genes associated with B-cell oncogenesis (Chiarle et al., 2011; Klein et al., 2011; Staszewski et al., 2011). Interestingly, many translocations were also observed in AID-deficient cells, indicating that certain genomic regions may be prone to DSB formation (Chiarle et al., 2011; Klein et al., 2011). This is consistent with an earlier observation that a small fraction of c-Myc:Igh translocations can also occur in the absence of AID (Unniraman, Zhou, & Schatz, et al., 2004). Both AID-dependent and - independent DSBs are associated with transcription, which could generate AID targets through collision with replication forks or through the formation of R-loops. It has also been proposed that collaboration between RAG1/2 and AID activities could promote chromosomal translocations. AID was shown to be expressed in developing B cells in the bone marrow (Han et al., 2007), and it has been proposed that AID deamination at certain CpG sites generates noncanonical RAG1/2 and Artemis substrates, leading to DSBs. The concerted activities of RAG1/2 and AID could potentially explain translocation hotspots in human cancers originating from developing B cells (Cui et al., 2013; Tsai et al., 2008).
It is important to note that not all translocations lead to B-cell lymphomas. This is best exemplified from AID overexpression studies using transgenic mouse models to assess the oncogenic ability of dysregulated AID expression. Mice with constitutive and ubiquitous expression of AID succumb to T-cell lymphomas along with developing lung adenocarcinoma (Okazaki et al., 2003). Strikingly, B cell tumors were not observed, either due to a longer latency of these tumors or due to a protective mechanism present in B cells. The idea that B cells may be protected from aberrant AID expression was given weight when a B cell-specific transgenic mouse line, driven by the CD19 promoter, was found to have reduced CSR and SHM, indicating that the transgenic AID expressed was being inactivated (Muto et al., 2006). In agreement with this finding, transgenic mice with AID expression controlled by the Igk regulatory elements developed lymphomas derived from B cells over a broad spectrum of stages only in the context of p53 deficiency (Robbiani et al., 2009). Interestingly, even though the Igk transgenic mice in a p53-proficient background did not generate B-cell lymphomas, B cells isolated from these animals possessed widespread genomic instability, including mutations and translocations at regions beyond the Ig loci (Robbiani et al., 2009).
Finally, spatial organization of the mouse genome might have a role in directing chromosomal translocations. A combination of high-resolution (Hi-C) depiction of the spatial organization of the mouse pro-B cell genome and high-throughput genome-wide translocation mapping of RAGinstigated DSBs showed that translocations were highly enriched in cis along a single chromosome, and that interchromosomal translocations occur in a manner directly related to pre-existing spatial proximity (Zhang et al., 2012). However, it is not entirely clear if AID-instigated interchromosomal translocations are determined by proximity between the Igh locus and translocation partners (Rocha et al., 2012), or by the extent and persistence of AID-induced damage (Hakim et al., 2012), or a combination of both scenarios. For a more comprehensive discussion of genomic integrity in cells undergoing antibody diversification, please read a recent review by Alt and colleagues (Alt et al., 2013).
Translocations are not the only transforming events arising from AID activity. SHM-like activity has also been implicated in contributing to transforming events that give rise to lymphomagenesis. A study assessing mutations within proto-oncogenes in diffuse large-cell lymphomas revealed that ~50% of the tumors analyzed contained mutations in Pim1, c-Myc, RhoH, or Pax5 genes and a mutation profile reminiscent of SHM (Pasqualucci et al., 2001). Mutations in the Bcl6 gene, whose expression is imperative for proper GC function, were also found within a region that was previously linked to chromosomal translocations in lymphoma cells and in B cells isolated from human tonsillar GCs (Migliazza et al., 1995; Pasqualucci et al., 1998). Similarly, the CD95/Fas gene, which is also actively expressed in GCs, was found to acquire somatic mutations during the GC reaction (Muschen et al., 2000). It is important to highlight that some of the mutations described above could be the result of increased mutagenicity upon transformation of the GC B cell and/or a global defect in DNA repair pathways. However, this does not seem to be the case with Bcl6 and CD95/Fas, as these genes appear to be directly targeted for mutation during the normal GC reaction (Muschen et al., 2000; Pasqualucci et al., 1998).
9.2. AID activity beyond B cells: Epigenetic reprogramming
While AID activity in SHM and CSR has been well studied, the observation that AID can deaminate methylated cytidines (Morgan, Dean, Coker, Reik, & Petersen-Mahrt, 2004) raised the possibility that AID could have roles beyond antibody diversification. It was discovered that AID is expressed in certain non-B cell populations, particularly in primordial germ cells (PGC), embryonic stem cells, oocytes, and somatic cells undergoing reprogramming to the pluripotent state (Bhutani et al., 2010; Kumar et al., 2013; Morgan et al., 2004). It was therefore proposed that AID mediates DNA demethylation through an indirect mechanism that involves deamination of 5′-methylated cytidines (5mC) with the subsequent replacement of the resulting thymidine with an unmethylated cytidine through the concerted activities of the BER pathway (Fig. 1.9).
Figure 1.9.

AID as an indirect demethylase. The deaminase activity of AID can lead to demethylation through two potential pathways. In one, AID can deaminate methylated cytidines (5mC) to generate thymidine. In the other, AID can deaminate hydroxymethylcytosine (5hmC) that is generated by the oxidation of 5mC by the ten-eleven translocation (TET) proteins. In both pathways, the mismatch is repaired by thymine DNA glycosylase (TDG) and BER components. The end-product is the replacement of methylated cytidines with unmethylated cytidines.
A rapidly accumulating body of work provided strong evidence that active DNA demethylation is quite prevalent in mammals. Following fertilization in mammals, the paternal pronuclei undergo extensive loss of 5mC in a process that is both rapid and independent of cell division, thus providing a classic example of active demethylation (Oswald et al., 2000). Genomewide epigenetic reprogramming also occurs in the PGCs after they have reached the embryonic gonads at embryonic days E10.5–13.5 (Popp et al., 2010). In this case, demethylation is truly global with genome-wide erasure of DNA demethylation marks at most promoters and genic, intergenic, and transposon sequences being hypomethylated at this stage (Popp et al., 2010). Active DNA demethylation has also been proposed as a gene-regulation mechanism in selected adult tissues, the most striking evidence being demethylation at the promoter of brain-derived neurotrophic factor and fibroblast growth factor 1 in postmitotic (and thus nonreplicating) neurons in animal models of emotional stress (Martinowich et al., 2003). Recent studies have implicated a role of AID and the ten-eleven translocation (TET) family of proteins in this active demethylation reaction (Guo, Su, Zhong,Ming,&Song, 2011). Finally, active demethylation appears to be a critical component of experimental reprogramming to a pluripotent stem cell state (Bhutani et al., 2010; Kumar et al., 2013).
Initial support of the requirement of AID in active demethylation came from genome-wide bisulphite-sequencing studies, which demonstrated a significant increase in global methylation in AID-deficient PGCs relative to wild-type cells (Popp et al., 2010). Additionally, in a heterokaryon-based reprogramming assay, AID was found to interact with the promoters of the pluripotency genes Oct4 and Nanog, which are rapidly demethylated during reprogramming (Bhutani et al., 2010). Short-hairpin-induced knock-down of AID led to a significant decrease in the demethylation at these promoters (Bhutani et al., 2010). In a compelling recent study, AID-deficient fibroblasts transduced to express the four pluripotency factors Oct4, Klf4, Sox2, and c-Myc were found to be defective in their ability to be stably reprogrammed into induced pluripotent stem (iPS) cells (Kumar et al., 2013). While the AID-deficient cells initiated the reprogramming process and expressed early markers of iPS cells, after 3 weeks in culture they were found to be significantly hypermethylated compared with wild-type cells and failed to express genes involved in the secondary pluripotency network, thereby leading to redifferentiation (Kumar et al., 2013).
While the above data seem to suggest a role for AID in demethylation, this activity might be limited to defined settings, such as during development and reprogramming of specific cells, for example, fibroblasts. AID is expressed most abundantly in stimulated B cells; however, reduced bisulphite-sequencing analysis from wild-type, AID-deficient, and AID overexpressing B cells revealed no differences in methylation status between the AID genotypes (Fritz et al., 2013). This highlighted the possibility that B cells can subvert the demethylase activity of AID as a protective mechanism during secondary antibody diversification reactions. It is also important to note that any role AID may have in demethylation is not absolutely required during development, as AID-deficient mice are viable, despite having smaller litter sizes (Kumar et al., 2013; Muramatsu et al., 2000; Popp et al., 2010).
A possible alternative pathway for AID-mediated demethylation involves a two-step process whereby the 5mC is first oxidized to hydroxymethylcytosine (5hmC) by the TET proteins and subsequently converted to 5-hydroxymethyluridine (5hmU), which can be repaired by thymine DNA glycosylase (TDG) (Cortellino et al., 2011). In support of this idea, TET1 overexpression increased demethylation through the formation of 5hmC and subsequent BER, while overexpression of AID led to a decrease in 5hmC levels (Guo et al., 2011). Expression of AID in the mouse brain also significantly reduced 5hmC levels. While endogenous AID expression in the brain was not detectable, Apobec1 was expressed in these neuronal populations and was similarly able to decrease 5hmC levels (Guo et al., 2011). Although these studies suggest a role for AID in this process, the activity of AID on 5hmC has not been demonstrated in a physiologically relevant setting. Furthermore, biochemical studies coupled with measurements of steric constraints using purified proteins have argued against AID or APOBEC1 acting on 5hmC residues (Nabel et al., 2012). Thus, the potential for AID to serve as a demethylase in this capacity, and as a demethylase in general, is an exciting new area of research.
10. PERSPECTIVES
The discovery of AID was one of the most significant advances in our understanding of the mechanism of CSR and SHM. Work over the past decade has provided us with a model that mechanistically links germline transcription and S region sequence to AID-mediatedDNAdeamination. We now have an understanding of how the BER, MMR, and C-NHEJ/A-EJ DNA repair pathways participate in the recombination reaction. There have also been significant advances made in our understanding of how potent a mutator AID is and how multiple processes come together to regulate the activity of this enzyme. Finally, we are beginning to unmask potential roles of AID beyond the immune system, such as in the maintenance of the pluripotent stem cell state. Yet, despite these advances, several key issues remain unresolved.
Several AID-interacting proteins that have the potential to recruit AID specifically to the relevant regions of the Ig loci have been identified, but there is still no coherent model that clearly explains how AID activity under physiological conditions is so exclusively specified for the Ig loci. Additionally, we know that multiple DNA repair pathways participate in CSR; however, these pathways are ubiquitous and have evolved to prevent the incorporation of mutations and deletions of genomic sequences. Thus, it is curious how the immune system manages to subvert normal repair machineries and instead hijacks components of these pathways to promote mutations and the persistence and synapsis of DSBs that are over 100 kb apart. The molecular basis underlying the balance between normal and aberrant repair requires further elucidation. Finally, with regard to the potential role of AID as a DNA demethylase, it remains to be resolved how this demethylase activity can be reconciled with the mutagenic abilities of AID. A combination of biochemical and novel in vivo genetic manipulations will be required to address these exciting issues, which will be at the forefront of AID research for the next several years.
Acknowledgments
We would like to thank all members of the Chaudhuri lab for discussion and useful suggestions. This work is supported by a grant from the US National Institutes of Health (1RO1AI072194) to JC.
References
- Alt FW, Rosenberg N, Casanova RJ, Thomas E, Baltimore D. Immunoglobulin heavy-chain expression and class switching in a murine leukaemia cell line. Nature. 1982;296(5855):325–331. doi: 10.1038/296325a0. [DOI] [PubMed] [Google Scholar]
- Alt FW, Rosenberg N, Enea V, Siden E, Baltimore D. Multiple immunoglobulin heavy-chain gene transcripts in Abelson murine leukemia virus-transformed lymphoid cell lines. Molecular and Cellular Biology. 1982;2(4):386–400. doi: 10.1128/mcb.2.4.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell. 2013;152(3):417–429. doi: 10.1016/j.cell.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoufouchi S, Faili A, Zober C, D’Orlando O, Weller S, Weill JC, et al. Proteasomal degradation restricts the nuclear lifespan of AID. The Journal of Experimental Medicine. 2008;205(6):1357–1368. doi: 10.1084/jem.20070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell PD, Woo CJ, Wei K, Li Z, Martin A, Sack SZ, et al. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nature Immunology. 2004;5(2):224–229. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- Barreto V, Reina-San-Martin B, Ramiro AR, McBride KM, Nussenzweig MC. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Molecular Cell. 2003;12(2):501–508. doi: 10.1016/s1097-2765(03)00309-5. [DOI] [PubMed] [Google Scholar]
- Basso K, Dalla-Favera R. Roles of BCL6 in normal and transformed germinal center B cells. Immunological Reviews. 2012;247(1):172–183. doi: 10.1111/j.1600-065X.2012.01112.x. [DOI] [PubMed] [Google Scholar]
- Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438(7067):508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- Basu U, Meng FL, Keim C, Grinstein V, Pefanis E, Eccleston J, et al. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 2011;144(3):353–363. doi: 10.1016/j.cell.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu U, Wang Y, Alt FW. Evolution of phosphorylation-dependent regulation of activation-induced cytidine deaminase. Molecular Cell. 2008;32(2):285–291. doi: 10.1016/j.molcel.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum NA, Izumi N, Nishikori M, Nagaoka H, Shinkura R, Honjo T. Requirement of non-canonical activity of uracil DNA glycosylase for class switch recombination. The Journal of Biological Chemistry. 2007;282(1):731–742. doi: 10.1074/jbc.M607439200. [DOI] [PubMed] [Google Scholar]
- Begum NA, Kinoshita K, Kakazu N, Muramatsu M, Nagaoka H, Shinkura R, et al. Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch. Science. 2004;305(5687):1160–1163. doi: 10.1126/science.1098444. [DOI] [PubMed] [Google Scholar]
- Begum NA, Kinoshita K, Muramatsu M, Nagaoka H, Shinkura R, Honjo T. De novo protein synthesis is required for activation-induced cytidine deaminase-dependent DNA cleavage in immunoglobulin class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(35):13003–13007. doi: 10.1073/pnas.0405219101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begum NA, Stanlie A, Doi T, Sasaki Y, Jin HW, Kim YS, et al. Further evidence for involvement of a noncanonical function of uracil DNA glycosylase in class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(8):2752–2757. doi: 10.1073/pnas.0813252106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Aebersold R, Shiloh Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Letters. 2011;585(11):1625–1639. doi: 10.1016/j.febslet.2011.05.013. [DOI] [PubMed] [Google Scholar]
- Betz BC, Jordan-Williams KL, Wang C, Kang SG, Liao J, Logan MR, et al. Batf coordinates multiple aspects of B and T cell function required for normal antibody responses. The Journal of Experimental Medicine. 2010;207(5):933–942. doi: 10.1084/jem.20091548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz AG, Milstein C, Gonzalez-Fernandez A, Pannell R, Larson T, Neuberger MS. Elements regulating somatic hypermutation of an immunoglobulin kappa gene: Critical role for the intron enhancer/matrix attachment region. Cell. 1994;77(2):239–248. doi: 10.1016/0092-8674(94)90316-6. [DOI] [PubMed] [Google Scholar]
- Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature. 2010;463(7284):1042–1047. doi: 10.1038/nature08752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annual Review of Biochemistry. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Advances in Immunology. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- Boboila C, Jankovic M, Yan CT, Wang JH, Wesemann DR, Zhang T, et al. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3034–3039. doi: 10.1073/pnas.0915067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Oksenych V, Gostissa M, Wang JH, Zha S, Zhang Y, et al. Robust chromosomal DNA repair via alternative end-joining in the absence of X-ray repair cross-complementing protein 1 (XRCC1) Proceedings of the National Academy of Sciences of the United States of America. 2012;109(7):2473–2478. doi: 10.1073/pnas.1121470109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C, Yan C, Wesemann DR, Jankovic M, Wang JH, Manis J, et al. Alternative end-joining catalyzes class switch recombination in the absence of both Ku70 and DNA ligase 4. The Journal of Experimental Medicine. 2010;207(2):417–427. doi: 10.1084/jem.20092449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchert GM, Holton NW, Larson ED. Repression of human activation induced cytidine deaminase by miR-93 and miR-155. BMC Cancer. 2011;11:347. doi: 10.1186/1471-2407-11-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottaro A, Lansford R, Xu L, Zhang J, Rothman P, Alt FW. S region transcription per se promotes basal IgE class switch recombination but additional factors regulate the efficiency of the process. The EMBO Journal. 1994;13(3):665–674. doi: 10.1002/j.1460-2075.1994.tb06305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar SS, Watson M, Diaz M. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. The Journal of Biological Chemistry. 2004;279(25):26395–26401. doi: 10.1074/jbc.M403503200. [DOI] [PubMed] [Google Scholar]
- Buis J, Wu Y, Deng Y, Leddon J, Westfield G, Eckersdorff M, et al. Mre11 nuclease activity has essential roles in DNA repair and genomic stability distinct from ATM activation. Cell. 2008;135(1):85–96. doi: 10.1016/j.cell.2008.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter MA, Rajagurubandara E, Wijesinghe P, Bhagwat AS. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair. 2010;9(5):579–587. doi: 10.1016/j.dnarep.2010.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R, Nussenzweig A, Wuerffel R, Pelanda R, Reichlin A, Suh H, et al. Ku80 is required for immunoglobulin isotype switching. The EMBO Journal. 1998;17(8):2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Molecular Cell. 2013;49(5):858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Alt FW. Class-switch recombination: Interplay of transcription, DNA deamination and DNA repair. Nature Reviews Immunology. 2004;4(7):541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Basu U, Zarrin A, Yan C, Franco S, Perlot T, et al. Evolution of the immunoglobulin heavy chain class switch recombination mechanism. Advances in Immunology. 2007;94:157–214. doi: 10.1016/S0065-2776(06)94006-1. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430(7003):992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, Alt FW. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422(6933):726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- Chen K, Xu W, Wilson M, He B, Miller NW, Bengten E, et al. Immunoglobulin D enhances immune surveillance by activating antimicrobial, proinflammatory and B cell-stimulating programs in basophils. Nature Immunology. 2009;10(8):889–898. doi: 10.1038/ni.1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HL, Vuong BQ, Basu U, Franklin A, Schwer B, Astarita J, et al. Integrity of the AID serine-38 phosphorylation site is critical for class switch recombination and somatic hypermutation in mice. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(8):2717–2722. doi: 10.1073/pnas.0812304106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147(1):107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cogne M, Lansford R, Bottaro A, Zhang J, Gorman J, Young F, et al. A class switch control region at the 3’ end of the immunoglobulin heavy chain locus. Cell. 1994;77(5):737–747. doi: 10.1016/0092-8674(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146(1):67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch EE, Li Z, Takizawa M, Fichtner-Feigl S, Gourzi P, Montano C, et al. Regulation of AID expression in the immune response. The Journal of Experimental Medicine. 2007;204(5):1145–1156. doi: 10.1084/jem.20061952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui X, Lu Z, Kurosawa A, Klemm L, Bagshaw AT, Tsai AG, et al. Both CpG methylation and activation-induced deaminase are required for the fragility of the human bcl-2 major breakpoint region: Implications for the timing of the breaks in the t(14;18) translocation. Molecular and Cellular Biology. 2013;33(5):947–957. doi: 10.1128/MCB.01436-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel JA, Santos MA, Wang Z, Zang C, Schwab KR, Jankovic M, et al. PTIP promotes chromatin changes critical for immunoglobulin class switch recombination. Science. 2010;329(5994):917–923. doi: 10.1126/science.1187942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels GA, Lieber MR. RNA:DNA complex formation upon transcription of immunoglobulin switch regions: Implications for the mechanism and regulation of class switch recombination. Nucleic Acids Research. 1995;23(24):5006–5011. doi: 10.1093/nar/23.24.5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson NO, Shelness GS. APOLIPOPROTEIN B: mRNA editing, lipoprotein assembly, and presecretory degradation. Annual Review of Nutrition. 2000;20:169–193. doi: 10.1146/annurev.nutr.20.1.169. [DOI] [PubMed] [Google Scholar]
- Davis MM, Kim SK, Hood LE. DNA sequences mediating class switching in alpha-immunoglobulins. Science. 1980;209(4463):1360–1365. doi: 10.1126/science.6774415. [DOI] [PubMed] [Google Scholar]
- de Yebenes VG, Belver L, Pisano DG, Gonzalez S, Villasante A, Croce C, et al. miR-181b negatively regulates activation-induced cytidine deaminase in B cells. The Journal of Experimental Medicine. 2008;205(10):2199–2206. doi: 10.1084/jem.20080579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedeoglu F, Horwitz B, Chaudhuri J, Alt FW, Geha RS. Induction of activation-induced cytidine deaminase gene expression by IL-4 and CD40 ligation is dependent on STAT6 and NFkappaB. International Immunology. 2004;16(3):395–404. doi: 10.1093/intimm/dxh042. [DOI] [PubMed] [Google Scholar]
- Dege C, Hagman J. Activation of Aicda gene transcription by Pax5 in plasmacytoma cells. Immunologic Research. 2013;55(1–3):155–161. doi: 10.1007/s12026-012-8357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delbos F, De Smet A, Faili A, Aoufouchi S, Weill JC, Reynaud CA. Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse. The Journal of Experimental Medicine. 2005;201(8):1191–1196. doi: 10.1084/jem.20050292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demorest ZL, Li M, Harris RS. Phosphorylation directly regulates the intrinsic DNA cytidine deaminase activity of activation-induced deaminase and APOBEC3G protein. The Journal of Biological Chemistry. 2011;286(30):26568–26575. doi: 10.1074/jbc.M111.235721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey LA, Sun H, Hanakahi LA, Maizels N. G4 DNA binding by LR1 and its subunits, nucleolin and hnRNP D, A role for G-G pairing in immunoglobulin switch recombination. The Journal of Biological Chemistry. 1999;274(2):1066–1071. doi: 10.1074/jbc.274.2.1066. [DOI] [PubMed] [Google Scholar]
- Deriano L, Roth DB. Modernizing the nonhomologous end-joining repertoire: Alternative and classical NHEJ share the stage. Annual Review of Genetics. 2013;47:433–455. doi: 10.1146/annurev-genet-110711-155540. [DOI] [PubMed] [Google Scholar]
- Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419(6902):43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annual Review of Biochemistry. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339(6120):711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson SK, Market E, Besmer E, Papavasiliou FN. AID mediates hypermutation by deaminating single stranded DNA. The Journal of Experimental Medicine. 2003;197(10):1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkelmann M, Spehalski E, Stoneham T, Buis J, Wu Y, Sekiguchi JM, et al. Multiple functions of MRN in end-joining pathways during isotype class switching. Nature Structural & Molecular Biology. 2009;16(8):808–813. doi: 10.1038/nsmb.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi T, Kinoshita K, Ikegawa M, Muramatsu M, Honjo T. De novo protein synthesis is required for the activation-induced cytidine deaminase function in class-switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(5):2634–2638. doi: 10.1073/pnas.0437710100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Sola D, Victora GD, Ying CY, Phan RT, Saito M, Nussenzweig MC, et al. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nature Immunology. 2012;13(11):1083–1091. doi: 10.1038/ni.2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorsett Y, McBride KM, Jankovic M, Gazumyan A, Thai TH, Robbiani DF, et al. MicroRNA-155 suppresses activation-induced cytidine deaminasemediated Myc-Igh translocation. Immunity. 2008;28(5):630–638. doi: 10.1016/j.immuni.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DD, Manis JP, Zarrin AA, Kaylor L, Tian M, Alt FW. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(15):9984–9989. doi: 10.1073/pnas.152333499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick W, Hertz GZ, Scappino L, Gritzmacher C. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Research. 1993;21(3):365–372. doi: 10.1093/nar/21.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunnick W, Rabbitts TH, Milstein C. An immunoglobulin deletion mutant with implications for the heavy-chain switch and RNA splicing. Nature. 1980;286(5774):669–675. doi: 10.1038/286669a0. [DOI] [PubMed] [Google Scholar]
- Dunnick W, Wilson M, Stavnezer J. Mutations, duplication, and deletion of recombined switch regions suggest a role for DNA replication in the immunoglobulin heavy-chain switch. Molecular and Cellular Biology. 1989;9(5):1850–1856. doi: 10.1128/mcb.9.5.1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenstein MR, Neuberger MS. Deficiency in Msh2 affects the efficiency and local sequence specificity of immunoglobulin class-switch recombination: Parallels with somatic hypermutation. The EMBO Journal. 1999;18(12):3484–3490. doi: 10.1093/emboj/18.12.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenstein MR, Rada C, Jones AM, Milstein C, Neuberger MS. Switch junction sequences in PMS2-deficient mice reveal a microhomology-mediated mechanism of Ig class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(25):14553–14558. doi: 10.1073/pnas.241525998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Molecular Cell. 2013;49(5):872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Faili A, Aoufouchi S, Weller S, Vuillier F, Stary A, Sarasin A, et al. DNA polymerase {eta} is involved in hypermutation occurring during immunoglobulin class switch recombination. The Journal of Experimental Medicine. 2004;199(2):265–270. doi: 10.1084/jem.20031831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1- mediated end resection during DNA repair. The Journal of Biological Chemistry. 2013;288(16):11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Ortiz M, Rodriguez L, Garcia A, Martinez D, Moreno de Alboran I. The proto-oncogene c-myc regulates antibody secretion and Ig class switch recombination. Journal of Immunology. 2013;190(12):6135–6144. doi: 10.4049/jimmunol.1300712. [DOI] [PubMed] [Google Scholar]
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Molecular Cell. 2006;21(2):201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Franco S, Murphy MM, Li G, Borjeson T, Boboila C, Alt FW. DNAPKcs and Artemis function in the end-joining phase of immunoglobulin heavy chain class switch recombination. The Journal of Experimental Medicine. 2008;205(3):557–564. doi: 10.1084/jem.20080044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz EL, Rosenberg BR, Lay K, Mihailovic A, Tuschl T, Papavasiliou FN. A comprehensive analysis of the effects of the deaminase AID on the transcriptome and methylome of activated B cells. Nature Immunology. 2013;14(7):749–755. doi: 10.1038/ni.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukita Y, Jacobs H, Rajewsky K. Somatic hypermutation in the heavy chain locus correlates with transcription. Immunity. 1998;9(1):105–114. doi: 10.1016/s1074-7613(00)80592-0. [DOI] [PubMed] [Google Scholar]
- Gazumyan A, Timachova K, Yuen G, Siden E, Di Virgilio M, Woo EM, et al. Amino-terminal phosphorylation of activation-induced cytidine deaminase suppresses c-myc/IgH translocation. Molecular and Cellular Biology. 2011;31(3):442–449. doi: 10.1128/MCB.00349-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gearhart PJ, Bogenhagen DF. Clusters of point mutations are found exclusively around rearranged antibody variable genes. Proceedings of the National Academy of Sciences of the United States of America. 1983;80(11):3439–3443. doi: 10.1073/pnas.80.11.3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annual Review of Biochemistry. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- Gonda H, Sugai M, Nambu Y, Katakai T, Agata Y, Mori KJ, et al. The balance between Pax5 and Id2 activities is the key to AID gene expression. The Journal of Experimental Medicine. 2003;198(9):1427–1437. doi: 10.1084/jem.20030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Yan CT, Bianco JM, Cogne M, Pinaud E, Alt FW. Longrange oncogenic activation of Igh-c-myc translocations by the Igh 3’ regulatory region. Nature. 2009;462(7274):803–807. doi: 10.1038/nature08633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Zou YR, Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73(6):1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- Guikema JE, Stavnezer J, Schrader CE. The role of Apex2 in class-switch recombination of immunoglobulin genes. International Immunology. 2010;22(3):213–214. doi: 10.1093/intimm/dxq003. [DOI] [PubMed] [Google Scholar]
- Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145(3):423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim O, Resch W, Yamane A, Klein I, Kieffer-Kwon KR, Jankovic M, et al. DNA damage defines sites of recurrent chromosomal translocations in B lymphocytes. Nature. 2012;484(7392):69–74. doi: 10.1038/nature10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Akira S, Calame K, Beutler B, Selsing E, Imanishi-Kari T. Class switch recombination and somatic hypermutation in early mouse B cells are mediated by B cell and Toll-like receptors. Immunity. 2007;27(1):64–75. doi: 10.1016/j.immuni.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L, Yu K. Altered kinetics of nonhomologous end joining and class switch recombination in ligase IV-deficient B cells. The Journal of Experimental Medicine. 2008;205(12):2745–2753. doi: 10.1084/jem.20081623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasham MG, Donghia NM, Coffey E, Maynard J, Snow KJ, Ames J, et al. Widespread genomic breaks generated by activation-induced cytidine deaminase are prevented by homologous recombination. Nature Immunology. 2010;11(9):820–826. doi: 10.1038/ni.1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasham MG, Snow KJ, Donghia NM, Branca JA, Lessard MD, Stavnezer J, et al. Activation-induced cytidine deaminase-initiated off-target DNA breaks are detected and resolved during S phase. Journal of Immunology. 2012;189(5):2374–2382. doi: 10.4049/jimmunol.1200414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honjo T. A memoir of AID, which engraves antibody memory on DNA. Nature Immunology. 2008;9(4):335–337. doi: 10.1038/ni0408-335. [DOI] [PubMed] [Google Scholar]
- Honjo T, Kinoshita K, Muramatsu M. Molecular mechanism of class switch recombination: Linkage with somatic hypermutation. Annual Review of Immunology. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- Hu Y, Ericsson I, Torseth K, Methot SP, Sundheim O, Liabakk NB, et al. A combined nuclear and nucleolar localization motif in activation-induced cytidine deaminase (AID) controls immunoglobulin class switching. Journal of Molecular Biology. 2013;425(2):424–443. doi: 10.1016/j.jmb.2012.11.026. [DOI] [PubMed] [Google Scholar]
- Huong le T, Kobayashi M, Nakata M, Shioi G, Miyachi H, Honjo T, et al. In vivo analysis of Aicda gene regulation: A critical balance between upstream enhancers and intronic silencers governs appropriate expression. PLoS One. 2013;8(4):e61433. doi: 10.1371/journal.pone.0061433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nature Immunology. 2003;4(10):1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- Incorvaia E, Sicouri L, Petersen-Mahrt SK, Schmitz KM. Hormones and AID: Balancing immunity and autoimmunity. Autoimmunity. 2013;46(2):128–137. doi: 10.3109/08916934.2012.748752. [DOI] [PubMed] [Google Scholar]
- Ise W, Kohyama M, Schraml BU, Zhang T, Schwer B, Basu U, et al. The transcription factor BATF controls the global regulators of class-switch recombination in both B cells and T cells. Nature Immunology. 2011;12(6):536–543. doi: 10.1038/ni.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Nagaoka H, Shinkura R, Begum N, Muramatsu M, Nakata M, et al. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):1975–1980. doi: 10.1073/pnas.0307335101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasato T, Shimizu A, Honjo T, Yamagishi H. Circular DNA is excised by immunoglobulin class switch recombination. Cell. 1990;62(1):143–149. doi: 10.1016/0092-8674(90)90248-d. [DOI] [PubMed] [Google Scholar]
- Jeevan-Raj BP, Robert I, Heyer V, Page A, Wang JH, Cammas F, et al. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. The Journal of Experimental Medicine. 2011;208(8):1649–1660. doi: 10.1084/jem.20110118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Z, Volpi SA, Hassan R, Martinez N, Giannini SL, Gold T, et al. Evidence for physical interaction between the immunoglobulin heavy chain variable region and the 3’ regulatory region. The Journal of Biological Chemistry. 2007;282(48):35169–35178. doi: 10.1074/jbc.M705719200. [DOI] [PubMed] [Google Scholar]
- Jung S, Rajewsky K, Radbruch A. Shutdown of class switch recombination by deletion of a switch region control element. Science. 1993;259(5097):984–987. doi: 10.1126/science.8438159. [DOI] [PubMed] [Google Scholar]
- Kao YP, Hsieh WC, Hung ST, Huang CW, Lieber MR, Huang FT. Detection and characterization of R-loops at the murine immunoglobulin Salpha region. Molecular Immunology. 2013;54(2):208–216. doi: 10.1016/j.molimm.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Kataoka T, Yamawaki-Kataoka Y, Yamagishi H, Honjo T. Cloning immunoglobulin gamma 2b chain gene of mouse: Characterization and partial sequence determination. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(9):4240–4244. doi: 10.1073/pnas.76.9.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenter AL, Feldman S, Wuerffel R, Achour I, Wang L, Kumar S. Threedimensional architecture of the IgH locus facilitates class switch recombination. Annals of the New York Academy of Sciences. 2012;1267:86–94. doi: 10.1111/j.1749-6632.2012.06604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamlichi AA, Glaudet F, Oruc Z, Denis V, Le Bert M, Cogne M. Immunoglobulin class-switch recombination in mice devoid of any S mu tandem repeat. Blood. 2004;103(10):3828–3836. doi: 10.1182/blood-2003-10-3470. [DOI] [PubMed] [Google Scholar]
- Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. 2013;153(1):126–138. doi: 10.1016/j.cell.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147(1):95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluin PM, Kayano H, Zani VJ, Kluin-Nelemans HC, Tucker PW, Satterwhite E, et al. IgD class switching: Identification of a novel recombination site in neoplastic and normal B cells. European Journal of Immunology. 1995;25(12):3504–3508. doi: 10.1002/eji.1830251244. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Aida M, Nagaoka H, Begum NA, Kitawaki Y, Nakata M, et al. AID-induced decrease in topoisomerase 1 induces DNA structural alteration and DNA cleavage for class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(52):22375–22380. doi: 10.1073/pnas.0911879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodgire P, Mukkawar P, Ratnam S, Martin TE, Storb U. Changes inRNA polymerase II progression influence somatic hypermutation of Ig-related genes by AID. The Journal of Experimental Medicine. 2013;210(7):1481–1492. doi: 10.1084/jem.20121523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, DiMenna L, Schrode N, Liu TC, Franck P, Munoz-Descalzo S, et al. AID stabilizes stem-cell phenotype by removing epigenetic memory of pluripotency genes. Nature. 2013;500:89–92. doi: 10.1038/nature12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuppers R. Mechanisms of B-cell lymphoma pathogenesis. Nature Reviews Cancer. 2005;5(4):251–262. doi: 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- Kuwahara K, Fujimura S, Takahashi Y, Nakagata N, Takemori T, Aizawa S, et al. Germinal center-associated nuclear protein contributes to affinity maturation of B cell antigen receptor in T cell-dependent responses. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(4):1010–1015. doi: 10.1073/pnas.0307609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzin II, Ugine GD, Wu D, Young F, Chen J, Bottaro A. Normal isotype switching in B cells lacking the I mu exon splice donor site: Evidence for multiple I mu-like germline transcripts. Journal of Immunology. 2000;164(3):1451–1457. doi: 10.4049/jimmunol.164.3.1451. [DOI] [PubMed] [Google Scholar]
- Lebecque SG, Gearhart PJ. Boundaries of somatic mutation in rearranged immunoglobulin genes: 5’ boundary is near the promoter, and 3’ boundary is approximately 1 kb from V(D)J gene. The Journal of Experimental Medicine. 1990;172(6):1717–1727. doi: 10.1084/jem.172.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117(2):171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- Lee-Theilen M, Chaudhuri J. Walking the AID tightrope. Nature Immunology. 2010;11(2):107–109. doi: 10.1038/ni0210-107. [DOI] [PubMed] [Google Scholar]
- Lee-Theilen M, Matthews AJ, Kelly D, Zheng S, Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nature Structural & Molecular Biology. 2011;18(1):75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennon GG, Perry RP. C mu-containing transcripts initiate heterogeneously within the IgH enhancer region and contain a novel 5’-nontranslatable exon. Nature. 1985;318(6045):475–478. doi: 10.1038/318475a0. [DOI] [PubMed] [Google Scholar]
- Li SC, Rothman PB, Zhang J, Chan C, Hirsh D, Alt FW. Expression of I mu-C gamma hybrid germline transcripts subsequent to immunoglobulin heavy chain class switching. International Immunology. 1994;6(4):491–497. doi: 10.1093/intimm/6.4.491. [DOI] [PubMed] [Google Scholar]
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual Review of Biochemistry. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, et al. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 2008;451(7180):841–845. doi: 10.1038/nature06547. [DOI] [PubMed] [Google Scholar]
- Lorenz M, Jung S, Radbruch A. Switch transcripts in immunoglobulin class switching. Science. 1995;267(5205):1825–1828. doi: 10.1126/science.7892607. [DOI] [PubMed] [Google Scholar]
- Luby TM, Schrader CE, Stavnezer J, Selsing E. The mu switch region tandem repeats are important, but not required, for antibody class switch recombination. The Journal of Experimental Medicine. 2001;193(2):159–168. doi: 10.1084/jem.193.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden JM, McCarty T, Petiniot LK, Shen R, Barlow C, Wynn TA, et al. Immunoglobulin class switch recombination is impaired in Atm-deficient mice. The Journal of Experimental Medicine. 2004;200(9):1111–1121. doi: 10.1084/jem.20041074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutzker S, Rothman P, Pollock R, Coffman R, Alt FW. Mitogen- and IL-4-regulated expression of germ-line Ig gamma 2b transcripts: Evidence for directed heavy chain class switching. Cell. 1988;53(2):177–184. doi: 10.1016/0092-8674(88)90379-0. [DOI] [PubMed] [Google Scholar]
- Maeda K, Singh SK, Eda K, Kitabatake M, Pham P, Goodman MF, et al. GANP-mediated recruitment of activation-induced cytidine deaminase to cell nuclei and to immunoglobulin variable region DNA. The Journal of Biological Chemistry. 2010;285(31):23945–23953. doi: 10.1074/jbc.M110.131441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai T, Pone EJ, Li G, Lam TS, Moehlman J, Xu Z, et al. Induction of activation-induced cytidine deaminase-targeting adaptor 14-3-3 gamma is mediated by NF-kappaB-dependent recruitment of CFP1 to the 5’-CpG-3’-rich 14-3-3 gamma promoter and is sustained by E2A. Journal of Immunology. 2013;191(4):1895–1906. doi: 10.4049/jimmunol.1300922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai T, Zan H, Zhang J, Hawkins JS, Xu Z, Casali P. Estrogen receptors bind to and activate the HOXC4/HoxC4 promoter to potentiate HoxC4-mediated activation-induced cytosine deaminase induction, immunoglobulin class switch DNA recombination, and somatic hypermutation. The Journal of Biological Chemistry. 2010;285(48):37797–37810. doi: 10.1074/jbc.M110.169086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak CH, Pham P, Afif SA, Goodman MF. A mathematical model for scanning and catalysis on single-stranded DNA, illustrated with activation-induced deoxycytidine deaminase. The Journal of Biological Chemistry. 2013;288:29786–29795. doi: 10.1074/jbc.M113.506550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Gu Y, Lansford R, Sonoda E, Ferrini R, Davidson L, et al. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. The Journal of Experimental Medicine. 1998;187(12):2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain classswitch recombination. Nature Immunology. 2004;5(5):481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- Manis JP, van der Stoep N, Tian M, Ferrini R, Davidson L, Bottaro A, et al. Class switching in B cells lacking 3’ immunoglobulin heavy chain enhancers. The Journal of Experimental Medicine. 1998;188(8):1421–1431. doi: 10.1084/jem.188.8.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, et al. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302(5646):890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- Masani S, Han L, Yu K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Molecular and Cellular Biology. 2013;33(7):1468–1473. doi: 10.1128/MCB.00026-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Yoshida K, Maeda T, Usuda S, Sakano H. Switch circular DNA formed in cytokine-treated mouse splenocytes: Evidence for intramolecular DNA deletion in immunoglobulin class switching. Cell. 1990;62(1):135–142. doi: 10.1016/0092-8674(90)90247-c. [DOI] [PubMed] [Google Scholar]
- Maul RW, Gearhart PJ. Women, autoimmunity, and cancer: A dangerous liaison between estrogen and activation-induced deaminase? The Journal of Experimental Medicine. 2009;206(1):11–13. doi: 10.1084/jem.20080086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul RW, Saribasak H, Martomo SA, McClure RL, Yang W, Vaisman A, et al. Uracil residues dependent on the deaminase AID in immunoglobulin gene variable and switch regions. Nature Immunology. 2011;12(1):70–76. doi: 10.1038/ni.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. The Journal of Experimental Medicine. 2004;199(9):1235–1244. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Barreto VM, Robbiani DF, Chait BT, et al. Regulation of hypermutation by activation-induced cytidine deaminase phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(23):8798–8803. doi: 10.1073/pnas.0603272103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, Nussenzweig MC. Regulation of class switch recombination and somatic mutation by AID phosphorylation. The Journal of Experimental Medicine. 2008;205(11):2585–2594. doi: 10.1084/jem.20081319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proceedings of the National Academy of Sciences of the United States of America. 1984;81(10):3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael N, Martin TE, Nicolae D, Kim N, Padjen K, Zhan P, et al. Effects of sequence and structure on the hypermutability of immunoglobulin genes. Immunity. 2002;16(1):123–134. doi: 10.1016/s1074-7613(02)00261-3. [DOI] [PubMed] [Google Scholar]
- Michaelson JS, Giannini SL, Birshtein BK. Identification of 3’ alpha-hs4, a novel Ig heavy chain enhancer element regulated at multiple stages of B cell differentiation. Nucleic Acids Research. 1995;23(6):975–981. doi: 10.1093/nar/23.6.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliazza A, Martinotti S, Chen W, Fusco C, Ye BH, Knowles DM, et al. Frequent somatic hypermutation of the 5’ noncoding region of the BCL6 gene in B-cell lymphoma. Proceedings of the National Academy of Sciences of the United States of America. 1995;92(26):12520–12524. doi: 10.1073/pnas.92.26.12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min IM, Schrader CE, Vardo J, Luby TM, D’Avirro N, Stavnezer J, et al. The Smu tandem repeat region is critical for Ig isotype switching in the absence of Msh2. Immunity. 2003;19(4):515–524. doi: 10.1016/s1074-7613(03)00262-0. [DOI] [PubMed] [Google Scholar]
- Misaghi S, Garris CS, Sun Y, Nguyen A, Zhang J, Sebrell A, et al. Increased targeting of donor switch region and IgE in Sgamma1-deficient B cells. Journal of Immunology. 2010;185(1):166–173. doi: 10.4049/jimmunol.1000515. [DOI] [PubMed] [Google Scholar]
- Mizuta R, Iwai K, Shigeno M, Mizuta M, Uemura T, Ushiki T, et al. Molecular visualization of immunoglobulin switch region RNA/DNA complex by atomic force microscope. The Journal of Biological Chemistry. 2003;278(7):4431–4434. doi: 10.1074/jbc.M209262200. [DOI] [PubMed] [Google Scholar]
- Morgan HD, Dean W, Coker HA, Reik W, Petersen-Mahrt SK. Activation-induced cytidine deaminase deaminates 5-methylcytosine in DNA and is expressed in pluripotent tissues: Implications for epigenetic reprogramming. The Journal of Biological Chemistry. 2004;279(50):52353–52360. doi: 10.1074/jbc.M407695200. [DOI] [PubMed] [Google Scholar]
- Muller JR, Giese T, Henry DL, Mushinski JF, Marcu KB. Generation of switch hybrid DNA between Ig heavy chain-mu and downstream switch regions in B lymphocytes. Journal of Immunology. 1998;161(3):1354–1362. [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, et al. Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. The Journal of Biological Chemistry. 1999;274(26):18470–18476. doi: 10.1074/jbc.274.26.18470. [DOI] [PubMed] [Google Scholar]
- Muschen M, Re D, Jungnickel B, Diehl V, Rajewsky K, Kuppers R. Somatic mutation of the CD95 gene in human B cells as a side-effect of the germinal center reaction. The Journal of Experimental Medicine. 2000;192(12):1833–1840. doi: 10.1084/jem.192.12.1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muto T, Okazaki IM, Yamada S, Tanaka Y, Kinoshita K, Muramatsu M, et al. Negative regulation of activation-induced cytidine deaminase in B cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(8):2752–2757. doi: 10.1073/pnas.0510970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel CS, Jia H, Ye Y, Shen L, Goldschmidt HL, Stivers JT, et al. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nature Chemical Biology. 2012;8(9):751–758. doi: 10.1038/nchembio.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel CS, Lee JW, Wang LC, Kohli RM. Nucleic acid determinants for selective deamination of DNA over RNA by activation-induced deaminase. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(35):14225–14230. doi: 10.1073/pnas.1306345110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Kondo S, Sugai M, Nazarea M, Imamura S, Honjo T. High frequency class switching of an IgMþ B lymphoma clone CH12F3 to IgAþ cells. International Immunology. 1996;8(2):193–201. doi: 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- Nambu Y, Sugai M, Gonda H, Lee CG, Katakai T, Agata Y, et al. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 2003;302(5653):2137–2140. doi: 10.1126/science.1092481. [DOI] [PubMed] [Google Scholar]
- Nikaido T, Nakai S, Honjo T. Switch region of immunoglobulin Cmu gene is composed of simple tandem repetitive sequences. Nature. 1981;292(5826):845–848. doi: 10.1038/292845a0. [DOI] [PubMed] [Google Scholar]
- Nowak U, Matthews AJ, Zheng S, Chaudhuri J. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nature Immunology. 2011;12(2):160–166. doi: 10.1038/ni.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell. 2010;141(1):27–38. doi: 10.1016/j.cell.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata M, Yamawaki-Kataoka Y, Takahashi N, Kataoka T, Shimizu A, Mano Y, et al. Immunoglobulin gamma 1 heavy chain gene: Structural gene sequences cloned in a bacterial plasmid. Gene. 1980;9(1–2):87–97. doi: 10.1016/0378-1119(80)90168-7. [DOI] [PubMed] [Google Scholar]
- Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248(4962):1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- Okazaki IM, Hiai H, Kakazu N, Yamada S, Muramatsu M, Kinoshita K, et al. Constitutive expression of AID leads to tumorigenesis. The Journal of Experimental Medicine. 2003;197(9):1173–1181. doi: 10.1084/jem.20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, et al. Active demethylation of the paternal genome in the mouse zygote. Current Biology. 2000;10(8):475–478. doi: 10.1016/s0960-9822(00)00448-6. [DOI] [PubMed] [Google Scholar]
- Papavasiliou FN, Schatz DG. Somatic hypermutation of immunoglobulin genes: Merging mechanisms for genetic diversity. Cell. 2002;109(Suppl):S35–S44. doi: 10.1016/s0092-8674(02)00706-7. [DOI] [PubMed] [Google Scholar]
- Park SR, Kim PH, Lee KS, Lee SH, Seo GY, Yoo YC, et al. APRIL stimulates NF-kappaB-mediated HoxC4 induction for AID expression in mouse B cells. Cytokine. 2013;61(2):608–613. doi: 10.1016/j.cyto.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SR, Zan H, Pal Z, Zhang J, Al-Qahtani A, Pone EJ, et al. HoxC4 binds to the promoter of the cytidine deaminase AID gene to induce AID expression, class-switch DNA recombination and somatic hypermutation. Nature Immunology. 2009;10(5):540–550. doi: 10.1038/ni.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, et al. AID is required for germinal center-derived lymphomagenesis. Nature Genetics. 2008;40(1):108–112. doi: 10.1038/ng.2007.35. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Guglielmino R, Houldsworth J, Mohr J, Aoufouchi S, Polakiewicz R, et al. Expression of the AID protein in normal and neoplastic B cells. Blood. 2004;104(10):3318–3325. doi: 10.1182/blood-2004-04-1558. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(2):395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, et al. BCL-6 mutations in normal germinal center B cells: Evidence of somatic hypermutation acting outside Ig loci. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(20):11816–11821. doi: 10.1073/pnas.95.20.11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Neumeister P, Goossens T, Nanjangud G, Chaganti RS, Kuppers R, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412(6844):341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- Patenaude AM, Orthwein A, Hu Y, Campo VA, Kavli B, Buschiazzo A, et al. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nature Structural & Molecular Biology. 2009;16(5):517–527. doi: 10.1038/nsmb.1598. [DOI] [PubMed] [Google Scholar]
- Pauklin S, Petersen-Mahrt SK. Progesterone inhibits activation-induced deaminase by binding to the promoter. Journal of Immunology. 2009;183(2):1238–1244. doi: 10.4049/jimmunol.0803915. [DOI] [PubMed] [Google Scholar]
- Pauklin S, Sernandez IV, Bachmann G, Ramiro AR, Petersen-Mahrt SK. Estrogen directly activates AID transcription and function. The Journal of Experimental Medicine. 2009;206(1):99–111. doi: 10.1084/jem.20080521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, et al. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 2010;143(1):122–133. doi: 10.1016/j.cell.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Nussenzweig MC. AID targeting in antibody diversity. Advances in Immunology. 2011;110:1–26. doi: 10.1016/B978-0-12-387663-8.00005-3. [DOI] [PubMed] [Google Scholar]
- Pech M, Hochtl J, Schnell H, Zachau HG. Differences between germ-line and rearranged immunoglobulin V kappa coding sequences suggest a localized mutation mechanism. Nature. 1981;291(5817):668–670. doi: 10.1038/291668a0. [DOI] [PubMed] [Google Scholar]
- Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, et al. The biochemistry of somatic hypermutation. Annual Review of Immunology. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- Perlot T, Li G, Alt FW. Antisense transcripts from immunoglobulin heavychain locus V(D)J and switch regions. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(10):3843–3848. doi: 10.1073/pnas.0712291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Storb U. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 1996;4(1):57–65. doi: 10.1016/s1074-7613(00)80298-8. [DOI] [PubMed] [Google Scholar]
- Petersen S, Casellas R, Reina-San-Martin B, Chen HT, Difilippantonio MJ, Wilson PC, et al. AID is required to initiate Nbs1/gamma-H2AX focus formation and mutations at sites of class switching. Nature. 2001;414(6864):660–665. doi: 10.1038/414660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, Neuberger MS. AIDmutates E.coli suggestinga DNA deamination mechanism for antibody diversification. Nature. 2002;418(6893):99–103. doi: 10.1038/nature00862. [DOI] [PubMed] [Google Scholar]
- Pham P, Bransteitter R, Petruska J, Goodman MF. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature. 2003;424(6944):103–107. doi: 10.1038/nature01760. [DOI] [PubMed] [Google Scholar]
- Pham P, Calabrese P, Park SJ, Goodman MF. Analysis of a single-stranded DNA-scanning process in which activation-induced deoxycytidine deaminase (AID) deaminates C to U haphazardly and inefficiently to ensure mutational diversity. The Journal of Biological Chemistry. 2011;286(28):24931–24942. doi: 10.1074/jbc.M111.241208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham P, Smolka MB, Calabrese P, Landolph A, Zhang K, Zhou H, et al. Impact of phosphorylation and phosphorylation-null mutants on the activity and deamination specificity of activation-induced cytidine deaminase. The Journal of Biological Chemistry. 2008;283(25):17428–17439. doi: 10.1074/jbc.M802121200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinaud E, Khamlichi AA, Le Morvan C, Drouet M, Nalesso V, Le Bert M, et al. Localization of the 3’ IgH locus elements that effect long-distance regulation of class switch recombination. Immunity. 2001;15(2):187–199. doi: 10.1016/s1074-7613(01)00181-9. [DOI] [PubMed] [Google Scholar]
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature. 2010;463(7284):1101–1105. doi: 10.1038/nature08829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu G, Harriman GR, Stavnezer J. Ialpha exon-replacement mice synthesize a spliced HPRT-C(alpha) transcript which may explain their ability to switch to IgA. Inhibition of switching to IgG in these mice. International Immunology. 1999;11(1):37–46. doi: 10.1093/intimm/11.1.37. [DOI] [PubMed] [Google Scholar]
- Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Molecular Cell. 2004;16(2):163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Rada C, Gonzalez-Fernandez A, Jarvis JM, Milstein C. The 5’ boundary of somatic hypermutation in a V kappa gene is in the leader intron. European Journal of Immunology. 1994;24(6):1453–1457. doi: 10.1002/eji.1830240632. [DOI] [PubMed] [Google Scholar]
- Rada C, Jarvis JM, Milstein C. AID-GFP chimeric protein increases hypermutation of Ig genes with no evidence of nuclear localization. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(10):7003–7008. doi: 10.1073/pnas.092160999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Current Biology. 2002;12(20):1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- Rajewsky K. Clonal selection and learning in the antibody system. Nature. 1996;381(6585):751–758. doi: 10.1038/381751a0. [DOI] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Callen E, Difilippantonio S, Chen HT, McBride KM, et al. Role of genomic instability and p53 in AID-induced c-myc-Igh translocations. Nature. 2006;440(7080):105–109. doi: 10.1038/nature04495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, et al. AID is required for c-myc/IgH chromosome translocations in vivo. Cell. 2004;118(4):431–438. doi: 10.1016/j.cell.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Ramiro AR, Stavropoulos P, Jankovic M, Nussenzweig MC. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nature Immunology. 2003;4(5):452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- Ranjit S, Khair L, Linehan EK, Ucher AJ, Chakrabarti M, Schrader CE, et al. AID binds cooperatively with UNG and Msh2-Msh6 to Ig switch regions dependent upon the AID C terminus. Journal of Immunology. 2011;187(5):2464–2475. doi: 10.4049/jimmunol.1101406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratmeyer L, Vinayak R, Zhong YY, Zon G, Wilson WD. Sequence specific thermodynamic and structural properties for DNA.RNA duplexes. Biochemistry. 1994;33(17):5298–5304. doi: 10.1021/bi00183a037. [DOI] [PubMed] [Google Scholar]
- Reaban ME, Griffin JA. Induction of RNA-stabilized DNA conformers by transcription of an immunoglobulin switch region. Nature. 1990;348(6299):342–344. doi: 10.1038/348342a0. [DOI] [PubMed] [Google Scholar]
- Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. The Journal of Experimental Medicine. 2004;200(9):1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Chen J, Nussenzweig A, Nussenzweig MC. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1(-/-) B cells. European Journal of Immunology. 2007;37(1):235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, et al. Activationinduced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102(5):565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Roa S, Li Z, Peled JU, Zhao C, Edelmann W, Scharff MD. Activationinduced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) PLoS One. 2010;5(6):e11182. [Google Scholar]
- Robbiani DF, Bothmer A, Callen E, Reina-San-Martin B, Dorsett Y, Difilippantonio S. AID is required for the chromosomal breaks in c-myc that lead to c-myc/IgH translocations. Cell. 2008;135(6):1028–1038. doi: 10.1016/j.cell.2008.09.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbiani DF, Bunting S, Feldhahn N, Bothmer A, Camps J, Deroubaix S, et al. AID produces DNA double-strand breaks in non-Ig genes and mature B cell lymphomas with reciprocal chromosome translocations. Molecular Cell. 2009;36(4):631–641. doi: 10.1016/j.molcel.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert I, Dantzer F, Reina-San-Martin B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. The Journal of Experimental Medicine. 2009;206(5):1047–1056. doi: 10.1084/jem.20082468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha PP, Micsinai M, Kim JR, Hewitt SL, Souza PP, Trimarchi T, et al. Close proximity to Igh is a contributing factor to AID-mediated translocations. Molecular Cell. 2012;47(6):873–885. doi: 10.1016/j.molcel.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Diaz M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. Journal of Immunology. 2004;172(6):3382–3384. doi: 10.4049/jimmunol.172.6.3382. [DOI] [PubMed] [Google Scholar]
- Rothenfluh HS, Blanden RV, Steele EJ. Evolution of V genes: DNA sequence structure of functional germline genes and pseudogenes. Immunogenetics. 1995;42(3):159–171. doi: 10.1007/BF00191221. [DOI] [PubMed] [Google Scholar]
- Rothman P, Lutzker S, Cook W, Coffman R, Alt FW. Mitogen plus interleukin 4 induction of C epsilon transcripts in B lymphoid cells. The Journal of Experimental Medicine. 1988;168(6):2385–2389. doi: 10.1084/jem.168.6.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush JS, Fugmann SD, Schatz DG. Staggered AID-dependent DNA double strand breaks are the predominant DNA lesions targeted to S mu in Ig class switch recombination. International Immunology. 2004;16(4):549–557. doi: 10.1093/intimm/dxh057. [DOI] [PubMed] [Google Scholar]
- Sakano H, Maki R, Kurosawa Y, Roeder W, Tonegawa S. Two types of somatic recombination are necessary for the generation of complete immunoglobulin heavy-chain genes. Nature. 1980;286(5774):676–683. doi: 10.1038/286676a0. [DOI] [PubMed] [Google Scholar]
- Saribasak H, Gearhart PJ. Does DNA repair occur during somatic hypermutation? Seminars in Immunology. 2012;24(4):287–292. doi: 10.1016/j.smim.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nature Immunology. 2003;4(6):586–593. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Baltimore D. Uncovering the V(D)J recombinase. Cell. 2004;116(Suppl. 2):S103–S106. doi: 10.1016/s0092-8674(04)00042-x. 102 p following S106. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nature Reviews Immunology. 2011;11(4):251–263. doi: 10.1038/nri2941. [DOI] [PubMed] [Google Scholar]
- Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59(6):1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- Schrader CE, Guikema JE, Linehan EK, Selsing E, Stavnezer J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. Journal of Immunology. 2007;179(9):6064–6071. doi: 10.4049/jimmunol.179.9.6064. [DOI] [PubMed] [Google Scholar]
- Schrader CE, Guikema JE, Wu X, Stavnezer J. The roles of APE1, APE2, DNApolymerase beta and mismatch repair in creating S region DNAbreaks during antibody class switch. Philosophical Transactions of the Royal Society of London, Series B Biological Sciences. 2009;364(1517):645–652. doi: 10.1098/rstb.2008.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. The Journal of Experimental Medicine. 2005;202(4):561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Vardo J, Linehan E, Twarog MZ, Niedernhofer LJ, Hoeijmakers JH, et al. Deletion of the nucleotide excision repair gene Ercc1 reduces immunoglobulin class switching and alters mutations near switch recombination junctions. The Journal of Experimental Medicine. 2004;200(3):321–330. doi: 10.1084/jem.20040052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Vardo J, Stavnezer J. Role for mismatch repair proteins Msh2, Mlh1, and Pms2 in immunoglobulin class switching shown by sequence analysis of recombination junctions. The Journal of Experimental Medicine. 2002;195(3):367–373. doi: 10.1084/jem.20011877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Vardo J, Stavnezer J. Mlh1 can function in antibody class switch recombination independently of Msh2. The Journal of Experimental Medicine. 2003;197(10):1377–1383. doi: 10.1084/jem.20022190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl KJ, Bottaro A, Vo A, Zhang J, Davidson L, Alt FW. An expressed neo(r) cassette provides required functions of the 1gamma2b exon for class switching. International Immunology. 1998;10(11):1683–1692. doi: 10.1093/intimm/10.11.1683. [DOI] [PubMed] [Google Scholar]
- Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. The Journal of Experimental Medicine. 2009;206(5):1073–1087. doi: 10.1084/jem.20082311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, Gilbert W. Formation of parallel four-stranded complexes by guaninerich motifs in DNA and its implications for meiosis. Nature. 1988;334(6180):364–366. doi: 10.1038/334364a0. [DOI] [PubMed] [Google Scholar]
- Shen HM, Storb U. Activation-induced cytidine deaminase (AID) can target both DNA strands when the DNA is supercoiled. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(35):12997–13002. doi: 10.1073/pnas.0404974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6(-/-)Ung(-/-) double-knockout mice. Journal of Immunology. 2006;177(8):5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- Shi T, Bunker RD, Mattarocci S, Ribeyre C, Faty M, Gut H, et al. Rif1 and Rif2 shape telomere function and architecture through multivalent Rap1 interactions. Cell. 2013;153(6):1340–1353. doi: 10.1016/j.cell.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Shinkura R, Tian M, Smith M, Chua K, Fujiwara Y, Alt FW. The influence of transcriptional orientation on endogenous switch region function. Nature Immunology. 2003;4(5):435–441. doi: 10.1038/ni918. [DOI] [PubMed] [Google Scholar]
- Snapper CM, Finkelman FD, Paul WE. Regulation of IgG1 and IgE production by interleukin 4. Immunological Reviews. 1988;102:51–75. doi: 10.1111/j.1600-065x.1988.tb00741.x. [DOI] [PubMed] [Google Scholar]
- Sohail A, Klapacz J, Samaranayake M, Ullah A, Bhagwat AS. Human activation-induced cytidine deaminase causes transcription-dependent, strand-biased C to U deaminations. Nucleic Acids Research. 2003;31(12):2990–2994. doi: 10.1093/nar/gkg464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulas-Sprauel P, Le Guyader G, Rivera-Munoz P, Abramowski V, Olivier-Martin C, Goujet-Zalc C, et al. Role for DNA repair factor XRCC4 in immunoglobulin class switch recombination. The Journal of Experimental Medicine. 2007;204(7):1717–1727. doi: 10.1084/jem.20070255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staszewski O, Baker RE, Ucher AJ, Martier R, Stavnezer J, Guikema JE. Activation-induced cytidine deaminase induces reproducible DNA breaks at many non-Ig Loci in activated B cells. Molecular Cell. 2011;41(2):232–242. doi: 10.1016/j.molcel.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J. Antibody class switching. Advances in Immunology. 1996;61:79–146. doi: 10.1016/s0065-2776(08)60866-4. [DOI] [PubMed] [Google Scholar]
- Stavnezer J. Complex regulation and function of activation-induced cytidine deaminase. Trends in Immunology. 2011;32(5):194–201. doi: 10.1016/j.it.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Guikema JE, Schrader CE. Mechanism and regulation of class switch recombination. Annual Review of Immunology. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Radcliffe G, Lin YC, Nietupski J, Berggren L, Sitia R, et al. Immunoglobulin heavy-chain switching may be directed by prior induction of transcripts from constant-region genes. Proceedings of the National Academy of Sciences of the United States of America. 1988;85(20):7704–7708. doi: 10.1073/pnas.85.20.7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer-Nordgren J, Sirlin S. Specificity of immunoglobulin heavy chain switch correlates with activity of germline heavy chain genes prior to switching. The EMBO Journal. 1986;5(1):95–102. doi: 10.1002/j.1460-2075.1986.tb04182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stivers JT. Comment on “Uracil DNA glycosylase activity is dispensable for immunoglobulin class switch”. Science. 2004;306(5704):2042. doi: 10.1126/science.1105225. [DOI] [PubMed] [Google Scholar]
- Sun J, Keim CD, Wang J, Kazadi D, Oliver PM, Rabadan R, et al. E3- ubiquitin ligase Nedd4 determines the fate of AID-associated RNA polymerase II in B cells. Genes & Development. 2013;27(16):1821–1833. doi: 10.1101/gad.210211.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ta VT, Nagaoka H, Catalan N, Durandy A, Fischer A, Imai K, et al. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nature Immunology. 2003;4(9):843–848. doi: 10.1038/ni964. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Ueda S, Obata M, Nikaido T, Nakai S, Honjo T. Structure of human immunoglobulin gamma genes: Implications for evolution of a gene family. Cell. 1982;29(2):671–679. doi: 10.1016/0092-8674(82)90183-0. [DOI] [PubMed] [Google Scholar]
- Tashiro J, Kinoshita K, Honjo T. Palindromic but not G-rich sequences are targets of class switch recombination. International Immunology. 2001;13(4):495–505. doi: 10.1093/intimm/13.4.495. [DOI] [PubMed] [Google Scholar]
- Teng G, Hakimpour P, Landgraf P, Rice A, Tuschl T, Casellas R, et al. MicroRNA-155 is a negative regulator of activation-induced cytidine deaminase. Immunity. 2008;28(5):621–629. doi: 10.1016/j.immuni.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng G, Papavasiliou FN. Immunoglobulin somatic hypermutation. Annual Review of Genetics. 2007;41:107–120. doi: 10.1146/annurev.genet.41.110306.130340. [DOI] [PubMed] [Google Scholar]
- Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al. Regulation of the germinal center response by microRNA-155. Science. 2007;316(5824):604–608. doi: 10.1126/science.1141229. [DOI] [PubMed] [Google Scholar]
- Tian M, Alt FW. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. The Journal of Biological Chemistry. 2000;275(31):24163–24172. doi: 10.1074/jbc.M003343200. [DOI] [PubMed] [Google Scholar]
- Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302(5909):575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, et al. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nature Immunology. 2010;11(2):148–154. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- Tsai AG, Lu H, Raghavan SC, Muschen M, Hsieh CL, Lieber MR. Human chromosomal translocations at CpG sites and a theoretical basis for their lineage and stage specificity. Cell. 2008;135(6):1130–1142. doi: 10.1016/j.cell.2008.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumas-Brundage K, Manser T. The transcriptional promoter regulates hypermutation of the antibody heavy chain locus. The Journal of Experimental Medicine. 1997;185(2):239–250. doi: 10.1084/jem.185.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura Y, Barton LF, Rada C, Neuberger MS. REG-gamma associates with and modulates the abundance of nuclear activation-induced deaminase. The Journal of Experimental Medicine. 2011;208(12):2385–2391. doi: 10.1084/jem.20110856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unniraman S, Zhou S, Schatz DG. Identification of an AID-independent pathway for chromosomal translocations between the Igh switch region and Myc. Nature Immunology. 2004;5(11):1117–1123. doi: 10.1038/ni1127. [DOI] [PubMed] [Google Scholar]
- Vasudevan AA, Smits SH, Hoppner A, Haussinger D, Koenig BW, Munk C. Structural features of antiviral DNA cytidine deaminases. Biological Chemistry. 2013;394:1357–1370. doi: 10.1515/hsz-2013-0165. [DOI] [PubMed] [Google Scholar]
- Vincent-Fabert C, Fiancette R, Pinaud E, Truffinet V, Cogne N, Cogne M, et al. Genomic deletion of the whole IgH 3’ regulatory region (hs3a, hs1,2, hs3b, and hs4) dramatically affects class switch recombination and Ig secretion to all isotypes. Blood. 2010;116(11):1895–1898. doi: 10.1182/blood-2010-01-264689. [DOI] [PubMed] [Google Scholar]
- Vuong BQ, Chaudhuri J. Combinatorial mechanisms regulating AIDdependent DNA deamination: Interacting proteins and post-translational modifications. Seminars in Immunology. 2012;24(4):264–272. doi: 10.1016/j.smim.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella J, Ucher AJ, Donghia NM, et al. A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin class switch recombination. Nature Immunology. 2013;394:1411–1423. doi: 10.1038/ni.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong BQ, Lee M, Kabir S, Irimia C, Macchiarulo S, McKnight GS, et al. Specific recruitment of protein kinase A to the immunoglobulin locus regulates class-switch recombination. Nature Immunology. 2009;10(4):420–426. doi: 10.1038/ni.1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner SD, Neuberger MS. Somatic hypermutation of immunoglobulin genes. Annual Review of Immunology. 1996;14:441–457. doi: 10.1146/annurev.immunol.14.1.441. [DOI] [PubMed] [Google Scholar]
- Wang M, Rada C, Neuberger MS. Altering the spectrum of immunoglobulin V gene somatic hypermutation by modifying the active site of AID. The Journal of Experimental Medicine. 2010;207(1):141–153. doi: 10.1084/jem.20092238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Whang N, Wuerffel R, Kenter AL. AID-dependent histone acetylation is detected in immunoglobulin S regions. The Journal of Experimental Medicine. 2006;203(1):215–226. doi: 10.1084/jem.20051774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wuerffel R, Feldman S, Khamlichi AA, Kenter AL. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. The Journal of Experimental Medicine. 2009;206(8):1817–1830. doi: 10.1084/jem.20081678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS. 53BP1 is required for class switch recombination. The Journal of Cell Biology. 2004;165(4):459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, et al. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. The Journal of Experimental Medicine. 2005;201(4):637–645. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter DB, Sattar N, Mai JJ, Gearhart PJ. Insertion of 2 kb of bacteriophage DNA between an immunoglobulin promoter and leader exon stops somatic hypermutation in a kappa transgene. Molecular Immunology. 1997;34(5):359–366. doi: 10.1016/s0161-5890(97)00073-4. [DOI] [PubMed] [Google Scholar]
- Wu X, Geraldes P, Platt JL, Cascalho M. The double-edged sword of activation-induced cytidine deaminase. Journal of Immunology. 2005;174(2):934–941. doi: 10.4049/jimmunol.174.2.934. [DOI] [PubMed] [Google Scholar]
- Wuerffel RA, Du J, Thompson RJ, Kenter AL. Ig Sgamma3 DNAspecific double strand breaks are induced in mitogen- activated B cells and are implicated in switch recombination. Journal of Immunology. 1997;159(9):4139–4144. [PubMed] [Google Scholar]
- Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, et al. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27(5):711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie A, Kwok A, Scully R. Role of mammalian Mre11 in classical and alternative nonhomologous end joining. Nature Structural & Molecular Biology. 2009;16(8):814–818. doi: 10.1038/nsmb.1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Fulop Z, Wu G, Pone EJ, Zhang J, Mai T, et al. 14-3-3 adaptor proteins recruit AID to 5’-AGCT-3’-rich switch regions for class switch recombination. Nature Structural & Molecular Biology. 2010;17(9):1124–1135. doi: 10.1038/nsmb.1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav A, Olaru A, Saltis M, Setren A, Cerny J, Livak F. Identification of a ubiquitously active promoter of the murine activation-induced cytidine deaminase (AICDA) gene. Molecular Immunology. 2006;43(6):529–541. doi: 10.1016/j.molimm.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, et al. Deepsequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nature Immunology. 2011;12(1):62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Robbiani DF, Resch W, Bothmer A, Nakahashi H, Oliveira T, et al. RPA accumulation during class switch recombination represents 5’-3’ DNA-end resection during the S-G2/M phase of the cell cycle. Cell Reports. 2013;3(1):138–147. doi: 10.1016/j.celrep.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, et al. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449(7161):478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Yancopoulos GD, DePinho RA, Zimmerman KA, Lutzker SG, Rosenberg N, Alt FW. Secondary genomic rearrangement events in pre-B cells: VHDJH replacement by a LINE-1 sequence and directed class switching. The EMBO Journal. 1986;5(12):3259–3266. doi: 10.1002/j.1460-2075.1986.tb04637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelamos J, Klix N, Goyenechea B, Lozano F, Chui YL, Gonzalez Fernandez A, et al. Targeting of non-Ig sequences in place of the V segment by somatic hypermutation. Nature. 1995;376(6537):225–229. doi: 10.1038/376225a0. [DOI] [PubMed] [Google Scholar]
- Yu K, Chedin F, Hsieh CL, Wilson TE, Lieber MR. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nature Immunology. 2003;4(5):442–451. doi: 10.1038/ni919. [DOI] [PubMed] [Google Scholar]
- Yu K, Lieber MR. Nucleic acid structures and enzymes in the immunoglobulin class switch recombination mechanism. DNA Repair. 2003;2(11):1163–1174. doi: 10.1016/j.dnarep.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Yu K, Roy D, Bayramyan M, Haworth IS, Lieber MR. Fine-structure analysis of activation-induced deaminase accessibility to class switch region R-loops. Molecular and Cellular Biology. 2005;25(5):1730–1736. doi: 10.1128/MCB.25.5.1730-1736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, et al. An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nature Immunology. 2004;5(12):1275–1281. doi: 10.1038/ni1137. [DOI] [PubMed] [Google Scholar]
- Zarrin AA, Goff PH, Senger K, Alt FW. Sgamma3 switch sequences function in place of endogenous Sgamma1 to mediate antibody class switching. The Journal of Experimental Medicine. 2008;205(7):1567–1572. doi: 10.1084/jem.20080451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrin AA, Tian M, Wang J, Borjeson T, Alt FW. Influence of switch region length on immunoglobulin class switch recombination. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2466–2470. doi: 10.1073/pnas.0409847102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrin AA, Vecchio CD, Tseng E, Gleason M, Zarrin P, Tian M, et al. Antibody class switching mediated by yeast endonuclease generated DNAbreaks. Science. 2007;315:377–381. doi: 10.1126/science.1136386. [DOI] [PubMed] [Google Scholar]
- Zhang J, Bottaro A, Li S, Stewart V, Alt FW. A selective defect in IgG2b switching as a result of targeted mutation of the I gamma 2b promoter and exon. The EMBO Journal. 1993;12(9):3529–3537. doi: 10.1002/j.1460-2075.1993.tb06027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Franklin A, Boboila C, McQuay A, Gallagher MP, Manis JP, et al. Downstream class switching leads to IgE antibody production by B lymphocytes lacking IgM switch regions. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):3040–3045. doi: 10.1073/pnas.0915072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, McCord RP, Ho YJ, Lajoie BR, Hildebrand DG, Simon AC, et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148(5):908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Lottenbach KR, Barenkamp SJ, Reason DC. Somatic hypermutation and diverse immunoglobulin gene usage in the human antibody response to the capsular polysaccharide of Streptococcus pneumoniae Type 6B. Infection and Immunity. 2004;72(6):3505–3514. doi: 10.1128/IAI.72.6.3505-3514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5’ end resection. Science. 2013;339(6120):700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]