

Abstract
Small nucleolar RNAs (snoRNAs) are among the first discovered and most extensively studied group of small non-coding RNA. However, most studies focused on a small subset of snoRNAs that guide the modification of ribosomal RNA. In this study, we annotated the expression pattern of all box C/D snoRNAs in normal and cancer cell lines independent of their functions. The results indicate that C/D snoRNAs are expressed as two distinct forms differing in their ends with respect to boxes C and D and in their terminal stem length. Both forms are overexpressed in cancer cell lines but display a conserved end distribution. Surprisingly, the long forms are more dependent than the short forms on the expression of the core snoRNP protein NOP58, thought to be essential for C/D snoRNA production. In contrast, a subset of short forms are dependent on the splicing factor RBFOX2. Analysis of the potential secondary structure of both forms indicates that the k-turn motif required for binding of NOP58 is less stable in short forms which are thus less likely to mature into a canonical snoRNP. Taken together the data suggest that C/D snoRNAs are divided into at least two groups with distinct maturation and functional preferences.
INTRODUCTION
Small nucleolar RNAs (snoRNAs) are ubiquitous ribonucleoprotein particles required for the processing, modification and assembly of ribosomal RNA (rRNA) (1–3). This abundant group of small RNA is divided into two families based on activity and structural features: the C/D snoRNAs are responsible for 2′-O-ribose methylation and the H/ACA snoRNAs catalyze pseudouridylation of their targets (4–6). snoRNAs of both families guide their associated protein catalytic subunit (the pseudouridylase dyskerin for H/ACA snoRNAs and the methyltransferase fibrillarin for C/D snoRNAs) by base pairing between the snoRNA guide region and the target sequence in rRNA and snRNA (6–8).
Box C/D snoRNAs are 50–100 nucleotides (nt)-long transcripts featuring conserved box C (RUGAUGA where R is a purine) and box D (CUGA) motifs that align to form a characteristic structural motif called the kink-turn or K-turn motif (4,7,9–11), as illustrated in Figure 1A. K-turn motifs are widespread in many classes of RNA and involve non-canonical G–A base pairing causing a tight kink in the axis of double-stranded RNA (12,13). The k-turn motif is typically flanked by a 5’ canonical stem composed of regular base pairs and by a 3’ non-canonical stem consisting of the G–A base pairs (11,13). The minor grooves of the canonical and non-canonical stems can interact, coordinated by metal ions or binding proteins, stabilizing the structure (11). In box C/D snoRNAs, constrained by the sequences of the boxes C and D, the non-canonical G–A stem is typically followed by one pair of U–U mismatched nucleotides and two canonical base pairs (Figure 1A). Both the canonical stem and the extended non-canonical stem appear to be important for proper processing of the snoRNA (14) and assembly of the snoRNP complex. In vertebrates, most snoRNAs are encoded in introns and co-transcribed from the promoter of their host genes. The assembly of the pre-snoRNP complex typically occurs on spliced and debranched introns and is initiated by the recognition of the k-turn structure by the core binding protein 15.5K (15–20). The binding of 15.5K to the k-turn provides a scaffold for the assembly of the box C/D snoRNP complex which includes additional core binding proteins NOP56, NOP58 and the methyltransferase fibrillarin (15,21–26). The resulting pre-snoRNPs are exonucleolytically trimmed from both the 5′ and 3′ ends, generating mature snoRNPs (19,20,27). The binding of the core proteins likely protects the snoRNAs from further trimming and determines their exact termini (14). Disruption of the k-turn and flanking stems prevents stable accumulation of snoRNAs (14,22), underlining the importance of these motifs for the appropriate processing and the stability of C/D snoRNAs and the proper structure and assembly C/D snoRNPs.
Figure 1.
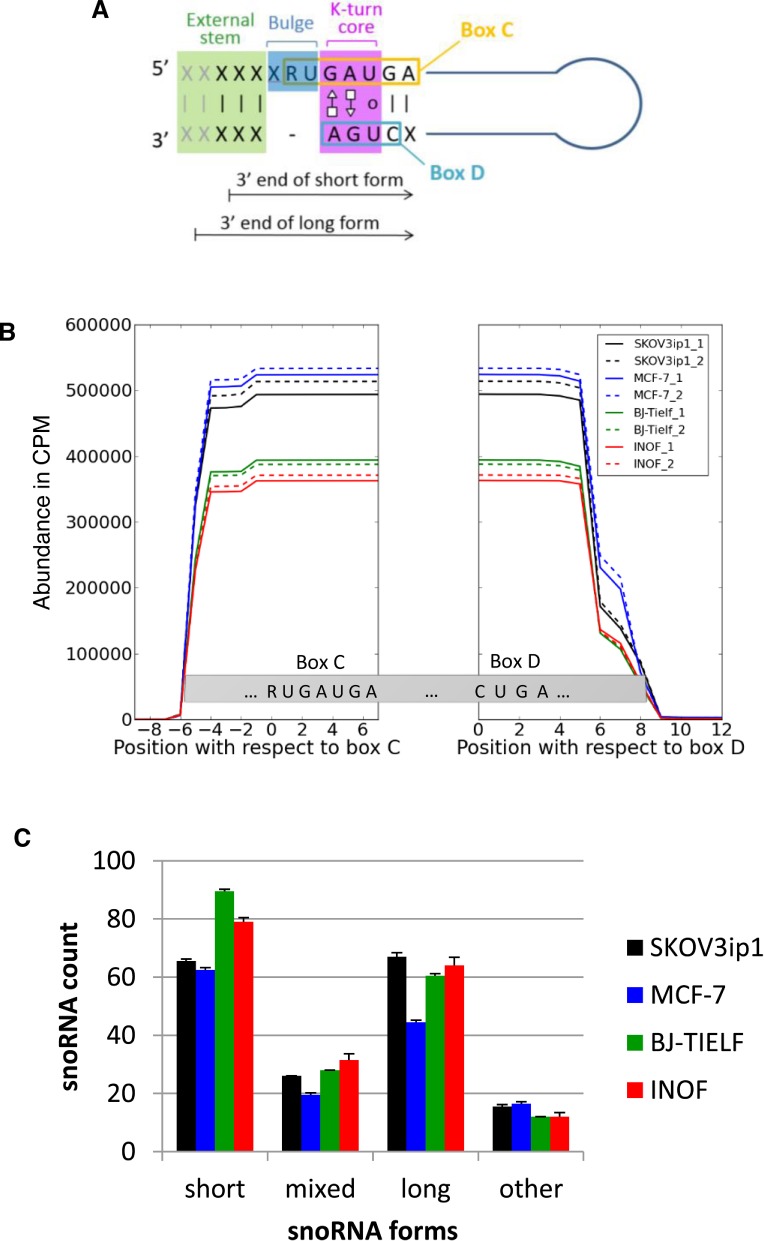
Comparison of box C/D snoRNA expression patterns in normal and cancer cell lines. (A) Schematic representation of box C/D snoRNA structure. Box C/D snoRNAs are small non-coding RNAs featuring two short sequence motifs (C: RUGAUGA and D: CUGA) that are aligned together through base pairing to form a characteristic structural k-turn motif. This motif typically involves a bulge upstream from the box C and non-canonical A–G and G–A base pairing between box C and D residues, preceded on the 5′ side by a stem involving canonical base pairing (12). X and R represent any nucleotide and purines, respectively. The 3′ end terminus of the short and long snoRNA forms detected in this study are indicated by arrows. (B) The processing pattern of box C/D snoRNA is conserved in normal and cancer cells. Sequence reads mapping to at least 77% of full-length box C/D snoRNAs in normal (BJ-Tielf, INOF), breast (MCF-7) and ovarian cancer cell lines (SKOV3ip) were counted and plotted with respect to their corresponding boxes C and D for every residue of all box C/D snoRNAs. CPM indicates count per million. All experiments were performed in duplicate. (C) Identification of two distinct forms of C/D snoRNA. Two general forms (long and short) of box C/D snoRNAs were identified according to the distance between their ends and their characteristic C/D motifs. The short forms (snoRNASH) start 4 or 5 nt upstream of their box C and end 2 or 3 nt downstream of their box D, while the long forms (snoRNAL) start 5 or 6 nt upstream of their box C and end 4 or 5 nt downstream of their box D. The number of snoRNAs displaying only short or only long forms, a mix of the two forms or neither long nor short forms (other) was counted in the different cell lines and presented in the form of a histogram. The standard deviation between the duplicate experiments is shown as error bars.
Most C/D snoRNAs in vertebrate genomes are located within the introns of genes that typically code for proteins involved in related functions like translation and ribosome biogenesis (22,23). However, in human only ∼20% of the 269 annotated box C/D snoRNAs in snoRNABase (28) are found in genes coding for proteins involved in ribosome biogenesis or translation. Strikingly, over 50% of human snoRNAs are not associated with established coding genes but instead are generated from long non-coding or hypothetical host genes. This suggests that some human snoRNAs may have functions other than rRNA modification. Indeed, recent studies have revealed non-canonical characteristics for some snoRNAs including members with no rRNA targets (referred to as orphan snoRNAs) (22,29), smaller stable and conserved RNA fragments derived from snoRNAs (30–32), non-canonical functionality for some snoRNAs including miRNA-like functions (33–36) and a role in the regulation of alternative splicing (30,32,37). In addition, despite the fundamental housekeeping role of many snoRNAs, their abundance is cell type and condition dependent (38–40) and specific snoRNAs display significantly altered expression in diverse cancers, some having been described as tumor suppressors or oncogenes (41–43).
Despite two decades of extensive studies, our knowledge of snoRNA biology in human remains limited to a small subset of canonical snoRNAs required for rRNA modification. The processing mechanism and protein factors required for the function of the majority of orphan snoRNAs and their contribution to cell- and tissue-specific function is largely unexplored. In this study, we provide an extensive analysis of the expression pattern, protein requirement and cell specificity of all known human box C/D snoRNAs. Paired-end sequencing of RNA extracted from a variety of normal and cancer cell lines indicates that while most snoRNAs are upregulated in cancer cell lines their overall processing patterns are conserved. Surprisingly, snoRNAs are not expressed as a homogenous population with randomly heterogeneous termini as previously believed. Instead, we found that box C/D snoRNAs fall into two groups, the first displaying long ends (snoRNAL) and the second with short ends (snoRNASH). The long forms exhibit the features of canonical snoRNAs while snoRNAs produced as short forms include more orphans and non-canonical snoRNAs. Strikingly, the snoRNAL are more sensitive to the depletion of the core snoRNP protein NOP58 while a subset of snoRNASH are affected by the depletion of the splicing factor RBFOX2 that was previously shown to bind snoRNA flanking long non-coding RNA (44). Together the data suggest that some snoRNAs are differentially processed to form snoRNP with distinct protein components.
MATERIALS AND METHODS
Cell culture and transfections
All cell lines were cultured in antibiotic- and antimycotic-free media (Wisent). The ovarian adenocarcinoma SKOV3ip1 cell line was grown in DMEM/F12 (50/50) medium (Wisent), the breast adenocarcinoma MCF-7 cell line was grown in EMEM (Wisent) supplemented with 1 mM sodium pyruvate and MEM nonessential amino acid (Wisent), the immortalized normal ovarian fibroblast cell line INOF was grown in OSE (ovarian surface epithelium) medium (Wisent) and normal immortalized skin fibroblast BJ-Tielf was grown in alpha-MEM (Wisent). All media were supplemented with 10% fetal bovine serum and 2 mM L-glutamine. Cell propagation and passaging were as recommended by American Type Culture Collection. For transfection, cells were seeded at 350 000 cells per well in 6-well plates (BD Biosciences). Transfections were performed in suspension using Lipofectamine 2000 (Life technologies) according to the manufacturer's protocol and 20 nM of 21-mer siRNA (Sigma) for 48 h. All cell lines were actively growing when harvested. All knockdowns (KD) were done in duplicate using different siRNAs for each sample and were validated by qPCR (Supplementary File 1, Figure S1). The sequences of the siRNAs used are the following: CUUCUACCGUUCAGAUUCU (NOP58 KD 1), GACAAGUCCCAAACACAAA (NOP58 KD 2), GAUGGUCACACCAUAUGCA (RBFOX2 KD 1), CACCUCCGCAGAAUGGAAU (RBFOX2 KD 2).
RNA extraction and quantitative PCR
Total RNA extractions were performed using miRNeasy kit (Qiagen) as recommended by the manufacturer. RNA integrity of each sample was assessed with an Agilent 2100 Bioanalyzer. Reverse transcription was performed at 55°C on a fixed volume (11 μl) of RNA sample with Transcriptor (Roche Diagnostics), random hexamers, dNTPs and 10 units of RNaseOUT (Invitrogen) in a total volume of 20 μl. All cDNAs were diluted in RNAse DNAse-free water (IDT). Quantitative PCR reactions (Supplementary File 1, Figure S1) were performed on 10 ng of cDNA as described elsewhere (45). Relative expression levels were calculated using the qBASE framework (46) using PSMC4 and SDHA as reference genes. Primer design and validation were evaluated as described (45). In every run, a no template control was performed for each primer pair. These controls were consistently negative. To measure the abundance of U31 (Supplementary File 1, Figure S6), real-time PCR quantification was performed as described (45). Fifty nanogram of total RNA was reverse transcribed using Transcriptor Reverse Transcriptase (Roche) and the PCRs were performed in a realplex (Eppendorf) using primers complementary to the sequence of U31 RNA (U31_forward: TGAGTTGAATACCGCCCCAG and U31_reverse: GCTCAGAAAA TACCTTTCAGTCAC).
Preparation of small RNA samples for RNA-seq
Small RNAs (<200 nt in length) were isolated from the different cell lines and transfected cell lines considered using a low molecular weight RNA extraction kit (mirVana, Invitrogen) and from these samples, cDNA libraries were prepared using the TruSeq small RNA Sample Prep kit (Illumina) which includes adapter ligation, reverse transcription and PCR amplification. The quality of the RNA and libraries were analyzed using an Agilent Bioanalyzer (Supplementary File 1, Figure S2). The resulting cDNA libraries were paired-end sequenced on Illumina HiSeq sequencers at the McGill University and Genome Quebec Innovation Centre. For these sequencing runs, eight samples were multiplexed per lane, resulting in between 15M and 24M paired reads per dataset. Paired-end sequencing was chosen because the length of some of the molecules of interest surpassed the read length (which was 100 nt). Additionally, the paired-end sequencing ensures high-quality sequences at both ends of the RNAs of interest. It should be noted that because only RNAs of <200 nt in length were kept and the paired-end read length used was 2 × 100 nt, all small RNAs present in our sequencing dataset were entirely sequenced (including the middle of the molecule). The sequencing datasets were deposited in GEO (GSE55946).
Processing of RNA-seq datasets
Because many small RNAs isolated using our procedure are shorter than the sequencing read length (100 nt), the 3′ adapters were still present in the fastq files for many read pairs. Cutadapt (47) was used to remove the adapters and control the quality of the sequences. Parameters were set to -minimum-length 2, -match-read-wildcards, -q 3 and the adapter sequences were TGGAATTCTCGGGTGCCAAGG (R1 file) and GATCGTCGGACTGTAGAACTCTGAAC (R2 file). The resulting reads were aligned to the build hg19 of the human genome using Bowtie2 (48) with parameters –local, -I 13. SAMtools (49) and BEDTools (50) were used to obtain the positions of all reads on the small RNAs of interest and calculate alignment statistics. Reference sequences of snoRNAs, miRNAs and tRNAs were obtained respectively from snoRNAbase (28), miRBase version 18 (51) and the genomic tRNA database (52). Sequences of snRNAs and rRNAs were obtained from Genbank (53) and sequences of Y RNAs were obtained from the supplementary material of (54).
Accumulation profiles of full-length snoRNAs in different cell lines and abundance of different forms
To be considered in these analyses, snoRNAs had to be represented by at least 10 reads covering at least 77% of the full-length snoRNA as defined in snoRNAbase (28). For analyses considering all reads mapping to full-length snoRNAs regardless of forms (Figures 3A and 4A), the normalized abundance value per snoRNA (one value per snoRNA) as well as per miRNA, per tRNA, per rRNA, per YRNA and per snRNA was calculated using DESeq (55). The fold change following the depletions (Figures 3A and 4A) was calculated as the average normalized abundance value in the two depletion samples divided by the average normalized abundance in the three mock-treated (Lipofectamine) samples.
Figure 3.
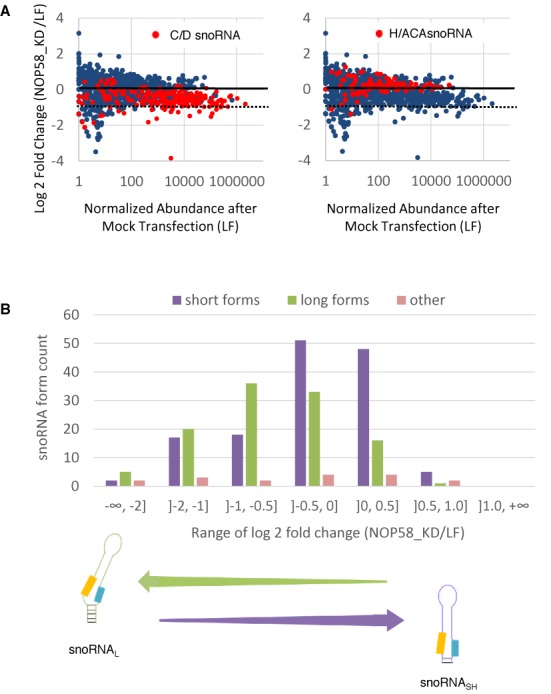
Depletion of the core box C/D snoRNA binding protein NOP58 preferentially downregulates the expression of the long snoRNA forms. (A) KD of NOP58 inhibits the expression of some box C/D and not H/ACA snoRNAs. NOP58 was knocked down in SKOV3ip1 using two independent siRNAs and the impact on the abundance of RNA shorter than 200 nt (snoRNA, miRNA, snRNA, YRNA and tRNA) was calculated relative to the level in mock-transfected cells (LF). The log2 fold change in RNA abundance is shown in the form of scatter plots. The different small RNA members are indicated by blue dots, while the box C/D (left panel) and H/ACA snoRNA (right panel) are highlighted in red. The dashed lines represent a decrease of 2-fold in the NOP58 depletion. (B) Effect of the NOP58 KD on the different forms of box C/D snoRNA. The number of long (snoRNAL), short (snoRNASH) and other (that differ from the snoRNAL and snoRNASH) predominant forms of box C/D snoRNAs was determined and plotted relative to binned values of the log2 fold change in expression after the NOP58 KD. The enrichment in snoRNAL downregulated by the KD is shown at bottom.
Figure 4.
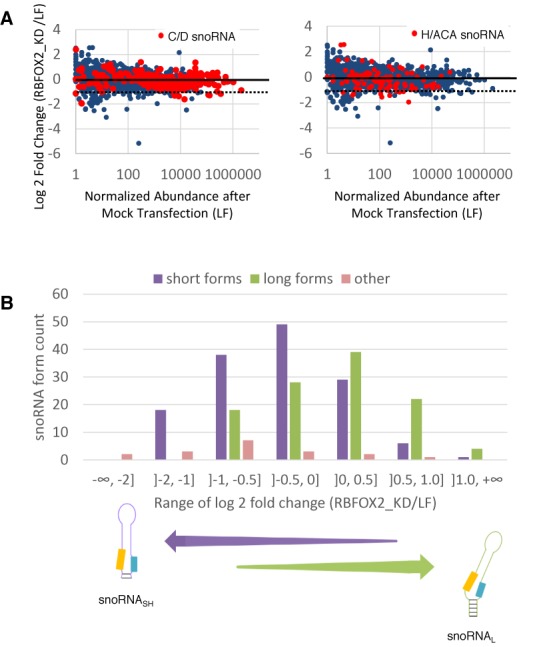
The KD of the RBFOX2 splicing factor preferentially downregulates the expression of short snoRNA forms. (A) KD of RBFOX2 inhibits the expression of select box C/D and H/ACA snoRNAs. RBFOX2 was knocked down in SKOV3ip1 using two independent siRNAs and the impact on the abundance of RNA shorter than 200 nt (snoRNA, miRNA, snRNA, YRNA and tRNA) was calculated relative to the level in mock-transfected cells (LF). The log2 fold change in RNA abundance is shown in the form of scatter plots. The different small RNA members are indicated by blue dots, while the box C/D (left panel) and H/ACA snoRNA (right panel) are highlighted in red. The dashed lines represent a decrease of 2-fold in the RBFOX2 depletion. (B) Effect of the RBFOX2 KD on the different forms of box C/D snoRNA. The number of long (snoRNAL), short (snoRNASH) and other (that differ from the snoRNAL and snoRNASH) forms of box C/D snoRNAs was determined and plotted relative to binned values of the log2 fold change in expression after the RBFOX2 KD. The enrichment in snoRNASH downregulated by the KD is shown at bottom.
For form-specific analyses (Figures 1, 2, 3B, 4B, 5 and Supplementary File 1, Figures S3–S9), for any given snoRNA, all sequences differing by as little as 1 nt but present with a count of at least 10 reads were considered as different forms of the same snoRNA. Predominant forms were defined as forms representing at least 20% of the total number of reads mapping to the full-length snoRNAs as defined above (i.e. these reads must cover at least 77% of the full-length snoRNA). The different snoRNA forms were compared to the positions of the boxes C and D as defined in snoRNAbase (28) and sno/scaRNAbase (56). Most box C/D snoRNA sequences start 4–6 nt before the box C and end 2–5 nt after the box D. Short snoRNA forms were defined as sequences starting 4 or 5 nt before the box C and ending 2 to 3 nt after the box D, while long snoRNA forms were defined as sequences starting 5 or 6 nt before the box C and ending 4 or 5 nt after the box D. The abundance of each different snoRNA form was calculated by normalizing the read count corresponding to the form by the total number of reads mapped to the genome for the given sample and multiplying by 1 000 000, obtaining abundance values in count per million (CPM). For all figures analyzing short and long snoRNA forms separately, only predominant forms with a minimum abundance of 1 CPM were considered. The fold change following the NOP58 depletion was calculated as the average normalized abundance for the NOP58 depletions divided by the average normalized abundance in the mock-treated samples. The fold change following the RBFOX2 depletion was calculated using the same procedure. The U3 family members were not considered in the analysis as their length exceeds the size cutoff of 200 nt used to isolate the RNA.
Figure 2.
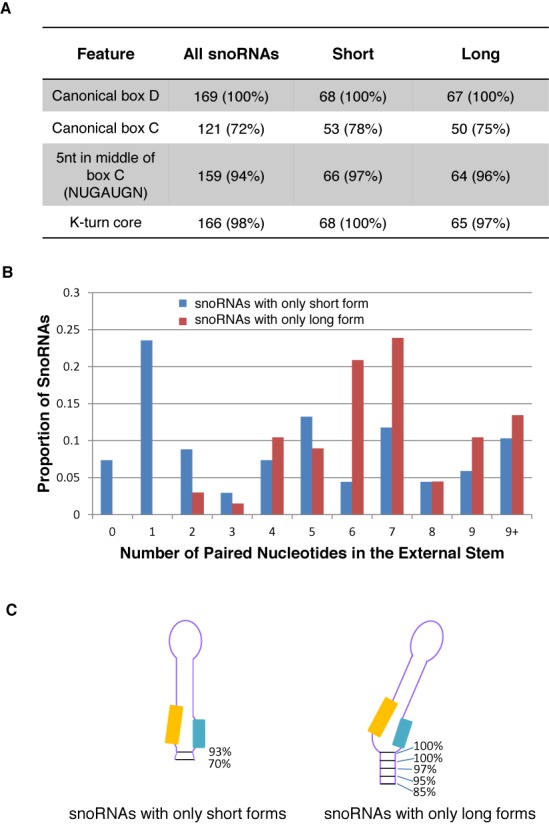
The long form of box C/D snoRNA (snoRNAL) features extended base pairing downstream of the k-turn structural motif. (A) The long and short snoRNA forms share the basic structural features of box C/D snoRNA. The characteristic box C, box D and k-turn were identified and their level of sequence conservation in each class of snoRNA was determined using the sequence obtained from the SKOV3ip1 cell line. The number and percent of snoRNAs displaying each feature are shown. Only predominant snoRNA forms expressed above 1 CPM in both replicates were counted. (B) The snoRNAL form features more stable k-turn structure. The length of the external stem was measured using SKOV3ip1 RNA for snoRNAs only expressed as short or only expressed as long forms. The proportion of snoRNAs from these two groups is expressed as a function of the number of paired nucleotides in their external stem. (C) Schematic representation of the external stem observed in snoRNAs only expressed as long or short snoRNA forms. The percentage of snoRNAs containing the different number of base pairs downstream of box D is shown on the right of each form. The orange and blue boxes indicate the position of the C and D motifs, respectively.
Figure 5.
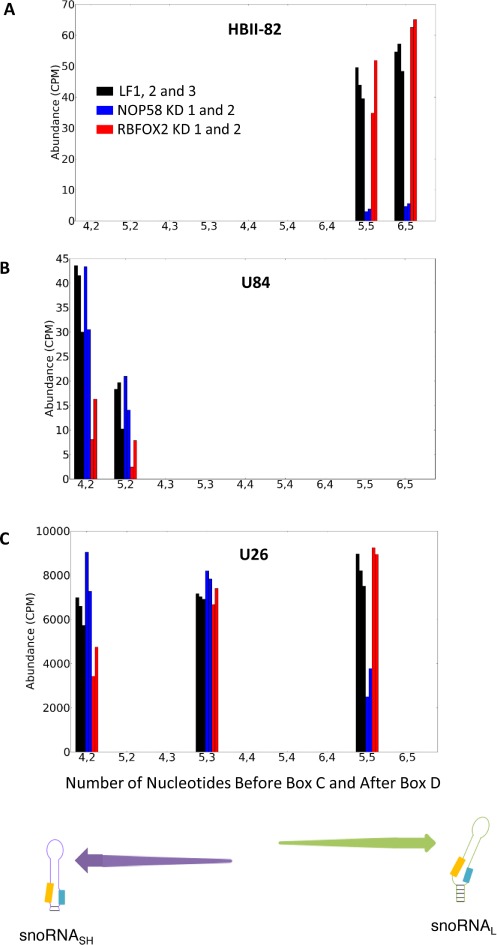
NOP58 and RBFOX2 snoRNA targets are form-dependent. The abundance of the different forms generated from each snoRNA was determined before and after the KD of either NOP58 or RBFOX2 and plotted relative to the number of nucleotides upstream of box C and downstream of box D. Shown are three examples representing snoRNAs producing only long (A), only short (B) or both long and short (C) forms. The snoRNA names are shown on top, while the enrichment of the short and long forms are illustrated at bottom. CPM and LF respectively indicate counts per million reads mapped and mock transfection (Lipofectamine). The data obtained after the transfection of two independent siRNAs targeting either NOP58 (blue bars) or RBFOX2 (red bars) and three mock transfections (black bars) are shown.
Measurement of the length of the external stem
To investigate whether the length of the external stem of snoRNAs differs for short and long snoRNAs, snoRNAs producing only short forms or only long forms were considered separately. The external stem length was calculated as the number of consecutive canonical base pairs and G–U pairs from the k-turn towards the end of the molecule. The maximum stem length considering a k-turn bulge (see Figure 1A) of two and three residues is reported.
Northern blot
Total RNA was isolated from siRNA-transfected cells grown 48 h using a commercial kit (Qiagen rneasy mini kit). DNase treatment (QIAGEN) was performed. The integrity of the treated RNA was examined using Agilent 2100 Bioanalyzer that calculates the rRNA ratio. Northern blots were performed as described (57) using 15 μg of total RNA and an 8% denaturing polyacrylamide gel. The RNA was visualized by autoradiography using 5′-end-labeled oligonucleotide probes complimentary to the mature sequence of the snoRNA (snR39B: 5′-TGAGTATTCTCTTCATTTCAGGTCA-3′; U31: 5′-CCTTTCAGTCACACATTGATCAGA-3′). The radiolabeled bands were visualized using a Storm 860 scanner (GE Healthcare) and analyzed using the Quantity One software (Bio-Rad).
RESULTS
Many box C/D snoRNAs are overexpressed in ovarian and breast cancer cell lines
To monitor the expression pattern and define the expressed sequence of human snoRNAs without predetermined structural or functional bias, we extracted and sequenced all small RNAs shorter than 200 nt from two immortalized normal fibroblasts (INOF and BJ-Tielf) and two cancer cell lines (ovarian: SKOV3ip1 and breast: MCF7). High-throughput paired-end sequencing of the RNA extracted from two biological replicates of each cell line generated between 15 and 24 million read pairs per sample. The read pairs were processed to remove adapters, controlled for quality and aligned to the human genome as detailed in the Materials and Methods section. Across the four cell lines, between 37% and 56% of the reads mapped to snoRNAs (Table 1), while the rest mapped to other small RNA including tRNA and microRNA. As expected, the read distribution and number of read per transcript were very close for the different biological replicates (Table 1) confirming the quality and reproducibility of the sequencing.
Table 1. Sequencing alignment statistics.
| Dataset | Number of aligned read pairs | Percentage of reads aligned to | |||||||
|---|---|---|---|---|---|---|---|---|---|
| C/D snoRNA | H/ACA snoRNA | scaRNA | miRNA | snRNA | YRNA | tRNA | rRNA | ||
| SKOV3ip1 rep1 | 16660014 | 50 | 0.82 | 0.13 | 12 | 0.13 | 0.14 | 2.6 | 29 |
| SKOV3ip1 rep2 | 14925302 | 52 | 0.82 | 0.12 | 12 | 0.14 | 0.15 | 2.0 | 28 |
| MCF7 rep1 | 21538622 | 53 | 0.79 | 0.28 | 6.1 | 0.089 | 0.16 | 1.4 | 35 |
| MCF7 rep2 | 19362058 | 54 | 0.78 | 0.27 | 5.7 | 0.092 | 0.15 | 1.4 | 35 |
| BJ-Tielf rep1 | 17557177 | 56 | 0.95 | 0.16 | 9.9 | 0.095 | 0.24 | 5.9 | 27 |
| BJ-Tielf rep2 | 17373969 | 54 | 0.95 | 0.17 | 10 | 0.084 | 0.23 | 5.9 | 29 |
| INOF rep1 | 16716777 | 37 | 0.59 | 0.11 | 14 | 0.099 | 0.20 | 2.9 | 42 |
| INOF rep2 | 16663938 | 38 | 0.63 | 0.12 | 14 | 0.097 | 0.19 | 2.7 | 40 |
As shown in Figure 1B, a higher proportion of reads were mapped to box C/D snoRNA in the ovarian and breast cancer cell lines than those in the normal fibroblast cell lines. Although this difference in expression levels may be caused by cell-type differences, it seems to be specific to cancer cells since it was also observed between the closely related ovarian fibroblast and serous ovarian cancer cell lines. This increase in the expression of snoRNA is consistent with a large body of literature showing that ribosome production and its associated factors are upregulated in cancers (43,58,59). Interestingly, the opposite trend is seen for orphan snoRNAs which are more abundant in normal cell lines than in cancer cell lines (Supplementary File 1, Figure S3). These data suggest that orphan snoRNAs might play antiproliferative roles in the cell and that cancer can induce both loss and gain of snoRNA functions.
The box C/D snoRNAs are expressed in discrete forms with varying ends
The analysis of all paired-end reads mapping to full-length snoRNAs reveals that the great majority of box C/D snoRNAs start sharply 4–6 nt upstream from the box C and end between 2 and 5 nt downstream from the box D (Figure 1B). This variation in snoRNA 5′ and 3′ ends was previously observed but was not further considered and was likely dismissed as processing heterogeneity (14,60). However, close scrutiny of individual snoRNA accumulation profiles suggests non-random distribution of snoRNA 5′ and 3′ ends. Indeed, sequence analysis reveals the presence of two prevalent forms of C/D snoRNA, a short form (snoRNASH) starting 4 or 5 nt upstream of the box C and finishing 2 or 3 nt downstream from the box D, and a long form (snoRNAL) starting 5 or 6 nt upstream from the box C and ending 4 or 5 nt downstream from the box D (Figure 1C and Supplementary File 1, Figure S4). Most snoRNAs were found expressed either as the short (e.g. U106) or the long form (e.g. HBII-295), while a minority was found expressed as both forms (e.g. U15B) or neither as long nor as short forms (Figure 1C and Supplementary File 1, Figure S4A). These findings were also seen by northern blot for two snoRNAs chosen for validation. U31 is detected only as a short form by RNA-seq and is present as one band by northern blot, and snR39B is detected as both a long and a short form by RNA-seq and is present as two bands by northern blot (Supplementary File 1, Figures S5 and S6). This snoRNA-specific distribution of 5′ and 3′ ends suggests that the difference between snoRNA forms is generated through specific processing pathways and not random processing heterogeneity. Indeed, the exact snoRNA forms are well conserved across the four cell lines considered regardless of their tissular origin and proliferative potential (Supplementary File 1, Figure S4B).
Extension of snoRNA 5′ and 3′ ends increases the stability of the external stem structure
To understand the relevance of the different snoRNA forms, we investigated their structural and functional differences and their impact if any on the basic features of box C/D snoRNA. As summarized in Figure 2A, no significant differences between the long and short forms were found in the consensus sequences of boxes C and D, or in their k-turn characteristics as defined in Figure 1A. However, the external stem, which is expected to increase the stability of the k-turn motif and believed to be important for canonical snoRNP assembly (17,61,62), is significantly shorter in snoRNASH (P-value < 10−4 by Kolmogorov–Smirnov goodness-of-fit test). As shown in Figure 2B and C, while 30% of snoRNAs produced only as short forms have no external stem (i.e. 0 or 1 consecutive paired residues in this region), over 75% of snoRNAs produced only as long forms have a stem of over five base pairs and many extend well passed the end of the actual mature snoRNA (but are present in the genomic DNA and could be present as long stems before the snoRNA is excised and processed from the intron). Thermodynamic analyses using mfold (63) on a small number of snoRNAs produced as both long and short forms indicate that the longer forms have an average 4 kcal/mol lower free energy than the shorter forms, when folding to form k-turns (e.g. compare Supplementary File1, Figure S7A and B, D and E as well as G and H). The short forms are often more likely to fold in a way that maximizes canonical base pairing, but makes k-turn formation unlikely (e.g. Supplementary File 1, Figure S7C, F and I). Therefore, the long snoRNA forms are more likely to preserve the structure of the k-turn motif needed for the assembly of core snoRNA proteins, such as NOP58, than the short form.
Depletion of NOP58 preferentially inhibits the expression of the box C/D snoRNA long form
Since the snoRNA forms appear to exhibit differences in the stability of the k-turn motif required for the assembly of the NOP58 associated protein complex, we investigated the impact of depleting this protein on the abundance of the different snoRNA forms. NOP58 is essential for the accumulation and stability of canonical box C/D snoRNAs (16,21,64,65) and thus its depletion should inhibit the expression of all box C/D snoRNAs. Accordingly, NOP58 was knocked down in the ovarian cancer model cell line SKOV3ip1 using two independent siRNAs and the resulting RNA was sequenced and compared to that extracted from mock-transfected cells. As expected, box H/ACA snoRNAs, miRNAs and tRNAs, which do not interact with NOP58, were not affected by the KD of NOP58 (Figure 3A). A subset of 13 box C/D snoRNAs are strongly affected by the KD of NOP58 (overall decrease in abundance of more than 2-fold following NOP58 depletion, considering all forms of the snoRNA together). However, a significant proportion of box C/D snoRNAs are not strongly affected by the KD of NOP58.
The effect of the NOP58 depletion is better understood when snoRNAL and snoRNASH are considered separately. The abundance distributions of snoRNAL and snoRNASH are significantly different following the depletion of NOP58 (P-value < 10−8 by one-tailed Kolmogorov–Smirnov test) and while most of the long snoRNA forms are affected by the NOP58 KD, the majority of the short forms are not (Figure 3B). Indeed, the expression of over 70% of snoRNASH is either increased or not affected by the NOP58 depletion, while the expression of 63% of snoRNAL is downregulated by 1.5-fold or more. Forty-five snoRNA forms are negatively affected more than 2-fold in the NOP58 depletion (listed in Supplementary File 1, Figure S8). The small number of other box C/D snoRNA forms that do not correspond to the definition of either the short or long forms did not show preferential NOP58 effects. The great majority of these other snoRNAs are either at least as short if not shorter than the snoRNASH for both of their ends, or at least as long if not longer than the snoRNAL for both of their ends.
The form-specific effect of the NOP58 depletion was confirmed by northern blot for snR39B, which is expressed as both a long and a short form. Corroborating the results obtained by RNA-seq analysis, northern blot analysis showed the long form to be negatively affected by the NOP58 KD while the short form is unaffected (Supplementary File 1, Figure S5). Collectively, these data indicate that NOP58 is important for the stability of some but not all box C/D snoRNAs and that snoRNAs most strongly affected by the depletion of NOP58 are predominantly produced with long ends.
The RNA binding protein RBFOX2 is required for the expression of some short form snoRNA
The insensitivity of snoRNASH to NOP58 depletion suggests that this group of snoRNAs uses a different or partially altered set of protein factors to form a stable snoRNA complex. To investigate this possibility, we examined the effect of proteins implicated in RNA processing and stability on snoRNASH expression by KD of these proteins. Surprisingly, out of eight different ribonucleases and RNA binding proteins tested (single KD of NOP58, RBFOX2, QKI, Drosha, Dicer and DGCR8, as well as double KD of XRN1 and XRN2, as well as of DDX47 and EXOSC10), only NOP58 and RBFOX2 showed a significant and widespread snoRNA-specific effect (Figures 3 and 4 and data not shown). RBFOX2 is a splicing factor (66–70) that is not considered to be a component of the canonical snoRNP complex but instead was shown to physically interact with and stabilize the precursors of the large HBII-85 box C/D snoRNA family (44). As shown in Figure 4A, the RBFOX2 depletion reduced the expression of a specific subset of box C/D and H/ACA snoRNAs without affecting the overall expression levels of most small RNAs. Interestingly, in complete opposite to the NOP58 depletion shown in Figure 3B, the RBFOX2 depletion preferentially decreases the abundance of snoRNASH (Figure 4B). Once again, snoRNAL and snoRNASH display significantly different abundance distributions following the depletion of RBFOX2 (P-value < 10−8 by one-tailed Kolmogorov–Smirnov test). The C/D snoRNAs with forms most strongly affected by the depletion of RBFOX2 are shown in Supplementary File 1, Figure S9. Close scrutiny of individual snoRNA forms indicates that the great majority are either affected by the RBFOX2 or NOP58 depletions or affected by neither, but very few are affected by both (for examples see Figure 5). In general, NOP58 affects snoRNA producing the long form (e.g. HBII-82, Figure 5A), while RBFOX2 affects snoRNA producing the short form (e.g. U84, Figure 5B). Strikingly, in the case of snoRNAs expressing both short and long forms, the NOP58 KD typically affects specifically only the long form, while the RBFOX2 depletion only affects the short form (e.g. U26 in Figure 5C). Three box C/D snoRNAs expressing both a long form strongly affected by the depletion of NOP58 and a short form strongly affected by the depletion of RBFOX2 were identified: U15B, U26 and SNORD126. The analysis of their predicted secondary structure (Supplementary File 1, Figure S7) indicates that the minimum free energy of the long form with likely k-turn formation is on average 3.9 +/−0.8 kcal/mol lower than the minimum free energy of the short form with likely k-turn formation. In all three cases, other predicted secondary structures of the short form were also predicted with strong canonical pairing in the box C and box D region and lower minimum free energy than the short k-turn form (Supplementary File 1, Figure S7), suggesting that the short forms are more likely to fold maximizing the canonical pairing than favoring the k-turn formation. We conclude that the differences between the forms of box C/D snoRNAs alter their protein interactions and possibly the nature of their mature snoRNP complex.
NOP58 and RBFOX2 affect snoRNAs with different structure and genomic position
The distinct effects of NOP58 and RBFOX2 on box C/D snoRNA suggest that snoRNAs displaying different genomic characteristics may form snoRNP complexes with distinct protein requirement. To further examine this possibility, we compared the genomic location and precursor structure of NOP58- and RBFOX2-dependent snoRNAs. In accordance with results described above, only 39% of the individual forms affected by the NOP58 depletion are short, while 95% of those affected by the RBFOX2 depletion are short (Supplementary File 1, Figure S10). This indicates that these proteins affect distinct subsets of box C/D snoRNAs. In general, NOP58-sensitive snoRNAs display higher count and form diversity than those affected by RBFOX2 (Supplementary File 1, Figures S7–S9). This supports the more general role of NOP58 in snoRNA biogenesis and suggests that RBFOX2 has more specialized functions affecting a small subset of snoRNA. In addition, NOP58 and RBFOX2 appear to affect snoRNAs produced via different processing pathways. NOP58-dependent snoRNAs typically form a strong terminal stem extending within the intronic sequence and are located in close proximity (<150 nt) to the nearest downstream exon, both characteristics of snoRNAs produced through the canonical processing pathway (18). In contrast, 71% of snoRNAs affected by the depletion of RBFOX2 either have short or no terminal stems (< = 4bp) or are found more than 150 nt away from the nearest downstream exon (Supplementary File 1, Figure S10 right column). Overall, the data suggest that the majority of RBFOX2- and NOP58-dependent snoRNAs have different structural requirements for processing and biogenesis.
DISCUSSION
Classical approaches for the study of snoRNA structure and function (e.g. northern blot, low-throughput sequencing of individual snoRNAs) have led us to believe that a given snoRNA is always recognized in the same way and processed/bound by the same interactors, generating a single functional molecule. In contrast, the genome scale analysis of snoRNA presented here suggests otherwise. Box C/D snoRNAs are produced as specific and distinct forms that are conserved in different cell lines (Figure 1 and Supplementary File 1, Figure S4) and affected by different factors (Figures 3 and 4), which cannot be explained by stochastic differences in processing events. Indeed, comparison between normal and cancer cell lines indicates that while snoRNAs are generally overexpressed in cancer, their processing pattern and the relative form distribution is highly conserved (Figure 1 and Supplementary File 1, Figures S3 and S4). Most box C/D snoRNAs detected in this study are found in one of two forms: (i) the long form, which consists of snoRNAs starting between 5 and 6 nt before the box C and ending between 4 and 5 nt after the box D and (ii) the short form, which is defined as snoRNAs starting 4 or 5 nt before the box C and ending 2 or 3 nt after the box D. The long forms are more likely to display canonical snoRNA features, such as long terminal stems, location near the 3′ splice site of the host intron and dependency on core snoRNA binding proteins (e.g. NOP58). In contrast, most short snoRNAs are NOP58 independent, fewer have validated targets and a subset requires non-core snoRNA binding proteins like RBFOX2 for expression. These observations indicate that snoRNAs are expressed in subclasses with varying processing and functional preference and underscore the importance of the 5′ and 3′ end sequences for snoRNA biogenesis and function.
The sequence and functions of most snoRNAs are conserved but their genomic location and expression patterns vary between species. In yeast most snoRNAs are transcribed from independent promoters, while in human they are excised from the introns of host genes (71). Studies using model snoRNAs have shown that the processing pathways of intronic snoRNAs depend on their location within the host gene (18,72,73). snoRNAs located ∼75 nt upstream of a 3′ splice site are efficiently processed in a splicing-dependent manner, while those further upstream require extended terminal stems for splicing-independent processing (18,72,73). Consistently, the results of this study indicate that all but four of the 45 snoRNAs following the canonical processing pathway, as judged by their dependency on NOP58, are either near the 3′ splice site, possess an extended terminal stem or exhibit both features (Supplementary Data File 2). In contrast, snoRNAs that do not exhibit these canonical features do not require NOP58 and instead some require the expression of the splicing factor RBFOX2.
Many snoRNAs have previously been found bound to RBFOX2 suggesting that RBFOX2 might affect their maturation. Indeed, an RBFOX2 cross-linking immunoprecipitation coupled with high-throughput sequencing (CLIP-seq) dataset (70) identified many reads mapping to snoRNAs (Supplementary File 1, Figure S11). The interaction between RBFOX2 and snoRNAs appears to be specific since a much larger proportion of CLIP-seq reads map to snoRNAs than to mRNAs or even miRNAs, which originate from precursors of a size similar to snoRNAs and like snoRNAs, are also highly abundant (Supplementary File 1, Figure S11). Furthermore, one of the most strongly enriched RBFOX2 consensus sequences, GUGAUG, (70) resembles the sequence of the box C (RUGAUGA). It is not clear at this stage whether RBFOX2 forms a stable complex with snoRNAs or transiently binds these snoRNAs during splicing and biogenesis. However, unlike NOP58, RBFOX2-dependent snoRNAs are more likely to be located away from splice sites (Supplementary File 1, Figure S10) and thus do not overlap with the regions harboring the RBFOX2 binding motifs affecting the splicing decision (70). In addition, RBFOX2-dependent snoRNAs are not enriched in introns flanking RBFOX2-regulated exons (data not shown). Together these observations suggest that the regulatory role of RBFOX2 in alternative splicing is not strictly linked to its role in snoRNA biogenesis.
The data obtained in this study suggest a snoRNA processing model (Figure 6) where snoRNAs forming a k-turn motif stabilized by a long terminal stem, and typically located close to a 3′ splice site, are likely to bind to the NOP58 complex leading to the formation of canonical snoRNPs that mostly target rRNA for modification. In cases where the k-turn is unstable (e.g. snoRNASH) and the snoRNA is positioned further upstream from the 3′ splice site, binding of the NOP58 complex becomes less efficient, making the snoRNA available for association with other protein factors like RBFOX2. According to this model, non-canonical snoRNAs that do not have conventional targets or activity are more likely to be generated by an alternative processing mechanism independent of the snoRNP core proteins that obligatorily associate with NOP58. This group of non-canonical snoRNAs may include those serving as precursors for miRNAs or involved in rRNA independent functions such as splicing regulation and chromatin modulation (30,32,35–37,74–76). Many of these snoRNAs are not affected by either NOP58 or RBFOX2 depletions, suggesting that other protein factors might be involved in forming specific subclasses of snoRNAs. Detailed analysis of the effects of different RNA binding proteins on snoRNA may shed light on the breadth and depth of the snoRNA super family. Meanwhile, the data presented here provide a clear example of snoRNA subclasses and challenges the notion that all box C/D snoRNAs are obligatorily dependent on core snoRNA binding proteins.
Figure 6.
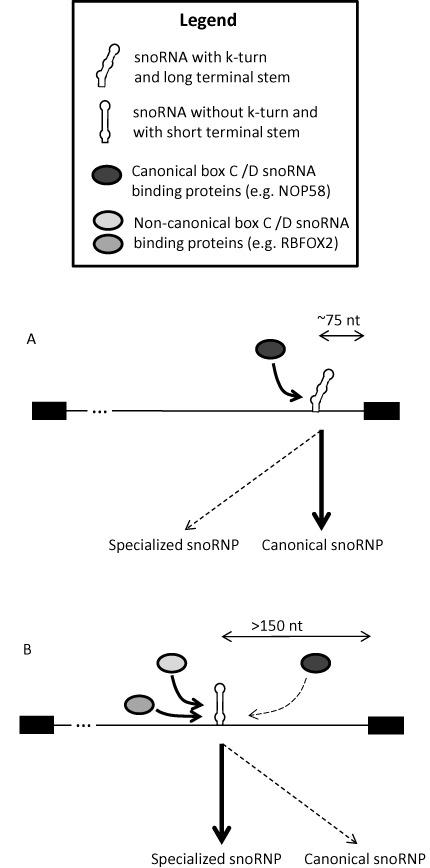
Intronic snoRNA processing model. (A) Intronic snoRNAs displaying canonical features including a strong k-turn and/or proximity to the downstream exon are more likely to follow the canonical processing pathway including dependency on core box C/D snoRNP proteins such as NOP58. (B) In contrast, snoRNAs displaying non-canonical features are more likely to depend on non-canonical snoRNA binding proteins such as RBFOX2.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
The authors are grateful to François Bachand for critical reading of the manuscript and Leandro Fequino for technical support. M.S.S. and S.A.E. are members of the RNA group and the Centre de recherche du Centre hospitalier universitaire de Sherbrooke (CRCHUS).
FUNDING
The Canadian Institutes of Health Research; the Banting Research Foundation/Rx&D Health Research Foundation [to M.S.S.]; a Canada Research Chair in RNA Biology and Cancer Genomics [to S.A.E.]; the Fonds de Recherche du Québec – Santé Research Scholar Junior 1 Career Award [to M.S.S.]. Source of Open Access funding: Canadian Institutes of Health Research grant.
Conflict of interest statement. None declared.
REFERENCES
- 1.Maden B.E. Mapping 2′-O-methyl groups in ribosomal RNA. Methods. 2001;25:374–382. doi: 10.1006/meth.2001.1250. [DOI] [PubMed] [Google Scholar]
- 2.Kiss A.M., Jady B.E., Bertrand E., Kiss T. Human box H/ACA pseudouridylation guide RNA machinery. Mol. Cell. Biol. 2004;24:5797–5807. doi: 10.1128/MCB.24.13.5797-5807.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Watkins N.J., Bohnsack M.T. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip. Rev. RNA. 2012;3:397–414. doi: 10.1002/wrna.117. [DOI] [PubMed] [Google Scholar]
- 4.Henras A.K., Dez C., Henry Y. RNA structure and function in C/D and H/ACA s(no)RNPs. Curr. Opin. Struct. Biol. 2004;14:335–343. doi: 10.1016/j.sbi.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Kiss T. Small nucleolar RNA-guided post-transcriptional modification of cellular RNAs. EMBO J. 2001;20:3617–3622. doi: 10.1093/emboj/20.14.3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bachellerie J.P., Cavaille J., Huttenhofer A. The expanding snoRNA world. Biochimie. 2002;84:775–790. doi: 10.1016/s0300-9084(02)01402-5. [DOI] [PubMed] [Google Scholar]
- 7.Reichow S.L., Hamma T., Ferre-D'Amare A.R., Varani G. The structure and function of small nucleolar ribonucleoproteins. Nucleic Acids Res. 2007;35:1452–1464. doi: 10.1093/nar/gkl1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tollervey D., Lehtonen H., Carmo-Fonseca M., Hurt E.C. The small nucleolar RNP protein NOP1 (fibrillarin) is required for pre-rRNA processing in yeast. EMBO J. 1991;10:573–583. doi: 10.1002/j.1460-2075.1991.tb07984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matera A.G., Terns R.M., Terns M.P. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat. Rev. Mol. Cell Biol. 2007;8:209–220. doi: 10.1038/nrm2124. [DOI] [PubMed] [Google Scholar]
- 10.Kiss-Laszlo Z., Henry Y., Kiss T. Sequence and structural elements of methylation guide snoRNAs essential for site-specific ribose methylation of pre-rRNA. EMBO J. 1998;17:797–807. doi: 10.1093/emboj/17.3.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schroeder K.T., McPhee S.A., Ouellet J., Lilley D.M. A structural database for k-turn motifs in RNA. RNA. 2010;16:1463–1468. doi: 10.1261/rna.2207910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lilley D.M. The structure and folding of kink turns in RNA. Wiley Interdiscip. Rev. RNA. 2012;3:797–805. doi: 10.1002/wrna.1136. [DOI] [PubMed] [Google Scholar]
- 13.Klein D.J., Schmeing T.M., Moore P.B., Steitz T.A. The kink-turn: a new RNA secondary structure motif. EMBO J. 2001;20:4214–4221. doi: 10.1093/emboj/20.15.4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Darzacq X., Kiss T. Processing of intron-encoded box C/D small nucleolar RNAs lacking a 5′,3′-terminal stem structure. Mol. Cell. Biol. 2000;20:4522–4531. doi: 10.1128/mcb.20.13.4522-4531.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watkins N.J., Dickmanns A., Luhrmann R. Conserved stem II of the box C/D motif is essential for nucleolar localization and is required, along with the 15.5K protein, for the hierarchical assembly of the box C/D snoRNP. Mol. Cell. Biol. 2002;22:8342–8352. doi: 10.1128/MCB.22.23.8342-8352.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watkins N.J., Segault V., Charpentier B., Nottrott S., Fabrizio P., Bachi A., Wilm M., Rosbash M., Branlant C., Luhrmann R. A common core RNP structure shared between the small nucleoar box C/D RNPs and the spliceosomal U4 snRNP. Cell. 2000;103:457–466. doi: 10.1016/s0092-8674(00)00137-9. [DOI] [PubMed] [Google Scholar]
- 17.Szewczak L.B., DeGregorio S.J., Strobel S.A., Steitz J.A. Exclusive interaction of the 15.5 kD protein with the terminal box C/D motif of a methylation guide snoRNP. Chem. Biol. 2002;9:1095–1107. doi: 10.1016/s1074-5521(02)00239-9. [DOI] [PubMed] [Google Scholar]
- 18.Hirose T., Shu M.D., Steitz J.A. Splicing-dependent and -independent modes of assembly for intron-encoded box C/D snoRNPs in mammalian cells. Mol. Cell. 2003;12:113–123. doi: 10.1016/s1097-2765(03)00267-3. [DOI] [PubMed] [Google Scholar]
- 19.Kiss T., Filipowicz W. Exonucleolytic processing of small nucleolar RNAs from pre-mRNA introns. Genes Dev. 1995;9:1411–1424. doi: 10.1101/gad.9.11.1411. [DOI] [PubMed] [Google Scholar]
- 20.Tycowski K.T., Shu M.D., Steitz J.A. A small nucleolar RNA is processed from an intron of the human gene encoding ribosomal protein S3. Genes Dev. 1993;7:1176–1190. doi: 10.1101/gad.7.7a.1176. [DOI] [PubMed] [Google Scholar]
- 21.Cahill N.M., Friend K., Speckmann W., Li Z.H., Terns R.M., Terns M.P., Steitz J.A. Site-specific cross-linking analyses reveal an asymmetric protein distribution for a box C/D snoRNP. EMBO J. 2002;21:3816–3828. doi: 10.1093/emboj/cdf376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filipowicz W., Pogacic V. Biogenesis of small nucleolar ribonucleoproteins. Curr. Opin. Cell Biol. 2002;14:319–327. doi: 10.1016/s0955-0674(02)00334-4. [DOI] [PubMed] [Google Scholar]
- 23.Weinstein L.B., Steitz J.A. Guided tours: from precursor snoRNA to functional snoRNP. Curr. Opin. Cell Biol. 1999;11:378–384. doi: 10.1016/S0955-0674(99)80053-2. [DOI] [PubMed] [Google Scholar]
- 24.Caffarelli E., Losito M., Giorgi C., Fatica A., Bozzoni I. In vivo identification of nuclear factors interacting with the conserved elements of box C/D small nucleolar RNAs. Mol. Cell. Biol. 1998;18:1023–1028. doi: 10.1128/mcb.18.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lafontaine D.L., Tollervey D. Nop58p is a common component of the box C+D snoRNPs that is required for snoRNA stability. RNA. 1999;5:455–467. doi: 10.1017/s135583829998192x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyman S.K., Gerace L., Baserga S.J. Human Nop5/Nop58 is a component common to the box C/D small nucleolar ribonucleoproteins. RNA. 1999;5:1597–1604. doi: 10.1017/s1355838299991288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cavaille J., Bachellerie J.P. Processing of fibrillarin-associated snoRNAs from pre-mRNA introns: an exonucleolytic process exclusively directed by the common stem-box terminal structure. Biochimie. 1996;78:443–456. doi: 10.1016/0300-9084(96)84751-1. [DOI] [PubMed] [Google Scholar]
- 28.Lestrade L., Weber M.J. snoRNA-LBME-db, a comprehensive database of human H/ACA and C/D box snoRNAs. Nucleic Acids Res. 2006;34:D158–D162. doi: 10.1093/nar/gkj002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huttenhofer A., Kiefmann M., Meier-Ewert S., O'Brien J., Lehrach H., Bachellerie J.P., Brosius J. RNomics: an experimental approach that identifies 201 candidates for novel, small, non-messenger RNAs in mouse. EMBO J. 2001;20:2943–2953. doi: 10.1093/emboj/20.11.2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scott M.S., Ono M., Yamada K., Endo A., Barton G.J., Lamond A.I. Human box C/D snoRNA processing conservation across multiple cell types. Nucleic Acids Res. 2012;40:3676–3688. doi: 10.1093/nar/gkr1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taft R.J., Glazov E.A., Lassmann T., Hayashizaki Y., Carninci P., Mattick J.S. Small RNAs derived from snoRNAs. RNA. 2009;15:1233–1240. doi: 10.1261/rna.1528909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kishore S., Khanna A., Zhang Z., Hui J., Balwierz P.J., Stefan M., Beach C., Nicholls R.D., Zavolan M., Stamm S. The snoRNA MBII-52 (SNORD 115) is processed into smaller RNAs and regulates alternative splicing. Hum. Mol. Genet. 2010;19:1153–1164. doi: 10.1093/hmg/ddp585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ender C., Krek A., Friedlander M.R., Beitzinger M., Weinmann L., Chen W., Pfeffer S., Rajewsky N., Meister G. A human snoRNA with microRNA-like functions. Mol. Cell. 2008;32:519–528. doi: 10.1016/j.molcel.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 34.Saraiya A.A., Wang C.C. snoRNA, a Novel Precursor of microRNA in Giardia lamblia. PLoS Pathog. 2008;4:e1000224. doi: 10.1371/journal.ppat.1000224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brameier M., Herwig A., Reinhardt R., Walter L., Gruber J. Human box C/D snoRNAs with miRNA like functions: expanding the range of regulatory RNAs. Nucleic Acids Res. 2011;39:675–686. doi: 10.1093/nar/gkq776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott M.S., Ono M. From snoRNA to miRNA: dual function regulatory non-coding RNAs. Biochimie. 2011;93:1987–1992. doi: 10.1016/j.biochi.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kishore S., Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 38.Jima D.D., Zhang J., Jacobs C., Richards K.L., Dunphy C.H., Choi W.W., Yan Au W., Srivastava G., Czader M.B., Rizzieri D.A., et al. Deep sequencing of the small RNA transcriptome of normal and malignant human B cells identifies hundreds of novel microRNAs. Blood. 2010;116:e118–e127. doi: 10.1182/blood-2010-05-285403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rogelj B. Brain-specific small nucleolar RNAs. J. Mol. Neurosci. 2006;28:103–109. doi: 10.1385/JMN:28:2:103. [DOI] [PubMed] [Google Scholar]
- 40.Liu Z.H., Yang G., Zhao T., Cao G.J., Xiong L., Xia W., Huang X., Wu L.Y., Wu K., Fan M., et al. Small ncRNA expression and regulation under hypoxia in neural progenitor cells. Cell. Mol. Neurobiol. 2011;31:1–5. doi: 10.1007/s10571-010-9556-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mannoor K., Liao J., Jiang F. Small nucleolar RNAs in cancer. Biochim. Biophys. Acta. 2012;1826:121–128. doi: 10.1016/j.bbcan.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen W.D., Zhu X.F. Small nucleolar RNAs (snoRNAs) as potential non-invasive biomarkers for early cancer detection. Chin. J. Cancer. 2013;32:99–101. doi: 10.5732/cjc.012.10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams G.T., Farzaneh F. Are snoRNAs and snoRNA host genes new players in cancer. Nat. Rev. Cancer. 2012;12:84–88. doi: 10.1038/nrc3195. [DOI] [PubMed] [Google Scholar]
- 44.Yin Q.F., Yang L., Zhang Y., Xiang J.F., Wu Y.W., Carmichael G.G., Chen L.L. Long noncoding RNAs with snoRNA ends. Mol. Cell. 2012;48:219–230. doi: 10.1016/j.molcel.2012.07.033. [DOI] [PubMed] [Google Scholar]
- 45.Brosseau J.P., Lucier J.F., Lapointe E., Durand M., Gendron D., Gervais-Bird J., Tremblay K., Perreault J.P., Elela S.A. High-throughput quantification of splicing isoforms. RNA. 2010;16:442–449. doi: 10.1261/rna.1877010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–12. [Google Scholar]
- 48.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kozomara A., Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2011;39:D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chan P.P., Lowe T.M. GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 2009;37:D93–D97. doi: 10.1093/nar/gkn787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benson D.A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D.J., Ostell J., Sayers E.W. GenBank. Nucleic Acids Res. 2013;41:D36–D42. doi: 10.1093/nar/gks1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Perreault J., Perreault J.P., Boire G. Ro-associated Y RNAs in metazoans: evolution and diversification. Mol. Biol. Evol. 2007;24:1678–1689. doi: 10.1093/molbev/msm084. [DOI] [PubMed] [Google Scholar]
- 55.Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xie J., Zhang M., Zhou T., Hua X., Tang L., Wu W. Sno/scaRNAbase: a curated database for small nucleolar RNAs and cajal body-specific RNAs. Nucleic Acids Res. 2007;35:D183–D187. doi: 10.1093/nar/gkl873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghazal G., Gagnon J., Jacques P.E., Landry J.R., Robert F., Elela S.A. Yeast RNase III triggers polyadenylation-independent transcription termination. Mol. Cell. 2009;36:99–109. doi: 10.1016/j.molcel.2009.07.029. [DOI] [PubMed] [Google Scholar]
- 58.Drygin D., Rice W.G., Grummt I. The RNA polymerase I transcription machinery: an emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010;50:131–156. doi: 10.1146/annurev.pharmtox.010909.105844. [DOI] [PubMed] [Google Scholar]
- 59.Ruggero D., Pandolfi P.P. Does the ribosome translate cancer. Nat. Rev. Cancer. 2003;3:179–192. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 60.Kishore S., Gruber A.R., Jedlinski D.J., Syed A.P., Jorjani H., Zavolan M. Insights into snoRNA biogenesis and processing from PAR-CLIP of snoRNA core proteins and small RNA sequencing. Genome Biol. 2013;14:R45. doi: 10.1186/gb-2013-14-5-r45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Charron C., Manival X., Clery A., Senty-Segault V., Charpentier B., Marmier-Gourrier N., Branlant C., Aubry A. The archaeal sRNA binding protein L7Ae has a 3D structure very similar to that of its eukaryal counterpart while having a broader RNA-binding specificity. J. Mol. Biol. 2004;342:757–773. doi: 10.1016/j.jmb.2004.07.046. [DOI] [PubMed] [Google Scholar]
- 62.Szewczak L.B., Gabrielsen J.S., Degregorio S.J., Strobel S.A., Steitz J.A. Molecular basis for RNA kink-turn recognition by the h15.5K small RNP protein. RNA. 2005;11:1407–1419. doi: 10.1261/rna.2830905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ye K., Jia R., Lin J., Ju M., Peng J., Xu A., Zhang L. Structural organization of box C/D RNA-guided RNA methyltransferase. Proc. Natl Acad. Sci. U.S.A. 2009;106:13808–13813. doi: 10.1073/pnas.0905128106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Venema J., Tollervey D. Ribosome synthesis in Saccharomyces cerevisiae. Annu. Rev. Genet. 1999;33:261–311. doi: 10.1146/annurev.genet.33.1.261. [DOI] [PubMed] [Google Scholar]
- 66.Underwood J.G., Boutz P.L., Dougherty J.D., Stoilov P., Black D.L. Homologues of the Caenorhabditis elegans Fox-1 protein are neuronal splicing regulators in mammals. Mol. Cell. Biol. 2005;25:10005–10016. doi: 10.1128/MCB.25.22.10005-10016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Braeutigam C., Rago L., Rolke A., Waldmeier L., Christofori G., Winter J. The RNA-binding protein Rbfox2: an essential regulator of EMT-driven alternative splicing and a mediator of cellular invasion. Oncogene. 2014;33:1082–1092. doi: 10.1038/onc.2013.50. [DOI] [PubMed] [Google Scholar]
- 68.Venables J.P., Brosseau J.P., Gadea G., Klinck R., Prinos P., Beaulieu J.F., Lapointe E., Durand M., Thibault P., Tremblay K., et al. RBFOX2 is an important regulator of mesenchymal tissue-specific splicing in both normal and cancer tissues. Mol. Cell. Biol. 2013;33:396–405. doi: 10.1128/MCB.01174-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brosseau J.P., Lucier J.F., Nwilati H., Thibault P., Garneau D., Gendron D., Durand M., Couture S., Lapointe E., Prinos P., et al. Tumor microenvironment-associated modifications of alternative splicing. RNA. 2014;20:189–201. doi: 10.1261/rna.042168.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yeo G.W., Coufal N.G., Liang T.Y., Peng G.E., Fu X.D., Gage F.H. An RNA code for the FOX2 splicing regulator revealed by mapping RNA-protein interactions in stem cells. Nat. Struct. Mol. Biol. 2009;16:130–137. doi: 10.1038/nsmb.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dieci G., Preti M., Montanini B. Eukaryotic snoRNAs: a paradigm for gene expression flexibility. Genomics. 2009;94:83–88. doi: 10.1016/j.ygeno.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 72.Hirose T., Steitz J.A. Position within the host intron is critical for efficient processing of box C/D snoRNAs in mammalian cells. Proc. Natl Acad. Sci. U.S.A. 2001;98:12914–12919. doi: 10.1073/pnas.231490998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang Z.P., Zhou H., He H.L., Chen C.L., Liang D., Qu L.H. Genome-wide analyses of two families of snoRNA genes from Drosophila melanogaster, demonstrating the extensive utilization of introns for coding of snoRNAs. RNA. 2005;11:1303–1316. doi: 10.1261/rna.2380905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ono M., Scott M.S., Yamada K., Avolio F., Barton G.J., Lamond A.I. Identification of human miRNA precursors that resemble box C/D snoRNAs. Nucleic Acids Res. 2011;39:3879–3891. doi: 10.1093/nar/gkq1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Scott M.S., Avolio F., Ono M., Lamond A.I., Barton G.J. Human miRNA precursors with box H/ACA snoRNA features. PLoS Comput. Biol. 2009;5:e1000507. doi: 10.1371/journal.pcbi.1000507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Schubert T., Pusch M.C., Diermeier S., Benes V., Kremmer E., Imhof A., Langst G. Df31 protein and snoRNAs maintain accessible higher-order structures of chromatin. Mol. Cell. 2012;48:434–444. doi: 10.1016/j.molcel.2012.08.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.