


Abstract
Heterogeneous nuclear ribonucleoprotein K (hnRNPK) is an RNA/DNA-binding protein involved in chromatin remodeling, RNA processing and the DNA damage response. In addition, increased hnRNPK expression has been associated with tumor development and progression. A variety of post-translational modifications of hnRNPK have been identified and shown to regulate hnRNPK function, including phosphorylation, ubiquitination, sumoylation and methylation. However, the functional significance of hnRNPK arginine methylation remains unclear. In the present study, we demonstrated that the methylation of two essential arginines, Arg296 and Arg299, on hnRNPK inhibited a nearby Ser302 phosphorylation that was mediated through the pro-apoptotic kinase PKCδ. Notably, the engineered U2OS cells carrying an Arg296/Arg299 methylation-defective hnRNPK mutant exhibited increased apoptosis upon DNA damage. While such elevated apoptosis can be diminished through addition with wild-type hnRNPK, we further demonstrated that this increased apoptosis occurred through both intrinsic and extrinsic pathways and was p53 independent, at least in part. Here, we provide the first evidence that the arginine methylation of hnRNPK negatively regulates cell apoptosis through PKCδ-mediated signaling during DNA damage, which is essential for the anti-apoptotic role of hnRNPK in apoptosis and the evasion of apoptosis in cancer cells.
INTRODUCTION
Function of heterogeneous nuclear ribonucleoprotein K (hnRNPK) has been implicated in various cellular events such as chromatin remodeling, transcription, RNA splicing, mRNA stability, translation and DNA damage response (1). In addition, hnRNPK interacts with diverse molecular partners including RNA, DNA and various proteins, contributing to its involvement in viral propagation (2–4), erythroid cell maturation (5–7) and other processes. Increasing evidences have indicated the elevation of hnRNPK in many cancers (8–15) and correlation of hnRNPK with aggressive metastasis (8,16) as well as poor prognosis (11–12,17), suggesting an important role for hnRNPK in tumorigenesis.
The involvement of hnRNPK in the DNA damage response and cell cycle arrest has been reported. Currently, it is known that hnRNPK is sumoylated (18,19) and phosphorylated (20) upon DNA damage, which is essential to the role of hnRNPK as a p53 co-activator to promote p53-dependent transcription. In addition, hnRNPK has been implicated in the p53-independent pathway for the regulation of apoptosis. For example, hnRNPK was down-regulated after 5-fluorouracil treatment in Hep3B cells, and the maintenance of endogenous caspase inhibitors was interrupted, resulting in cellular apoptosis (21). Because the aggressive knockdown of endogenous hnRNPK promotes cellular apoptosis (12,21–23), it has been suggested that hnRNPK might play a critical role in DNA damage-induced apoptosis.
Several post-translational modifications (PTMs) of hnRNPK have been identified including phosphorylation (20,24–26), ubiquitination (27), sumoylation (18,19) and arginine methylation (28,29). Some of these PTMs have been shown to regulate hnRNPK function in several molecular processes. Besides ubiquitination and sumoylation, hnRNPK phosphorylation at Ser284 and Ser353 induces hnRNPK cytoplasmic accumulation during erythroid cell maturation (25), and hnRNPK Ser302 phosphorylation regulates VEGF mRNA translation during angiotensin II-mediated renal injury (30). Currently, little is known regarding the functional role of arginine methylation on hnRNPK.
Arginine methylation is an abundant PTM in mammals and mediated through the protein arginine methyltransferase (PRMT) family. In humans, PRMTs are classified into type I (PRMT1, PRMT2, PRMT3, PRMT4 and PRMT6), type II (PRMT5 and PRMT7) and type III (PRMT7) methyltransferases, based on their corresponding asymmetric dimethylation, symmetric dimethylation and monomethylation activities, respectively (31). Of these PRMTs, PRMT1 is the predominant type I enzyme involved in signal transduction, transcriptional regulation and the DNA damage response (31,32). It has been suggested that the PRMT1-mediated arginine methylation of hnRNPK regulates the protein–protein interaction of hnRNPK such as the oncogenic protein Src (29) and tumor suppressor p53 (33). However, the functional consequence of hnRNPK arginine methylation in cancer progression remains poorly understood.
There are five major arginine methylation sites in hnRNPK (28,29). Interestingly, our investigation showed that PRMT1 methylates hnRNPK preferentially on Arg296 and Arg299 in vitro and in vivo, suggesting that hnRNPK methylation at Arg296 and Arg299 might play distinct roles in the biological function of hnRNPK. Accumulating evidence has shown that certain arginine methylations could inhibit nearby modifications to regulate protein function. For example, PRMT1 methylates FOXO1 on Arg248 and Arg250 to block the Akt-mediated phosphorylation of FOXO1 at Ser253, which inhibits pro-survival signaling through Akt (34). In the present study, we investigated whether the methylation of hnRNPK at Arg296 and Arg299 regulates hnRNPK functions. Our results demonstrated that hnRNPK methylation at Arg296 and Arg299 directly abrogated the PKCδ-mediated phosphorylation of Ser302 on hnRNPK in vitro and in vivo, resulting in the negative modulation of cellular apoptosis. In addition, methylation-defective hnRNPK promoted apoptosis upon DNA damage through both intrinsic and extrinsic pathways, which was partially p53 independent. Evasion of apoptosis is considered a hallmark of cancer, and impairment of apoptotic pathways is one of the underlying mechanisms of chemoresistance in cancer cells. Our study provides an evidence for apoptosis regulation through crosstalk between hnRNPK arginine methylation and phosphorylation, which supports the previous observation that antiapoptotic effect of hnRNPK promotes tumorigenicity.
MATERIALS AND METHODS
Plasmids and antibodies
Wild-type (WT) pET23a-Trx-hnRNPK and pGEX4T-1-PRMT1 were prepared in a previous study (28). All mutant hnRNPK constructs were generated using a site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA). A total of nine arginine-to-lysine mutants were established: 2RK (R296K/R299K); 3RK-1 (R268K/R296K/R299K); 3RK-2 (R256K/R258K/R268K); 4RK-1 (R258K/R268K/R296K/R299K); 4RK-2 (R256K/R268K/R296K/R299K); 4RK-3 (R256K/R258K/R296K/R299K); 4RK-4 (R256K/R258K/R268K/R299K); 4RK-5 (R256K/R258K/R268K/R296K) and 5RK (R256K/R258K/R268K/R296K/R299K) and are listed in Supplementary Table S1. Full-length cDNAs encoding human PKCδ were constructed in the pCDNA4-myc/His vector. The recombinant catalytic fragment PKCδ was constructed in the pGEX4T1 vector. Pre-methylated hnRNPK was constructed in the pETDuet vector as described below.
The following antibodies were used in this study and purchased from vendors as indicated: anti-Myc tag (#115046) and GAPDH (#100118) (GeneTex lnc.); anti-hnRNPK/J (3C2) and anti-Flag (M2; Sigma-Aldrich); anti-mono and dimethyl arginine (AB412; Abcam); anti-Ser302-phosphorylation-hnRNPK (#18361; Santa Cruz Biotechnology); and anti-cleaved caspase3 (#9661), anti-cleaved caspase8 (#9496) and anti-cleaved caspase9 (#7237; Cell Signaling).
In vitro methylation
In vitro PRMT1-mediated methylation was performed as previously described (28). Briefly, 1.5 μg of His-tagged hnRNPK was incubated with 0.75 μg of GST-PRMT1 and 1.65 μCi of [methyl-3H]-S-adenosyl methionine (SAM) in 25 mM Tris–HCl buffer (pH 8.0) at 30°C for the indicated times. The reaction was terminated with the addition of sodium dodecylsulphate (SDS) sample buffer, analyzed through sodium dodecyl sulphate-polyacryl amide gel electrophoresis (SDS-PAGE) and detected for fluorography.
Preparation of pre-methylated hnRNPK
cDNA encoding the full-length human hnRNPK fragment from pET23a-Trx-hnRNPK was subcloned into the EcoRI/XhoI sites of pGEX4T-1 to establish a GST-hnRNPK fusion protein. The GST-hnRNPK fragment with NcoI and XhoI sites were obtained through polymerase chain reaction (PCR) and further subcloned into the NcoI and SalI sites of the pET-DUET vector at multiple cloning site 1. The resulting control plasmid was named pET-DUET-GST-hnRNPK. In addition, cDNA encoding the full-length human PRMT1 fragment from pCDNA3HA2-PRMT1 (35) was subcloned into the NdeI/XhoI site of the pET-DUET vector at multiple cloning site 2. Moreover, the lipoprotein (lpp) promoter fragment, carrying the BsrGI and NdeI sites, was PCR amplified from pACYC184-lpp (36). The PRMT1 and lpp promoter fragments were annealed and subcloned into the pET-DUET-GST-hnRNPK to construct pET-Duet-GST-hnRNPK-lpp-PRMT1 to generate pre-methylated hnRNPK.
Escherichia coli BL21(DE3) cells harboring pETDUET-GST-hnRNPK or pETDUET-GST-hnRNPK-lpp-PRMT1 plasmids were cultured in LB medium. The expression of GST-hnRNPK or pre-methylated GST-hnRNPK was induced using 0.2 μM IPTG, and the recombinant proteins were purified using glutathione-Sepharose 4 Fast Flow beads (GE Healthcare Bio-Sciences, Uppsala, Sweden) according to the manufacturer's instructions.
In vitro kinase assay
Recombinant GST-hnRNPK or pre-methylated GST-hnRNPK were pre-incubated with the GST-catalytic fragment PKCδ (CF-PKCδ) in kinase buffer (50 mM Tris–HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl2 and 1 mM dithiothreitol) on ice for 10 min. Subsequently, 0.25 mCi/ml [γ-32P]-ATP was added to the solution, and the reaction was incubated at 30°C for 15 min. The reactions were terminated upon the addition of SDS sample buffer. The samples were analyzed through SDS-PAGE and autoradiography.
RNA interference and the establishment of stable methylation-defective hnRNPK cell line
A lentivirus for hnRNPK knockdown was packaged in HEK293T cells according to the manufacturer's instructions (National RNAi Core Facility, Taipei, Taiwan). For virus production, 4 μg of packaging pCMVΔR8.91 and 0.4 μg of envelope VSV-G pMD.G were co-transfected with 4 μg of pLKO.1-shhnRNPK.puro (puromycin resistance and hnRNPK knockdown through shRNA targeting TGATGTTTGATGACCGTCGCG) or PLKO.AS3w-hnRNPK.hyg (hygromycin resistance and exogenous expression of shRNA-resistant hnRNPK) into 2.4 × 106 cells using the JetPEI™ Transfection Reagent. The virus particles were collected at 24 and 36 h post-transfection. U2OS cells were simultaneously infected with lentivirus carrying pLKO.1-shhnRNPK.puro and PLKO.AS3w-hnRNPK.hyg in the presence of 10 μg/ml polybrene. After 24 h, the infected cells were selected with 3 μg/ml puromycin and 100 μg/ml hygromycin for 48 h. The infected puromycin- and hygromycin-resistant cells were collected and named according to the types of infected hnRNPKs, namely, the U2OS-K-WT and U2OS-K-2RK cells.
Cell culture and transfection
U2OS and HEK293T cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (HyClone, Logan, UT, USA), L-glutamine, 100 units/ml of penicillin and 100 μg/ml of streptomycin (Gibco BRL, Grand Island, NY) in a 5% CO2-humidified incubator at 37°C. H1299 cells were cultured in Roswell Park Memorial Institute 1640 Medium (Gibco BRL, Grand Island, NY) supplemented with 10% fetal bovine serum, L-glutamine, 100 units/ml penicillin and 100 μg/ml streptomycin in a 5% CO2-humidified incubator at 37°C. In addition, U2OS-K-WT and U2OS-K-2RK cells were cultured in DMEM supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml puromycin and 100 μg/ml hygromycin B (Sigma, St Louis, USA) in a 5% CO2-humidified incubator at 37°C. Transient transfections were performed using TurboFect™ (Fermentas, Carlsbad, CA, USA) according to the manufacturer's instructions.
Immunoprecipitation and western blot analysis
The cell pellets were washed with ice-cold phosphate buffered saline (PBS) (pH 7.4) and re-suspended with PBS buffer containing 0.5% Triton X-100 with general protease inhibitor (Sigma–Aldrich). The cell lysate was lysed after freeze-thawing twice. The total cell lysate was incubated with primary antibodies and protein G Sepharose beads (GE Healthcare Bio-Sciences, Uppsala, Sweden) at 4°C for 3 h. The beads were washed three times with binding buffer, and the bound proteins were analyzed through SDS–PAGE and subsequently transferred to a Polyvinylidene fluoride (PVDF) membrane (Millipore, Bedford, MA, USA). The membrane was incubated with blocking solution (5% non-fat milk in TBS/0.05% Tween-20) for 1 h and incubated with primary antibody overnight at 4°C. The membrane was subsequently incubated with horseradish peroxidase-conjugated secondary antibodies for 1 h at ambient temperature. The protein signals were detected by exposing the membrane to X-ray film after treatment with ECL Western Blotting Detection Reagent (Millipore, Bedford, MA, USA) and exposure to X-ray film. The western blot analysis was repeated three times using different lysates. The images from three independent experiments were analyzed using ImageJ software for quantitation (National Institutes of Health, USA). A P-value of <0.05 was considered significant using Student's t-test.
Immunofluorescence microscopy
Cells were grown on glass coverslips for 24 h, followed by transfection with PKCδ for 24 h and further treated with etoposide at the indicated times. The collected cells were washed with PBS, fixed in 4% paraformaldehyde/PBS for 10 min and followed by permeabilization with 0.5% Triton X-100/PBS for 10 min at room temperature. The permeabilized cells were washed with PBS and blocked for 1 h in 4% bovine serum albumin/PBS prior to incubation with a myc-tag (PKCδ) or flag-tag (hnRNPK) primary antibodies for 1 h. The resulting cells were washed with PBS and then incubated with a conjugated-Alexa Fluor 488 or conjugated-Rhodamine 123 secondary antibodies (Chemicon, Temecula, CA, USA). Further incubation of these cells with 10 mg/ml DAPI (Sigma, St Louis, USA) was carried out for 15 min. The resulting cells were washed with PBS and coverslips were mounted with 1,4-diazabicyclo[2.2.2]octane (Sigma, St Louis, USA). Locations of hnRNPK and PKCδ in these cells were detected using a laser scanning confocal microscope (ZEISS LSM 700, Carl Zeiss MicroImaging GmbH, Jena, Germany) at 100× magnification. Each slide was examined for different stains at three excitation wavelengths (405, 488 and 555 nm).
Caspase-3 activity assay
Caspase-3 activity was measured using a caspase-3 assay kit (BD Biosciences, San Jose, CA, USA). Briefly, the cell lysates were incubated with the caspase-3 substrate Ac-DEVD-7-amino-4-methylcoumarin at 37°C for 1 h, and the 7-amino-4-methylcoumarin fluorescence was measured using AutoLumat LB953 (Berthold Technologies, Bad Wildbad, Germany).
Analysis of the sub-G1 population
U2OS-K-WT and U2OS-K-2RK cells were trypsinized, collected and washed with PBS buffer. The cells were fixed overnight in 70% cold ethanol with PBS buffer at −20°C. The cells were further resuspended in PBS buffer supplemented with 0.1% Triton X-100 and 0.2 mg/ml RNase A followed by staining with 20 μg/ml propidium iodide. The cell cycle distribution was determined through flow cytometry, and sub-G1 cells were regarded as apoptotic cells.
TUNEL assay
U2OS-K-WT and U2OS-K-2RK cells were transfected with PKCδ or empty vector for 24 h and subsequently incubated with either Dimethylsulfoxide (DMSO) or 50 μM etoposide for the indicated times. The cells were fixed with 2% paraformaldehyde. Apoptotic cancer cells were detected using the in situ cell death detection kit (Roche Applied Science), followed by flow cytometry.
RESULTS
PRMT1 preferentially methylates Arg296 and Arg299 on hnRNPK in vitro and in vivo
PRMT1 catalyzes the transfer of methyl groups onto hnRNPK at five major sites including Arg256, Arg258, Arg268, Arg296 and Arg299 (28,29). Whether these different arginines exhibit similar efficiency and preference toward PRMT1-mediated methylation remains unknown. To this end, we established diverse hnRNPK arginine mutants exhibiting limited arginine methylation, as described in the ‘Materials and Methods’ section and Supplementary Table S1. Upon in vitro methylation, catalyzed through PRMT1 in the presence of [3H]-SAM, the 2RK, 3RK-1 and 5RK mutants all exhibited reduced methylation compared with WT hnRNPK (Figure 1a). Notably, the mutation of Arg296/Arg299 in 2RK hnRNPK resulted in a significant reduction of methylation, up to 60% of WT hnRNPK methylation. Next, we examined the in vivo methylation levels of WT, 2RK and 5RK hnRNPKs in HEK293T cells using an anti-methyl arginine antibody (7E6). As shown in Figure 1b, a similar loss of 60% WT methylation was observed for the Arg296/Arg299 mutation in 2RK hnRNPK. Accordingly, these results suggest that different arginines on hnRNPK are not chemically equivalent in PRMT1-mediated methylation. To explore this hypothesis, we further determined the efficiency and time dependence of the in vitro methylation on all five 4RK mutants containing only one arginine for methylation. The results showed that Arg299 and Arg296 exhibited the most and second-most efficient methylation, respectively, through either recombinant PRMT1 or the cellular PRMT1 complex (Figure 2a and b). In addition, time-dependent methylation of the five 4RK mutants revealed that Arg296 and Arg299 also had higher methylation rates than other arginines (Figure 3a and b). Taken together, these results demonstrated that the known methylable arginines in hnRNPK exhibit non-equivalent reactivity toward the PRMT1-catalyzed methylation. Moreover, Arg296 and Arg299 had higher methylation potential than other residues in vitro and in vivo, suggesting that these two residues might play distinct roles in the regulation of hnRNPK functions.
Figure 1.
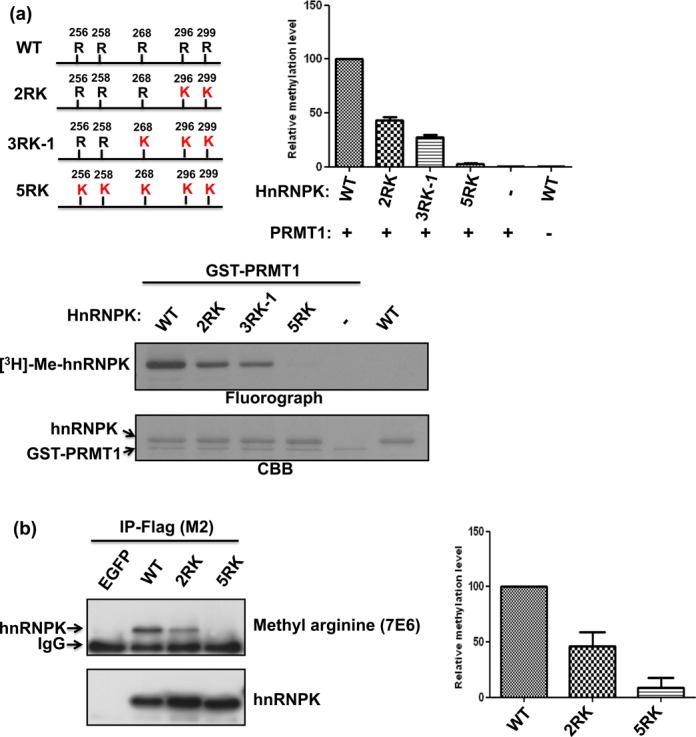
Arg296 and Arg299 are the major methylation sites of hnRNPK in vitro and in vivo. (a) Detection and quantification of in vitro hnRNPK methylation levels and the arginine mutants (2RK, 3RK-1 and 5RK, as shown in the schematic diagram). HnRNPK and diverse mutants were incubated with GST-PRMT1 in the presence of [3H] S-adenosylmethionine (SAM), followed by SDS-PAGE. The proteins were stained with Coomassie blue (bottom). Methylation was detected through fluorography (top) and quantified using a liquid scintillation counter. The methylation levels of all arginine mutants relative to wild-type (WT) hnRNPK are shown as a bar graph. (b) Detection and quantification of in vivo methylation levels of WT hnRNPK and arginine mutants (2RK, 5RK). Flag-hnRNPKs were immunoprecipitated from the DF-1 cells expressing Flag-hnRNPKs using an anti-Flag antibody (M2). Protein expression was determined using an anti-hnRNPK antibody (bottom). Methylation levels were detected using an antibody against anti-asymmetric di-methylated arginine (top) and quantified using ImageJ software. The relative methylation levels of 2RK and 5RK mutants compared with WT hnRNPK are shown as a bar graph.
Figure 2.
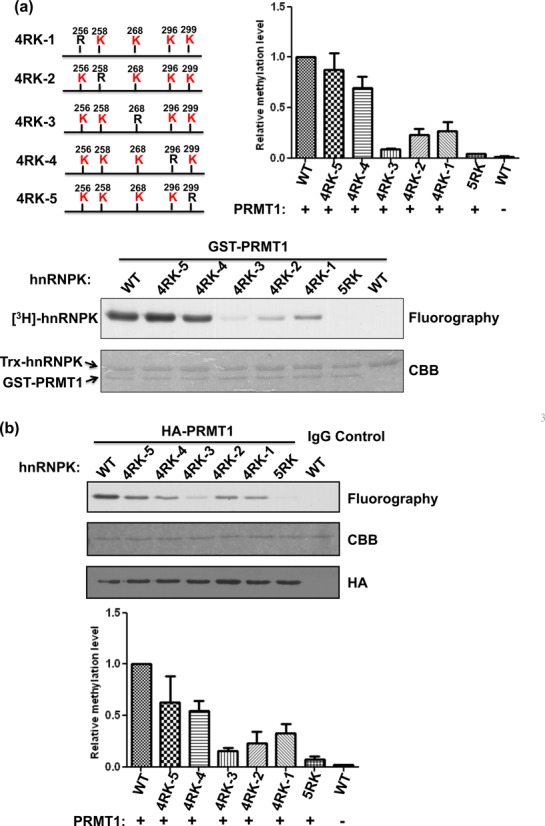
PRMT1 preferentially methylates Arg296 and Arg299 of hnRNPK. (a) Detection and quantification of the recombinant PRMT1-mediated methylation of WT hnRNPK, the 5RK mutant and five 4RK mutants, containing only one remaining arginine for methylation (as shown in the schematic diagram). HnRNPKs were incubated with GST-PRMT1 in the presence of [3H] S-adenosylmethionine (SAM), followed by SDS-PAGE. The proteins were stained with Coomassie blue (bottom). Methylation was detected through fluorography (top) and quantified using a liquid scintillation counter. The relative methylation levels of all mutants to WT hnRNPK are shown as a bar graph. (b) Detection and quantification of the PRMT1 complex-mediated methylation of WT hnRNPK, the 5RK mutant and five 4RK mutants in vitro. HA-PRMT1 complex were immunoprecipitated from the HEK293 cells using an anti-HA antibody and incubated with diverse hnRNPKs in the presence of [3H] S-adenosylmethionine (SAM). After SDS-PAGE analysis, the proteins were stained with Coomassie blue (bottom). Methylation was detected through fluorography (top) and quantified using a liquid scintillation counter. The relative methylation levels of all mutants compared with WT hnRNPK are shown as a bar graph. CBB, Coomassie Brilliant Blue.
Figure 3.
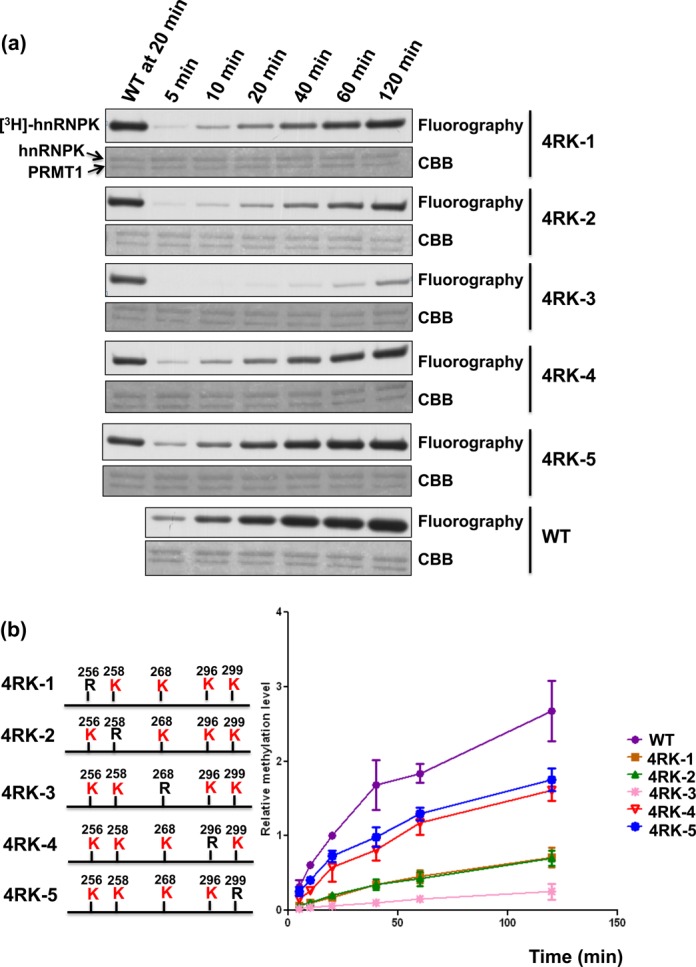
Kinetics analysis of PRMT-mediated methylation of hnRNPK and its 4RK mutants. (a) Detection and quantification of PRMT1-mediated methylation of hnRNPK and diverse 4RK mutants over 120 min. HnRNPKs were incubated with GST-PRMT1 in the presence of [3H] S-adenosylmethionine (SAM), followed by SDS-PAGE. Proteins were stained with Coomassie blue (bottom). Methylation was detected through fluorography (top) and quantified using a liquid scintillation counter. (b) The methylation levels of all mutants relative to WT hnRNPK at the indicated times were determined and are shown as a stacked line chart.
PKCδ-mediated Ser302 phosphorylation of hnRNPK is suppressed by its own arginine methylation in vitro
The interplay between diverse PTMs, including methylation, phosphorylation and acetylation, has been shown to regulate histone protein functions (37,38). In addition, arginine methylation has also been proposed to regulate protein–protein interactions (39–41). Accordingly, we further investigated the methylated arginine-containing sequences of hnRNPK via bioinformatics to determine whether Arg296 and Arg299 methylation have functions distinct from other methylable arginine residues. Several studies have revealed that arginine methylation inhibits the nearby phosphorylation level on the same protein and thus regulates phosphorylation-mediated functions (34,42–44), i.e. human FOXO1 and Bcl2-associated agonist of cell death (BAD) . Further comparison of the methylated arginine-containing sequences among FOXO1, BAD and hnRNPK revealed that Arg296 and Arg299 in hnRNPK are only three residues away from Ser302, similar to the locations of these residues in FOXO1 and BAD (Figure 4a). Ser302 phosphorylation is mediated through PKCδ (24). In addition, it has been suggested that Arg299 is an essential residue in the consensus substrate sequence of PKCδ (RXXSXSR), as shown in Figure 4b (24). Therefore, we further determined whether hnRNPK methylation at Arg296 and Arg299 regulates the interaction of this protein with PKCδ. The results of the co-immunoprecipitation of WT or 2RK hnRNPK with PKCδ in U2OS cells revealed that the loss of hnRNPK arginine methylation on Arg296 and Arg299 increased the interaction of this protein with PKCδ (Supplementary Figure S1).
Figure 4.

Arg296 and Arg299 are conserved methylation sites located near the PKCδ-mediated phosphorylation site of hnRNPK (a) Alignment of the arginine methylation sites within the Akt consensus phosphorylation motif (RXRXXS/T) in two known Akt substrates, FOXO1 and BAD. The conserved arginine methylation sites by PRMT1 in AKT substrates are shaded in gray, and an asterisk indicates the Akt phosphorylation sites. (b) Alignment of Arg296 and Arg299 methylation sites within the PKCδ consensus phosphorylation motif (RXXSRXR) of human hnRNPK with two other vertebrate hnRNPKs. The conserved arginine methylation sites by PRMT1 in hnRNPK are shaded in gray, and an asterisk indicates the known PKCδ phosphorylation sites.
We next investigated whether arginine methylation of hnRNPK interferes with Ser302 phosphorylation. To this end, we generated M-GST-K, a recombinant GST-hnRNPK fusion protein carrying pre-methylated arginines, in E. coli as described in the ‘Materials and Methods’ section. The pre-methylation of this M-GST-K was verified using a methyl arginine-specific antibody, and the control GST-K showed no methylation signal (Figure 5a). Next, we performed the in vitro phosphorylation of pre-methylated M-GST-K and the un-methylated control (GST-K) using constitutively active PKCδ (catalytic fragment, CF-PKCδ) in the presence of [γ-32P]-ATP. A comparison of the phosphorylation levels showed that GST-K exhibited higher phosphorylation levels than M-GST-K (Figure 5b), suggesting that the pre-methylation of hnRNPK suppressed subsequent PKCδ-mediated phosphorylation in vitro.
Figure 5.
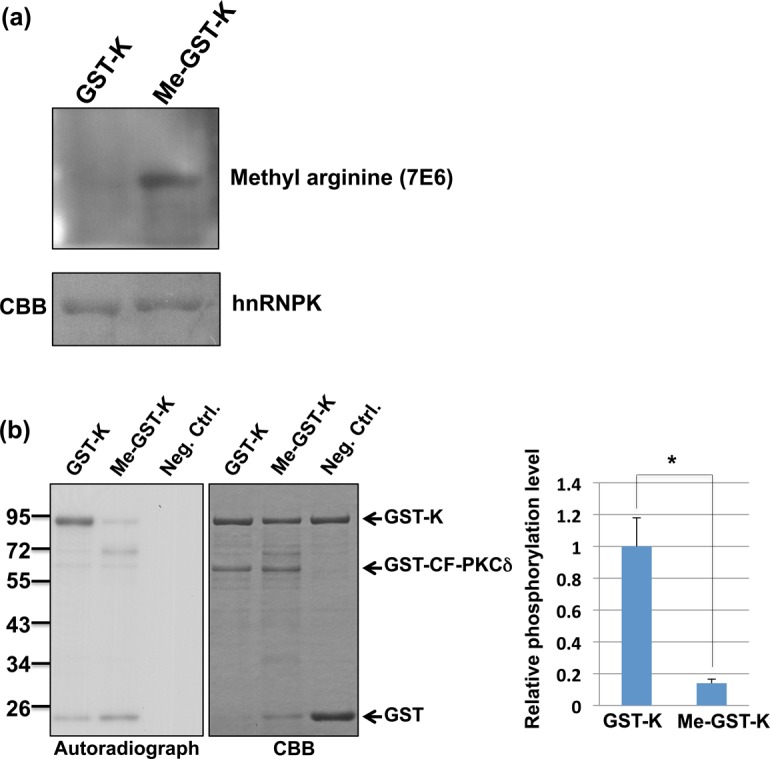
PRMT1-mediated methylation of hnRNPK suppresses its own PKCδ-mediated phosphorylation in vitro. (a) WT (GST-K) and pre-methylated hnRNPK (Me-GST-K) were purified from Escherichia coli, as described in the ‘Materials and Methods’ section. The methylation levels of both hnRNPKs were determined using an asymmetric dimethylarginine-specific antibody (7E6). (b) In vitro phosphorylation of GST-K and Me-GST-K. GST-K and Me-GST-K were incubated with the GST-catalytic fragment PKCδ GST-CF-PKCδ in the presence of [32P]-ATP, followed by SDS-PAGE analysis. The proteins were stained with Coomassie blue (right). The phosphorylation levels were analyzed through autoradiography (left). The relative phosphorylation levels of Me-GST-K to GST-K were quantified using ImageJ software (NIH) and shown as a bar graph. Single asterisk indicates statistical significance (p < 0.05).
Etoposide treatment in U2OS cells elevated hnRNPK Ser302 phosphorylation while arginine methylation of hnRNPK was reduced
PKCδ, a serine/threonine kinase, is involved in both intrinsic and extrinsic apoptotic pathways and is activated during apoptosis upon treatment with cytotoxic agents such as etoposide (45,46). In addition, PKCδ knockdown abolishes DNA damage-induced apoptosis in various cells (47–49). Thus, we further investigated whether etoposide treatment induces PKCδ-mediated phosphorylation of hnRNPK at Ser302. PKCδ is strongly activated after 4 h of etoposide treatment based on the maximal amount of PKCδ catalytic fragment (Supplementary Figure S2). Subsequently, hnRNPK was immunoprecipitated from U2OS cells with or without etoposide treatment, and Ser302 phosphorylation levels were determined using a phosphorylation-specific antibody. As shown in Figure 6a, the Ser302 phosphorylation of hnRNPK in etoposide-treated cells was higher than in untreated cells, suggesting that PKCδ-mediated phosphorylation is inducible through the etoposide treatment of U2OS cells. In addition, we further determined whether etoposide treatment affects the endogenous hnRNPK methylation status. The U2OS cells were treated with etoposide, and followed by immunoprecipitation of hnRNPK. Subsequent methylation detection of the precipitated hnRNPK was carried out using pan-asymmetric methyl-arginine antibody. As shown in Figure 6b, the methylation level of endogenous hnRNPK in U2OS cells was decreased after etoposide treatment, suggesting that the reduced methylation of hnRNPK is correlated with elevation of its Ser302 phosphorylation during etoposide treatment.
Figure 6.
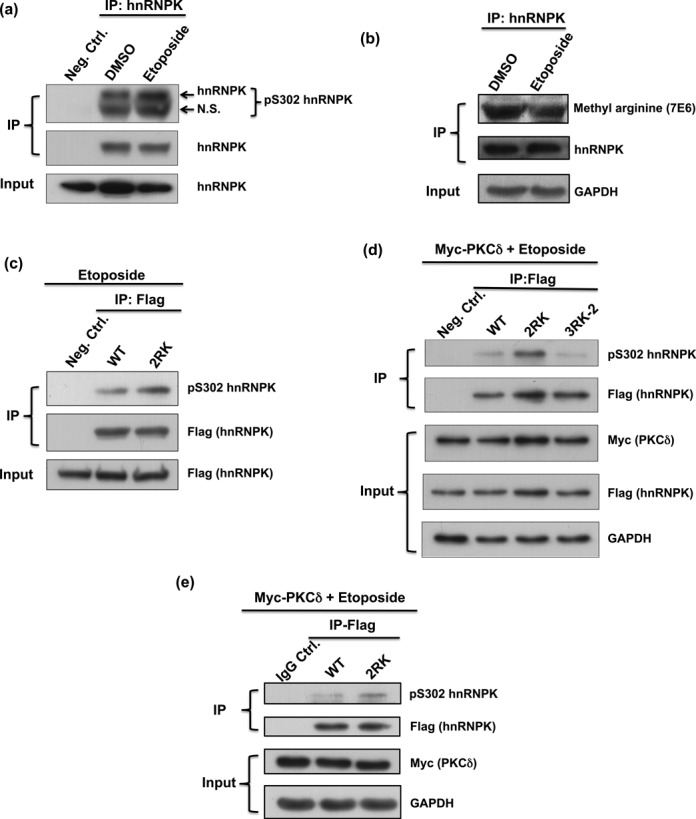
Loss of Arg296 and Arg299 methylation on hnRNPK promotes the nearby Ser302 phosphorylation under etoposide treatment in vivo. (a) Ser302 phosphorylation of endogenous hnRNPK was up-regulated upon etoposide treatment. U2OS cells were treated with or without etoposide for 4 h. Endogenous hnRNPKs were immunoprecipitated from U2OS lysates, followed by western blot analysis to reveal the Ser302 phosphorylation of hnRNPK. N.S., non-specific. (b) Etoposide treatment reduced methylation level of endogenous hnRNPK. The U2OS cells were treated with or without etoposide, and followed by immunoprecipitation of hnRNPK. Subsequent methylation detection and expression of the precipitated hnRNPKs were carried out using pan-asymmetric methyl-arginine antibody and hnRNPK antibody, respectively. (c) Arg296 and Arg299 mutations increased the Ser302 phosphorylation of hnRNPK in U2OS cells treated with etoposide. U2OS cells were transfected with Flag-WT or Flag-2RK (R296K/R299K) hnRNPK, followed by treatment with etoposide for 4 h. The exogenous hnRNPKs were immunoprecipitated from U2OS lysates using a Flag antibody followed by western blot analysis to determine the hnRNPK Ser302 phosphorylation status. (d) hnRNPK methylation at Arg296 and Arg299, but not the other arginines, affects Ser302 phosphorylation. U2OS cells carrying overexpressed Myc-tag PKCδ were transfected with Flag-WT or Flag-2RK (R296K/R299K) or Flag-3RK-2 (R256K/R258K/R268K) hnRNPK, followed by etoposide treatment for 4 h. The exogenous hnRNPKs were immunoprecipitated from U2OS lysates using a Flag antibody followed by western blot analysis to determine the hnRNPK Ser302 phosphorylation state. (e) Mutations at Arg296 and Arg299 increased hnRNPK Ser302 phosphorylation in H1299 cells treated with etoposide. H1299 cells carrying overexpressed Myc-tag PKCδ were transfected with Flag-WT or Flag-2RK (R296K/R299K) hnRNPK, followed by treatment with etoposide for 4 h. The exogenous hnRNPKs were immunoprecipitated from H1299 lysates, followed by western blot analysis to reveal the hnRNPK Ser302 phosphorylation status.
Mutation of Arg296 and Arg299 to lysine in hnRNPK elevates Ser302 phosphorylation
We next determined whether etoposide-induced Ser302 phosphorylation was regulated through Arg296 and Arg299 methylation. U2OS cells were transfected with either WT or Arg296/Arg299-mutated hnRNPK (R296K/R299K, 2RK) followed by etoposide treatment. The results showed that 2RK hnRNPK exhibited higher Ser302 phosphorylation levels than WT hnRNPK upon etoposide treatment (Figure 6c). In addition, the difference in S302 phosphorylation levels was further increased in the etoposide-treated U2OS cells with overexpression of PKCδ (Figure 6d). Similarly, the inhibition of Ser302 phosphorylation through the Arg296/Arg299 methylation of hnRNPK was also observed in H1299 cells (Figure 6e). However, hnRNPK mutants with Arg256/Arg258/Arg268 mutations (3RK-2) exhibited similar Ser302 phosphorylation levels of WT hnRNPK, indicating that the methylation of Arg296 and Arg299, but not other residues, plays a regulatory role in the PKCδ-mediated phosphorylation of hnRNPK (Figure 6d).
Establishment of stable cell lines carrying Arg296/Arg299 methylation-defective hnRNPK
HnRNPK is highly abundant in cells, and its gene knockout is lethal in several species (1,50–52). In addition, the aggressive knockdown of endogenous hnRNPK in cells leads to cell death (12,21–23). To investigate the putative function of Arg296/Arg299 methylation of hnRNPK in vivo, we first overexpressed exogenous hnRNPK carrying diverse arginine mutations in U2OS cells. However, an insignificant phenotype was observed, likely reflecting the abundant presence of endogenous hnRNPK. We thus further established stable cell lines expressing exogenous WT hnRNPK or hnRNPK-2RK (R296K/R299K) and knocked down endogenous hnRNPK through lentivirus-based RNA interference, as described in the ‘Materials and Methods’ section. The resulting cells, U2OS-K-WT and U2OS-K-2RK cells, expressed a significant amount of exogenous hnRNPK-WT or hnRNPK-2RK over the endogenous hnRNPK (Figure 7a). Despite the arginine mutations in hnRNPK, the growth rate and morphology of these two cell lines were nearly identical (data not shown).
Figure 7.
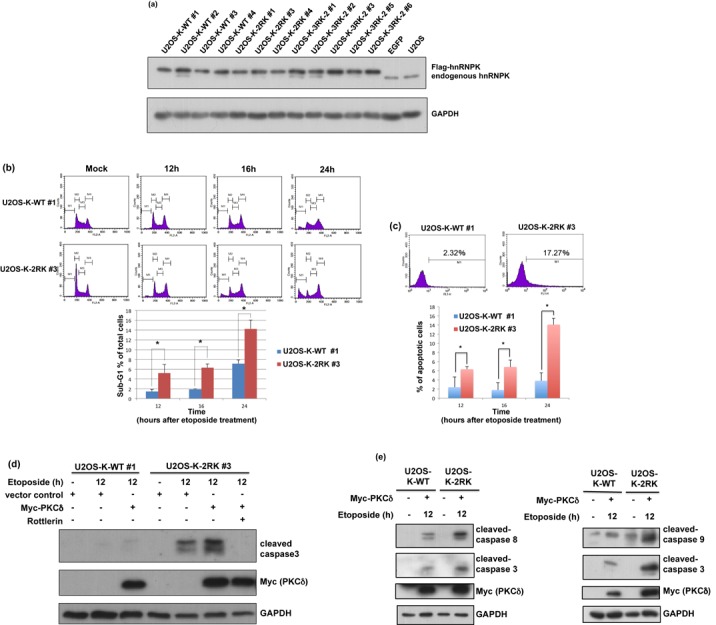
Blockage of Arg299 and Arg296 methylation in hnRNPK promotes U2OS cell apoptosis upon DNA damage. (a) Establishment of stable cell lines carrying Arg296 and Arg299 methylation-defective hnRNPK. U2OS cells were simultaneously infected with lentivirus carrying shRNA against endogenous hnRNPK and lentivirus carrying shRNA-resistant WT or 2RK mutant hnRNPKs. The efficiency of knockdown and ectopic expression were determined according to the protein levels of endogenous and exogenous hnRNPKs, measured using hnRNPK and GAPDH antibodies. (b) U2OS-K-WT and U2OS-K-2RK (R296K/R299K) cells were transfected with Myc-PKCδ for 24 h and treated with etoposide for the indicated times. The cell lysates were collected, stained with propidium iodide and measured through FACS to calculate the percentages of sub-G1 cells. The data are shown as the mean value and SD from three independent experiments. (c) Under the same treatment as described above, the cells were collected at the indicated times and analyzed using a TUNEL assay to determine the percentages of apoptotic cells through FACS. (d) U2OS-K-WT and U2OS-K-2RK (R296K/R299K) cells under the same treatment at 12 h were collected and analyzed for the expression levels of Myc-PKCδ, GAPDH and cleaved caspase 3 using specific antibodies. Pretreatment with the PKCδ inhibitor rottlerin in U2OS-K-2RK cells prior to etoposide treatment was also performed. (e) Arg296 and Arg299 methylation-defective hnRNPK promotes apoptosis via both extrinsic and intrinsic pathways. U2OS-K-WT and U2OS-K-2RK (R296K/R299K) cells were transfected with Myc-PKCδ, followed by etoposide treatment for 12 h. The expression levels of Myc-PKCδ and GAPDH and cleaved caspases 3, 8 and 9 were measured using specific antibodies.
Down-regulation of hnRNPK upon DNA damage to trigger apoptosis has been previously demonstrated in several cells (12,21,23). It is important to determine whether induction of DNA damage in the U2OS-K-WT and U2OS-K-2RK cells also down-regulates the total hnRNPK level. Our data showed that both WT and Arg296/Arg299-mutated (2RK) hnRNPKs were decreased after etoposide treatment alone or treatment combined with PKCδ overexpression (Supplementary Figure S3). This result suggests that the response of hnRNPK to stress induction in both U2OS-K-WT and U2OS-K-2RK cells coincides to those in previous studies. In addition, the U2OS-K-WT cells exhibited similar reduction of hnRNPK methylation as the parental U2OS cells upon etoposide treatment (Figure 6b, Supplementary Figure S4). Therefore, these paired cells provide an adequate tool to investigate the biological consequence induced through the manipulation of the arginine methylation of hnRNPK, but not its expression.
Methylation-defective hnRNPK induces caspase 3 activity and promotes cell apoptosis in U2OS cells after etoposide-induced DNA damage
We demonstrated that suppression of Arg296 and Arg299 methylation in the hnRNPK-2RK mutant increased PKCδ-mediated Ser302 phosphorylation. Because hnRNPK phosphorylation has previously been demonstrated as important for apoptosis induction (45,46), we further examined whether the methylation defects in the hnRNPK mutant affect cell apoptosis. Both U2OS-K-WT and U2OS-K-2RK cells were induced to activate PKCδ and analyzed for the degree of apoptosis through flow cytometry. Upon etoposide treatment and PKCδ overexpression, U2OS-K-2RK cells showed a higher ratio of sub-G1 cells compared with U2OS-K-WT cells, as shown in Figure 7b. In addition, a TUNEL assay was performed to measure the apoptosis percentage in both U2OS-K-WT and U2OS-K-2RK cells. As shown in Figure 7c, U2OS-K-2RK cells exhibited a higher degree of DNA fragmentation than U2OS-K-WT cells.
It has been reported that PKCδ activation upon etoposide treatment leads to its translocation into the nucleus, where it can regulate apoptosis initiation (53). In addition, hnRNPK is ubiquitously found in many subcellular locations, but majorly locate in nucleus for its transcriptional regulation in DNA damage response (1,27). We thus further determined whether hnRNPK and PKCδ co-localize in nucleus following etoposide treatment. Both U2OS-K-WT and U2OS-K-2RK cells were transfected with PKCδ and treated with etoposide. Immunofluorescence detection of the resulting cells was carried out. As shown in the Supplementary Figure S5, PKCδ majorly localized in cytoplasm of U2OS-K-WT and U2OS-K-2RK cells before treatment, whereas the activated PKCδ was translocated into nucleus and co-localized with hnRNPK in both cells after etoposide treatment.
Caspase 3 plays a critical role in apoptosis (54). In addition, caspase 3 cleaves PKCδ and produces constitutively active PKCδ (catalytic fragment), which further promotes caspase 3 activation and apoptosis (55–57). Accordingly, caspase 3 activity levels were measured in these cells after DNA damage to determine whether the methylation defect in Arg296 and Arg299 of hnRNPK mutant affects PKCδ-mediated apoptosis. After a 12-h etoposide treatment, the U2OS-K-2RK cells exhibited a higher degree of caspase 3 cleavage compared with U2OS-K-WT cells, as shown in Figure 7d. In addition, PKCδ overexpression together with etoposide treatment further increased the difference in caspase 3 cleavage between U2OS-K-WT and U2OS-K-2RK cells (Figure 7d and Supplementary Figure S6). Similarly, caspase 3 activity levels (based on the presence of cleaved substrate) also showed a clear difference between U2OS-K-WT and U2OS-K-2RK cells upon etoposide treatment and PKCδ overexpression (Supplementary Figure S7). In contrast, pre-treatment with rottlerin (a PKCδ inhibitor) significantly attenuated the increased caspase 3 cleavage induced through etoposide treatment and PKCδ overexpression in U2OS-K-2RK cells (Figure 7d). Because cleaved caspase 3 can be activated via both extrinsic and intrinsic pathways, we further measured the levels of cleaved caspase 8 and caspase 9 in U2OS-K-WT and U2OS-K-2RK cells upon apoptosis (Figure 7e). The results showed that U2OS-K-2RK cells exhibited increased caspase 8 and 9 cleavage compared with U2OS-K-WT cells after etoposide treatment and PKCδ overexpression, suggesting that the methylation deficiency in hnRNPK promotes cell apoptosis via both extrinsic and intrinsic pathways.
Supplementing with additional hnRNPKs carrying R296/R299 methylation reduces apoptosis in U2OS-K-2RK cells after DNA damage
To further validate the negative role of hnRNPK methylation on apoptosis induction, we expressed either the shRNA-resistant WT hnRNPK or shRNA-resistant 3RK-2 (R256K/R258K/R268K) hnRNPK mutants in the etoposide-treated U2OS-K-2RK cells. As shown in Figure 8, supplementing these cells with Arg296/Arg299-methylable hnRNPKs resulted in reduced caspase 3 cleavage compared with the vector control. This result indicates that methylation at arginine residues 296 and 299 of hnRNPK indeed suppresses PKCδ-mediated apoptosis upon DNA damage.
Figure 8.
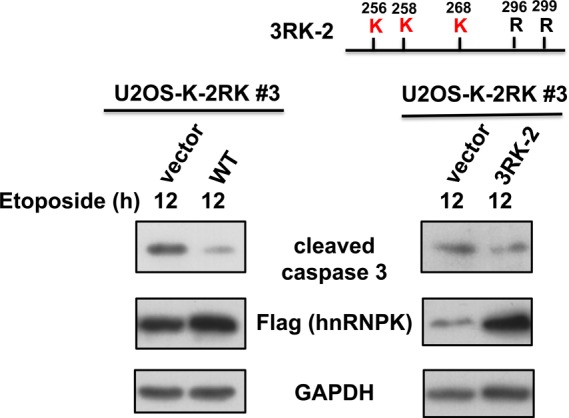
Supplementing with exogenous hnRNPK carrying R296/R299 methylation reduced U2OS-K-2RK cell apoptosis after DNA damage. The U2OS-K-2RK cells were transfected with shRNA-resistant WT or 3RK-2 mutant (R256K/ R258K/ R268K) hnRNPKs, followed by etoposide treatment for 12 h. The expression levels of hnRNPK, GAPDH and cleaved caspase 3 were determined using specific antibodies.
Blockage of Arg296 and Arg299 methylation in hnRNPK promotes apoptosis in a p53-independent manner
hnRNPK physically interacts with p53 as a co-transcriptional activator to regulate the downstream genes responsible for cell cycle arrest and apoptosis (27). However, co-immunoprecipitation experiments showed that the interaction of p53 and hnRNPK is not affected by the methylation defects of hnRNPK in U2OS cells after etoposide treatment and PKCδ overexpression (Supplementary Figure S8). Notably, p53-independent hnRNPK-mediated apoptosis has recently been reported (21). To determine whether the observed apoptotic induction through methylation-defective hnRNPK is p53 independent, we further established p53-null SaOS2 cells carrying WT and 2RK hnRNPKs, namely SaOS2-K-WT and SaOS2-K-2RK cells (Figure 9a). hnRNPK immunoprecipitation experiments in SaOS2-K-WT and SaOS2-K-2RK cells treated with etoposide and PKCδ overexpression showed that 2RK hnRNPK exhibited higher Ser302 phosphorylation than WT hnRNPK, as shown in Figure 9b. Further analysis showed that, after a 12-h etoposide treatment, the SaOS2-K-2RK cells exhibited a higher degree of caspase 3 cleavage than SaOS2-K-WT cells. In addition, PKCδ overexpression and etoposide treatment together in both cells further increased the differences in caspase 3 cleavage between SaOS2-K-WT and SaOS2-K-2RK cells (Figure 9b). Taken together, these results suggest that the methylation defect in hnRNPK promotes caspase 3 activity and cellular apoptosis during etoposide-induced DNA damage in a p53-independent manner.
Figure 9.
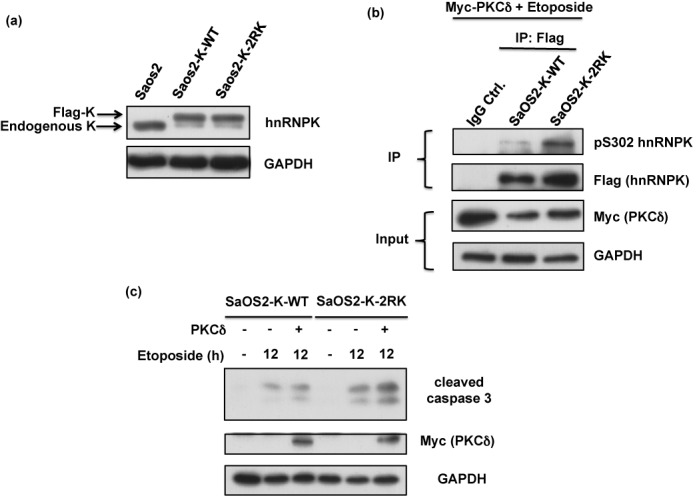
Blockage of Arg296 and Arg299 methylation in hnRNPK promotes apoptosis in a p53-independent manner. (a) SaOS-2 cells were simultaneously infected with lentivirus carrying shRNA against endogenous hnRNPK and lentivirus carrying shRNA-resistant WT or 2RK mutant hnRNPKs. The efficiency of knockdown and ectopic expression was determined after measuring the protein levels of endogenous and exogenous hnRNPKs using hnRNPK and GAPDH antibodies. (b) The SaOS2-K-WT and SaOS2-K-2RK (R296K/R299K) cells were transfected with Myc-PKCδ for 24 h and subsequently treated with etoposide for 12 h. The cell lysates were collected and analyzed for Myc-PKCδ, GAPDH and cleaved caspase 3 expression levels using specific antibodies.
DISCUSSION
We herein demonstrated the functional crosstalk between the PRMT1-catalyzed arginine 296/299 methylations and PKCδ-mediated phosphorylation of hnRNPK. In addition, replacing endogenous hnRNPK with methylation-defective hnRNPK in different cells induced caspase3 activation and apoptosis upon DNA damage, i.e. etoposide treatment. Moreover, the etoposide-induced apoptosis in cells carrying methylation-defective hnRNPK was suppressed through the ectopic expression of WT hnRNPK. These findings provide strong evidence that the PRMT1-induced methylation of hnRNPK plays an important role in guiding the cell fate toward apoptosis. This study is the first to demonstrate that the interplay between PTMs of hnRNPK modulates cell apoptosis.
The functional roles of protein arginine methylation remain largely unknown (58). The current understanding of such protein modification is based on the functional knowledge of proteins that have been identified as methyl acceptors during PRMTs-mediated methylation (31,32). We previously reported that hnRNPK undergoes Arg296 and Arg299 methylation in vitro in the presence of PRMT1 (28), and a total of five major methylated arginines have now been identified on cellular hnRNPK (29) including Arg256, Arg258, Arg268, Arg296 and Arg299. Whether these arginine methylations are either chemically or functionally equivalent has never been explored. In the present study, we showed that Arg296 and Arg299 are the dominant sites for hnRNPK methylation, mediated through PRMT1 in vitro and in vivo (Figures 1–3). Moreover, methylation of these specific arginine residues, but not the other arginines, negatively regulates the phosphorylation of hnRNPK at Ser302 (Figures 5 and 6). Arg299 has been previously reported to be involved in the consensus substrate sequence (RXXSRXR) of PKCδ-mediated phosphorylation in hnRNPK (24). It is speculated that Arg296 and Arg299 methylation might increase steric hindrance and interfere with the catalytic site of PKCδ.
However, we also demonstrated that the suppression of Arg296 and Arg299 methylation increased PKCδ-mediated Ser302 phosphorylation on hnRNPK (Figure 6). Accordingly, upon induction of PKCδ activity, the U2OS cells showed that only the Arg296 and Arg299 methylation-defective hnRNPK increased Ser302 phosphorylation, and this effect was not observed for WT or mutant hnRNPK carrying other arginine mutations. Based on the difference in methylation efficiency and arginine locations, it is suggested that the methylation of hnRNPK at Arg296 and Arg299 has a function distinct from the other methylated arginines. More importantly, Arg296, Arg299 and Ser302 are located at highly conserved regions of hnRNPK throughout vertebrates, suggesting that the interplay between arginine methylation and nearby serine phosphorylation is conserved in evolution and might play an important role.
PKCδ is essential for apoptosis induction upon DNA damage (45,46). In addition, activated PKCδ interacts with and/or phosphorylates downstream pro-apoptotic proteins such as p53 (59), c-Abl (60,61), DNA-dependent protein kinase (62) and Rad9 (63) to trigger apoptosis upon DNA damage. Recently, it has been shown that hnRNPK and activated PKCδ might co-regulate apoptosis induction via hnRNPK ubiquitination during DNA damage (23). Indeed, hnRNPK is stabilized via the reduction of MDM2-mediated ubiquitination during DNA damage and thus acts together with p53 to promote the transcription of the cell cycle-related and pro-apoptotic genes (27). In contrast, hnRNPK suppression through RNA interference down-regulates anti-apoptotic proteins such as XIAP and FLIP to promote apoptosis (21). Because the PKCδ-mediated degradation of hnRNPK has been implied in apoptosis induction (23), it is possible that elevated hnRNPK Ser302 phosphorylation, upon loss of Arg296 and Arg299 methylation, might increase the proteasome-mediated degradation of hnRNPK to promote apoptosis. On the other hand, hnRNPK is a polycytosine RNA binding protein and it is interesting to know whether methylation of Arg296 and Arg299 alters the RNA binding capacity of hnRNPK upon etoposide treatment. A luciferase reporter plasmid containing CU-rich element sequence was used to monitor the RNA binding of hnRNPK in cells (64). As shown in Supplementary Figure S9, a similar decrease of luciferase activity was observed in both U2OS-K-WT and U2OS-K-2RK cells after etoposide treatment, which is due to reduction of total hnRNPK level upon etoposide treatment in both U2OS-K-WT and U2OS-K-2RK cells (Supplementary Figure S3). Therefore, it is suggested that Arg296 and Arg299 methylations of hnRNPK may not affect its RNA binding capacity.
hnRNPK up-regulation has been observed in many cancers (9–10,13–15) and is associated with increased metastasis (8,16) and poor prognosis (11–12,17), suggesting an important role for hnRNPK in tumorigenesis. In addition, hnRNPK knockdown suppresses the survival of various cancer cells (12,22). The present evidence demonstrating that the methylation-defective hnRNPK promoted apoptosis upon DNA damage suggests the putative correlation of hnRNPK arginine methylation with cancer survival. Thus, the elevation of PKCδ-mediated phosphorylation on methylation-defective hnRNPK might facilitate the downstream signaling of PKCδ-dependent apoptosis induction. In addition, hnRNPK is known to interact with p53 to act as a transcriptional co-activator during DNA damage to promote the signaling of p53 downstream genes, such as p21, and induce cell-cycle arrest (27). Moreover, the elevated arginine methylation of hnRNPK is reported to enhance p53 transcriptional activity during the early stages of DNA damage (33). Thus, we further determined whether elevated Ser302 phosphorylation on methylation-defective hnRNPK affects the interaction of this protein with p53. Notably, the results of co-immunoprecipitation experiments showed that WT and methylation-defective hnRNPKs exhibited similar p53-interacting ability upon DNA damage (Supplementary Figure S8). Recently, it has been shown that hnRNPK suppresses apoptosis in a p53-independent manner by maintaining the expression levels of endogenous caspase inhibitors, including XIAP and FLIP (21). Thus, we further proposed that the observed apoptosis mediated through methylation-defective hnRNPK during DNA damage is p53-independent. The comparison between SaOS2 cells expressing either WT or 2RK hnRNPKs showed that methylation-defective hnRNPK also promoted apoptosis in p53-null SaOS2 cells, similar to U2OS cells carrying 2RK mutant hnRNPK (Figure 9). This result suggested that the methylation-defective hnRNPK-mediated induction of apoptosis is partially p53 independent.
Alternatively, it has been previously demonstrated that the p53 family protein, p73 or p63, share high sequence identity and functional similarities with p53 (65,66). In addition, these proteins showed many p53-like transcription activities to induce p53-reponsive genes and trigger apoptosis (67,68). Moreover, p73 can compensate p53 functions to response DNA damage in the p53-deficient cells like H1299 and SaOS2 cells (65,69–70). Therefore, the present observation that SaOS2-K-2RK cells exhibit higher apoptosis than SaOS2-K-WT cells upon etoposide treatment may involve p73-mediated regulation of cell death. Whether p73 participates in the increased apoptosis by methylation-defective hnRNPK in SaOS2-K-2RK cells requires further investigation.
Studies have shown that PTMs might be responsible for functional role of hnRNPK in the DNA damage response (19–20,27). PTMs of hnRNPK involved in the DNA damage response include phosphorylation (20), ubiquitination (27) and sumoylation (18,19). For example, the mutation of four ATM-mediated phosphorylation sites of hnRNPK suppresses the ubiquitination of this protein and inhibits p53-induced cell-cycle arrest upon DNA damage (20). Similarly, the UV radiation-induced elevation of hnRNPK sumoylation on Lys422 also suppresses the ubiquitination of this protein and induces cell-cycle arrest (18,19). In the present study, the inhibition of PKCδ-mediated phosphorylation through Arg296/Arg299 methylation indicated that hnRNPK methylation regulates cell apoptosis. Because methylation-defective hnRNPK exhibited higher PKCδ-mediated phosphorylation and promoted apoptosis, it is implied that hnRNPK accommodates distinct protein modifications to regulate the role of this protein in response to DNA damage. Extensive studies of methylation-mediated regulation on other PTMs of hnRNPK are suggested.
In addition to cell apoptosis, whether the interplay between arginine methylation and hnRNPK phosphorylation regulates other hnRNPK-mediated function is interesting. Previous studies have shown that the PKCδ-mediated Ser302 phosphorylation of hnRNPK is associated with the regulation of VEGF mRNA translation upon angiotensin II-induced renal injury (30). It has been suggested that hnRNPK phosphorylation positively regulates RNA binding toward VEGF mRNA. Therefore, whether the arginine methylation of hnRNPK also contributes to renal injury through this regulatory mechanism remains unknown.
Other than the present study, only four other reports have shown a similar interplay between arginine methylation and phosphorylation (Supplementary Table S2). Two Akt substrates, Foxo1 and Bad, demonstrated PRMT1-mediated methylation that inhibits nearby phosphorylation and thereby downstream Akt-mediated signaling (34,44). Notably, these methylation sites are also located within the consensus substrate motif of Akt kinase. However, FEN1 and EGFR exhibit the distinct regulation of phosphorylation upon PRMT5-mediated methylation (42,43). Similar to Foxo1 and Bad, hnRNPK methylation interferes with PKCδ-mediated phosphorylation. Taken together, accumulating evidence of crosstalk between arginine methylation, and phosphorylation might establish a new functional interpretation of protein arginine methylation.
In summary, the present study demonstrated that the functional crosstalk between the arginine methylation and PKCδ-mediated phosphorylation of hnRNPK regulates cell apoptosis upon DNA damage. Notably, hnRNPK has been correlated with cancer progression and resistance to etoposide-induced DNA damage (71). This is the first report to establish a correlation between hnRNPK methylation and cell apoptosis. Notably, the induced apoptosis by methylation-defective hnRNPK occurred through both intrinsic and extrinsic pathways in a partially p53-independent manner. As shown in Figure 10, we hypothesized that the frequently found Arg296 and Arg299 methylation maintains a low Ser302 phosphorylation level in hnRNPK to support its anti-apoptotic role after DNA damage. However, loss of Arg296 and Arg299 methylation in hnRNPK elevates Ser302 phosphorylation level and promotes apoptosis, implicating that methylation-defective hnRNPK might sensitize cancer cells to etoposide-induced apoptosis. Therefore, the aggressive regulation of arginine methylation may serve as a putative anticancer strategy.
Figure 10.
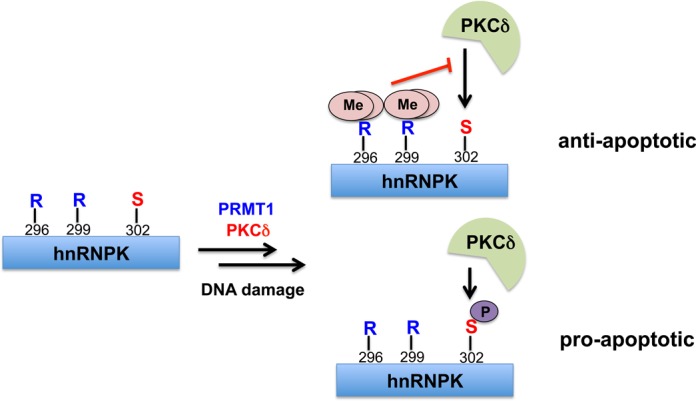
Crosstalk between Arg296/Arg299 methylation and Ser302 phosphorylation results in role switch of hnRNPK in DNA damage response. Frequently occurred methylation of Arg296/Arg299 suppresses the PKCδ-mediated Ser302 phosphorylation, which results in the anti-apoptotic effect of hnRNPK. Alternatively, low level of Arg296/Arg299 methylation promotes the nearby Ser302 phosphorylation and pro-apoptotic effect of hnRNPK after DNA damage.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Prof. Yu-Sun Chang at Graduate Institute of Biomedical Sciences, Chang Gung University for the reporter plasmid of 3'-UTR of thymidine phosphorylase (pMIR-CURE).
FUNDING
National Science Council [101-2311-B-010-006, 101-2321-B-010-016, 102-2321-B-010-014]; Taipei Veteran's General Hospital [100-G7-5-1]; Cheng Hsin General Hospital [101F195CY21]; Ministry of Education, Aiming for the Top University Plan in Taiwan. Funding for open access charge: Personal expense.
Conflict of interest statement. None declared.
REFERENCES
- 1.Bomsztyk K., Denisenko O., Ostrowski J. hnRNP K: one protein multiple processes. Bioessays. 2004;26:629–638. doi: 10.1002/bies.20048. [DOI] [PubMed] [Google Scholar]
- 2.van Domselaar R., de Poot S.A., Remmerswaal E.B., Lai K.W., ten Berge I.J., Bovenschen N. Granzyme M targets host cell hnRNP K that is essential for human cytomegalovirus replication. Cell Death Differ. 2013;20:419–429. doi: 10.1038/cdd.2012.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng L.F., Chan M., Chan S.H., Cheng P.C., Leung E.H., Chen W.N., Ren E.C. Host heterogeneous ribonucleoprotein K (hnRNP K) as a potential target to suppress hepatitis B virus replication. PLoS Med. 2005;2:e163. doi: 10.1371/journal.pmed.0020163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolf D., Witte V., Clark P., Blume K., Lichtenheld M.G., Baur A.S. HIV Nef enhances Tat-mediated viral transcription through a hnRNP-K-nucleated signaling complex. Cell Host Microbe. 2008;4:398–408. doi: 10.1016/j.chom.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 5.Ostareck D.H., Ostareck-Lederer A., Wilm M., Thiele B.J., Mann M., Hentze M.W. mRNA silencing in erythroid differentiation: hnRNP K and hnRNP E1 regulate 15-lipoxygenase translation from the 3’ end. Cell. 1997;89:597–606. doi: 10.1016/s0092-8674(00)80241-x. [DOI] [PubMed] [Google Scholar]
- 6.Ostareck D.H., Ostareck-Lederer A., Shatsky I.N., Hentze M.W. Lipoxygenase mRNA silencing in erythroid differentiation: the 3’UTR regulatory complex controls 60S ribosomal subunit joining. Cell. 2001;104:281–290. doi: 10.1016/s0092-8674(01)00212-4. [DOI] [PubMed] [Google Scholar]
- 7.Naarmann-de Vries I.S., Urlaub H., Ostareck D.H., Ostareck-Lederer A. Caspase-3 cleaves hnRNP K in erythroid differentiation. Cell Death Dis. 2013;4:e548. doi: 10.1038/cddis.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoue A., Sawata S.Y., Taira K., Wadhwa R. Loss-of-function screening by randomized intracellular antibodies: identification of hnRNP-K as a potential target for metastasis. Proc. Natl. Acad. Sci. U.S.A. 2007;104:8983–8988. doi: 10.1073/pnas.0607595104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roychoudhury P., Chaudhuri K. Evidence for heterogeneous nuclear ribonucleoprotein K overexpression in oral squamous cell carcinoma. Br. J. Cancer. 2007;97:574–575. doi: 10.1038/sj.bjc.6603911. author reply 576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen L.C., Hsueh C., Tsang N.M., Liang Y., Chang K.P., Hao S.P., Yu J.S., Chang Y.S. Heterogeneous ribonucleoprotein k and thymidine phosphorylase are independent prognostic and therapeutic markers for nasopharyngeal carcinoma. Clin. Cancer Res. 2008;14:3807–3813. doi: 10.1158/1078-0432.CCR-08-0155. [DOI] [PubMed] [Google Scholar]
- 11.Barboro P., Repaci E., Rubagotti A., Salvi S., Boccardo S., Spina B., Truini M., Introini C., Puppo P., Ferrari N., et al. Heterogeneous nuclear ribonucleoprotein K: altered pattern of expression associated with diagnosis and prognosis of prostate cancer. Br. J. Cancer. 2009;100:1608–1616. doi: 10.1038/sj.bjc.6605057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L.C., Chung I.C., Hsueh C., Tsang N.M., Chi L.M., Liang Y., Chen C.C., Wang L.J., Chang Y.S. The antiapoptotic protein, FLIP, is regulated by heterogeneous nuclear ribonucleoprotein K and correlates with poor overall survival of nasopharyngeal carcinoma patients. Cell Death Differ. 2010;17:1463–1473. doi: 10.1038/cdd.2010.24. [DOI] [PubMed] [Google Scholar]
- 13.Wang F., Zhang P., Shi C., Yang Y., Qin H. Immunohistochemical detection of HSP27 and hnRNP K as prognostic and predictive biomarkers for colorectal cancer. Med. Oncol. 2012;29:1780–1788. doi: 10.1007/s12032-011-0037-3. [DOI] [PubMed] [Google Scholar]
- 14.Zhou R., Shanas R., Nelson M.A., Bhattacharyya A., Shi J. Increased expression of the heterogeneous nuclear ribonucleoprotein K in pancreatic cancer and its association with the mutant p53. Int. J. Cancer. 2010;126:395–404. doi: 10.1002/ijc.24744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang P., Huang L., Ma Y.L., Peng J.Y., Shen T.Y., Chen H.Q., Zhou Y.K., Zhang M., Chu Z.X., Qin H.L. HnRNP K and PDI marked response to chemotherapy to human colorectal cancer cells. Electrophoresis. 2010;31:1731–1738. doi: 10.1002/elps.200900495. [DOI] [PubMed] [Google Scholar]
- 16.Gao R., Yu Y., Inoue A., Widodo N., Kaul S.C., Wadhwa R. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement and angiogenesis. J. Biol. Chem. 2013;288:15046–15056. doi: 10.1074/jbc.M113.466136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matta A., Tripathi S.C., DeSouza L.V., Grigull J., Kaur J., Chauhan S.S., Srivastava A., Thakar A., Shukla N.K., Duggal R., et al. Heterogeneous ribonucleoprotein K is a marker of oral leukoplakia and correlates with poor prognosis of squamous cell carcinoma. Int. J. Cancer. 2009;125:1398–1406. doi: 10.1002/ijc.24517. [DOI] [PubMed] [Google Scholar]
- 18.Pelisch F., Pozzi B., Risso G., Munoz M.J., Srebrow A. DNA damage-induced heterogeneous nuclear ribonucleoprotein K sumoylation regulates p53 transcriptional activation. J. Biol. Chem. 2012;287:30789–30799. doi: 10.1074/jbc.M112.390120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S.W., Lee M.H., Park J.H., Kang S.H., Yoo H.M., Ka S.H., Oh Y.M., Jeon Y.J., Chung C.H. SUMOylation of hnRNP-K is required for p53-mediated cell-cycle arrest in response to DNA damage. EMBO J. 2012;31:4441–4452. doi: 10.1038/emboj.2012.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moumen A., Magill C., Dry K.L., Jackson S.P. ATM-dependent phosphorylation of heterogeneous nuclear ribonucleoprotein K promotes p53 transcriptional activation in response to DNA damage. Cell Cycle. 2013;12:698–704. doi: 10.4161/cc.23592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Z., Ko H.L., Goh E.H., Wang B., Ren E.C. hnRNP K suppresses apoptosis independent of p53 status by maintaining high levels of endogenous caspase inhibitors. Carcinogenesis. 2013;34:1458–1467. doi: 10.1093/carcin/bgt085. [DOI] [PubMed] [Google Scholar]
- 22.van Domselaar R., Quadir R., van der Made A.M., Broekhuizen R., Bovenschen N. All human granzymes target hnRNP K that is essential for tumor cell viability. J. Biol. Chem. 2012;287:22854–22864. doi: 10.1074/jbc.M112.365692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao F.H., Wu Y.L., Zhao M., Liu C.X., Wang L.S., Chen G.Q. Protein kinase C-delta mediates down-regulation of heterogeneous nuclear ribonucleoprotein K protein: involvement in apoptosis induction. Exp. Cell Res. 2009;315:3250–3258. doi: 10.1016/j.yexcr.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 24.Schullery D.S., Ostrowski J., Denisenko O.N., Stempka L., Shnyreva M., Suzuki H., Gschwendt M., Bomsztyk K. Regulated interaction of protein kinase Cdelta with the heterogeneous nuclear ribonucleoprotein K protein. J. Biol. Chem. 1999;274:15101–15109. doi: 10.1074/jbc.274.21.15101. [DOI] [PubMed] [Google Scholar]
- 25.Habelhah H., Shah K., Huang L., Ostareck-Lederer A., Burlingame A.L., Shokat K.M., Hentze M.W., Ronai Z. ERK phosphorylation drives cytoplasmic accumulation of hnRNP-K and inhibition of mRNA translation. Nat. Cell Biol. 2001;3:325–330. doi: 10.1038/35060131. [DOI] [PubMed] [Google Scholar]
- 26.Messias A.C., Harnisch C., Ostareck-Lederer A., Sattler M., Ostareck D.H. The DICE-binding activity of KH domain 3 of hnRNP K is affected by c-Src-mediated tyrosine phosphorylation. J. Mol. Biol. 2006;361:470–481. doi: 10.1016/j.jmb.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 27.Moumen A., Masterson P., O'Connor M.J., Jackson S.P. hnRNP K: an HDM2 target and transcriptional coactivator of p53 in response to DNA damage. Cell. 2005;123:1065–1078. doi: 10.1016/j.cell.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 28.Chiou Y.Y., Lin W.J., Fu S.L., Lin C.H. Direct mass-spectrometric identification of Arg296 and Arg299 as the methylation sites of hnRNP K protein for methyltransferase PRMT1. Protein J. 2007;26:87–93. doi: 10.1007/s10930-006-9049-9. [DOI] [PubMed] [Google Scholar]
- 29.Ostareck-Lederer A., Ostareck D.H., Rucknagel K.P., Schierhorn A., Moritz B., Huttelmaier S., Flach N., Handoko L., Wahle E. Asymmetric arginine dimethylation of heterogeneous nuclear ribonucleoprotein K by protein-arginine methyltransferase 1 inhibits its interaction with c-Src. J. Biol. Chem. 2006;281:11115–11125. doi: 10.1074/jbc.M513053200. [DOI] [PubMed] [Google Scholar]
- 30.Feliers D., Lee M.J., Ghosh-Choudhury G., Bomsztyk K., Kasinath B.S. Heterogeneous nuclear ribonucleoprotein K contributes to angiotensin II stimulation of vascular endothelial growth factor mRNA translation. Am. J. Physiol. Renal Physiol. 2007;293:F607–F615. doi: 10.1152/ajprenal.00497.2006. [DOI] [PubMed] [Google Scholar]
- 31.Yang Y., Bedford M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 32.Bedford M.T., Clarke S.G. Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y., Zhou X., Liu N., Wang C., Zhang L., Mo W., Hu G. Arginine methylation of hnRNP K enhances p53 transcriptional activity. FEBS Lett. 2008;582:1761–1765. doi: 10.1016/j.febslet.2008.04.051. [DOI] [PubMed] [Google Scholar]
- 34.Yamagata K., Daitoku H., Takahashi Y., Namiki K., Hisatake K., Kako K., Mukai H., Kasuya Y., Fukamizu A. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell. 2008;32:221–231. doi: 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 35.Chang Y.I., Hua W.K., Yao C.L., Hwang S.M., Hung Y.C., Kuan C.J., Leou J.S., Lin W.J. Protein-arginine methyltransferase 1 suppresses megakaryocytic differentiation via modulation of the p38 MAPK pathway in K562 cells. J. Biol. Chem. 2010;285:20595–20606. doi: 10.1074/jbc.M109.092411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Su P.T., Yen P.W., Wang S.H., Lin C.H., Chiou A., Syu W.J. Factors affecting daughter cells’ arrangement during the early bacterial divisions. PLoS One. 2010;5:e9147. doi: 10.1371/journal.pone.0009147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suganuma T., Workman J.L. Crosstalk among histone modifications. Cell. 2008;135:604–607. doi: 10.1016/j.cell.2008.10.036. [DOI] [PubMed] [Google Scholar]
- 38.Daujat S., Bauer U.M., Shah V., Turner B., Berger S., Kouzarides T. Crosstalk between CARM1 methylation and CBP acetylation on histone H3. Curr. Biol. 2002;12:2090–2097. doi: 10.1016/s0960-9822(02)01387-8. [DOI] [PubMed] [Google Scholar]
- 39.Sikorsky T., Hobor F., Krizanova E., Pasulka J., Kubicek K., Stefl R. Recognition of asymmetrically dimethylated arginine by TDRD3. Nucleic acids research. 2012;40:11748–11755. doi: 10.1093/nar/gks929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen C., Nott T.J., Jin J., Pawson T. Deciphering arginine methylation: Tudor tells the tale. Nat. Rev. Mol. Cell Biol. 2011;12:629–642. doi: 10.1038/nrm3185. [DOI] [PubMed] [Google Scholar]
- 41.Liu K., Chen C., Guo Y., Lam R., Bian C., Xu C., Zhao D.Y., Jin J., MacKenzie F., Pawson T., et al. Structural basis for recognition of arginine methylated Piwi proteins by the extended Tudor domain. Proc. Natl. Acad. Sci. U.S.A. 2010;107:18398–18403. doi: 10.1073/pnas.1013106107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo Z., Zheng L., Xu H., Dai H., Zhou M., Pascua M.R., Chen Q.M., Shen B. Methylation of FEN1 suppresses nearby phosphorylation and facilitates PCNA binding. Nat. Chem. Biol. 2010;6:766–773. doi: 10.1038/nchembio.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsu J.M., Chen C.T., Chou C.K., Kuo H.P., Li L.Y., Lin C.Y., Lee H.J., Wang Y.N., Liu M., Liao H.W., et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011;13:174–181. doi: 10.1038/ncb2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakamaki J., Daitoku H., Ueno K., Hagiwara A., Yamagata K., Fukamizu A. Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc. Natl. Acad. Sci. U.S.A. 2011;108:6085–6090. doi: 10.1073/pnas.1015328108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshida K. PKCdelta signaling: mechanisms of DNA damage response and apoptosis. Cell. Signal. 2007;19:892–901. doi: 10.1016/j.cellsig.2007.01.027. [DOI] [PubMed] [Google Scholar]
- 46.Zhao M., Xia L., Chen G.Q. Protein kinase cdelta in apoptosis: a brief overview. Arch. Immunol. Ther. Exp. (Warsz) 2012;60:361–372. doi: 10.1007/s00005-012-0188-8. [DOI] [PubMed] [Google Scholar]
- 47.LaGory E.L., Sitailo L.A., Denning M.F. The protein kinase Cdelta catalytic fragment is critical for maintenance of the G2/M DNA damage checkpoint. J. Biol. Chem. 2010;285:1879–1887. doi: 10.1074/jbc.M109.055392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leitges M., Mayr M., Braun U., Mayr U., Li C., Pfister G., Ghaffari-Tabrizi N., Baier G., Hu Y., Xu Q. Exacerbated vein graft arteriosclerosis in protein kinase Cdelta-null mice. J. Clin. Invest. 2001;108:1505–1512. doi: 10.1172/JCI12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watanabe T., Ono Y., Taniyama Y., Hazama K., Igarashi K., Ogita K., Kikkawa U., Nishizuka Y. Cell division arrest induced by phorbol ester in CHO cells overexpressing protein kinase C-delta subspecies. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10159–10163. doi: 10.1073/pnas.89.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piano F., Schetter A.J., Morton D.G., Gunsalus K.C., Reinke V., Kim S.K., Kemphues K.J. Gene clustering based on RNAi phenotypes of ovary-enriched genes in C. elegans. Curr. Biol. 2002;12:1959–1964. doi: 10.1016/s0960-9822(02)01301-5. [DOI] [PubMed] [Google Scholar]
- 51.Gates J., Thummel C.S. An enhancer trap screen for ecdysone-inducible genes required for Drosophila adult leg morphogenesis. Genetics. 2000;156:1765–1776. doi: 10.1093/genetics/156.4.1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Charroux B., Angelats C., Fasano L., Kerridge S., Vola C. The levels of the bancal product, a Drosophila homologue of vertebrate hnRNP K protein, affect cell proliferation and apoptosis in imaginal disc cells. Mol. Cell. Biol. 1999;19:7846–7856. doi: 10.1128/mcb.19.11.7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeVries T.A., Neville M.C., Reyland M.E. Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J. 2002;21:6050–6060. doi: 10.1093/emboj/cdf606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Porter A.G., Janicke R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 55.Reyland M.E. Protein kinase Cdelta and apoptosis. Biochem. Soc. Trans. 2007;35:1001–1004. doi: 10.1042/BST0351001. [DOI] [PubMed] [Google Scholar]
- 56.Ghayur T., Hugunin M., Talanian R.V., Ratnofsky S., Quinlan C., Emoto Y., Pandey P., Datta R., Huang Y., Kharbanda S., et al. Proteolytic activation of protein kinase C delta by an ICE/CED 3-like protease induces characteristics of apoptosis. J. Exp. Med. 1996;184:2399–2404. doi: 10.1084/jem.184.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Emoto Y., Manome Y., Meinhardt G., Kisaki H., Kharbanda S., Robertson M., Ghayur T., Wong W.W., Kamen R., Weichselbaum R., et al. Proteolytic activation of protein kinase C delta by an ICE-like protease in apoptotic cells. EMBO J. 1995;14:6148–6156. doi: 10.1002/j.1460-2075.1995.tb00305.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Di Lorenzo A., Bedford M.T. Histone arginine methylation. FEBS Lett. 2011;585:2024–2031. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshida K., Liu H., Miki Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J. Biol. Chem. 2006;281:5734–5740. doi: 10.1074/jbc.M512074200. [DOI] [PubMed] [Google Scholar]
- 60.Yoshida K. Nuclear trafficking of pro-apoptotic kinases in response to DNA damage. Trends Mol. Med. 2008;14:305–313. doi: 10.1016/j.molmed.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 61.Yuan Z.M., Utsugisawa T., Ishiko T., Nakada S., Huang Y., Kharbanda S., Weichselbaum R., Kufe D. Activation of protein kinase C delta by the c-Abl tyrosine kinase in response to ionizing radiation. Oncogene. 1998;16:1643–1648. doi: 10.1038/sj.onc.1201698. [DOI] [PubMed] [Google Scholar]
- 62.Bharti A., Kraeft S.K., Gounder M., Pandey P., Jin S., Yuan Z.M., Lees-Miller S.P., Weichselbaum R., Weaver D., Chen L.B., et al. Inactivation of DNA-dependent protein kinase by protein kinase Cdelta: implications for apoptosis. Mol. Cell. Biol. 1998;18:6719–6728. doi: 10.1128/mcb.18.11.6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoshida K., Wang H.G., Miki Y., Kufe D. Protein kinase Cdelta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 2003;22:1431–1441. doi: 10.1093/emboj/cdg134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen L.C., Liu H.P., Li H.P., Hsueh C., Yu J.S., Liang C.L., Chang Y.S. Thymidine phosphorylase mRNA stability and protein levels are increased through ERK-mediated cytoplasmic accumulation of hnRNP K in nasopharyngeal carcinoma cells. Oncogene. 2009;28:1904–1915. doi: 10.1038/onc.2009.55. [DOI] [PubMed] [Google Scholar]
- 65.Melino G., De Laurenzi V., Vousden K.H. p73: friend or foe in tumorigenesis. Nat. Rev. Cancer. 2002;2:605–615. doi: 10.1038/nrc861. [DOI] [PubMed] [Google Scholar]
- 66.Yang A., McKeon F. P63 and P73: P53 mimics, menaces and more. Nat. Rev. Mole. Cell Biol. 2000;1:199–207. doi: 10.1038/35043127. [DOI] [PubMed] [Google Scholar]
- 67.Allocati N., Di Ilio C., De Laurenzi V. p63/p73 in the control of cell cycle and cell death. Exp. Cell Res. 2012;318:1285–1290. doi: 10.1016/j.yexcr.2012.01.023. [DOI] [PubMed] [Google Scholar]
- 68.Pietsch E.C., Sykes S.M., McMahon S.B., Murphy M.E. The p53 family and programmed cell death. Oncogene. 2008;27:6507–6521. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zaika A., Irwin M., Sansome C., Moll U.M. Oncogenes induce and activate endogenous p73 protein. J. Biol. Chem. 2001;276:11310–11316. doi: 10.1074/jbc.M005737200. [DOI] [PubMed] [Google Scholar]
- 70.Gong J.G., Costanzo A., Yang H.Q., Melino G., Kaelin W.G., Jr, Levrero M., Wang J.Y. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature. 1999;399:806–809. doi: 10.1038/21690. [DOI] [PubMed] [Google Scholar]
- 71.Urbani A., Poland J., Bernardini S., Bellincampi L., Biroccio A., Schnolzer M., Sinha P., Federici G. A proteomic investigation into etoposide chemo-resistance of neuroblastoma cell lines. Proteomics. 2005;5:796–804. doi: 10.1002/pmic.200401147. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.