

Abstract
Epithelial-mesenchymal transition (EMT) is associated with characteristics of breast cancer stem cells, including chemoresistance and radioresistance. However, it is unclear whether EMT itself or specific EMT regulators play causal roles in these properties. Here we identify an EMT-inducing transcription factor, zinc finger E-box binding homeobox 1 (ZEB1), as a regulator of radiosensitivity and DNA damage response (DDR). Radioresistant subpopulations of breast cancer cells derived from ionizing radiation exhibit hyperactivation of ATM and upregulation of ZEB1, and ZEB1 promotes tumor cell radioresistance in vitro and in vivo. Mechanistically, ATM kinase phosphorylates and stabilizes ZEB1 in response to DNA damage, and ZEB1 in turn directly interacts with USP7 and enhances its ability to deubiquitinate and stabilize CHK1, thereby promoting homologous recombination-dependent DNA repair and resistance to radiation. These findings identify ZEB1 as an ATM substrate linking ATM to CHK1 and as the mechanism underlying the association between EMT and radioresistance.
Radiation therapy causes cell death by inducing single- and double-strand DNA breaks1, 2. The rationale for treating tumor tissues with radiation without damaging normal tissues is that compared with normal cells, tumor cells are actively dividing and often have defects in DNA damage repair machinery, and thus are less able to repair DNA damage3. A major cause of the failure in radiation treatment is intrinsic and therapy-induced radioresistant tumor cells, which exhibit increased DNA repair ability4.
The DNA damage response (DDR) pathway consists of sensors, transducers and effectors5, 6. In response to genotoxic damage, the RAD9-HUS1-RAD1 (9-1-1) complex is recruited to the DNA damage sites by a RAD17-containing protein complex and then facilitates ATR-mediated phosphorylation and activation of CHK1, an effector protein kinase that regulates S phase progression and G2/M cell cycle arrest5–7. Another sensor complex, the MRE11-RAD50-NBS1 (MRN) complex, detects double-strand breaks (DSBs), recruits ATM and promotes ATM-mediated phosphorylation of histone H2AX (γH2AX) surrounding the DNA breaks8, 9. Subsequently, a number of signaling and repair proteins accumulate at DNA lesions and form discrete foci10–12.
Recently, cancer stem cells (CSCs) have been shown to promote radioresistance through activation of DDR13, 14. Moreover, the epithelial-mesenchymal transition (EMT) trans-differentiation program can generate cells with stem-like properties15. EMT can be induced by various transcription factors, including Twist, Snail, Slug, ZEB1 and ZEB216, 17. However, it is unclear whether EMT itself or specific EMT regulators cause CSC-associated properties such as chemoresistance and radioresistance.
In the present study, we found that the EMT regulator ZEB1 promotes DDR and tumor radioresistance. This regulation is initiated by phosphorylation and stabilization of ZEB1 by ATM and is mediated by stabilization of CHK1 by a ZEB1-interacting deubiquitinase, USP7.
RESULTS
ZEB1 underlies the association between EMT and radioresistance
To examine the association between EMT and radioresistance, we overexpressed Snail, Twist or ZEB1 in the experimentally immortalized, non-transformed human mammary epithelial cells18, termed HMLE cells. Each of these transcription factors induced EMT – as evidenced by changes in morphology (Supplementary Fig. 1a), downregulation of E-cadherin and upregulation of Vimentin (Fig. 1a), and increased clonogenic survival upon irradiation (Fig. 1b and Supplementary Fig. 1b). In each case, expression of Snail, Twist and ZEB1 was upregulated; in particular, overexpression of either Snail or Twist increased ZEB1 expression to the level as high as that of ZEB1-overexpressing cells (Fig. 1a). Next, we silenced each of the three transcription factors in HMLE cells overexpressing Snail, Twist or ZEB1, which did not cause reversal of EMT (Fig. 1c). Notably, only knockdown of ZEB1 reduced radioresistance (Fig. 1d), suggesting that ZEB1 underlies the association between EMT and radioresistance. Consistent with this notion, we observed upregulation of ZEB1 in the survival fraction of mock-infected HMLE cells (Supplementary Fig. 1c); moreover, the survival fraction of ZEB1-depleted HMLE cells re-expressed ZEB1 (Supplementary Fig. 1c).
Figure 1. ZEB1 confers radioresistance on mammary epithelial cells.

(a) Immunoblotting of E-cadherin, Vimentin, Snail, Twist, ZEB1 and GAPDH in HMLE cells transduced with Snail, Twist or ZEB1.
(b) Clonogenic survival assays of HMLE cells transduced with Snail, Twist or ZEB1. n = 3 wells per group.
(c) Immunoblotting of Snail, Twist, ZEB1, E-cadherin, Vimentin and GAPDH in HMLE cells transduced with Snail, Twist or ZEB1 alone or in combination with the siRNA targeting Snail, Twist or ZEB1.
(d) Clonogenic survival assays of HMLE cells transduced with Snail, Twist or ZEB1 alone or in combination with the siRNA targeting Snail, Twist or ZEB1. n = 3 wells per group.
(e) Immunoblotting of Snail, Twist, ZEB1, E-cadherin, Vimentin and GAPDH in MCF7 cells transduced with Snail, Twist or ZEB1.
(f) Clonogenic survival assays of MCF7 cells transduced with Snail, Twist or ZEB1. n = 3 wells per group.
Data in b, d and f are the mean of biological replicates from a representative experiment, and error bars indicate s.e.m. Statistical significance was determined by a two-tailed, unpaired Student’s t-test. The experiments were repeated 3 times. The source data can be found in Supplementary Table 3. Uncropped images of blots are shown in Supplementary Figure 7.
We then overexpressed these three transcription factors in the MCF7 human breast cancer cell line. Unlike HMLE cells, MCF7 cells express intact p53 which acts as a barrier to EMT induction19, 20. Indeed, none of these three transcription factors induced EMT in MCF7 cells (Fig. 1e and data not shown). Moreover, only ZEB1, but not Snail or Twist, conferred radioresistance on these cells (Fig. 1f). Taken together, it may not be EMT itself that causes radioresistance; instead, it is a specific EMT regulator, ZEB1, that plays a causal role in regulating the response to radiation.
ZEB1 is upregulated in radioresistant cancer cells and promotes tumor radioresistance
To determine whether ZEB1 is indeed upregulated in radioresistant tumor cells, we employed γ-ionizing radiation (IR) to select the radioresistant subpopulation from the SUM159 human breast cancer cells which express moderate levels of ZEB1. After a 6 Gray (Gy) dose, survived cells formed colonies. We pooled the colonies and repeated the dose one more time (Fig. 2a). Cells derived from this selection, named SUM159-P2 cells, displayed increased clonogenic survival upon irradiation compared with the parental SUM159 cells (SUM159-P0) (Fig. 2b). Irradiation causes DSBs resulting in the formation of γH2AX foci, and the persistence of γH2AX foci marks delayed repair and correlates with radiosensitivity21–23. At 24 hours after irradiation, γH2AX remained in SUM159-P0 cells but disappeared in SUM159-P2 cells (Fig. 2c), indicating that this radioresistant subline has enhanced clearance of DNA breaks.
Figure 2. ZEB1 is upregulated in radioresistant cancer cells and promotes tumor radioresistance.

(a) Schematic representation of generation of a radioresistant subline (SUM159-P2) from parental SUM159 cells (SUM159-P0).
(b) Clonogenic survival assays of SUM159-P0 and SUM159-P2 cells. n = 3 wells per group.
(c) Immunoblotting of γH2AX and HSP90 in SUM159-P0 and SUM159-P2 cells treated with 6 Gy IR.
(d) Immunoblotting of Snail, Twist, ZEB1 and GAPDH in SUM159-P0 and SUM159-P2 cells. SUM159-P0 cells transfected with Snail or Twist were used as positive controls.
(e) Immunoblotting of Snail, Twist, ZEB1 and GAPDH in SUM159-P0 cells transduced with Snail, Twist or ZEB1.
(f) Clonogenic survival assays of SUM159-P0 cells transduced with Snail, Twist or ZEB1. n = 3 wells per group.
(g) Clonogenic survival assays of SUM159-P2 cells transduced with ZEB1 shRNA (sh-ZEB1). Inset: immunoblotting of ZEB1 and GAPDH. n = 3 wells per group.
(h, i) Tumor size of mice bearing control (scramble) or ZEB1 shRNA-transduced SUM159-P2 xenografts. Tumors were locally irradiated with 15 Gy single dose (h) or 2 Gy fractionated dose (XRT) twice per day for 7 consecutive days (i). n = 5 mice per group. General linear model multivariate analysis was performed to determine statistical significance.
(j) Immunoblotting of ZEB1 and HSP90 in tumor lysates.
Data in b, f, g, h and i are the mean of biological replicates from a representative experiment, and error bars indicate s.e.m. Statistical significance in b, f and g was determined by a two-tailed, unpaired Student’s t-test. The experiments were repeated 3 times. The source data can be found in Supplementary Table 3. Uncropped images of blots are shown in Supplementary Figure 7.
Next, we examined the protein levels of Snail, Twist and ZEB1. Only one factor, ZEB1, was significantly upregulated in SUM159-P2 cells (Fig. 2d). On the other hand, ZEB1 mRNA levels showed no increase (Supplementary Fig. 2a), suggesting that the observed ZEB1 upregulation was due to post-transcriptional or post-translational regulation.
We overexpressed Snail, Twist or ZEB1 in parental SUM159 cells (Fig. 2e) and found that ZEB1 was much more powerful than Snail and Twist in promoting radioresistance (Fig. 2f). Since ZEB1 was upregulated in SUM159-P2 cells, we silenced its expression, which markedly inhibited clonogenic survival at 6 Gy and higher doses (Fig. 2g). In addition, knockdown of ZEB1 rendered the U2OS human osteosarcoma cells more sensitive to IR (Supplementary Fig. 2b). In each case, the EMT status was not altered, further confirming that it is ZEB1 rather than EMT itself that causes radioresistance.
We validated our findings in mice bearing SUM159-P2 xenograft tumors. When the tumor diameter reached 8 mm, we locally irradiated the tumor with 15 Gy single dose or 2 Gy fractionated dose twice a day for 7 consecutive days. Knockdown of ZEB1 had no effect on tumor growth without irradiation. In contrast, treatment with either 15 Gy single dose or 2 Gy dose twice daily for 7 days led to sustained growth inhibition of tumors formed by ZEB1-depleted cells, whereas tumors formed by the control cells showed a short initial response and then re-grew at a rate similar to the non-irradiated tumors (Fig. 2h, i). The knockdown effect of ZEB1 shRNA was retained throughout this tumor radiosensitivity study (Fig. 2j). Taken together, ZEB1 is required for the radioresistance of these breast cancer cells in vitro and in vivo.
ZEB1 regulates DNA damage repair
After IR treatment, γH2AX foci persist longer in radiosensitive cell lines than in radioresistant lines23. In ZEB1 shRNA-expressing SUM159-P2 cells but not cells infected with a scrambled control, we observed persistence of γH2AX foci 24 hours after IR (Fig. 3a, b), indicating that ZEB1-depleted cells were less able to repair DNA lesions. To directly gauge damaged DNA, we performed a comet assay to detect both single- and double-strand DNA breaks. 24 hours after IR, ZEB1-depleted SUM159-P2 cells exhibited a 4.5-fold increase in the comet ‘tail moment’ (= percentage of the DNA in the tail × length of the tail in μm) – a previously described measure of DNA damage24, compared with the control cells (Fig. 3c, d).
Figure 3. ZEB1 regulates DNA damage repair.

(a) γH2AX and DAPI staining of SUM159-P2 cells transduced with ZEB1 shRNA, 24 hr after 6 Gy IR. Scale bar: 10 μm.
(b) Immunoblotting of ZEB1, γH2AX, H2AX and GAPDH in SUM159-P2 cells transduced with ZEB1 shRNA, at the indicated time points after 6 Gy IR.
(c, d) Images (c) and data quantification (d) of comet assays of SUM159-P2 cells transduced with ZEB1 shRNA, at the indicated time points after 6 Gy IR. n = 62 cells per group. Scale bar in (c): 50 μm.
(e) Immunoblotting of ZEB1 and GAPDH in U2OS_DR-GFP cells transfected with ZEB1 siRNA alone or in combination with ZEB1.
(f) HR repair assays of U2OS_DR-GFP cells transfected with ZEB1 siRNA alone or in combination with ZEB1. n = 3 wells per group.
Data in d and f are the mean of biological replicates from a representative experiment, and error bars indicate s.e.m. Statistical significance was determined by a two-tailed, unpaired Student’s t-test. The experiments were repeated 3 times. The source data can be found in Supplementary Table 3. Uncropped images of blots are shown in Supplementary Figure 7.
Our results demonstrate that ZEB1 is required for DSB clearance. In mammalian cells, a key conserved pathway involved in DSB repair is the homologous recombination (HR) pathway25. To determine the effect of ZEB1 on HR repair, we utilized a U2OS cell clone with chromosomal integration of an HR repair reporter consisting of two differentially mutated GFP genes (SceGFP and iGFP) oriented as direct repeats (DR-GFP); in this assay, expression of I-SceI endonuclease generates a site-specific DSB in the SceGFP coding region, and when this DSB is repaired by HR, the expression of GFP is restored and can be analyzed by flow cytometry to gauge the efficiency of HR repair26, 27. We found that upon I-SceI expression, ZEB1-depleted U2OS cells displayed a significant decrease (~50%) in the percentage of GFP-positive cells, indicating defective HR repair (Fig. 3e, f). Moreover, re-expression of ZEB1 in ZEB1 siRNA-expressing U2OS cells restored HR-based repair (Fig. 3e, f). Collectively, these results suggest that ZEB1 is required for HR-mediated DNA damage repair and the clearance of DNA breaks.
ZEB1 regulates radiosensitivity through USP7-mediated stabilization of CHK1
We reasoned that ZEB1 regulates radiosensitivity by modulating DDR pathways. CHK1 and CHK2 are two critical effector kinases in DDR and checkpoint control28–30, which prompted us to examine their status in ZEB1-depleted breast cancer cells. Interestingly, knockdown of ZEB1 in SUM159-P2 cells resulted in a significant reduction in CHK1 protein levels in the presence or absence of IR (Fig. 4a); in contrast, neither CHK2 total protein level nor its phosphorylation was affected (Fig. 4a). Moreover, expression of an RNAi-resistant ZEB1 mutant completely reversed the effect of ZEB1 shRNA on CHK1, γH2AX and clonogenic survival (Supplementary Fig. 3a, b). Conversely, overexpression ZEB1 in MCF7 cells significantly upregulated CHK1 and promoted the clearance of DNA breaks post-IR (gauged by γH2AX) (Fig. 4b). In addition, Zeb1-deficient mouse embryonic fibroblasts (MEFs) displayed downregulation of Chk1 (Fig. 4c).
Figure 4. CHK1 mediates ZEB1 regulation of radiosensitivity.

(a) Immunoblotting of p-CHK1, CHK1, p-CHK2, CHK2 and GAPDH in SUM159-P2 cells transduced with ZEB1 shRNA, at the indicated time points after 6 Gy IR.
(b) Immunoblotting of ZEB1, CHK1, γH2AX, H2AX and GAPDH in MCF7 cells transduced with ZEB1, at the indicated time points after 6 Gy IR.
(c) Immunoblotting of Zeb1, Chk1 and Gapdh in Zeb1+/+, Zeb1+/− and Zeb1−/− MEFs.
(d) Immunoblotting of ZEB1, CHK1, Cyclin A, p-H3 (S10) and GAPDH in SUM159-P2 cells transfected with ZEB1 siRNA or the scramble control. Cells were arrested overnight with 0.5 μg/ml nocodazole. Mitotic cells were “shaken off” and then released into normal medium. Samples were collected at the indicated time points and analyzed by western blotting. Cell cycle distribution was gauged by Cyclin A and p-H3 (S10).
(e) Clonogenic survival assays of SUM159-P2 cells transfected with CHK1 siRNA. Inset: immunoblotting of CHK1 and GAPDH. n = 3 wells per group.
(f) Immunoblotting of CHK1 and GAPDH in ZEB1 shRNA-transduced SUM159-P2 cells with or without ectopic expression of CHK1.
(g) Clonogenic survival assays of ZEB1 shRNA-transduced SUM159-P2 cells with or without ectopic expression of CHK1. n = 3 wells per group.
(h) Immunoblotting of ZEB1, CHK1 and GAPDH in SUM159-P0 cells transfected with ZEB1 alone or in combination with CHK1 siRNA.
(i) Clonogenic survival assays of SUM159-P0 cells transfected with ZEB1 alone or in combination with CHK1 siRNA. n = 3 wells per group.
Data in e, g and i are the mean of biological replicates from a representative experiment, and error bars indicate s.e.m. Statistical significance was determined by a two-tailed, unpaired Student’s t-test. The experiments were repeated 3 times. The source data can be found in Supplementary Table 3. Uncropped images of blots are shown in Supplementary Figure 7.
CHK1 activates the G2 checkpoint in response to stalled replication forks or DNA damage31. As anticipated, irradiation resulted in the arrest of SUM159-P2 cells in the G2/M phase, and knockdown of ZEB1 led to a moderate but significant decrease in the G2/M population (Supplementary Fig. 3c). CHK1 levels are known to vary depending on the cell cycle phase32. To exclude the indirect effect due to the difference in cell cycle, we synchronized scramble-transfected or ZEB1 siRNA-transfected SUM159-P2 cells in the G2/M phase by nocodazole treatment and released the cells at different time points. As expected, CHK1 was detected in S and G2/M phases (Fig. 4d). We found that in these synchronized cells, CHK1 levels correlated with ZEB1 levels, and that knockdown of ZEB1 led to downregulation of CHK1 at each cell cycle stage (Fig. 4d), which suggested that the downregulation of CHK1 caused by ZEB1 depletion is not an indirect effect of cell cycle changes.
We assessed the effect of CHK1 on radioresistance. Silencing CHK1 expression recapitulated the effect of ZEB1 shRNA on sensitizing SUM159-P2 cells to IR (Fig. 4e). Conversely, re-expression of CHK1 in ZEB1-depleted SUM159-P2 cells rescued radioresistance (Fig. 4f, g). Moreover, knockdown of CHK1 reversed ZEB1-induced radioresistance in SUM159-P0 cells (Fig. 4h, i). These data suggest that ZEB1 regulates tumor cell radioresistance through, at least in part, CHK1.
Because depletion of ZEB1 downregulated CHK1 protein (Fig. 4a) but not CHK1 mRNA (Supplementary Fig. 4a), and because CHK1 is subject to ubiquitin-dependent degradation following replication stress33–35, we reasoned that ZEB1 may regulate CHK1 protein levels through ubiquitin-dependent mechanisms. Indeed, knockdown of ZEB1 significantly induced the poly-ubiquitination of endogenous CHK1 protein with or without IR (Fig. 5a).
Figure 5. ZEB1 interacts with USP7 which deubiquitinates and stabilizes CHK1.

(a) SUM159-P2 cells transduced with ZEB1 shRNA were treated with 10 μM MG132, irradiated with 6 Gy IR and harvested 6 hr later. Lysates were immunoprecipitated with the CHK1 antibody and immunoblotted with antibodies indicated.
(b) A partial list of ZEB1-associated proteins.
(c, d) 293T cells were transfected with SFB-ZEB1 (c) or SFB-USP7 (d), followed by pull-down with streptavidin-sepharose beads (s-s beads) and immunoblotting with antibodies indicated.
(e) Top: bacterially purified GST-USP7 was incubated with amylose resin conjugated with bacterially expressed MBP-GFP or MBP-ZEB1. Proteins retained on the amylose resin were immunoblotted with the USP7 antibody. Bottom: bacterially purified recombinant proteins were analyzed by SDS-PAGE and Coomassie blue staining. * indicates the predicted position.
(f) 293T cells were transfected with SFB-USP7 and treated with cycloheximide (CHX). Cells were harvested at different time points and immunoblotted with antibodies indicated.
(g, h) SUM159-P2 cells were transfected with USP7 siRNA (si-USP7, g) or transduced with ZEB1 shRNA (sh-ZEB1, h), and treated with cycloheximide. Cells were harvested at different time points and immunoblotted with antibodies indicated.
(i) HA-ubiquitin was co-transfected with SFB-GFP or SFB-USP7 into 293T cells. Lysates from cells with or without 6 Gy IR treatment were immunoprecipitated with the CHK1 antibody and immunoblotted with the HA antibody. Cells were treated with MG132 (10 μM) for 6 hr before harvest.
(j) Top: ubiquitinated CHK1 was incubated with SFB-GFP control or SFB-USP7 purified with streptavidin-sepharose beads from 293T cells with or without ZEB1 co-transfection. The reaction mixture was then immunoprecipitated with the FLAG antibody and immunoblotted with the CHK1 antibody. Bottom: purified SFB-USP7 was immunoblotted with antibodies to ZEB1 and USP7.
(k) Clonogenic survival assays of USP7 siRNA-transfected SUM159-P2 cells. n = 3 wells per group.
Data in k are the mean of biological replicates from a representative experiment, and error bars indicate s.e.m. Statistical significance was determined by a two-tailed, unpaired Student’s t-test. The experiments were repeated 3 times. The source data can be found in Supplementary Table 3. Uncropped images of blots are shown in Supplementary Figure 7.
To further investigate the mechanism by which ZEB1 regulates CHK1 ubiquitination, we attempted to identify ZEB1-interacting proteins using a triple-epitope (S-protein, FLAG tag and streptavidin-binding peptide)-tagged version of ZEB1 (SFB-ZEB1). Tandem affinity purification using streptavidin-sepharose beads (s-s beads) and S-protein-agarose beads followed by mass spectrometric analysis identified several reported ZEB1 interactors including CTBP2, CTBP1 and SIRT136–38, as well as a previously undescribed ZEB1 interactor, USP7 (Supplementary Table 1 and Fig. 5b).
USP7 is a deubiquitinating enzyme with several reported substrates, such as p5339, Mdm240, 41, HLTF42, PTEN43 and Claspin44. Co-immunoprecipitation assays confirmed that both USP7 and CHK1 could be detected in ZEB1 immunoprecipitates (Fig. 5c), and that both ZEB1 and CHK1 were present in USP7 immunoprecipitates (Fig. 5d). Moreover, purified GST-USP7 could bind to purified MBP-tagged ZEB1 under cell-free conditions (Fig. 5e), demonstrating direct interaction between ZEB1 and USP7.
To investigate whether USP7 regulates the stability of CHK1 protein, we examined CHK1 proteins levels in the presence of cycloheximide (CHX), an inhibitor of translation. Notably, overexpression of USP7 in 293T cells led to a pronounced increase in CHK1 protein stability (Fig. 5f and Supplementary Fig. 4b). Conversely, knockdown of USP7 in SUM159-P2 cells reduced CHK1 stability (Fig. 5g and Supplementary Fig. 4c) but not ZEB1 stability (Supplementary Fig. 4d). Interestingly, knockdown of ZEB1 in SUM159-P2 cells destabilized CHK1, but not other USP7 substrates such as HLTF, p53 or Claspin (Fig. 5h and Supplementary Fig. 4e, f).
Consistent with stabilization of CHK1, overexpression of USP7 markedly reduced the poly-ubiquitination level of CHK1 in 293T cells (Fig. 5i). To directly examine the deubiquitinating activity of USP7 toward CHK1, we purified USP7 and ubiquitinated CHK1 and then incubated them in a cell-free system. USP7 purified from 293T cells transfected with USP7 alone decreased CHK1 poly-ubiquitination by 25% in vitro, and USP7 and ZEB1 co-purified from 293T cells with co-transfection of USP7 and ZEB1 reduced CHK1 poly-ubiquitination by 43% (Fig. 5j). Similar to the knockdown effect of ZEB1 and CHK1, depletion of USP7 also radiosensitized SUM159-P2 cells (Fig. 5k). We conclude from these experiments that CHK1 is a USP7 substrate, and that ZEB1 directly interacts with USP7 and enhances its ability to deubiquitinate and stabilize CHK1, which in turn promotes radioresistance.
To further understand why ZEB1 regulates the stability of CHK1 but not the stability of other USP7 substrates (Fig. 5h and Supplementary Fig. 4f), we examined the effect of ZEB1 on the interaction between USP7 and its various substrates. As expected, CHK1, HLTF, p53 and Claspin could be detected in USP7 immunoprecipitates (Fig. 6a and Supplementary Fig. 4g). Interestingly, ectopic expression of ZEB1 markedly enhanced the interaction of USP7 with CHK1, but not its association with HLTF, p53 or Claspin (Fig. 6a and Supplementary Fig. 4g). Conversely, knockdown of ZEB1 dramatically decreased the interaction between USP7 (either overexpressed or endogenous) and CHK1, but not HLTF and p53 (Fig. 6b, c). Therefore, ZEB1 specifically promotes the interaction between USP7 and CHK1.
Figure 6. ZEB1 specifically promotes the interaction between USP7 and CHK1.
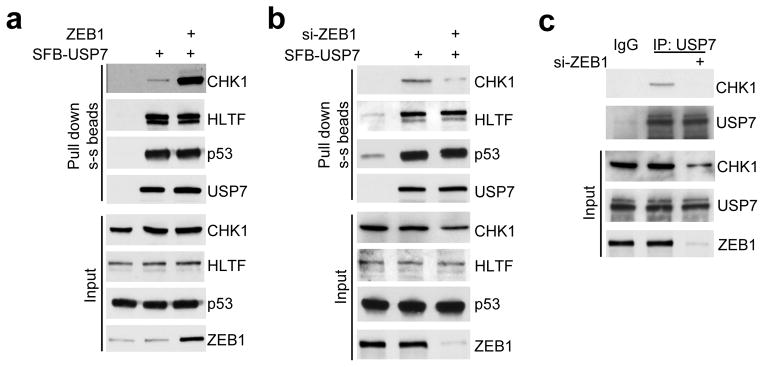
(a) 293T cells were transfected with SFB-USP7 alone or in combination with ZEB1, followed by pull-down with streptavidin-sepharose beads and immunoblotting with antibodies to CHK1, HLTF, p53 and USP7.
(b) 293T cells were transfected with ZEB1 siRNA alone or in combination with SFB-USP7, followed by pull-down with streptavidin-sepharose beads and immunoblotting with antibodies to CHK1, HLTF, p53 and USP7.
(c) SUM159-P2 cells were transfected with ZEB1 siRNA, followed by immunoprecipitation with the USP7 antibody and immunoblotting with antibodies to CHK1 and USP7. Uncropped images of blots are shown in Supplementary Figure 7.
ZEB1 is phosphorylated and stabilized by ATM
We sought to determine the mechanism by which ZEB1 protein is upregulated in radioresistant cells derived from irradiation. A central component in the DNA repair pathway is ATM45: upon exposure to IR, ATM kinase is rapidly activated, leading to phosphorylation of a number of key players in DDR, cell cycle arrest and apoptosis, such as γH2AX8, CHK246, BRCA147 and p5348, 49.
We asked whether ZEB1 is regulated by ATM. Co-immunoprecipitation revealed physical interaction of ZEB1 with ATM (Fig. 7a), whereas ATR showed no association with ZEB1 (Supplementary Fig. 5a). Moreover, depletion of ATM in SUM159-P2 cells significantly downregulated ZEB1 and CHK1 proteins (Fig. 7b); in contrast, neither knockdown of ATR or treatment with the ATR inhibitor ETP-4646450 affected ZEB1 protein levels (Supplementary Fig. 5b, c). ATM substrates have a common S/T-Q motif. Analysis of the ZEB1 protein sequence revealed one evolutionarily conserved S/T-Q motif encompassing serine 585. An inhibitor of ATM kinase, Ku55933, significantly decreased the stability of ZEB1 and reduced S/T-Q phosphorylation of ZEB1, as gauged by a phospho-S/TQ (p-S/TQ) antibody (Fig. 7c). Moreover, this phospho-S/TQ antibody detected much higher signals in ZEB1 purified from irradiated 293T cells than that purified from non-irradiated cells (Fig. 7d). Consistently, S/T-Q phosphorylation of endogenous ZEB1 was significantly upregulated in SUM159-P2 cells which displayed much higher levels of ATM phosphorylation than SUM159-P0 cells (Fig. 7e).
Figure 7. ATM phosphorylates and stabilizes ZEB1.

(a) 293T cells were transfected with SFB-ZEB1 and treated with IR, followed by pull-down with streptavidin-sepharose beads and immunoblotting with antibodies to ATM and FLAG.
(b) SUM159-P2 cells were transduced with ATM shRNA and treated with IR. Lysates were immunoblotted with antibodies to p-ATM, ATM, ZEB1, CHK1 and GAPDH.
(c) SUM159-P2 cells with or without Ku55933 pretreatment (10 μM, 1 hr) were treated with IR (6 Gy) and CHX (50 μg/ml), harvested at different time points, immunoprecipitated with the ZEB1 antibody and immunoblotted with antibodies to p-S/TQ and ZEB1.
(d) 293T cells were transfected with SFB-ZEB1 and treated with IR, followed by pull-down with streptavidin-sepharose beads and immunoblotting with antibodies to p-S/TQ and ZEB1.
(e) Endogenous ZEB1 was immunoprecipitated from SUM159-P0 and SUM159-P2 cells and immunoblotted with antibodies to p-S/TQ and ZEB1.
(f) Consensus ATM phosphorylation site on human ZEB1 (S585) and alignment with the conserved site on mouse, rat and Xenopus Zeb1.
(g) 293T cells were transfected with wild-type, the S585A or S585D mutant of SFB-ZEB1 and treated with IR, followed by pull-down with streptavidin-sepharose beads and immunoblotting with antibodies to p-S/TQ and ZEB1.
(h) Immunopurified wild-type ZEB1 or the S585A mutant was incubated with immunopurified wild-type ATM or the kinase-dead (KD) mutant in kinase buffer containing 32P-ATP. After reaction, proteins were resolved by SDS-PAGE and subjected to autoradiography and immunoblotting with antibodies to ZEB1 and p-ATM. Purified GST-p53 was used as a positive control for ATM kinase activity.
(i) HeLa cells were co-transfected with SFB-GFP and wild-type, the S585A or S585D mutant of SFB-ZEB1, treated with CHX with or without IR, harvested at different time points and immunoblotted with antibodies to FLAG. SFB-GFP serves as the control for transfection.
(j) Clonogenic survival assays of SUM159-P0 cells transfected with wild-type ZEB1 or the mutant. n = 3 wells per group.
Data in j are the mean of biological replicates from a representative experiment, and error bars indicate s.e.m. Statistical significance was determined by a two-tailed, unpaired Student’s t-test. The experiments were repeated 3 times. The source data can be found in Supplementary Table 3. Uncropped images of blots are shown in Supplementary Figure 7.
Substitution of serine 585 with either alanine (S585A) or aspartic acid (S585D) resulted in a 70–80% decrease in S/T-Q phosphorylation of ZEB1 in irradiated 293T cells (Fig. 7f, g), suggesting that this serine residue accounts for the majority of ZEB1 S/T-Q phosphorylation in cells with activated ATM. In order to determine whether ZEB1 is a direct substrate of ATM, we purified ZEB1 and ATM and then performed in vitro kinase assays. As a positive control, the known ATM substrate p53 was phosphorylated by wild-type ATM, but not the kinase-dead mutant48, 49 (Fig. 7h). Notably, ATM exhibited robust kinase activity toward wild-type ZEB1, whereas the phosphorylation of the S585A mutant was reduced by 60% (Fig. 7h), which suggested that ATM can directly phosphorylate ZEB1 at S585, but other phosphorylation sites may also exist.
To determine whether ATM can stabilize ZEB1 through phosphorylating it at S585, we compared wild-type ZEB1 with the phosphodeficient (S585A) and phosphomimetic (S585D) mutants. Mutation at S585 did not alter the physical association between ZEB1 and USP7 (Supplementary Fig. 6a) but did affect ZEB1 protein stability: in the absence of IR, the stability of wild-type ZEB1 was much higher than that of the S585A mutant but much lower than that of the S585D mutant (Fig. 7i and Supplementary Fig. 6b); in the presence of IR, the stability of wild-type ZEB1 was markedly increased to the level as high as that of the S585D mutant, whereas the S585A mutant was much less stable (Fig. 7i and Supplementary Fig. 6c). Therefore, ATM-dependent phosphorylation of ZEB1 at S585 is crucial for IR-induced stabilization of ZEB1 but not the interaction between ZEB1 and USP7. This reveals the underlying mechanism by which ZEB1 protein is upregulated in radioresistant breast cancer cells with hyperactivation of ATM. Finally, in SUM159-P0 cells, the S585A mutant was less able to promote radioresistance than wild-type ZEB1 or the S585D mutant (Fig. 7j), suggesting that ATM-dependent phosphorylation of ZEB1 is important for the regulation of radiation response.
ZEB1 correlates with CHK1 protein levels and poor clinical outcome in human breast cancer
To validate the association between CHK1 and ZEB1 in breast cancer patients, we performed immunohistochemical staining of these two proteins (Fig. 8a) on the breast cancer progression tissue microarrays from the National Cancer Institute51. A highly significant positive correlation (R = 0.43, P < 1 × 10−6) between CHK1 and ZEB1 was observed in these breast carcinomas, in which 69% (89 of 129) of the tumors with high ZEB1 expression exhibited high CHK1 expression, and 77% (47 of 61) of the tumors with low ZEB1 expression showed low CHK1 expression (Fig. 8b).
Figure 8. ZEB1 correlates with CHK1 protein levels and poor clinical outcome in human breast cancer.

(a) Immunohistochemical staining of ZEB1 and CHK1 in representative carcinoma specimens on the NCI breast cancer progression tissue microarrays. Brown staining indicates positive immunoreactivity. Scale bar: 50 μm.
(b) Correlation between ZEB1 and CHK1 protein levels in human breast tumors. Statistical significance was determined by a χ2 test. R is the correlation coefficient.
(c) Kaplan-Meier curves showing the distant relapse-free survival of patients with high or low expression of ZEB1 in their breast tumors. Statistical significance was determined by a log-rank test.
(d) The working model of regulation of radiosensitivity and DDR by ZEB1.
Tumor cells with therapy resistance including radioresistance are likely to be a source of tumor recurrence and metastatic relapse52. To determine the correlation of ZEB1 expression with clinical outcome, we analyzed a cohort of human breast cancer patients in which transcriptomic profiling was obtained from 286 tumor samples; 87% of these patients received radiotherapy53. This analysis revealed that patients with high ZEB1 expression levels (defined as the top 5%) in their tumors had much worse distant relapse-free survival than those with low ZEB1 expression levels (defined as the bottom 5%; P = 0.02, Fig. 8c). Collectively, these data suggest that upregulation of ZEB1 may contribute to overexpression of CHK1 in human breast tumors, which may lead to radioresistance and eventually metastatic relapse.
DISCUSSION
Radiation therapy plays an important role in breast cancer management, and one of the major barriers in curing breast cancer is the intrinsic and therapy-induced radioresistant behavior of tumor cells4. Combining chemotherapy with radiation improves outcomes in many cases, but this strategy also increases toxicity54. To overcome this obstacle, it is important to identify the critical determinants of radioresistance and to develop safe and effective tumor radiosensitizers.
Recently, a growing body of evidence implicated EMT and cancer stem cells in the acquisition of radioresistance and drug resistance13, 14, 55. Here we identified ZEB1 as an ATM substrate and the mechanism underlying the association between EMT and radioresistance (Fig. 8d): in response to IR, ATM kinase is activated, which phosphorylates and stabilizes ZEB1; ZEB1 in turn interacts with and promotes the activity of USP7, which deubiquitinates and stabilizes CHK1.
Cul1- and Cul4-containing E3 ubiquitin ligase complexes target CHK1 for poly-ubiquitination and degradation during periods of replicative and genotoxic stress34, 35. However, whether this ubiquitination is reversible and can be antagonized by deubiquitinases remains elusive. In this study, we identified CHK1 as a substrate of a ZEB1-associated deubiquitinating enzyme, USP7. How exactly ZEB1 specifically promotes the interaction of USP7 with CHK1 but not with other USP7 substrates warrants future investigation.
ATM kinase is constitutively activated in radioresistant breast cancer cells (Fig. 7b, e), which could explain upregulation of ZEB1 protein in these cells. It should be noted that checkpoint activation and DNA repair normally occur within minutes or hours after DNA damage56, whereas the half-life of ZEB1 protein is approximately 24 hours (Fig. 7i and Supplementary Fig. 6b, c). Therefore, ATM-mediated stabilization of ZEB1 may not play a major role in the acute response to IR, but instead is important for the enhanced DNA repair ability of radioresistant tumor cells with hyperactivated ATM.
Overexpression of ZEB1 has been observed in human breast tumors57, 58 and other cancer types59, 60. Our findings raise the caution that radiation treatment may lead to upregulation of ZEB1 and therapy-induced radioresistance. Because depletion of ZEB1 can radiosensitize breast cancer cells in vitro and in vivo, we envision that ZEB1-targeting agents have the potential to be used as tumor radiosensitizers. Moreover, various CHK1 inhibitors are being tested in anti-cancer clinical trials61, which warrant investigation as candidate radiosensitizing agents for breast tumors with high levels of ZEB1.
METHODS
Cell culture
Mouse embryo fibroblasts were isolated from Zeb1-deficient embryos, genotyped and cultured as previously described62. The 293T, MCF7 and HeLa cell lines were from ATCC and cultured under conditions specified by the manufacturer. The SUM159 cell line was from S. Ethier and cultured as described (http://www.asterand.com/Asterand/human_tissues/159PT.htm). The HMLE cell line was from R. A. Weinberg’s lab stock and cultured in complete Mammary Epithelial Cell Growth Medium (MEGM from Lonza). The DR-GFP-expressing U2OS cell line was from Maria Jasin and cultured in RPMI 1640 medium supplemented with 10% FBS and 1% penicillin and streptomycin.
Plasmids and shRNA
The Snail, Twist and ZEB1 expression constructs were from R. A. Weinberg. Wild-type ATM and the kinase-dead mutant constructs were described previously48. The following shRNA and ORF clones were from Open Biosystems through MD Anderson’s shRNA and ORFeome Core: human ZEB1 shRNA, V3LHS-356186 (5′-AGATTTACTGTGCTGTCCT-3′); human ATM shRNA, V3LHS-350469 (5′-TCAAGAACACCACTTCGCT-3′) and V3LHS-350471 (5′-AGTTTTACAAACATCTTGG-3′); human CHK1 ORF, PLOHS-100005537; human USP7 ORF, PLOHS-100066416. The ZEB1 and USP7 ORFs were subcloned into the pBabe-SFB vector using the Gateway system (Invitrogen). The RNAi-resistant ZEB1 mutant (ZEB1-RE) was generated using a QuikChange Site-Directed Mutagenesis Kit (Stratagene). The vectors used in this study are listed in Supplementary Table 2.
siRNA oligonucleotides
The following siRNA oligonucleotides were purchased from Sigma: CHK1 siRNA, SASI_Hs02_00326305 (5′-GGAGAGAAGGCAAUAUCCAdTdT-3′); Snail siRNA, SASI_Hs01_00039785 (5′-GGACUGUACGCUAUTUGCAdTdT-3′); Twist siRNA, SASI_Mm01_00043024 (5′-GGUCACUAGCCAAUCGCCAdTdT-3′). The on-target plus siRNA that targets ZEB1 was purchased from Dharmacon (J-006564-10-0005, 5′-CUGUAAGAGAGAAGCGGAA-3′). Cells were transfected with 150 nM of the indicated oligonucleotide using the Oligofectamine reagent (Invitrogen). 48 hr after siRNA transfection, cells were used for functional assays, and the remaining cells were harvested for Western blot analysis.
RNA isolation and real-time RT-PCR
Total RNA was isolated using the mirVana RNA Isolation Kit (Ambion) and was then reverse transcribed with an iScript cDNA Synthesis Kit (Bio-Rad). The resulting cDNA was used for qPCR using the TaqMan Gene Expression Assays (Applied Biosystems), and data were normalized to an endogenous control GAPDH. Real-time PCR and data collection were performed on a CFX96 instrument (Bio-Rad).
Lentiviral and retroviral transduction
The production of lentivirus and amphotropic retrovirus and infection of target cells were performed as described previously63.
Immunoblotting
Western blot analysis was performed with precast gradient gels (Bio-Rad) using standard methods. Briefly, cells were lysed in the RIPA buffer containing protease inhibitors (Roche) and phosphatase inhibitors (Sigma). Proteins were separated by SDS-PAGE and blotted onto a nitrocellulose membrane (Bio-Rad). Membranes were probed with the specific primary antibodies, followed by peroxidase-conjugated secondary antibodies. The bands were visualized by chemiluminescence (Denville Scientific). The following antibodies were used: antibodies to ZEB1 (1:1000, Bethyl Laboratories, A301-922A), CHK1 (1:1000, Santa Cruz Biotechnology, sc-8408, clone G-4), p-CHK1 (S317, 1:1000, Cell Signaling Technology, 2344), CHK2 (1:1000, Cell Signaling Technology, 2662), p-CHK2 (T68, 1:1000, Cell Signaling Technology, 2661), H2AX (1:1000, Cell Signaling Technology, 2595), γH2AX (1:1000, Cell Signaling Technology, 2577), Snail (1:1000, Cell Signaling Technology, 3879), Twist (1:50, Abcam, ab50887), p-S/TQ (1:1000, Cell Signaling Technology, 9607), p-ATM (S1981, 1:1000, Cell Signaling Technology, 5883), ATM (1:1000, Santa Cruz Biotechnology, sc-23921, clone 2C1), USP7 (1:2000, Bethyl Laboratories, A300-033A), p53 (HRP conjugate, 1:2000, Santa Cruz Biotechnology, sc-126 HRP, clone DO-1), HLTF (1:1000, Santa Cruz Biotechnology, sc-27542, clone Y-20), Cyclin A (1:1000, Santa Cruz Biotechnology, sc-751, clone H-432), p-H3 (1:1000, S10, Cell Signaling Technology, 9701), Claspin (1:1000, Bethyl Laboratories, A300-265), p-ATR (S428, 1:1000, Cell Signaling Technology, 2853), ATR (1:1000, Abcam, ab10312), HSP90 (1:3000, BD Transduction Laboratories, 610419, clone 68) and GAPDH (1:3000, Thermo, MA5-15738, clone GA1R). The ImageJ program (http://rsbweb.nih.gov/ij/download.html) was used for densitometric analysis of Western blots, and the quantification results were normalized to the loading control.
Immunofluorescence
Cells were cultured in chamber slides overnight and fixed with 3.7% formaldehyde in PBS for 20 min at 4°C, followed by permeabilization with 0.5% Triton X-100 in PBS for 30 min. Cells were then blocked for non-specific binding with 10% goat serum in PBS and Tween-20 (PBST) overnight, and incubated with the γH2AX antibody (1:300, Millipore, 07-164) for 1 hr at 37°C, followed by incubation with Alexa Fluor 594 goat anti-mouse IgG (1:400, Invitrogen, A11005) for 1 hr at 37 °C. Cover slips were mounted on slides using anti-fade mounting medium with DAPI. Immunofluorescence images were acquired on a Zeiss Axio Observer Z1 fluorescence microscope.
Immunoprecipitation and pull-down assays
Cells were lysed in NETN buffer (100 mM NaCl, 1 mM EDTA, 20 mM Tris-HCl [pH 8.0], 0.5% Nonidet P-40) containing protease inhibitors (Roche). For immunoprecipitation of protein complexes, cell extracts were pre-cleared with protein-A/G beads and incubated with the antibody to CHK1 (1:100, Santa Cruz Biotechnology, sc-8048) or ZEB1 (1:100, Bethyl Laboratories, A301-922A) for 2 hr at 4 °C. For pull-down of SFB-tagged proteins, cell extracts were incubated with streptavidin-sepharose beads (Amersham Biosciences) for 2 hr at 4 °C. For in vitro binding assays, bacterially purified GST-USP7 was eluted with glutathione (Amersham Biosciences) and then incubated with amylose resin (New England BioLabs) conjugated with bacterially expressed MBP-GFP or MBP-ZEB1. The amylose resin was washed with NETN buffer and the bound proteins were eluted by boiling in 1× Laemmli buffer.
Tandem affinity purification and mass spectrometry (TAP-MS)
293T cells were transfected with SFB-tagged ZEB1. The expression of exogenous protein was confirmed by immunoblotting. For affinity purification, a total of twenty 10-cm dishes of 293T cells expressing SFB-tagged ZEB1 were lysed in NETN buffer containing protease inhibitors for 20 min at 4°C. Crude lysates were cleared by centrifugation, and the supernatants were incubated with 300 μl streptavidin-sepharose beads (Amersham Biosciences) for 2 hr at 4°C. The beads were washed three times with NETN buffer, and the bound proteins were eluted with NETN buffer containing 2 mg/ml biotin (Sigma) for 2 hr at 4°C. The eluates were incubated with 100 μl S-protein agarose beads (Novagen) for 2 hr at 4°C, and the beads were washed three times with NETN buffer. The bound proteins were eluted by boiling in SDS sample buffer, resolved by SDS-PAGE, visualized by Coomassie Blue staining and subjected to mass spectrometric analysis (Taplin Biological Mass Spectrometry Facility at Harvard).
Deubiquitination of CHK1 in vivo and in vitro
For the in vivo deubiquitination assay, transfected 293T cells were treated with a proteasome inhibitor MG132 (10 μM) for 6 hr. The cell extracts were subjected to immunoprecipitation and western blot analysis with the indicated antibodies. For preparation of ubiquitinated CHK1 as the substrate for the in vitro deubiquitination assay, 293T cells were co-transfected with HA-ubiquitin and SFB-CHK1 and were treated with MG132 for 6 hr. Ubiquitinated CHK1 was purified from the cell extracts with streptavidin-sepharose beads. After extensive washing with NETN buffer, the bound proteins were eluted with biotin. SFB-USP7 was transfected into 293T cells alone or in combination with ZEB1, purified with streptavidin-sepharose beads and eluted with biotin. In vitro deubiquitination reaction was performed as described previously64. Briefly, ubiquitinated CHK1 protein was incubated with purified USP7 in deubiquitination buffer (50 mM Tris-HCl [pH 8.0], 50 mM NaCl, 1 mM EDTA, 10 mM DTT, and 5% glycerol) for 2 hr at 37 °C. After reaction, CHK1 was immunoprecipitated with FLAG antibody-conjugated beads. The beads were washed with deubiquitination buffer, and the bound proteins were eluted by boiling in 1 × Laemmlibuffer and subjected to Western blot analysis with the indicated antibodies.
In vitro kinase assay
293T cells were transfected with 10 μg of wild-type FLAG-ATM or the kinase-dead mutant and then irradiated. Activated or kinase-dead ATM was immunopurified from the cell extracts with FLAG beads (Sigma, M8823). Wild-type SFB-ZEB1 or the S585A mutant was transfected into 293T cells and immunopurified with FLAG beads. Kinase reactions were initiated by incubating purified ATM with purified ZEB1 or GST-p53 (Millipore, 14–865) in kinase buffer (Upstate, 20–108) containing 10 μCi [γ-32P] ATP for 30 minutes at 30 °C in a hybridization oven-shaker (Thermo Scientific). After reaction, proteins were resolved by SDS-PAGE, transferred to nitrocellulose membrane and analyzed by autoradiography. The membrane was then subjected to immunoblotting with the antibodies to p-ATM, ZEB1 and p53.
Clonogenic survival assay
Equal numbers of cells were plated in 10-cm tissue culture dishes at a clonogenic density (500 cells per dish) and irradiated by using a JL Shepherd Mark I-68A 137Cs irradiator with the indicated doses. Cells were incubated for 10–14 days. Colonies were stained with crystal violet and quantitated using a Gel Doc EZ Imager instrument (Bio-Rad) with the Quantity One software. Survival fraction was calculated as: (number of colonies/number of cells plated)irradiated/(number of colonies/number of cells plated)non-irradiated.
Comet assay
DNA damage was assessed by a single-cell gel electrophoresis assay using a CometAssay Kit (Trevigen, 4250–050-K) according to the manufacturer’s protocol. Briefly, cells were harvested at the indicated times after 6 Gy irradiation, mixed with low-melting-point agarose and plated on the CometSlide. Cells on the slides were lysed for 30 min at 4°C, subjected to electrophoresis at 21 V for 30 min under alkaline conditions, and then neutralized and stained with SYBR Green. The presence of comet tails was examined with a Zeiss Axio Observer Z1 fluorescence microscope. Tail moment was calculated as previously described24: (percentage of the DNA in the tail) × (length of the tail in μm), where percentage of the DNA in the tail and length of the tail were quantitated by using a software from Trevigen (Comet Assay IV, http://www.perceptive.co.uk/downloads/getcomet.php?a=588EAFB5&c=dss).
Homologous recombination (HR) repair assay
A U2OS cell clone stably expressing an HR repair reporter was described previously27. Briefly, 2 days after transfection with the ZEB1 siRNA, 1 × 106 U2OS cells expressing the HR repair reporter were electroporated with 10 μg of pCBASce, an I-SceI expression vector described previously65. Cells were harvested 2 days after electroporation and subjected to flow cytometry analysis to determine the percentage of GFP-positive cells resulting from HR-based repair of I-SceI-induced DSBs.
Tumor radiosensitivity study
Animal experiments were performed as previously described66 in accordance with a protocol approved by the Institutional Animal Care and Use Committee of MD Anderson Cancer Center, and mice were euthanized when they met the institutional euthanasia criteria for tumor size and overall health condition. When used in a power calculation, our sample size predetermination experiments indicated that 5 mice per group can identify the expected effect of ZEB1 on tumor radiosensitivity (P < 0.05) with 100% power. Solitary tumor xenografts were produced in the muscle of the right hind limb of twelve-week-old female nude mice (NCR Nu/Nu) by inoculation of 3 × 106 ZEB1-depleted (shZEB1) or control (scramble) SUM159-P2 cells. Mice were randomly assigned to no treatment or treatment groups consisting of 5 mice per group. Radiation treatment was initiated when tumors grew to approximately 8.0 mm (range: 7.7–8.2 mm) in diameter. 15 Gy single dose or fractionated dose (2 Gy per fraction, twice daily for 7 consecutive days) was delivered to the tumor-bearing limb of mice using a small-animal irradiator (Co-V, Theratron 780; MDS Nordion, Ottawa, Ontario) with a cobalt-60 source (field size, 10 × 10 cm; source axis distance, 64.9 cm), at a dose rate of 0.955 Gy/min. During irradiation, unanesthetized mice were mechanically immobilized in a jig so that the tumor was exposed in radiation field and the animal’s body was shielded from radiation exposure. Three mutually orthogonal diameters of the tumor were measured every other day with a vernier caliper, and the mean value was calculated and used as the tumor diameter. An investigator (Li Wang) who measured tumor size was blinded to the group allocation during all animal experiments and outcome assessment. General linear model multivariate analysis was performed to determine statistical significance using the SPSS 14.0 software package.
Patient study
The breast cancer progression TMAs were purchased from the NCI Cancer Diagnosis Program. These TMAs consist of three different case sets, including 190 analyzable cases of breast carcinoma. Samples were deparaffinized and rehydrated. Antigen retrieval was done by using 0.01 M sodium-citrate buffer (pH 6.0) in a microwave oven. To block endogenous peroxidase activity, the sections were treated with 1% hydrogen peroxide in methanol for 30 min. After 1 h pre-incubation in 10% normal serum to prevent nonspecific staining, the samples were incubated with the antibodies against ZEB1 (1:400, Bethyl Laboratories, A301–922A) and CHK1 (1:150, Santa Cruz Biotechnology, sc-7234) at 4 °C overnight. The sections were then incubated with a biotinylated secondary antibody, followed by incubation with avidin-biotin peroxidase complex solution (1:100) for 1 h at room temperature. Color was developed with the 3-amino-9-ethylcarbazole (AEC) solution. Counterstaining was carried out using Mayer’s hematoxylin. All immunostained slides were scanned on the Automated Cellular Image System III (ACIS III) for quantification by digital image analysis. A total score of protein expression was calculated from both the percentage of immunopositive cells and immunostaining intensity. High and low protein expression was defined using the mean score of all samples as a cutoff point. The χ2 test was used for statistical analysis of the correlation between ZEB1 and CHK1.
Statistical analysis
Each experiment was repeated three times or more. Unless otherwise noted, data are presented as mean ± s.e.m, and Student’s t test (unpaired, two-tailed) was used to compare two groups for independent samples. The data analyzed by t-test meet normal distribution; we used an F-test to compare variances, and the variances are not significantly different. Therefore, when using an unpaired t-test, we assumed equal variance, and no samples were excluded from the analysis. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank the shRNA and ORFeome Core at MD Anderson Cancer Center and Drs. Z. Gong, A. Lin, J. Wang, W. Wang, G. Wan and X. Lu for reagents and technical assistance. We thank Drs. A. Postigo, H-L. Piao and J. Kim for discussion. This work is supported by the NIH grants R00CA138572, R01CA166051 and R01CA181029 (to L.M.) and a CPRIT Scholar Award R1004 (to L.M.). L.M. is a R. Lee Clark Fellow of The University of Texas MD Anderson Cancer Center. Y.H. is supported in part by NIH U54CA151668. We wish to dedicate this work to the memory of Dr. K. Kian Ang.
Footnotes
AUTHOR CONTRIBUTIONS
P.Z. and L.M. conceived and designed the project. P.Z. performed and analyzed most of the experiments. Y.W. and M-C.H. performed studies on tissue microarrays of human patient samples. L.W. and K.K.A performed tumor radiosensitivity studies. B.G.D. and W.A.W. established the radioresistant subline. Y.Y. and H.L. performed computational data analysis. J.Z. and D.C. made some constructs. J.Y. and J.C. provided DR-GFP-expressing U2OS cells and performed TAP-MS analysis. M.W. maintained mice. Y.S maintained shRNA and ORF clones. Y.L. and D.C.D. provided Zeb1-deficient MEFs. Y.H. contributed to discussion and revision of the manuscript. P.Z. and L.M. wrote the manuscript with input from all other authors.
COMPETING FINANCIAL INTEREST
The authors declare no competing financial interests.
References
- 1.Bedford JS. Sublethal damage, potentially lethal damage, and chromosomal aberrations in mammalian cells exposed to ionizing radiations. Int J Radiat Oncol Biol Phys. 1991;21:1457–1469. doi: 10.1016/0360-3016(91)90320-4. [DOI] [PubMed] [Google Scholar]
- 2.Frankenberg-Schwager M, Frankenberg D, Blocher D, Adamczyk C. Effect of dose rate on the induction of DNA double-strand breaks in eucaryotic cells. Radiat Res. 1981;87:710–717. [PubMed] [Google Scholar]
- 3.Buchholz TA. Radiation therapy for early-stage breast cancer after breast-conserving surgery. N Engl J Med. 2009;360:63–70. doi: 10.1056/NEJMct0803525. [DOI] [PubMed] [Google Scholar]
- 4.Jameel JK, Rao VS, Cawkwell L, Drew PJ. Radioresistance in carcinoma of the breast. Breast. 2004;13:452–460. doi: 10.1016/j.breast.2004.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 6.Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem. 2004;73:39–85. doi: 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 7.Parrilla-Castellar ER, Arlander SJ, Karnitz L. Dial 9-1-1 for DNA damage: the Rad9-Hus1-Rad1 (9-1-1) clamp complex. DNA Repair (Amst) 2004;3:1009–1014. doi: 10.1016/j.dnarep.2004.03.032. [DOI] [PubMed] [Google Scholar]
- 8.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273:5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 9.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–255. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 11.Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961–966. doi: 10.1038/nature01446. [DOI] [PubMed] [Google Scholar]
- 12.Stucki M, et al. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 13.Bao S, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 14.Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer. 2008;8:545–554. doi: 10.1038/nrc2419. [DOI] [PubMed] [Google Scholar]
- 15.Mani SA, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 17.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 18.Elenbaas B, et al. Human breast cancer cells generated by oncogenic transformation of primary mammary epithelial cells. Genes Dev. 2001;15:50–65. doi: 10.1101/gad.828901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CJ, et al. p53 regulates epithelial-mesenchymal transition and stem cell properties through modulating miRNAs. Nat Cell Biol. 2011;13:317–323. doi: 10.1038/ncb2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim T, et al. p53 regulates epithelial-mesenchymal transition through microRNAs targeting ZEB1 and ZEB2. J Exp Med. 2011;208:875–883. doi: 10.1084/jem.20110235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banath JP, Macphail SH, Olive PL. Radiation sensitivity, H2AX phosphorylation, and kinetics of repair of DNA strand breaks in irradiated cervical cancer cell lines. Cancer Res. 2004;64:7144–7149. doi: 10.1158/0008-5472.CAN-04-1433. [DOI] [PubMed] [Google Scholar]
- 22.Olive PL, Banath JP. Phosphorylation of histone H2AX as a measure of radiosensitivity. Int J Radiat Oncol Biol Phys. 2004;58:331–335. doi: 10.1016/j.ijrobp.2003.09.028. [DOI] [PubMed] [Google Scholar]
- 23.Taneja N, et al. Histone H2AX phosphorylation as a predictor of radiosensitivity and target for radiotherapy. J Biol Chem. 2004;279:2273–2280. doi: 10.1074/jbc.M310030200. [DOI] [PubMed] [Google Scholar]
- 24.Bauer E, et al. The distribution of the tail moments in single cell gel electrophoresis (comet assay) obeys a chi-square (chi2) not a gaussian distribution. Mutat Res. 1998;398:101–110. doi: 10.1016/s0027-5107(97)00246-7. [DOI] [PubMed] [Google Scholar]
- 25.Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7:739–750. doi: 10.1038/nrm2008. [DOI] [PubMed] [Google Scholar]
- 26.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13:2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weinstock DM, Nakanishi K, Helgadottir HR, Jasin M. Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol. 2006;409:524–540. doi: 10.1016/S0076-6879(05)09031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010;108:73–112. doi: 10.1016/B978-0-12-380888-2.00003-0. [DOI] [PubMed] [Google Scholar]
- 29.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 30.Sorensen CS, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol. 2005;7:195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 31.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends in molecular medicine. 2011;17:88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lukas C, et al. DNA damage-activated kinase Chk2 is independent of proliferation or differentiation yet correlates with tissue biology. Cancer Res. 2001;61:4990–4993. [PubMed] [Google Scholar]
- 33.Collis SJ, et al. HCLK2 is essential for the mammalian S-phase checkpoint and impacts on Chk1 stability. Nat Cell Biol. 2007;9:391–401. doi: 10.1038/ncb1555. [DOI] [PubMed] [Google Scholar]
- 34.Leung-Pineda V, Huh J, Piwnica-Worms H. DDB1 targets Chk1 to the Cul4 E3 ligase complex in normal cycling cells and in cells experiencing replication stress. Cancer Res. 2009;69:2630–2637. doi: 10.1158/0008-5472.CAN-08-3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang YW, et al. Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol Cell. 2005;19:607–618. doi: 10.1016/j.molcel.2005.07.019. [DOI] [PubMed] [Google Scholar]
- 36.Furusawa T, Moribe H, Kondoh H, Higashi Y. Identification of CtBP1 and CtBP2 as corepressors of zinc finger-homeodomain factor deltaEF1. Mol Cell Biol. 1999;19:8581–8590. doi: 10.1128/mcb.19.12.8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Postigo AA, Dean DC. ZEB represses transcription through interaction with the corepressor CtBP. Proc Natl Acad Sci U S A. 1999;96:6683–6688. doi: 10.1073/pnas.96.12.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byles V, et al. SIRT1 induces EMT by cooperating with EMT transcription factors and enhances prostate cancer cell migration and metastasis. Oncogene. 2012;31:4619–4629. doi: 10.1038/onc.2011.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li M, et al. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- 40.Cummins JM, et al. Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature. 2004;428:1–486. doi: 10.1038/nature02501. [DOI] [PubMed] [Google Scholar]
- 41.Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13:879–886. doi: 10.1016/s1097-2765(04)00157-1. [DOI] [PubMed] [Google Scholar]
- 42.Qing P, Han L, Bin L, Yan L, Ping WX. USP7 regulates the stability and function of HLTF through deubiquitination. J Cell Biochem. 2011;112:3856–3862. doi: 10.1002/jcb.23317. [DOI] [PubMed] [Google Scholar]
- 43.Song MS, et al. The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature. 2008;455:813–817. doi: 10.1038/nature07290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Faustrup H, Bekker-Jensen S, Bartek J, Lukas J, Mailand N. USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. The Journal of cell biology. 2009;184:13–19. doi: 10.1083/jcb.200807137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savitsky K, et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–1753. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- 46.Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000;60:5934–5936. [PubMed] [Google Scholar]
- 47.Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- 48.Canman CE, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- 49.Banin S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 50.Toledo LI, et al. A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat Struct Mol Biol. 2011;18:721–727. doi: 10.1038/nsmb.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen D, et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat Med. 2012;18:1511–1517. doi: 10.1038/nm.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brabletz T, Lyden D, Steeg PS, Werb Z. Roadblocks to translational advances on metastasis research. Nature medicine. 2013;19:1104–1109. doi: 10.1038/nm.3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 54.Horsman MR, et al. Tumor radiosensitizers--current status of development of various approaches: report of an International Atomic Energy Agency meeting. Int J Radiat Oncol Biol Phys. 2006;64:551–561. doi: 10.1016/j.ijrobp.2005.09.032. [DOI] [PubMed] [Google Scholar]
- 55.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bartek J, Lukas J. Mammalian G1- and S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol. 2001;13:738–747. doi: 10.1016/s0955-0674(00)00280-5. [DOI] [PubMed] [Google Scholar]
- 57.Graham TR, et al. Reciprocal regulation of ZEB1 and AR in triple negative breast cancer cells. Breast Cancer Res Treat. 2010;123:139–147. doi: 10.1007/s10549-009-0623-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Karihtala P, et al. Vimentin, zeb1 and Sip1 are up-regulated in triple-negative and basal-like breast cancers: association with an aggressive tumour phenotype. Breast Cancer Res Treat. 2013;138:81–90. doi: 10.1007/s10549-013-2442-0. [DOI] [PubMed] [Google Scholar]
- 59.Kenney PA, et al. Novel ZEB1 expression in bladder tumorigenesis. BJU Int. 2011;107:656–663. doi: 10.1111/j.1464-410X.2010.09489.x. [DOI] [PubMed] [Google Scholar]
- 60.Spoelstra NS, et al. The transcription factor ZEB1 is aberrantly expressed in aggressive uterine cancers. Cancer Res. 2006;66:3893–3902. doi: 10.1158/0008-5472.CAN-05-2881. [DOI] [PubMed] [Google Scholar]
- 61.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends Pharmacol Sci. 2011;32:308–316. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 62.Liu Y, El-Naggar S, Darling DS, Higashi Y, Dean DC. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development. 2008;135:579–588. doi: 10.1242/dev.007047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stewart SA, et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan J, Luo K, Zhang L, Cheville JC, Lou Z. USP10 regulates p53 localization and stability by deubiquitinating p53. Cell. 2010;140:384–396. doi: 10.1016/j.cell.2009.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Richardson C, Moynahan ME, Jasin M. Double-strand break repair by interchromosomal recombination: suppression of chromosomal translocations. Genes Dev. 1998;12:3831–3842. doi: 10.1101/gad.12.24.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, et al. MK-4827, a PARP-1/-2 inhibitor, strongly enhances response of human lung and breast cancer xenografts to radiation. Invest New Drugs. 2012;30:2113–2120. doi: 10.1007/s10637-011-9770-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.