

Abstract
Purpose of review
Based on interim results from an ongoing study, we have reported that consumption of a high-fructose diet, but not a high-glucose diet, promotes the development of three of the pathological characteristics associated with metabolic syndrome: visceral adiposity, dyslipidemia, and insulin resistance. From these results and a review of the current literature, we present two potential sequences of events by which fructose consumption may contribute to metabolic syndrome.
Recent findings
The earliest metabolic perturbation resulting from fructose consumption is postprandial hypertriglyceridemia, which may increase visceral adipose deposition. Visceral adiposity contributes to hepatic triglyceride accumulation, novel protein kinase C activation, and hepatic insulin resistance by increasing the portal delivery of free fatty acids to the liver. With insulin resistance, VLDL production is upregulated and this, along with systemic free fatty acids, increase lipid delivery to muscle. It is also possible that fructose initiates hepatic insulin resistance independently of visceral adiposity and free fatty acid delivery. By providing substrate for hepatic lipogenesis, fructose may result in a direct lipid overload that leads to triglyceride accumulation, novel protein kinase C activation, and hepatic insulin resistance.
Summary
Our investigation and future studies of the effects of fructose consumption may help to clarify the sequence of events leading to development of metabolic syndrome.
Keywords: dyslipidemia, free fatty acids, fructose consumption, hepatic steatosis, insulin resistance, metabolic syndrome
Introduction
Studies investigating the effects of fructose consumption in humans and animals have been comprehensively reviewed [1-3,4••,5••]. These reviews are in agreement in their conclusions that, while there is strong evidence that diets high in fructose can produce obesity, insulin resistance/glucose intolerance, and dyslipidemia in animals, direct experimental evidence that consumption of fructose promotes the development of metabolic syndrome in humans is equivocal. We are currently conducting an investigation comparing the metabolic effects of consuming beverages sweetened with fructose or glucose providing 25% of energy requirements for 10 weeks in older, overweight and obese men and women. Based on interim results [6], we have reported that consumption of the high-fructose diet promotes the development of three of the pathological characteristics associated with metabolic syndrome: dyslipidemia, insulin resistance, and increased visceral adiposity. These observations have potentially important public health implications. In addition, these results suggest that such investigations may help to illuminate the causes and sequence of events leading to the development of metabolic syndrome. Rutledge and Adeli [7••] have recently outlined a potential sequence of events by which fructose consumption may contribute to development of the metabolic syndrome. They suggest that increased VLDL production induced by fructose increases visceral adiposity, which leads to insulin resistance in adipose tissue and, subsequently, to hepatic insulin resistance. We present this sequence of events, along with supporting evidence from the literature and from our current study. We then present another scenario in which overconsumption of fructose may result in a lipid overload within the liver that contributes to hepatic insulin resistance independently of visceral adiposity.
Fructose and hepatic lipogenesis/VLDL production
Both our current study [6] and earlier data [8] demonstrate that 10 weeks of fructose consumption markedly increases circulating postprandial triglyceride concentrations in older adults. In short-term studies in younger adults, we demonstrated that fructose consumption increases postprandial triglyceride concentrations within 24 h [9,10], which suggests that postprandial hypertriglyceridemia is the earliest metabolic perturbation associated with fructose consumption. The most likely mechanism for the postprandial hypertriglyceridemia is increased hepatic de-novo lipogenesis (DNL), which in turn upregulates VLDL production and secretion. Fructose consumption can promote hepatic lipogenesis because, first, the liver is the main site of fructose metabolism [11]; second, entry of fructose into glycolysis via fructose-1-phosphate bypasses the main rate-controlling step of glycolysis catalyzed by phosphofructokinase, thus providing unregulated amounts of lipogenic substrates acetyl-CoA and glycerol-3-phosphate [11], and, third, fructose can activate sterol receptor element binding protein-1c (SREBP-1c) independently of insulin, which then activates genes involved in DNL [12,13].
VLDL production and secretion are mainly regulated by the availability of lipid substrate [14]. Apolipoprotein B100 (ApoB) is essential for the intracellular assembly of triglyceride into VLDL. ApoB undergoes co-translational and posttranslational degradation, and its degradation is dramatically reduced when hepatic lipid content is increased [15]. In subjects consuming fructose, plasma ApoB concentrations were increased by more than 25% [6].
Recently it was reported that the contribution of de novo lipogenesis to fructose-induced hypertriacylglyceridemia is small [16•]. In this acute study of 14 healthy men and women, fructose contributed only 0.4% of the circulating VLDL-triglyceride measured 6 h after consumption of a high-fat (~60%), high-fructose (~40%) meal that contained 250 mg d-[U13C]fructose. It is possible, however, that the 6-h measurement of the incorporation of the 13C label into VLDL may not accurately reflect the rate of DNL. Fatty acids produced via DNL appear to be partitioned into the liver cytosolic triglyceride storage pool rather than immediately assembled into VLDL and secreted [17••]. Several studies have demonstrated that the measured contribution of DNL-derived free fatty acids (FFA) to VLDL-triglyceride increases progressively with longer periods of labeled precursor infusion (+24 h) that allows for equilibration of newly produced fatty acids into the liver triglyceride storage pool [17••,18-20].
Fructose and visceral adiposity
Rutledge and Adeli [7••] suggest that the increased VLDL production induced by fructose promotes obesity, although currently there is little experimental evidence to support this suggestion. In our current study, subjects consumed their usual ad-libitum diets along with either fructose-sweetened or glucose-sweetened beverages. Within 8 weeks, both groups gained an average of 1.5 kg. Intra-abdominal fat measured by computerized tomography, however, was significantly increased in subjects consuming fructose but unchanged in subjects consuming glucose (K. Stanhope, P. Havel, unpublished data). These results suggest that postprandial hypertriglyceridemia may specifically promote lipid deposition in visceral adipose tissue. As recently reviewed by Votruba and Jensen [21••], fat uptake is higher in abdominal subcutaneous fat than in subcutaneous fat in the thigh region [22,23], and higher in omental than in abdominal subcutaneous fat [24,25] following consumption of high-fat meals. Whether adipose uptake of meal-derived chylomicron-fatty acids differs from that of VLDL-fatty acids derived from fructose-induced DNL is unknown, however. It was recently shown that there was uptake of both chylomicron-fatty acids and VLDL-fatty acids by subcutaneous abdominal adipose following a mixed meal; however, the fractional extraction of chylomicronderived fatty acids was greater, especially during the first 2 h after the meal [26••].
Visceral adiposity and portal free fatty acids concentrations
There is considerable evidence that visceral adiposity is associated with insulin resistance [27-32]. An important potential mediator of this association is the direct delivery of portal blood flow from visceral fat to the liver. Owing to the portal connection, FFAs released from visceral fat are more likely to contribute to disturbances in hepatic metabolism than FFAs released from other adipose depots [33-35]. Another important mechanism is the greater lipolytic capacity of visceral than peripheral adipocytes. Visceral adipocytes have been demonstrated to be more sensitive than subcutaneous fat cells to the lipolytic effect of catecholamines [36,37] and, importantly, less sensitive to the antilipolytic and fatty acid re-esterifying effects of insulin [38,39]. Furthermore, as visceral adiposity develops, visceral adipocytes enlarge. Large adipocytes are more insulin resistant than smaller adipocytes [40,41•], and therefore less sensitive to the effects of insulin to suppress lipolysis and promote re-esterification of fatty acids [42-44]. Visceral adiposity is also closely associated with reduced circulating levels of the adipocyte hormone adiponectin, perhaps because enlarged visceral adipocytes are also likely to produce less adiponectin. Adiponectin increases hepatic lipid oxidation and improves insulin sensitivity by activating AMP kinase (see review, [45]).
Free fatty acid in the liver and hepatic triglyceride deposition
With increasing visceral adiposity, there is increased portal FFA delivery to the liver. It has been demonstrated that the fraction of FFA delivered to the liver from visceral fat is positively related to the visceral fat area, and is approximately 5–10% in normal-weight subjects and 20–30% in obese subjects [46,47]. Hepatic uptake of FFA is proportional to the rate of delivery [48-50]. In the liver, FFA is either oxidized or esterified to form triglyceride. The triglyceride is stored in the cytosol prior to being incorporated into VLDL and secreted [51]. Recent studies suggest that triglyceride turnover through the cytosolic pool and incorporation into VLDL can be rapid or delayed [17••]. It has been suggested that plasma FFA entering the liver may be routed through a more rapid turnover pool than fatty acids from dietary sources or those produced from DNL [18]. When triglyceride production exceeds FFA oxidation and VLDL production and secretion, triglyceride accumulates in the liver [51]. Triglyceride accumulation in the liver (i.e. non-alcoholic fatty liver disease—NAFLD) is positively associated with visceral adiposity [52]. Several studies of patients with type-2 diabetes and insulin resistance indicate that liver triglyceride content is also a strong correlate of hepatic insulin resistance [53-57] and the relationship is independent of visceral adiposity in both type 2 diabetic [56] and nondiabetic subjects [58].
Liver triglyceride content and hepatic insulin resistance
It has been suggested that hepatic triglyceride accumulation is a major mediator of hepatic insulin resistance [58,59••]. The Shulman group [60] has provided support for the hypothesis that lipid accumulation within the liver induces hepatic insulin resistance with evidence of a dose–response relationship between hepatic lipid content and insulin action and by demonstrating that prevention of hepatic fat accumulation abrogates the development of hepatic insulin resistance. Morino et al. [59••] suggest that the mechanism by which intracellular lipid causes insulin resistance in both liver and muscle is through diacylglycerol (DAG)-induced activation of novel protein kinase C (nPKC). DAG is a known activator of nPKC [61] and both DAG and nPKC are associated with lipid-induced insulin resistance in human muscle [62,63]. Several reports suggest that nPKC activation is associated with decreased insulin receptor or insulin receptor substrate 1 (IRS1) tyrosine phosphorylation [64-66], and other reports more specifically implicate nPKC in serine phosphorylation of insulin receptor, which impairs insulin signaling [67,68]. Studies conducted in 3T3-L1 adipocytes suggest that inhibitor kappa B kinase and c-JUN NH2-terminal kinase (JNK1) may mediate the serine phosphorylation induced by nPKC [69].
Hepatic insulin resistance and lipogenesis
With impaired insulin signaling in the liver, there is decreased glycogen synthesis, and increased glycogenolysis and gluconeogenesis. As a compensatory response, insulin secretion increases. It has been suggested that the increased insulin secretion is a direct response to increased FFA levels rather than increased glucose production [70••]. Both fasting glucose and insulin concentrations were increased, however, in subjects consuming fructose within 2 weeks (K. Stanhope, P. Havel, unpublished data). As hyperinsulinemia develops, DNL is increased due to insulin activation of SREBP1-c [71]. Although the insulin-resistant liver is resistant to the effects of insulin to stimulate glycogen synthesis and inhibit gluconeogenesis and glycogenolysis, it does not appear to develop resistance to insulin’s effect to promote lipogenesis [72].
Hepatic insulin resistance and VLDL production
VLDL production is also increased in the insulin-resistant liver due to mechanisms independent of hepatic lipid supply. With insulin resistance, there is reduced ApoB degradation and increased VLDL production [73]. The mechanism by which insulin directly inhibits VLDL production is unknown [15], but it has been suggested that insulin promotes ApoB degradation by inhibiting lipid transfer to VLDL-precursor ApoB [74] and by regulating a protease enzyme implicated in ApoB degradation [75]. Insulin also inhibits microsomal triglyceride transfer protein (MTP) expression via an insulin response element on the MTP gene [76]. MTP is essential for assembly of triglyceride and ApoB into VLDL and secretion of VLDL [77]. Lewis et al. [78] suggested that, in insulin-resistant states, there may be sustained upregulation of MTP expression and protein levels as a result of resistance to insulin’s inhibitory effect on MTP.
Hypertriglyceridemia and cardiovascular disease risk
Upregulation of VLDL production leads to increased plasma triglyceride. Reduced clearance of triglyceride can also contribute to hypertriglyceridemia [79,80]. Insulin stimulates adipose lipoprotein lipase (LPL) and LPL activity is decreased in subjects with insulin resistance [81]. There is growing evidence linking postprandial hypertriglyceridemia with proatherogenic conditions [82••,83,84,85•,86••]. The relationship between nonfasting triglyceride and cardiovascular disease is most likely mediated by effects of postprandial hypertriglyceridemia to promote lipid remodeling to a more atherogenic lipid profile consisting of increased concentrations of triglyceride-rich remnant lipoproteins and small dense LDL, and decreased concentrations of HDL [87,88••,89••]. In subjects consuming fructose, we have reported significantly increased circulating levels of remnant lipoproteins, small dense LDL, and oxidized LDL [6].
Peripheral insulin resistance
Elevated triglyceride, along with elevated levels of plasma FFA released from insulin-resistant adipose tissue, lead to increased flux of FFA and triglyceride to other tissues. In skeletal muscle, increased FFA availability can lead to increased muscle triglyceride content and intramyocellular lipid (IMCL) deposition. IMCL is closely related to insulin resistance in skeletal muscle [90-93]. IMCL, or associated lipid metabolites such as DAG, appear to inhibit insulin signaling, leading to a reduction in insulin-stimulated glucose transport [59••,94,95•] and systemic insulin resistance.
Free fatty acid: link between visceral adiposity and hepatic insulin resistance
Strong support for the hypothesis that FFA release from enlarged visceral adipocytes is an important link between visceral adiposity and hepatic insulin resistance has been provided by Bergman et al. [96•], who conclude, first, that FFAs per se are among the most important products of the visceral adipocyte contributing to insulin resistance and hence metabolic syndrome; second, that the anatomical position of the visceral adipose depot (i.e. portal drainage to the liver) plays an important role in the pathogenesis of metabolic syndrome. When considering evidence that does not support these conclusions, it is important to be aware of the following. Bergman et al. [96•] reported that feeding dogs a 6-week hypercaloric high-fat diet resulted in a 76% increase in trunk fat, but fasting FFA concentrations were not affected. Twenty-four-hour systemic FFA profiles, however, determined from hourly blood sampling, were increased by 50% [96•]. This suggests that linking increases of FFA with visceral adiposity and insulin resistance may not be possible in studies that measure circulating metabolites only in the fasting state. In human subjects consuming a highfructose diet for 10 weeks, we found no change in 24-h systemic FFA profiles (36 samples collected over 24 h, K. Stanhope, P. Havel, unpublished data), despite modest increases in visceral adiposity and insulin resistance. This does not exclude the possibility that portal concentrations and hepatic extraction of FFAs were increased in these subjects. Parallel measurements, however, of arterial and portal FFA concentrations in conscious dogs under experimental conditions that resulted in a wide range of FFA release demonstrated that, whereas portal vein FFA levels tended to be higher than arterial levels (~5–6%), the values obtained were highly correlated (r2 = 0.96) [97]. These observations led us to consider the possibility that hepatic lipid overload, independent of visceral adiposity and FFA levels, may be an important mediator of insulin resistance in subjects consuming fructose.
Hepatic lipid overload may initiate liver triglyceride accumulation and hepatic insulin resistance independently of visceral adiposity and free fatty acid
The suggestion that fructose induces insulin resistance independently of visceral adiposity and FFA levels is supported by work from the Shulman group [59••,60]. These investigators have built on the work by Kraegen and colleagues [98], who demonstrated that 3 days of high-fat feeding results in hepatic insulin resistance prior to the development of peripheral insulin resistance. Shulman and colleagues also fed rats a high-fat diet (69%) for 3 days and reported a three-fold increase in liver triglyceride content without any significant changes in visceral fat weight [60]. The hepatic fat accumulation was associated with impaired IRS tyrosine phosphorylation, PKC-ε (a novel PKC) and JNK1 activation, decreased insulin stimulation of glycogen synthase and decreased insulin suppression of gluconeogenesis [60].
It has been proposed that obesity per se is not the main contributor to insulin resistance, but rather it is the accumulation of intracellular lipid metabolites (e.g. DAG) [58,99••]. As presented in Fig. 1, in addition to FFA, there are other additional sources of triglyceride that can lead to hepatic lipid accumulation: triglyceride generated by hepatic lipogenesis, and triglyceride derived from FFA released from VLDL and chylomicron remnants within hepatic lysosomes [51]. Therefore, by increasing the delivery of chylomicron remnant-triglyceride to the liver, feeding rats a high-fat diet for only 3 days resulted in impaired insulin signaling prior to increases in visceral adiposity [60]. We propose that a high-fructose diet, which provides substrate for de-novo lipogenesis, can also produce a lipid overload in the liver that results in hepatic insulin resistance independently of visceral adiposity and FFA levels. This suggestion is not mutually exclusive of the ‘portal’ FFA hypothesis. A sustained, moderate positive energy balance may indeed promote hepatic insulin resistance as a result of increased visceral fat accumulation and increased portal delivery of FFA. During consumption of a high-fructose diet, however, a contributing and possibly major mechanism may be a more direct intra-hepatic lipid oversupply via fructose-induced lipogenesis.
Figure 1. A high-fructose diet increases hepatic de-novo lipogenesis and a high-fat diet increases hepatic chylomicron remnant uptake.
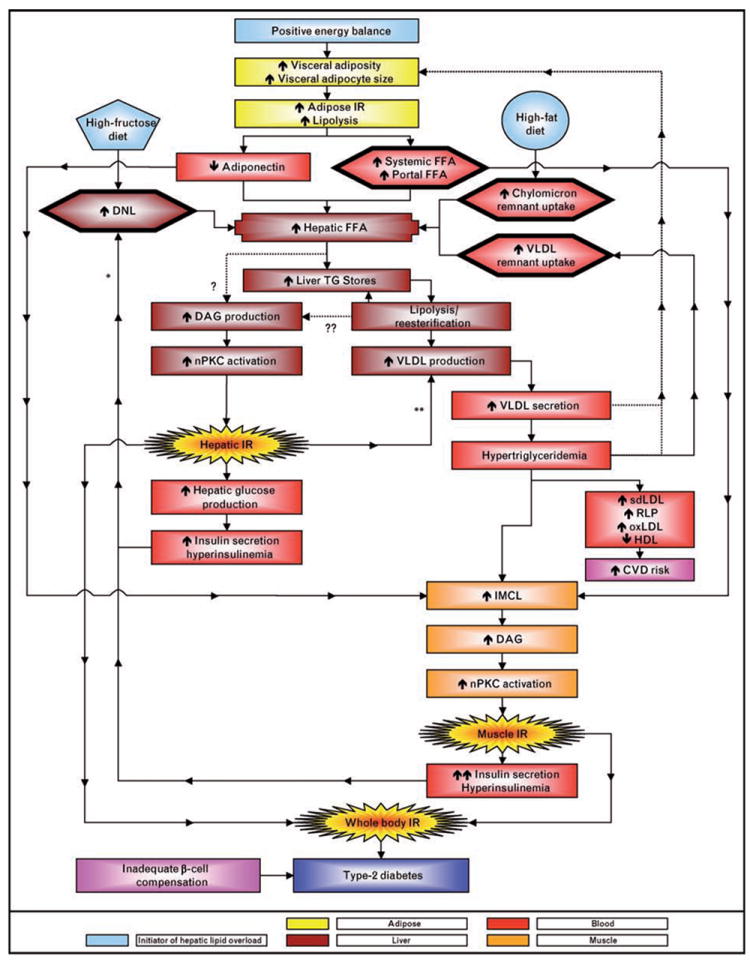
Either diet can produce a hepatic lipid overload along with, or independently of, visceral adiposity and increased portal free fatty acid (FFA) delivery. Visceral adiposity with adipocyte hypertrophy has been hypothesized to reduce adiponectin production and delivery to the liver which would be expected to promote hepatic lipid accumulation. The esterification of hepatic FFA to triglycerides (TG) stored in hepatocytes can increase diacylglycerol (DAG) levels (?); as can the lipolysis, re-esterification, recycling of cytosolic TG that is associated with VLDL assembly (??). Either or both of these sources of DAG may lead to activation of nPKC and impaired hepatic insulin signaling/insulin resistance. VLDL production is regulated by the hepatic lipid supply and is further upregulated by insulin resistance. Hyperinsulinemia may increase DNL because of insulin’s ability to activate SREBP1-c (*). Insulin promotes ApoB degradation and inhibits MTP(**), and both of these processes are likely to be downregulated in the insulin-resistant liver. Increased VLDL production and secretion lead to hypertriglyceridemia. Whether increased VLDL secretion and elevated triglyceride levels directly promote visceral adiposity warrants further investigation (???). Postprandial hypertriglyceridemia increases cardiovascular disease (CVD) risk by promoting lipid/lipoprotein remodeling leading to increased circulating concentrations of small dense LDL (sdLDL), remnant lipoproteins (RLP), and oxidized LDL (oxLDL) and decreased concentrations of HDL. Hypertriglyceridemia and increased levels of circulating FFA can promote accumulation of IMCL, DAG production, and nPKC activation and impaired insulin signaling in skeletal muscle. The end result is whole body insulin resistance, which, when accompanied by inadequate pancreatic beta cell compensation, leads to type 2 diabetes.
Liver lipid accumulation and insulin resistance are not always associated
Although there is much support for the hypothesis that hepatic lipid accumulation initiates insulin resistance, there is also contradictory evidence. Lonardo et al. [100••] investigated the association of hepatic steatosis with insulin resistance in patients with, first, familial heterozygous hypobetalipoproteinemia (FHBL), second, NAFLD, third, hepatitis C virus infection (HCV), and fourth, healthy subjects without steatosis. Data from subjects with NAFLD and HCV supported the association between liver steatosis and insulin resistance. FHBL subjects, however, did not have significantly increased HOMA-IR compared with healthy subjects, and the 17 FHBL subjects with liver steatosis did not have higher HOMA-IR than the five FHBL subjects without liver steatosis. Subjects with FHBL have mutations in the ApoB gene that lead to triglyceride accumulation in the liver due to impaired VLDL production and secretion. The lack of insulin resistance in these subjects suggests that the mechanism by which hepatic triglyceride stores are increased is key to the development of insulin resistance [100••]. It also suggests that there may be steps downstream of liver triglyceride accumulation, for example VLDL production or secretion, that are associated with the induction of insulin resistance. The very low fasting triglyceride concentrations observed in the subjects with FHBL (mean = 34 mg/dl) are consistent with reduced rates of VLDL production and secretion. Conversely, it has been reported that subjects heterozygous for a mutation that increases ApoB transcription (−516C/T) exhibited increased postprandial triglyceride concentrations [101] and insulin resistance [102•].
Data from other studies also indicate a lack of association between liver triglyceride and insulin resistance. Patients with glycogen storage disease type 1 have severe steatosis without insulin resistance [103,104]. In mice lacking hepatic MTP [105] and in transgenic mice overexpressing acyl-CoA:diacylglycerol acyltransferase 2 (DGAT2) in the liver [106•], there were increased liver triglyceride accumulation and reduced circulating triglyceride levels in the absence of insulin resistance. Rats administered antisense to stearyl CoA desaturase-1 (SCD1) and fed a lard-supplemented diet had increased liver triglyceride, reduced circulating triglyceride, but normal insulin sensitivity. Control rats treated with scrambled antisense exhibited the expected decrease of insulin sensitivity on the lard diet, yet had only one-third the hepatic triglyceride content [107•]. The dissociation between hepatic triglyceride content and insulin resistance noted in these studies again suggests that mechanisms operating downstream of liver triglyceride accumulation, which are connected to the formation or secretion of VLDL, may be involved in the development of hepatic insulin resistance.
Linking hepatic insulin resistance with VLDL production
A detailed model for production of VLDL has been proposed [51,108]. To briefly summarize, the liver triglyceride synthesized from extracelluar and endogenous sources of FFA does not serve as a direct precursor of VLDL, but rather is stored in the cytosolic triglyceride pool. This cytosolic triglyceride is not incorporated into VLDL en bloc, but rather is first hydrolyzed to FFA, monoacylglycerol, and DAG. These lipolytic products are then re-esterified in the vicinity of ApoB-VLDL precursor. Not all of this resynthesized triglyceride is incorporated into VLDL; instead, as much as 50% is recycled back to the cytosolic pool [51,108]. A possible explanation for the disconnect between liver triglyceride accumulation and hepatic insulin resistance may be that the DAG production responsible for the induction of hepatic insulin resistance results from the triglyceride lypolysis, re-esterification and recycling associated with VLDL assembly, rather than from the DAG associated with the initial synthesis of triglyceride from extra-hepatic and endogenous sources of FFA. Accordingly, when the assembly of VLDL is inhibited, as in the examples described above (FHBL, MTP blockade and administration of SCD1 antisense), the resulting high liver triglyceride content does not result in DAG/nPKC-induced hepatic insulin resistance [100••,105,107•].
Possibly contradicting this suggestion is the report that the transgenic mice overexpressing DGAT2 in the liver, described above as having increased liver triglyceride accumulation and normal insulin sensitivity, also had increased hepatic DAG content [106•]. The increased DAG accumulation, however, may have resulted from the upregulation of the initial synthesis of triglyceride, which increased triglyceride stores, rather than from lipolysis, re-esterification and recycling associated with VLDL assembly. The circulating triglyceride concentrations of the DGAT2 transgenic mice were reduced compared with the wild-type control mice, which suggests VLDL assembly was not upregulated by DGAT2 overexpression [106•]. Another recent study also reported that mice overexpressing DGAT2, after injection of adenovirus containing DGAT2 transgene, had increased liver triglyceride, but levels of plasma triglyceride and the hepatic production rate of VLDL were not affected [109]. It has been recently reported that hepatic levels of DAG were increased in patients with NAFLD and nonalcoholic steatohepatitis compared with control subjects; however, the authors noted that the impact of the location of DAG within the hepatocyte requires investigation [110•].
Conclusion
A sustained and moderate positive energy balance is likely to promote metabolic syndrome by increasing visceral-fat accumulation, resulting in increased portal delivery of FFA to the liver. A high-fructose diet may more directly and rapidly produce a lipid oversupply within the liver via increased DNL. An oversupply of hepatic lipid results in liver triglyceride deposition and increased VLDL assembly and secretion. It has been proposed [59••] that the liver triglyceride accumulation is associated with increased levels of DAG that activate nPKC and disrupt insulin signaling. Several recent studies, however, reporting a disconnect between liver triglyceride accumulation and insulin resistance [100••,107•] provide support for our hypothesis that there may be steps downstream of liver triglyceride accumulation (for example, VLDL production or secretion) that are associated with the induction of hepatic insulin resistance.
Acknowledgments
The authors would like to acknowledge and thank Dr Richard J. Havel for his expert guidance and editing, Dr Elizabeth Parks for her helpful advice, and James Graham for his assistance with the artwork in the figure. This work was supported in part by research funding from The American Diabetes Association, the United States Department of Agriculture, and the National Institutes of Health (HL-075675, AT-002599, AT-002993, and AT-003645). Dr Havel’s research programme also receives support from the University of California, Davis Clinical and Translational Science Center (Grant Number UL1 RR024146) from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research. The content of this article is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH. Information on NCRR is available at http://www.ncrr.nih.gov. Information on Re-engineering the Clinical Research Enterprise can be obtained from http://nihroadmap.nih.gov/clinicalre-search/overview-translational.asp.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
-
•
of special interest
-
••
of outstanding interest
Additional references related to this topic can also be found in the Current World Literature section in this issue (pp. 74–75).
- 1.Bizeau ME, Pagliassotti MJ. Hepatic adaptations to sucrose and fructose. Metabolism. 2005;54:1189–1201. doi: 10.1016/j.metabol.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 2.Elliott SS, Keim NL, Stern JS, et al. Fructose, weight gain, and the insulin resistance syndrome. Am J Clin Nutr. 2002;76:911–922. doi: 10.1093/ajcn/76.5.911. [DOI] [PubMed] [Google Scholar]
- 3.Havel PJ. Dietary fructose: implications for dysregulation of energy homeostasis and lipid/carbohydrate metabolism. Nutr Rev. 2005;63:133–157. doi: 10.1301/nr.2005.may.133-157. [DOI] [PubMed] [Google Scholar]
- 4••.Le KA, Tappy L. Metabolic effects of fructose. Curr Opin Clin Nutr Metab Care. 2006;9:469–475. doi: 10.1097/01.mco.0000232910.61612.4d. An informative summary of the potential metabolic outcomes of fructose consumption and possible mechanisms involved in mediating these effects. [DOI] [PubMed] [Google Scholar]
- 5••.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Fructose-mediated stress signaling in the liver: implications for hepatic insulin resistance. J Nutr Biochem. 2007;18:1–9. doi: 10.1016/j.jnutbio.2006.03.013. This paper presents the evidence and mechanisms related to the hypothesis that overconsumption of fructose induces stress signaling via JNK activation in the liver which impairs hepatic insulin action. [DOI] [PubMed] [Google Scholar]
- 6.Stanhope KL, Griffen S, Krauss RM, et al. Consumption of fructose-, but not glucose-sweetened beverages produces an atherogenic lipid profile in overweight/obese men and women. Diabetes. 2007;56 [Google Scholar]
- 7••.Rutledge AC, Adeli K. Fructose and the metabolic syndrome: pathophysiology and molecular mechanisms. Nutr Rev. 2007;65:S13–23. doi: 10.1111/j.1753-4887.2007.tb00322.x. In addition to proposing that fructose consumption can induce obesity and increase portal delivery of FFA to the liver, this review focuses on the role of fructose-induced defects in the insulin signaling cascade and hepatic inflammation. [DOI] [PubMed] [Google Scholar]
- 8.Havel PJ, Elliott S, Keim NL, et al. Short-term and long-term consumption of high fructose, but not high glucose, diets increases postprandial triglycerides and apo-lipoprotein-B in women. J Invest Med. 2003;51:S163. [Google Scholar]
- 9.Teff KL, Elliott SS, Tschop M, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab. 2004;89:2963–2972. doi: 10.1210/jc.2003-031855. [DOI] [PubMed] [Google Scholar]
- 10.Teff KL, Keim NL, Townsend RR, Havel PJ. Fructose-sweetened beverages decrease circulating leptin levels and increase postprandial triglycerides in obese men and women. Diabetes. 2005;54:A385. [Google Scholar]
- 11.Mayes PA. Intermediary metabolism of fructose. Am J Clin Nutr. 1993;58:754S–765S. doi: 10.1093/ajcn/58.5.754S. [DOI] [PubMed] [Google Scholar]
- 12.Matsuzaka T, Shimano H, Yahagi N, et al. Insulin-independent induction of sterol regulatory element-binding protein-1c expression in the livers of streptozotocin-treated mice. Diabetes. 2004;53:560–569. doi: 10.2337/diabetes.53.3.560. [DOI] [PubMed] [Google Scholar]
- 13.Nagai Y, Nishio Y, Nakamura T, et al. Amelioration of high fructose-induced metabolic derangements by activation of PPARalpha. Am J Physiol Endocrinol Metab. 2002;282:E1180–E1190. doi: 10.1152/ajpendo.00471.2001. [DOI] [PubMed] [Google Scholar]
- 14.Lewis GF. Fatty acid regulation of very low density lipoprotein production. Curr Opin Lipidol. 1997;8:146–153. doi: 10.1097/00041433-199706000-00004. [DOI] [PubMed] [Google Scholar]
- 15.Olofsson SO, Boren J. Apolipoprotein B: a clinically important apolipoprotein which assembles atherogenic lipoproteins and promotes the development of atherosclerosis. J Intern Med. 2005;258:395–410. doi: 10.1111/j.1365-2796.2005.01556.x. [DOI] [PubMed] [Google Scholar]
- 16•.Chong MF, Fielding BA, Frayn KN. Mechanisms for the acute effect of fructose on postprandial lipemia. Am J Clin Nutr. 2007;85:1511–1520. doi: 10.1093/ajcn/85.6.1511. An interesting result from this acute study was that 6 h after consumption of a high-fructose/fat meal, 38% of the glycerol in the VLDL-TG was derived from labeled fructose, whereas following consumption of a high-glucose/fat meal, none of the glycerol in the VLDL-TG was derived from labeled glucose. [DOI] [PubMed] [Google Scholar]
- 17••.Vedala A, Wang W, Neese RA, et al. Delayed secretory pathway contributions to VLDL-triglycerides from plasma NEFA, diet, and de novo lipogenesis in humans. J Lipid Res. 2006;47:2562–2574. doi: 10.1194/jlr.M600200-JLR200. Hellerstein and colleagues provide direct kinetic evidence for a hepatic TG storage pool in human subjects and document its metabolic sources. [DOI] [PubMed] [Google Scholar]
- 18.Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–1351. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudgins LC, Hellerstein MK, Seidman CE, et al. Relationship between carbohydrate-induced hypertriglyceridemia and fatty acid synthesis in lean and obese subjects. J Lipid Res. 2000;41:595–604. [PubMed] [Google Scholar]
- 20.Timlin MT, Parks EJ. Temporal pattern of de novo lipogenesis in the post-prandial state in healthy men. Am J Clin Nutr. 2005;81:35–42. doi: 10.1093/ajcn/81.1.35. [DOI] [PubMed] [Google Scholar]
- 21••.Votruba SB, Jensen MD. Regional fat deposition as a factor in FFA metabolism. Annu Rev Nutr. 2007;27:149–163. doi: 10.1146/annurev.nutr.27.061406.093754. An excellent summary of the important work conducted by Jensen and others on the regional disposal of meal-derived lipids and regional FFA kinetics. [DOI] [PubMed] [Google Scholar]
- 22.Marin P, Rebuffe-Scrive M, Bjorntorp P. Uptake of triglyceride fatty acids in adipose tissue in vivo in man. Eur J Clin Invest. 1990;20:158–165. doi: 10.1111/j.1365-2362.1990.tb02263.x. [DOI] [PubMed] [Google Scholar]
- 23.Romanski SA, Nelson RM, Jensen MD. Meal fatty acid uptake in adipose tissue: gender effects in nonobese humans. Am J Physiol Endocrinol Metab. 2000;279:E455–E462. doi: 10.1152/ajpendo.2000.279.2.E455. [DOI] [PubMed] [Google Scholar]
- 24.Jensen MD, Sarr MG, Dumesic DA, et al. Regional uptake of meal fatty acids in humans. Am J Physiol Endocrinol Metab. 2003;285:E1282–E1288. doi: 10.1152/ajpendo.00220.2003. [DOI] [PubMed] [Google Scholar]
- 25.Marin P, Andersson B, Ottosson M, et al. The morphology and metabolism of intraabdominal adipose tissue in men. Metabolism. 1992;41:1242–1248. doi: 10.1016/0026-0495(92)90016-4. [DOI] [PubMed] [Google Scholar]
- 26••.Bickerton AS, Roberts R, Fielding BA, et al. Preferential uptake of dietary fatty acids in adipose tissue and muscle in the postprandial period. Diabetes. 2007;56:168–176. doi: 10.2337/db06-0822. Using novel methodology which included a test meal containing [U-13C]palmitate combined with an IV infusion of [2H2]palmitate, this study showed differential adipose tissue uptake of fatty acids derived from chylomicrons compared with those from VLDL. [DOI] [PubMed] [Google Scholar]
- 27.Abate N, Garg A, Peshock RM, et al. Relationships of generalized and regional adiposity to insulin sensitivity in men. J Clin Invest. 1995;96:88–98. doi: 10.1172/JCI118083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bjorntorp P. Regional fat distribution: implications for type II diabetes. Int J Obes Relat Metab Disord. 1992;16:S19–S27. [PubMed] [Google Scholar]
- 29.Goodpaster BH, Kelley DE, Wing RR, et al. Effects of weight loss on regional fat distribution and insulin sensitivity in obesity. Diabetes. 1999;48:839–847. doi: 10.2337/diabetes.48.4.839. [DOI] [PubMed] [Google Scholar]
- 30.Kissebah AH, Vydelingum N, Murray R, et al. Relation of body fat distribution to metabolic complications of obesity. J Clin Endocrinol Metab. 1982;54:254–260. doi: 10.1210/jcem-54-2-254. [DOI] [PubMed] [Google Scholar]
- 31.Pouliot MC, Despres JP, Nadeau A, et al. Visceral obesity in men. Associations with glucose tolerance, plasma insulin, and lipoprotein levels. Diabetes. 1992;41:826–834. doi: 10.2337/diab.41.7.826. [DOI] [PubMed] [Google Scholar]
- 32.Vague J. The degree of masculine differentiation of obesities: a factor determining predisposition to diabetes, atherosclerosis, gout, and uric calculous disease. Am J Clin Nutr. 1956;4:20–34. doi: 10.1093/ajcn/4.1.20. [DOI] [PubMed] [Google Scholar]
- 33.Bergman RN, Van Citters GW, Mittelman SD, et al. Central role of the adipocyte in the metabolic syndrome. J Investig Med. 2001;49:119–126. doi: 10.2310/6650.2001.34108. [DOI] [PubMed] [Google Scholar]
- 34.Bjorntorp P. Obesity and adipose tissue distribution as risk factors for the development of disease. A review Infusionstherapie. 1990;17:24–27. doi: 10.1159/000222436. [DOI] [PubMed] [Google Scholar]
- 35.Montague CT, O’Rahilly S. The perils of portliness: causes and consequences of visceral adiposity. Diabetes. 2000;49:883–888. doi: 10.2337/diabetes.49.6.883. [DOI] [PubMed] [Google Scholar]
- 36.Arner P, Hellstrom L, Wahrenberg H, Bronnegard M. Beta-adrenoceptor expression in human fat cells from different regions. J Clin Invest. 1990;86:1595–1600. doi: 10.1172/JCI114880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engfeldt P, Arner P. Lipolysis in human adipocytes, effects of cell size, age and of regional differences. Horm Metab Res Suppl. 1988;19:26–29. [PubMed] [Google Scholar]
- 38.Bolinder J, Kager L, Ostman J, Arner P. Differences at the receptor and postreceptor levels between human omental and subcutaneous adipose tissue in the action of insulin on lipolysis. Diabetes. 1983;32:117–123. doi: 10.2337/diab.32.2.117. [DOI] [PubMed] [Google Scholar]
- 39.Mittelman SD, Van Citters GW, Kirkman EL, Bergman RN. Extreme insulin resistance of the central adipose depot in vivo. Diabetes. 2002;51:755–761. doi: 10.2337/diabetes.51.3.755. [DOI] [PubMed] [Google Scholar]
- 40.Foley JE, Laursen AL, Sonne O, Gliemann J. Insulin binding and hexose transport in rat adipocytes. Relation to cell size Diabetologia. 1980;19:234–241. doi: 10.1007/BF00275275. [DOI] [PubMed] [Google Scholar]
- 41•.Franck N, Stenkula KG, Ost A, et al. Insulin-induced GLUT4 translocation to the plasma membrane is blunted in large compared with small primary fat cells isolated from the same individual. Diabetologia. 2007;50:1716–1722. doi: 10.1007/s00125-007-0713-1. This study is the first to demonstrate a difference in insulin sensitivity between large and small human adipocytes obtained from the same individual. [DOI] [PubMed] [Google Scholar]
- 42.Karnieli E, Barzilai A, Rafaeloff R, Armoni M. Distribution of glucose transporters in membrane fractions isolated from human adipose cells. Relation to cell size. J Clin Invest. 1986;78:1051–1055. doi: 10.1172/JCI112660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olefsky JM. Insensitivity of large rat adipocytes to the antilipolytic effects of insulin. J Lipid Res. 1977;18:459–464. [PubMed] [Google Scholar]
- 44.Olefsky JM. Mechanisms of decreased insulin responsiveness of large adipocytes. Endocrinology. 1977;100:1169–1177. doi: 10.1210/endo-100-4-1169. [DOI] [PubMed] [Google Scholar]
- 45.Havel PJ. Update on adipocyte hormones: regulation of energy balance and carbohydrate/lipid metabolism. Diabetes. 2004;53(Suppl 1):S143–S151. doi: 10.2337/diabetes.53.2007.s143. [DOI] [PubMed] [Google Scholar]
- 46.Koutsari C, Jensen MD. Thematic review series: patient-oriented research. Free fatty acid metabolism in human obesity. J Lipid Res. 2006;47:1643–1650. doi: 10.1194/jlr.R600011-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Nielsen S, Guo Z, Johnson CM, et al. Splanchnic lipolysis in human obesity. J Clin Invest. 2004;113:1582–1588. doi: 10.1172/JCI21047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Havel RJ, Kane JP, Balasse EO, et al. Splanchnic metabolism of free fatty acids and production of triglycerides of very low density lipoproteins in normotriglyceridemic and hypertriglyceridemic humans. J Clin Invest. 1970;49:2017–2035. doi: 10.1172/JCI106422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soler-Argilaga C, Infante R, Polonovski J. Lipid metabolism of rat liver isolated and perfused in hypoxia. Biomedicine. 1974;20:154–159. [PubMed] [Google Scholar]
- 50.Wahren J, Sato Y, Ostman J, et al. Turnover and splanchnic metabolism of free fatty acids and ketones in insulin-dependent diabetics at rest and in response to exercise. J Clin Invest. 1984;73:1367–1376. doi: 10.1172/JCI111340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibbons GF, Islam K, Pease RJ. Mobilisation of triacylglycerol stores. Biochim Biophys Acta. 2000;1483:37–57. doi: 10.1016/s1388-1981(99)00182-1. [DOI] [PubMed] [Google Scholar]
- 52.Nakao K, Nakata K, Ohtsubo N, et al. Association between nonalcoholic fatty liver, markers of obesity, and serum leptin level in young adults. Am J Gastroenterol. 2002;97:1796–1801. doi: 10.1111/j.1572-0241.2002.05846.x. [DOI] [PubMed] [Google Scholar]
- 53.Marceau P, Biron S, Hould FS, et al. Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab. 1999;84:1513–1517. doi: 10.1210/jcem.84.5.5661. [DOI] [PubMed] [Google Scholar]
- 54.Marchesini G, Brizi M, Bianchi G, et al. Nonalcoholic fatty liver disease: a feature of the metabolic syndrome. Diabetes. 2001;50:1844–1850. doi: 10.2337/diabetes.50.8.1844. [DOI] [PubMed] [Google Scholar]
- 55.Marchesini G, Brizi M, Morselli-Labate AM, et al. Association of nonalcoholic fatty liver disease with insulin resistance. Am J Med. 1999;107:450–455. doi: 10.1016/s0002-9343(99)00271-5. [DOI] [PubMed] [Google Scholar]
- 56.Ryysy L, Hakkinen AM, Goto T, et al. Hepatic fat content and insulin action on free fatty acids and glucose metabolism rather than insulin absorption are associated with insulin requirements during insulin therapy in type 2 diabetic patients. Diabetes. 2000;49:749–758. doi: 10.2337/diabetes.49.5.749. [DOI] [PubMed] [Google Scholar]
- 57.Tiikkainen M, Tamminen M, Hakkinen AM, et al. Liver-fat accumulation and insulin resistance in obese women with previous gestational diabetes. Obes Res. 2002;10:859–867. doi: 10.1038/oby.2002.118. [DOI] [PubMed] [Google Scholar]
- 58.Seppala-Lindroos A, Vehkavaara S, Hakkinen AM, et al. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002;87:3023–3028. doi: 10.1210/jcem.87.7.8638. [DOI] [PubMed] [Google Scholar]
- 59••.Morino K, Petersen KF, Shulman GI. Molecular mechanisms of insulin resistance in humans and their potential links with mitochondrial dysfunction. Diabetes. 2006;55(Suppl 2):S9–S15. doi: 10.2337/db06-S002. This review presents evidence in support of the hypothesis that the accumulation of intracellular lipid may be related to mitochondrial density and mitochondrial dysfunction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samuel VT, Liu ZX, Qu X, et al. Mechanism of hepatic insulin resistance in nonalcoholic fatty liver disease. J Biol Chem. 2004;279:32345–32353. doi: 10.1074/jbc.M313478200. [DOI] [PubMed] [Google Scholar]
- 61.Newton AC. Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J. 2003;370:361–371. doi: 10.1042/BJ20021626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes. 2002;51:2005–2011. doi: 10.2337/diabetes.51.7.2005. [DOI] [PubMed] [Google Scholar]
- 63.Yu C, Chen Y, Cline GW, et al. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 64.Dey D, Basu D, Roy SS, et al. Involvement of novel PKC isoforms in FFA induced defects in insulin signaling. Mol Cell Endocrinol. 2006;246:60–64. doi: 10.1016/j.mce.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 65.Greene MW, Morrice N, Garofalo RS, Roth RA. Modulation of human insulin receptor substrate-1 tyrosine phosphorylation by protein kinase Cdelta. Biochem J. 2004;378:105–116. doi: 10.1042/BJ20031493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kellerer M, Mushack J, Mischak H, Haring HU. Protein kinase C (PKC) epsilon enhances the inhibitory effect of TNF alpha on insulin signaling in HEK293 cells. FEBS Lett. 1997;418:119–122. doi: 10.1016/s0014-5793(97)01357-4. [DOI] [PubMed] [Google Scholar]
- 67.Braiman L, Alt A, Kuroki T, et al. Insulin induces specific interaction between insulin receptor and protein kinase C delta in primary cultured skeletal muscle. Mol Endocrinol. 2001;15:565–574. doi: 10.1210/mend.15.4.0612. [DOI] [PubMed] [Google Scholar]
- 68.Strack V, Stoyanov B, Bossenmaier B, et al. Impact of mutations at different serine residues on the tyrosine kinase activity of the insulin receptor. Biochem Biophys Res Commun. 1997;239:235–239. doi: 10.1006/bbrc.1997.7457. [DOI] [PubMed] [Google Scholar]
- 69.Gao Z, Zhang X, Zuberi A, et al. Inhibition of insulin sensitivity by free fatty acids requires activation of multiple serine kinases in 3T3-L1 adipocytes. Mol Endocrinol. 2004;18:2024–2034. doi: 10.1210/me.2003-0383. [DOI] [PubMed] [Google Scholar]
- 70••.Bergman RN, Kim SP, Hsu IR, et al. Abdominal obesity: role in the pathophysiology of metabolic disease and cardiovascular risk. Am J Med. 2007;120:S3–S8. doi: 10.1016/j.amjmed.2006.11.012. discussion S29–S32. This review summarizes evidence that the sympathetic nervous system is an additional factor that may contribute to an increased flux of FFA from adipose tissue to the liver, and also presents the hypothesis that increased concentrations of plasma FFA are important in mediating compensatory insulin secretion/hyperinsulinemia in insulin resistance associated with obesity. [DOI] [PubMed] [Google Scholar]
- 71.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shimomura I, Matsuda M, Hammer RE, et al. Decreased IRS-2 and increased SREBP-1c lead to mixed insulin resistance and sensitivity in livers of lipodystrophic and ob/ob mice. Mol Cell. 2000;6:77–86. [PubMed] [Google Scholar]
- 73.Lewis GF, Uffelman KD, Szeto LW, Steiner G. Effects of acute hyperinsulinemia on VLDL triglyceride and VLDL apoB production in normal weight and obese individuals. Diabetes. 1993;42:833–842. doi: 10.2337/diab.42.6.833. [DOI] [PubMed] [Google Scholar]
- 74.Brown AM, Gibbons GF. Insulin inhibits the maturation phase of VLDL assembly via a phosphoinositide 3-kinase-mediated event. Arterioscler Thromb Vasc Biol. 2001;21:1656–1661. doi: 10.1161/hq1001.096640. [DOI] [PubMed] [Google Scholar]
- 75.Taghibiglou C, Rashid-Kolvear F, Van Iderstine SC, et al. Hepatic very low density lipoprotein-ApoB overproduction is associated with attenuated hepatic insulin signaling and overexpression of protein-tyrosine phosphatase 1B in a fructose-fed hamster model of insulin resistance. J Biol Chem. 2002;277:793–803. doi: 10.1074/jbc.M106737200. [DOI] [PubMed] [Google Scholar]
- 76.Lin MC, Gordon D, Wetterau JR. Microsomal triglyceride transfer protein (MTP) regulation in HepG2 cells: insulin negatively regulates MTP gene expression. J Lipid Res. 1995;36:1073–1081. [PubMed] [Google Scholar]
- 77.Hussain MM, Shi J, Dreizen P. Microsomal triglyceride transfer protein and its role in apoB-lipoprotein assembly. J Lipid Res. 2003;44:22–32. doi: 10.1194/jlr.r200014-jlr200. [DOI] [PubMed] [Google Scholar]
- 78.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23:201–229. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 79.Brunzell JD, Hazzard WR, Porte D, Jr, Bierman EL. Evidence for a common, saturable, triglyceride removal mechanism for chylomicrons and very low density lipoproteins in man. J Clin Invest. 1973;52:1578–1585. doi: 10.1172/JCI107334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Havel RJ, Gordon RS., Jr Idiopathic hyperlipemia: metabolic studies in an affected family. J Clin Invest. 1960;39:1777–1790. doi: 10.1172/JCI104202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eckel RH, Yost TJ, Jensen DR. Alterations in lipoprotein lipase in insulin resistance. Int J Obes Relat Metab Disord. 1995;19(Suppl 1):S16–S21. [PubMed] [Google Scholar]
- 82••.Bansal S, Buring JE, Rifai N, et al. Fasting compared with nonfasting triglycerides and risk of cardiovascular events in women. JAMA. 2007;298:309–316. doi: 10.1001/jama.298.3.309. In the Women’s Health Study, nonfasting TG concentrations were associated with incident cardiovascular events, independent of traditional cardiac risk factors, levels of other lipid parameters, and markers of insulin resistance. In contrast, fasting TG levels showed little independent relationship with incident cardiovascular events. [DOI] [PubMed] [Google Scholar]
- 83.Hyson D, Rutledge JC, Berglund L. Postprandial lipemia and cardiovascular disease. Curr Atheroscler Rep. 2003;5:437–444. doi: 10.1007/s11883-003-0033-y. [DOI] [PubMed] [Google Scholar]
- 84.Karpe F. Postprandial lipoprotein metabolism and atherosclerosis. J Intern Med. 1999;246:341–355. doi: 10.1046/j.1365-2796.1999.00548.x. [DOI] [PubMed] [Google Scholar]
- 85•.Lopez-Miranda J, Perez-Martinez P, Marin C, et al. Postprandial lipoprotein metabolism, genes and risk of cardiovascular disease. Curr Opin Lipidol. 2006;17:132–138. doi: 10.1097/01.mol.0000217894.85370.c2. A useful summary of the genetic polymorphisms that are associated with post-prandial lipid/lipoprotein responses. [DOI] [PubMed] [Google Scholar]
- 86••.Nordestgaard BG, Benn M, Schnohr P, Tybjaerg-Hansen A. Nonfasting triglycerides and risk of myocardial infarction, ischemic heart disease, and death in men and women. JAMA. 2007;298:299–308. doi: 10.1001/jama.298.3.299. In a 26-year prospective study, elevated nonfasting TG concentrations were associated with increased risk of myocardial infarction, ischemic heart disease and overall mortality. The subjects were divided into six groups within each gender based on baseline nonfasting TG concentrations. Even within the mode group (nonfasting TG =88.5–176.2 mg/dl) the hazard ratios for myocardial infarction were significantly higher (2.2 for women, 1.6 for men) than the reference group (nonfasting TG <88.5 mg/dl) [DOI] [PubMed] [Google Scholar]
- 87.Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002;43:1363–1379. doi: 10.1194/jlr.r200004-jlr200. [DOI] [PubMed] [Google Scholar]
- 88••.Marcovina S, Packard CJ. Measurement and meaning of apolipoprotein AI and apolipoprotein B plasma levels. J Intern Med. 2006;259:437–446. doi: 10.1111/j.1365-2796.2006.01648.x. An excellent review of the roles of circulating apolipoprotein AI and B concentrations in the development of an atherogenic lipoprotein phenotype. [DOI] [PubMed] [Google Scholar]
- 89••.Packard CJ. Small dense low-density lipoprotein and its role as an independent predictor of cardiovascular disease. Curr Opin Lipidol. 2006;17:412–417. doi: 10.1097/01.mol.0000236367.42755.c1. An informative summary of the evidence linking increased plasma concentrations of small dense LDL cholesterol to cardiovascular disease risk. [DOI] [PubMed] [Google Scholar]
- 90.Boden G, Lebed B, Schatz M, et al. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes. 2001;50:1612–1617. doi: 10.2337/diabetes.50.7.1612. [DOI] [PubMed] [Google Scholar]
- 91.Jacob S, Machann J, Rett K, et al. Association of increased intramyocellular lipid content with insulin resistance in lean nondiabetic offspring of type 2 diabetic subjects. Diabetes. 1999;48:1113–1119. doi: 10.2337/diabetes.48.5.1113. [DOI] [PubMed] [Google Scholar]
- 92.Krssak M, Falk Petersen K, Dresner A, et al. Intramyocellular lipid concentrations are correlated with insulin sensitivity in humans: a 1H NMR spectroscopy study. Diabetologia. 1999;42:113–116. doi: 10.1007/s001250051123. [DOI] [PubMed] [Google Scholar]
- 93.Virkamaki A, Korsheninnikova E, Seppala-Lindroos A, et al. Intramyocellular lipid is associated with resistance to in vivo insulin actions on glucose uptake, antilipolysis, and early insulin signaling pathways in human skeletal muscle. Diabetes. 2001;50:2337–2343. doi: 10.2337/diabetes.50.10.2337. [DOI] [PubMed] [Google Scholar]
- 94.Boden G. Interaction between free fatty acids and glucose metabolism. Curr Opin Clin Nutr Metab Care. 2002;5:545–549. doi: 10.1097/00075197-200209000-00014. [DOI] [PubMed] [Google Scholar]
- 95•.Holland WL, Knotts TA, Chavez JA, et al. Lipid mediators of insulin resistance. Nutr Rev. 2007;65:S39–46. doi: 10.1111/j.1753-4887.2007.tb00327.x. This review summarizes potential lipid mediators of impaired insulin signaling including DAG, glycerolipids, sphingolipids, ceramide, glucosylceramide, cholesterol, FFA, and their proposed mechanisms of action. [DOI] [PubMed] [Google Scholar]
- 96•.Bergman RN, Kim SP, Catalano KJ, et al. Why visceral fat is bad: mechanisms of the metabolic syndrome. Obesity (Silver Spring) 2006;14(Suppl 1):16S–19S. doi: 10.1038/oby.2006.277. A summary of the evidence supporting the ‘portal theory’ and the ‘overflow hypothesis’ as contributors to insulin hepatic and peripheral resistance. [DOI] [PubMed] [Google Scholar]
- 97.Jensen MD, Cardin S, Edgerton D, Cherrington A. Splanchnic free fatty acid kinetics. Am J Physiol Endocrinol Metab. 2003;284:E1140–E1148. doi: 10.1152/ajpendo.00268.2002. [DOI] [PubMed] [Google Scholar]
- 98.Kraegen EW, Clark PW, Jenkins AB, et al. Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes. 1991;40:1397–1403. doi: 10.2337/diab.40.11.1397. [DOI] [PubMed] [Google Scholar]
- 99••.Petersen KF, Shulman GI. Etiology of insulin resistance. Am J Med. 2006;119:S10–S16. doi: 10.1016/j.amjmed.2006.01.009. An excellent summary of the work of Shulman and colleagues, and others, supporting the hypothesis that accumulation of intracellular lipid metabolites rather than obesity per se is the major driver of insulin resistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100••.Lonardo A, Lombardini S, Scaglioni F, et al. Hepatic steatosis and insulin resistance: does etiology make a difference? J Hepatol. 2006;44:190–196. doi: 10.1016/j.jhep.2005.06.018. This study compares patients with hepatic steatosis resulting from three different pathological conditions with each other and with healthy control subjects and concludes that the etiology of hepatic fat accumulation affects its association with insulin resistance. [DOI] [PubMed] [Google Scholar]
- 101.Perez-Martinez P, Perez-Jimenez F, Ordovas JM, et al. Postprandial lipemia is modified by the presence of the APOB-516C/T polymorphism in a healthy Caucasian population. Lipids. 2007;42:143–150. doi: 10.1007/s11745-007-3027-7. [DOI] [PubMed] [Google Scholar]
- 102•.Perez-Martinez P, Perez-Jimenez F, Ordovas JM, et al. The APOB -516C/T polymorphism is associated with differences in insulin sensitivity in healthy males during the consumption of diets with different fat content. Br J Nutr. 2007;97:622–627. doi: 10.1017/S0007114507659005. In contrast to the FHBL patients studied by Lonardo et al. [100••], this study demonstrates that an ApoB polymorphism that increases the transcription rate of the ApoB gene is associated with increased plasma triglyceride concentrations and insulin resistance. [DOI] [PubMed] [Google Scholar]
- 103.Bandsma RH, Smit GP, Kuipers F. Disturbed lipid metabolism in glycogen storage disease type 1. Eur J Pediatr. 2002;161(Suppl 1):S65–S69. doi: 10.1007/s00431-002-1007-8. [DOI] [PubMed] [Google Scholar]
- 104.Havel RJ, Balasse EO, Williams HE, et al. Splanchnic metabolism in von Gierke’s disease (glycogenosis type I) Trans Assoc Am Physicians. 1969;82:305–323. [PubMed] [Google Scholar]
- 105.Bjorkegren J, Beigneux A, Bergo MO, et al. Blocking the secretion of hepatic very low density lipoproteins renders the liver more susceptible to toxin-induced injury. J Biol Chem. 2002;277:5476–5483. doi: 10.1074/jbc.M108514200. [DOI] [PubMed] [Google Scholar]
- 106•.Monetti M, Levin MC, Watt MJ, et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6:69–78. doi: 10.1016/j.cmet.2007.05.005. These investigators demonstrate a dissociation between hepatic steatosis and insulin resistance, as well as a dissociation between hepatic DAG accumulation and insulin resistance, in mice overexpressing DGAT in the liver. [DOI] [PubMed] [Google Scholar]
- 107•.Gutierrez-Juarez R, Pocai A, Mulas C, et al. Critical role of stearoyl-CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance. J Clin Invest. 2006;116:1686–1695. doi: 10.1172/JCI26991. In this study, hepatic fat accumulation induced by targeted disruption of SCD-1 activity is not associated with insulin resistance in overfed mice. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gibbons GF, Wiggins D, Brown AM, Hebbachi AM. Synthesis and function of hepatic very-low-density lipoprotein. Biochem Soc Trans. 2004;32:59–64. doi: 10.1042/bst0320059. [DOI] [PubMed] [Google Scholar]
- 109.Millar JS, Stone SJ, Tietge UJ, et al. Short-term overexpression of DGAT1 or DGAT2 increases hepatic triglyceride but not VLDL triglyceride or apoB production. J Lipid Res. 2006;47:2297–2305. doi: 10.1194/jlr.M600213-JLR200. [DOI] [PubMed] [Google Scholar]
- 110•.Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–1090. doi: 10.1002/hep.21763. This paper reports that the amounts of TG and DAG are increased in the livers of NAFLD patients compared with healthy patients, but the amount of FFA is not. [DOI] [PubMed] [Google Scholar]