

Summary
Protein arginine methylation regulates multiple biological processes. Deregulation of protein arginine methyltransferase (PRMT) activities has been observed in many disease phenotypes. Small molecule probes that target PRMTs with strong affinity and selectivity can be used as valuable tools to dissect biological mechanisms of arginine methylation and establish the role of PRMT proteins in a disease process. In this work, we report synthesis and evaluation of a class of carbocyanine compounds containing indolium, benz[e]indolium or benz[c,d]indolium heterocyclic moieties that bind to the predominant arginine methyltransferase PRMT1 and inhibit its methyltransferase activity at low micromolar potencies. In particular, the developed molecules have long wavelength colorimetric and fluorometric photoactivities, which can be used for optical and near-infrared fluorescence imaging in cells or biological tissues. Together, these new chemical probes have potential application in PRMT studies both as enzyme inhibitors and as fluorescent dyes for microscope imaging.
Introduction
Arginine methylation is a prevalent protein post-translational modification in cells, found in both nuclear and cytoplasmic proteins. This biochemical reaction is mediated by protein arginine methyltransferases (PRMTs) that utilize S-adenosyl-L-methionine (AdoMet, SAM) as the methyl donor to modify the guanidinium side chain of specific arginine residues, resulting in mono and di-methylated arginine residues and the coproduct S-adenosyl-L-homocysteine (AdoHcy, SAH). PRMTs are involved in the regulation of diverse biological processes ranging from structural remodeling of chromatins, gene transcription, RNA processing, DNA repair, translation, nucleocytoplasmatic protein shuttling, nuclear hormone receptor-mediated signaling, to cell proliferation and differentiation (Di Lorenzo et al., 2011; Wolf, 2009; Krause et al., 2007). Deregulation of PRMTs and arginine methylation has been reported to associate with a variety of human diseases including several types of tumors (e.g. breast cancer, prostate cancer, and leukemia) (Pal and Sif, 2007; Cheung et al., 2007), pulmonary disorders (Zakrzewicz and Eickelberg, 2009), and cardiovascular diseases (Beltowski and Kedra, 2006; Maas, 2005) etc. In particular, the predominant PRMT member, PRMT1, has been shown to impact a number of disease pathways. PRMT1 is an essential component of the mixed lineage leukemia (MLL) oncogenic transcriptional complex and confers an aberrant transcriptional activation property critical for the induction of leukemia (Cheung et al., 2007). Prominent roles of PRMT1 include the coactivation of nuclear receptors and regulation of hormone-dependent cancers (Koh et al., 2001; Wagner et al., 2006). Indeed, PRMT1 was found up-regulated in breast cancer concomitantly with a change in substrate methylation (Goulet et al., 2007). PRMT1 variant 1 expression may be used as a marker of unfavorable prognosis for breast cancer patients (Mathioudaki et al., 2011). PRMT1 variant 2 was regarded as a marker of unfavorable prognosis in colon cancer patients (Papadokostopoulou et al., 2009). Thus, targeting PRMT activities with small molecule inhibitors could be an effective therapeutic strategy in new drug development (Krause et al., 2007; Zheng et al., 2008; Spannhoff et al., 2009). Indeed, quite a few research efforts have been investigated in the past years in attempt to develop potent and selective PRMT inhibitors (Spannhoff et al., 2007a; Spannhoff et al., 2007b; Ragno et al., 2007; Mai et al, 2008; Cheng et al., 2004; Bedford et al., 2005; Li et al., 2010; Bissinger et al., 2011a; Feng et al., 2010; Cheng et al., 2011a; Cheng and Bedford, 2011b; Bissinger et al., 2011b; Heinke et al., 2009; Spannhoff et al., 2007c; Spannhoff et al., 2007d; Dowden, et al., 2011).
Carbocyanine dyes are unique organic molecules that contain a conjugated electron-deficient system between two heterocyclic nitrogen atoms that provides characteristically long absorption and emission wavelengths; the associated wavelengths of carbocyanine dyes have been exploited in many applications (Strekowski et al., 2003; Mishra et al., 2000). Using various synthetic methods, the conjugated system of these compounds can be altered to assume specific absorption and fluorescence spectra in the range of 400 to 1000 nm (Narayanan and Patonay, 1995; Mojzych and Henary, 2008). The number of carbon atoms between the heterocyclic nitrogen atoms in the polymethine chain determines the common name for the dyes. In particular, penta- and heptacarbocyanine dyes (5 and 7 carbon atoms, respectively) have been extensively utilized for binding analyses; however, tricarbocyanine dyes (3 carbon atoms) remain, until recently, unexplored (Losytskyy et al., 2005). Tricarbocyanine dyes commonly absorb and emit light in the visible region; however, varying the conjugated heterocyclic moieties greatly shifts the maximum absorption and emission wavelengths. Their binding properties to biomolecules can be studied using relative spectrophotometric changes either in absorption or emission spectra (Flanagan et al., 1997). In this study, we synthesized and identified a set of tricarbocyanine dyes that specifically inhibit the enzymatic activity of PRMT1. Also, a proof-of-principle study has demonstrated that these molecules are well suited for colorimetric and fluorescent imaging of PRMT activities in living cells. Overall, the identified carbocyanine molecules with unique photoactive properties could be powerful chemical probes in elucidating mechanisms and functions of PRMTs underlying different biological or pathological contexts.
Results and Discussion
Chemistry
Toward the development of PRMT inhibitors and imaging agents, we have concentrated on hydrophobic trimethine cyanine derivatives utilizing ethyl, butyl and phenylpropyl halides for nitrogen alkylation of the heterocyclic indolium, benz[e]indolium and benz[c,d]indolium moieties (Schemes 1 and 2). The synthetic sequence to NIR trimethine cyanine dyes 11-13 involves 6 steps as shown in Scheme 1. The key step is the formation of 5-(benz[c,d]indol-1H-2-ylidene)-1,3-dioxane-4,6-dione 4. The alkylation of 4 provides an easy access to various 1-alkyl-substituted 2-methylbenz[c,d]-indolium salts, the important precursors to a set of various carbocyanine dyes.
Scheme 8.

Scheme 9.

Benz[c,d]indole derivatives 2-4 were synthesized starting with the commercial substrate 1. Compound 4 was reacted with various alkyl iodides under basic conditions to afford esters 5-7, which were correspondingly transformed into various quaternary salts 8-10. Salts 8-10 were purified by crystallization from methanol. The condensation of 8-10 with triethyl orthoformate in Ac2O furnished the benz[c,d]indolium tri-methine cyanine dyes 11-13 in very good yield.
Additionally, tricarbocyanines containing slightly varied heterocyclic end units, indolium 14 and benz[e]indolium 15, were synthesized as depicted in Scheme 2. Using similar chemistry, as outlined in Scheme 1, quaternization of commercially obtained indolium and benz[e]indolium 14-15 proceeds by a SN2 reaction using corresponding alkyl halides in boiling acetonitrile to yield salts 16-21 corresponding to starting heterocyclic subunits. Tricarbocyanines 22-27 were synthesized in good yield under identical reaction conditions depicted in Scheme 1. Final trimethine cyanines were purified by recrystallization from methanol or column chromatography using 2% MeOH in DCM as eluting solvent. Experimental procedures for novel compound preparation, chemical and physical spectroscopic data are reported in the Supplemental Information.
Biochemical identification of the carbocyanine inhibitors of PRMT1
In an effort to discover potent and selective PRMT inhibitors, we synthesized and investigated several tricarbocyanine compounds by screening their anti-methyltransferase activity. Our initial hypothesis developed from the rationale that carbocyanine molecules typically display heteroatomic and cationic structures, which could potentially resemble positively charged arginine residues in protein substrates, thereby leading to competitive PRMT inhibitors. One of the characteristic features of carbocyanine molecules is their absorption and fluorescence emission in the near-IR range which offers a great advantage for in vivo fluorescent imaging with minimal background interference from biomolecular autofluorescence. In the inhibitor screening study, we selected PRMT1 as the primary target because it is the predominant PRMT isoform found in mammalian cells and is involved in many key biological processes. In a typical anti-arginine methyltransferase screening assay, the reaction mixture involved carbocyanine compounds, recombinantly expressed His-tagged PRMT1, [14C]-labeled AdoMet, and a histone H4 peptide containing the 20-amino acid sequence on the N-terminal tail of histone H4, i.e. H4-20. The reaction was conducted at 30°C in a volume of 30 μl. The degree of diminishment in PRMT1 activity was used as a parameter to evaluate the potency of the compounds in inhibiting PRMT1-mediated arginine methylation. In a more quantitative analysis of the inhibition potency, we measured the methyltransferase activity of the enzyme at a range of concentrations of inhibitors and IC50 values were derived from the dose response curve as the half-inhibition concentration. For all the enzymatic assays, the methylation reaction was controlled under the initial condition so that the reaction yields of the limiting substrates were lower than 15%, which was to ensure that the concentrations of AdoMet and peptide substrate did not decrease significantly over the time course of methylation reaction. This procedure also prevented potential product inhibition from SAH.
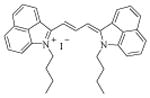
From the biochemical screening, we found three structurally related tricarbocyanine compounds that show desired anti-PRMT1 activity; the chemical structures and corresponding IC50 values are shown in Table 1 (Group A). Clearly these compounds are structurally analogous, all of which contain a delocalized positive charge on the nitrogen atom and two benz[c,d]indolium moieties at the ends of the trimethine cyanine main chain. The inhibition potency is in the order of 11 > 12 > 13. This could possibly suggest that increasing the size of the N-substituent causes increasing steric hindrance, which prevents the compound from entering the inhibitor-binding site of the enzyme. Interestingly, 11 (MHI-21) shows the smallest size but best inhibition potency with IC50 of 4.1 μM, which warrants further investigation.
Table 1.
The Names, Structures and IC50 Data of Trimethine Cyanine Compounds.
| Type of compound | Name | Structure | IC50 (μM) |
|---|---|---|---|
|
Group A Trimethine cyanine compounds containing benzo[c,d]indolium groups in the two ends |
11 [MHI-21] |
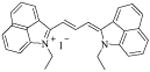
|
4.1 ± 0.1 |
| 12 [MHI-06] |
|
12.0 ± 0.5 | |
| 13 [MHI-36] |
|
15.5 ± 0.2 | |
|
Group B Trimethine cyanine compounds containing indolium groups in the two ends. |
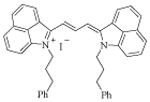
22 [E-14] |
|
284.2 ± 3.5 |
| 23 [E-6] |
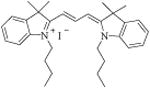
|
160.8 ± 1.2 | |
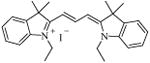
| 24 [E-4] |
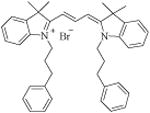
|
6.3 ± 0.3 | |
| 28 [A-C3] |
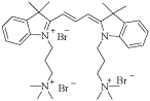
|
368.3 ± 25.9 | |
| 29 [A-N3] |
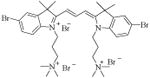
|
291.7 ± 40.1 | |
| 30 [MHI-106] |
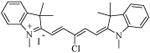
|
At 100 μM concentration 80% activity retained | |
| 31 [MHI-128] |
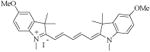
|
At 100 μM concentration 95% activity retained | |
|
Group C Trimethine cyanine compounds containing benzo[e]indolium group as the end units |
25 [E-5] |
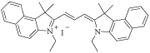
|
20.3 ± 0.2 |
| 26 [E-18] |
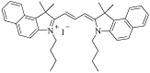
|
18.0 ± 0.5 | |
| 27 [E-8] |
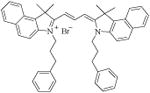
|
14.3 ± 0.2 |
To further this study, we synthesized and tested another set of carbocyanine compounds containing an indolium group as the heterocyclic moiety in order to screen for more potent inhibitors and explore possible structure-activity relationship (SAR) (Group B in Table 1). Compared to the above three benz[c,d]indolium-containing dyes, this set of compounds with the exception of 24 presented significantly weaker activity. IC50 values of 22, 23, 28 and 29 were all greater than 150 μM. For 30 and 31, at 100 μM concentration, PRMT1 still retained 80-95% of its activity. These data demonstrate that reducing the heterocyclic size generally lowers the binding of the carbocyanine molecules to PRMT1. Notably, the two molecules that harness additional positive charges on the N-substitution group, i.e. 28 and 29, do not offer any advantage for enhancing the inhibition potency. This indicates that charge alone is not the sole factor that determines the specific inhibitor-enzyme interactions, as a certain degree of hydrophobicity of the N-substitution is required for efficient inhibition. Contrary to the benz[c,d]indolium trimethine compounds (Group A in Table 1), longer and bulky substituents on the heterocyclic ring nitrogen increase inhibition potency. In particular, 24 having a bulky phenylpropyl substituent showed the strongest potency, with IC50 of 6.3 μM. This may suggest that a bulky hydrophobic group either on the heterocyclic ring moiety or on the heterocyclic ring nitrogen substitution is required for effective binding to PRMT1 protein.
To provide more information on the effect of the heterocyclic moiety, we synthesized and tested three other trimethine cyanine analogs bearing the benz[e]indolium end unit (Group C in Table 1) while maintaining the identical set of nitrogen substituents, i.e., ethyl, butyl, and phenylpropyl group. The potency of benz[e]indolium cyanines becomes weaker than the benz[c,d]indolium cyanines that contain the same substituent. For instance, comparing the ethyl-substituted compounds bearing the benz[e]indole 25 (IC50 of 20.3 μM) and the benz[c,d]indole 11 (IC50 of 4.1 μM) indicates that the position of the benz group attached on the heterocyclic ring affects inhibitor-enzyme interaction. Also, the weaker binding of benz[e]indolium is rescued by the incorporation of a bulky phenylpropyl substituent on the heterocyclic nitrogen; this is observed as the inhibition potency increases from 25 to 27. In contrast, in benz[c,d]indolium-containing cyanine compounds, the end head group seems to dominate the inhibitor-PRMT1 binding and the bulky substitutions on the heterocyclic nitrogen do not offer any positive contributions (i.e. 36).
Noncompetitive inhibition by 11
The finding that certain trimethine cyanine compounds exhibit low micromolar anti-PRMT1 activity is encouraging. Especially, 11 shows the smallest molecule size but the strongest potency in the tested compounds; the other one having similar potency to 11 is 24 (E-4). Thus, we next carried out more detailed analyses of PRMT1 inhibition by 11 in order to understand the inhibitory mechanism of this type of carbocyanine molecules.
First, we conducted steady-state enzymatic measurements to analyze the kinetic pattern of inhibition by 11. The initial velocities of PRMT1 were measured at several selected concentrations of the inhibitor over a range of varied concentrations of one substrate while fixing the concentration of the other. The kinetic inhibition data points were analyzed by fitting to the linear competitive or noncompetitive inhibition equations. The data were plotted in the double reciprocal format with 1/velocity versus 1/concentration of the varied substrate (Figure 1). It is seen that in both double-reciprocal plots, a series of straight lines intersect at a point in the second quadrant. These data demonstrate that 11 is a noncompetitive inhibitor with regard to both H4 and AdoMet substrates.
Figure 1.
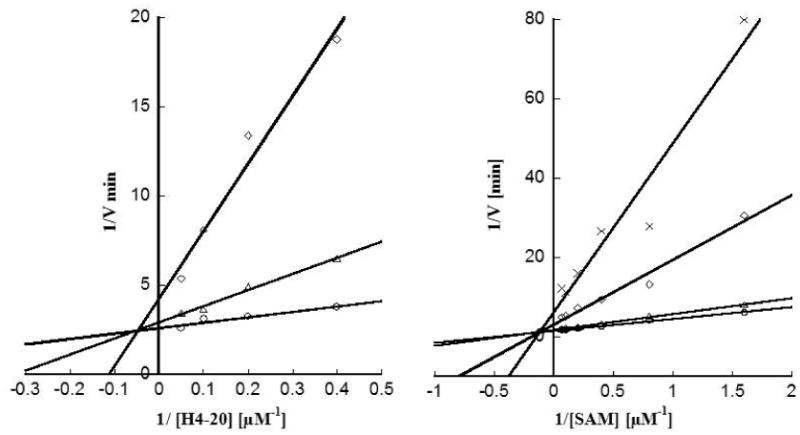
Steady-State Kinetic Analysis of PRMT1 Inhibition by 11. (A) Double-reciprocal plot of the initial velocities versus H4 peptide with AdoMet concentration fixed at 5 μM and with varied concentrations of 11 at 2.5 μM (O), 5 μM (Δ), and 7.5 μM (◊). (B) Double-reciprocal plot of the initial velocities versus AdoMet with H4-20 concentration fixed at 2 μM and with varied concentrations of 11 at 0 μM (O), 2.5 μM (Δ), 5 μM (◊), and 6.5 μM (×).
Reversible vs irreversible inhibition
To determine whether 11 reversibly or irreversibly inhibits PRMT1 enzyme, a dialysis experiment was performed. For each assay, fixed amounts of enzyme and inhibitor were incubated at 30°C for 1 h to allow for enzyme inactivation. Then the solution was dialyzed against the assay buffer for 72 h. The methyltransferase activity of PRMT1 before and after dialysis was measured using the filter binding method. As shown in Figure 2A, in the presence of 20 μM 11, PRMT1 only retained 10% activity prior to dialysis. After dialysis, the enzyme activity was restored to 66% suggesting that PRMT1 inhibition by 11 is reversible. To confirm this result, we measured time-dependent inhibition at several concentrations of the inhibitor (0, 1.5, 2.5, 4.0 μM). In the time vs product plot (Figure 2B), straight lines were seen for all the measurements instead of hyperbolic curves. Lack of any nonlinear time-dependent inhibition indicates that the inhibition is reversible and there is no covalent interaction between PRMT1 and the inhibitor.
Figure 2.

Test of the Reversibility of PRMT1 Inhibition by 11. (a) Dialysis experiment in presence and absence of 11. (b) Progression assay of PRMT1 inhibition at different concentrations (1.5, 2.5, and 4.0 μM) of 11.
Binding Model Analysis of Inhibitor-Enzyme Interaction
To understand the structural basis of PRMT1 inhibition by 11, we performed a docking study of the compound with PRMT1 structure using the program Autodock. A homology model of human PRMT1 structure was created from rat PRMT3. In the docking, a grid box was created to cover the whole protein structure. The docking result was analyzed according to the lowest energy order. As it turned out, the docked ligand was found to distribute predominantly in three regions on the protein surface (Figure 3A); none of them overlap with the arginine and SAM binding sites. Therefore 11 is likely an allosteric inhibitor. These results are in agreement with the kinetic results, which showed noncompetitive inhibition in respect to H4-20 and AdoMet. For a more accurate analysis of the binding mode, the compound was individually docked in each of the three regions by choosing a grid box covering only that selected region. A close inspection on region 1 revealed that 11 binds to a pocket formed by Glu 19, Tyr 35, Ser 38, Tyr 39, Glu 47, Met 48, Tyr 148, Tyr 152, Val 206, Pro 217, His 293, Lys 295, Thr 297, Arg 327, Met 352 and Arg 353 residues (Figure 3B). The inhibitor electrostatically interacts with residues Glu 19 and Glu 47, and makes hydrogen bond contacts with Ser 38, Tyr 39, Tyr 148, Tyr 152, Lys 295, Thr 297, Met 352 and Arg 353. The hydroxyl group of Tyr 148 seems to have a strong H-bonding interaction with two nitrogens of 11 (3.7 Å and 4.3 Å) and a moderate interaction with Tyr 152 (6.7 Å and 7.0 Å). A detailed study on region 2 shows that the compound binds to the pocket formed by residues Leu 49, His 45, Gly 80, Thr 81, Gly 82, Ile 83, Met 86, Phe 87, Asn 102, Tyr 107, and Ile111 and makes hydrogen bond interactions with Phe 42, His 45, Gly 80, Thr 81, Gly 82, Ile 83, Met 86 and Tyr 107 residue (Figure 3C). There is a moderate H-bonding interaction between the heterocyclic nitrogen of the compound and the side chain and backbone oxygen atoms of Thr 81, with 5.8 Å, 5.7 Å, respectively. Similar docking study on region 3 revealed that the compound binds to the pocket formed by Glu 47, Met 48, Leu 49, Lys 50, Glu 52, Val 53, Tyr 148, Pro 290, Tyr 291, Thr 292 and His 293 residue and makes several electrostatic interactions with residues Glu 47, Asp 51, Glu 52, and hydrogen bond interaction with Lys 50, Met 48, Leu 49, Glu 52, Val 53, Pro 290, Tyr 291, and Thr 292 (Figure 3D). Together, the inhibitory activity of 11 may come from its single or combined interactions at the three major binding sites of PRMT1.
Figure 3.
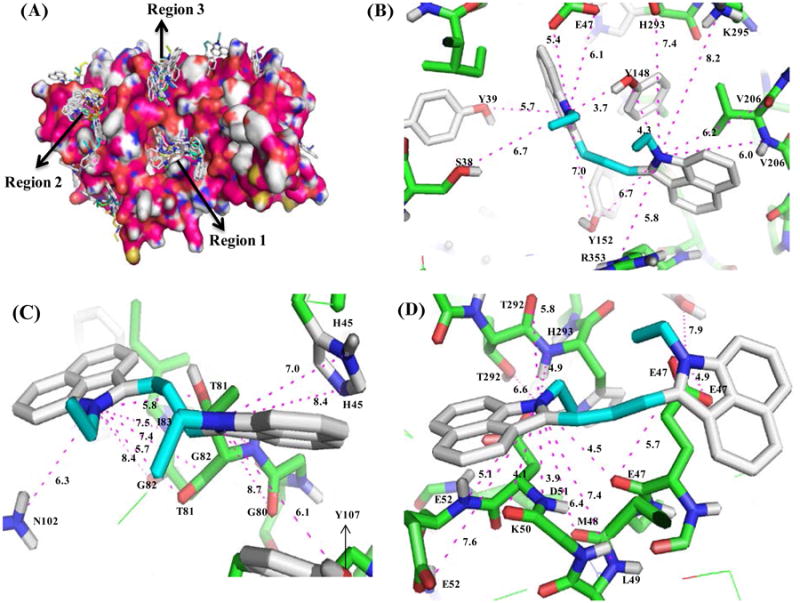
Docking Results. A) Probable binding regions of 11. Binding mode of the inhibitor in Region 1 (B), in Region 2 (C), and in Region 3 (D).
Investigation of Interaction of 11 and PRMT1 Using Site-Directed Mutagenesis
To test the validity of the docking results, site-directed mutagenesis was conducted to mutate the residues predicted to interact with the compound in the docking model. Since multiple hydrogen bonding interactions occur between the side chains of Tyr 148, Tyr 152, Thr 81 and Thr 292 residues and the heterocyclic ring nitrogen atom of 11, we introduced four mutations, i.e., Y148A, Y152A, T81A, T292A. All these PRMT1 mutants were produced using QuikChange site-directed mutagenesis protocol, expressed in E. coli, and purified by Ni-NTA affinity chromatography. Activity of each mutant protein was examined by the radioactive methyltransferase assay in which recombinant PRMT 1, [14C]AdoMet, and H4-20 were incubated at 30°C in the presence or absence of 4 μM 11. The retained fractional activity was quantified to evaluate the inhibition potency of the inhibitor on each PRMT mutant (Figure 4). It is seen that the activities of Y148A, T81A and T292A protein were inhibited by 11 by ≈ 60%, 65% and 70% respectively, less than that of wild type PRMT1 (92%). These results support that Y148, T81 and T292 residues might interact with 11 and thus mutation of these residues decreased the binding and inhibition. 11 inhibited Y152A mutant with same potency as is for wt-PRMT1, which signifies that Y152 quite likely is not involved in interaction with 11.
Figure 4.
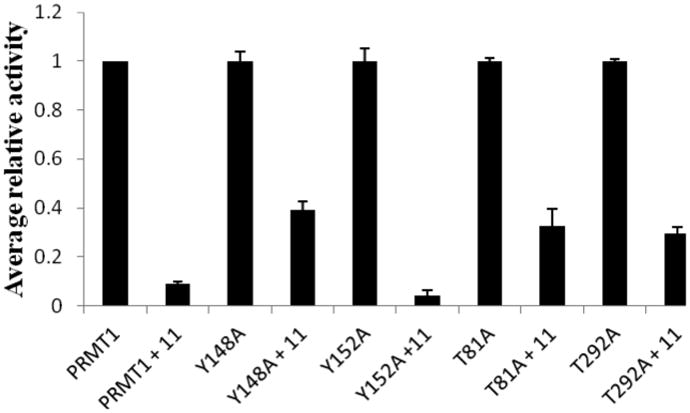
Inhibition of the Activity of WT- and Mutant- PRMT1 by 11. The reaction contained 2 μM H4-20, 5 μM [14C] AdoMet and 4 μM inhibitor. Activities of all PRMT1 mutants were normalized to the same level as that of wt-PRMT1 for clear comparison.
Selectivity of 11
We then investigated whether 11 is selective for PRMT1 inhibition. For this study, we examined the inhibition by 11 of several other PRMT isoforms, PRMT3, PRMT4/CARM1, PRMT5, and PRMT6, using the radioactive methyltransferase assays. The IC50 values are shown in Table 2. PRMT3 was inhibited at IC50 of 8.3 ± 0.1 μM, about 2.0 fold higher than PRMT1. 11 inhibits PRMT5 at IC50 9.1 ± 1.0 μM which is about 2.2 fold higher than PRMT1. The IC50 of 11 for CARM1 is particularly higher, ∼100 μM. 11 inhibits PRMT6 with an IC50 of 12.6 μM, 3.1-fold higher than PRMT1. Overall, 11 seems to inhibit all the tested PRMT proteins, with a little preference observed for PRMT1. The variation in inhibition potency likely is caused by the subtle differences between the structures of individual PRMT members.
Table 2.
Comparison of the Inhibition of PRMT-1, -3, -4, -5 and -6 by 11.a
| Peptide substrate | IC50 (μM) | |
|---|---|---|
| His6x-rPRMT1 | H4(1-20) | 4.1 ± 0.1 |
| His6x-PRMT3 | R4 | 8.3 ± 0.1 |
| GST-CARM1 | H3(1-31) | ≈ 100 |
| His6x-PRMT5 | H4(1-20) | 9.1 ± 1.0 |
| His6x-PRMT6 | H3(1-31) | 12.6 ± 1.1 |
Data obtained from the results of the radioactive methylation assays with varied concentrations of 11. For inhibition of His6x-PRMT1, 2 μM H4(1-20), 5 μM [14C]-SAM, and 0.05 μM enzyme were used. For inhibition of His6x-PRMT3, 2 μM R4, 5 μM [14C]-SAM, and 0.1 μM enzyme were used. For inhibition of GST-CARM1, 1 mM H3(1-31), 30 μM [14C]-SAM, and 5 μM enzyme were used. For inhibition of His6x-PRMT6, 10 μM H3(1-31), 5 μM [14C]-SAM, and 0.5 μM enzyme were used.
Evaluation of the Cellular Activity of 11
After the biochemical characterizations, we moved on to examine possible cellular effects of 11. We first examined subcellular distribution of 11 in HeLa cells. A marked feature of carbocyanine compounds is their spectral absorption and fluorescent emission in the near-IR region. Measured in methanol, 11 has absorption maximum at 758 nm and emission maximum at 774 nm. Interestingly, after incubation with 11 for 6 h, cells showed bright, blue-colored staining and can be viewed directly under bright field miscroscope. Microscopic images shown in Figure 5A and 5B and 5C demonstrated that 11 was concentrated in the nucleus of HeLa cells in a dose dependent manner and a small amount of 11 was also found in the cytoplasmic vesicles. Since carbocyanine compounds have fluorescent emission in the long wavelength region, we performed near-infrared fluorescent imaging of HeLa cells by confocal microscopy using a 633 nm laser for fluorescence excitation and a 715 to 785 nm Long Pass for emission. As can be seen in Figure 5E and 5F, after 6-h incubation in HeLa cells, the compound was concentrated around nucleus or in the nucleus and a low level was in the cytoplasmic vesicles. The result is consistent with the light microscopic images. As expected, the control HeLa cells treated with DMSO did not show any fluorescent signal (Figure 5D).
Figure 5.
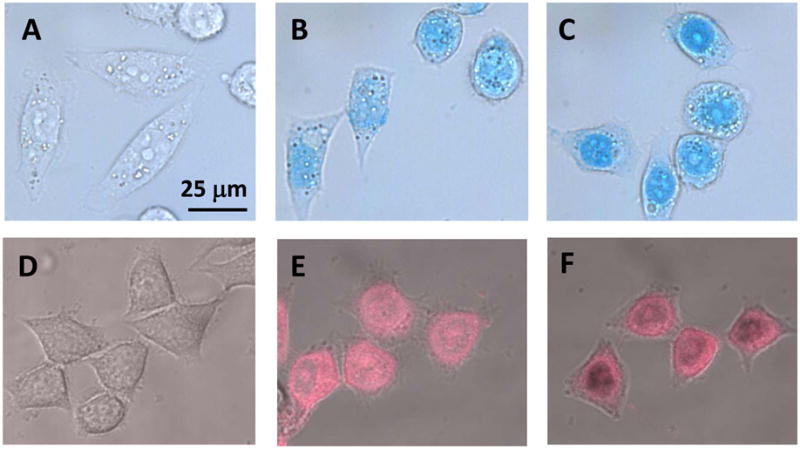
Microscopic Imagin g of 11 in HeLa Cells. HeLa cells are treated with 0 (control), 2, and 5 μM of 11. Figures A, B and C are the optical images of HeLa cells captured under visible light by CoolSNAP CF camera attached to a Nikon Ti-80 microscope. Figures D, E and F are fluorescent images of HeLa cel ls captured by fluorescence confocal microscopy.
We next studied the growth inhibitory effect of 11 in HeLa cells by using the MTT assay. The HeLa cells were cultured for 40 h in the presence of different doses of 11 (Figure 6). From the data, EC50, the concentration of the compound required to reduce the cell survival level to 50% at 48 h of exposure, was determined to be 0.94 ± 0.09 μM. This value is remarkably lower than the IC50 value (i.e. 4.1 μM) measured in the biochemical assay, likely suggesting that 11 easily penetrates the plasma membrane and subsequently affects the cell proliferation. To better understand the mechanism underlying the observed proliferation inhibition, we examined the effect of 11 on cell cycle of HeLa cells. The subpopulations of propidium iodide-stained HeLa cell were determined using flow cytometry at varying concentrations of the compound in a time dependent manner. Untreated HeLa cells showed a pattern of cell growth phase distribution at G1 (57.2%), S (12.2%) and G2/M (29.2%). With increasing doses (e.g. 0.3, 1, 2.5 and 5 μM) at 24 h, S phase arrest was shown to occur with a concomitant decrease of cell populations in the other phases of the cell cycle (Figure 7) In particular, S phase cell population increased from 12.2% to 56.5% at 1 μM 11. Under this condition, the G1 cells decreased from 57.2% in the control to 23.6% and the G2/M phase cells decreased from 29.2% to 13.9%. The increase in S phase likely suggests that 11 enters the nucleus and causes chromatin dysfunction, so that the spindles fail to assemble which requires DNA damage to be repaired before proceeding to the G2/M phase. After 48 h of treatment at 2.5 and 5 μM 11, appearance of a hypodiploid peak (Sub-G1) was observed, indicative of apoptosis (data not shown).
Figure 6.
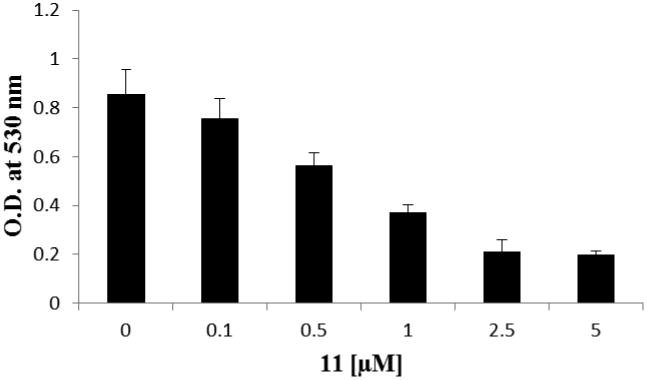
The Antiproliferation Effect of 11 in HeLa Cells. Cells were treated with different concentrations of 11 as indicated for 48 h and O.D. at 530 nm was measured using MTT assay. Data are presented as the average of three independent experiments.
Figure 7.
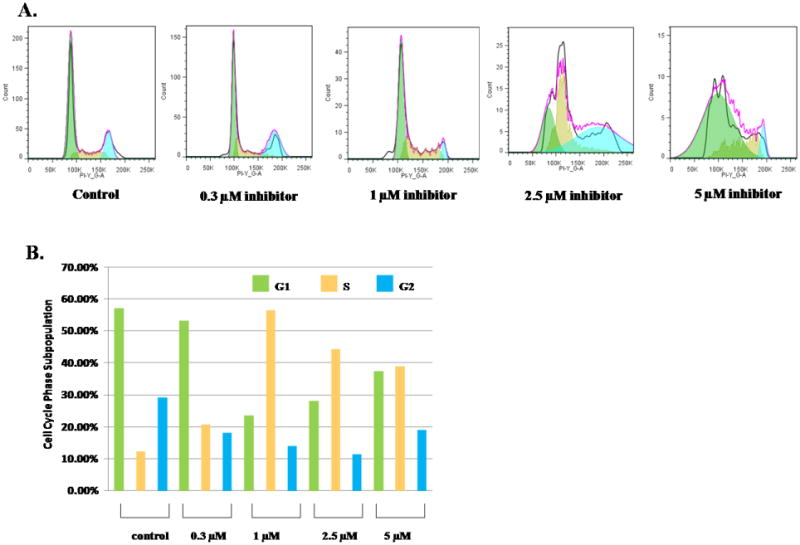
Cell Cycle Analysis of HeLa Cells Treated with Different Doses of 11. Data are presented as the relative fluorescence intensity of cell subpopulations in the FACS profile (panel A) and as percentage of cell subpopulations under each condition (panel B).
Significance
PRMT proteins play functional roles in a wide variety of diseased phenotypes, and therefore the development of novel potent small molecule inhibitors of PRMTs is highly desired for both basic and applied pharmacology. We synthesized carbocyanine molecules containing different heterocyclic moieties and hydrophobic substituents, and identified several possessing micromolar potency for PRMT1 inhibition. In particular, compound 11 presents potent inhibitory activity on PRMT1, with IC50 of 4.1 μM. We have demonstrated that this compound is a noncompetitive inhibitor, and combined with docking simulation and site mutagenesis analysis, we suggest 11 likely targets an allosteric site(s) in PRMT1. Selectivity study showed that the compound has certain specificity toward PRMT1 inhibition. The cyanine molecules typically possess long-wavelength absorption and fluorescence properties which allow them to be used for visualization in cells and tissues. By using the optical and fluorescent microscopies, cell proliferation, and flow cytometry assays, we found that 11 is capable of crossing the plasma membrane, staying in the nucleus and interfering with the chromatin function. Together, this study provides unique photoactive chemical probes for both PRMT inhibition and microscopic imaging.
Expermental Methods
Chemicals
Most chemical reagents were obtained from Sigma Aldrich. Melting points (open Pyrex capillary) were measured on a Mel-Temp apparatus and are uncorrected. 1H NMR and 13C NMR spectra were recorded on either Bruker Avance (400 MHz) or a Varian Unity+300 (300 MHz) spectrometer using DMSO-d6 or MeOD-d4 containing tetramethylsilane (TMS) as an internal standard. Vis/NIR absorption spectrum was recorded on a Varian Cary 50 spectrophotometer. High-resolution accurate mass spectra (HRMS) were obtained using a Waters Q-TOF micro (ESI-Q-TOF) mass spectrometer.
Benz[c,d]indole-2(1H)-thione (2)
This compound was obtained in an 93% yield; mp 146-148 °C; (reported: yield 82%, mp 156 °C.) (Vasilenko et al., 1981; Ficken and Kendall 1960; Lakshmikantham et al., 1989).
2-Methylthiobenz[c,d]indole hydroiodide (3)
This compound was prepared using the reported procedures (Ficken and Kendall 1960; Deligeorgiev and Gadjev 1991). Since the product was unstable, it is used in the next step without further purification.
2-(2,2-Dimethyl-4,6-dioxo-1,3-dioxane-5-yliden)1Hbenz[c,d]indole (4)
This compound was obtained in an 85% yield; mp 220 °C; (reported: yield 94%, mp 223 °C) (Ficken and Kendall 1960; Lakshmikantham et al., 1989).
General Procedures of the Synthesis of Compounds 5-7
A mixture of compound 4 (10.2 mmol), alkyl iodide (30.6 mmol) and K2CO3 (30.6 mmol) were heated in DMF (40 mL) at 90 °C for 18 hr under a nitrogen atmosphere. The mixture was cooled to room temperature and then filtered. The filtrate was concentrated under reduced pressure. The residue was purified on silica gel (flash column chromatography, using EtOAc-hexanes, 1:2 as an eluent).
1-Ethyl-2-(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)-1H-benz[c,d]indole (5)
Yield 65 %,mp 183-184° C; 1H NMR (300 MHz, DMSO-d6) δ 1.46 (t, J = 7 Hz, 3 H), 1.70 (s, 6 H), 4.39 (q, J = 7 Hz, 2 H), 7.78 (m, 1 H), 7.91 (m, 2 H), 8.01 (m, 1 H), 8.40 (d, J = 8 Hz, 1 H), 8.90 (d, J = 8 Hz, 1 H).
1-Butyl- 2-(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)-1H-benz[c,d]indole (6)
Yield 74 %, 1H NMR (300 MHz, DMSO-d6) δ 0.861 (t, J = 7.5 Hz, 3 H), 1.270 (q, J = 7.2, 2 H), 1.755 (s, 6 H), 1.826 (p, J = 6.6 Hz, 2 H), 3.394 (t, J = 7.5 Hz, 2 H), 7.790 (t, J =7.5 Hz, 1 H), 7.948 (t, J = 7.8 Hz, 1 H), 8.027 (d, J = 7.2 Hz, 1 H), 8.068 (d, J = 8.4 Hz, 1 H), 8.455 (d, J = 8.1 Hz, 1 H), 8.911(d, J = 7.2 Hz, 1 H).
1-Phenylpropyl-2-(2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)-1H-benz[c,d]indole (7)
Yield 92 %, mp 155-157 ° C 1H NMR (400 MHz, DMSO-d6) δ 2.17 (m, 2 H), 2.56 (t, J = 7 Hz, 2 H), 4.35 (t, J = 7 Hz, 2 H), 7.17 (m, 5 H), 7.71(d, J = 7.5 Hz, 1 H), 7.86 (m, 2 H), 7.88 (t, J = 7.5 Hz, 1 H), 8.38 (d, J = 8 Hz, 1 H), 8.88 (d, J = 8 Hz, 1 H).
General Procedures of the Synthesis of Salts 8-10
Ester 5-7 (2.9 mmol) was dissolved in acetic acid (4 mL) and the mixture was refluxed for 20 min. Concentrated HCl (4 mL) was added dropwise to the refluxing mixture until the color changed from red to green. The mixture was cooled to room temperature, and saturated KI solution was added until the product started to precipitate. The product was filtered off, washed with ether, and dried in vacuo affording quaternary ammonium salts 8-10 in good yield.
1-Ethyl-2-(2-dimethylaminovinyl)benz[c,d]indolium iodide (8)
Yield 76 %, mp 242-244 °C; 1H NMR (300 MHz, DMSO-d6) δ 1.56 (t, J = 7 Hz, 3 H), 4.46 (s, 3 H), 4.71 (q, J = 7 Hz, 2 H), 8.0 (m, 1 H), 8.19 (m, 1 H), 8.45 (d, J = 8 Hz, 1 H), 8.54 (d, J = 8 Hz, 1 H), 8.69 (d, J = 8 Hz, 1 H), 8.79 (d, J =8 Hz, 1 H).
1-Butyl-2-(2-dimethylaminovinyl)benz[c,d]indolium iodide, (9)
Yield 82 %, 1H NMR (400 MHz, DMSO-d6) δ 0.861 (t, J = 7.5 Hz 3 H), 1.270 (m, 2 H), 1.755 (s, 6 H), 1.826 (p, J = 6.6 Hz, 2 H), 4.394 (t, J = 7.5 Hz, 2 H), 7.790 (t, J = 8.1 Hz, 1 H), 7.948 (t, J = 7.5 Hz, 1 H), 8.027 (d, J = 7.2 Hz, 1 H), 8.073 (d, J = 8.4 Hz, 1 H), 8.455 (d, J = 8.1 Hz, 1 H), 8.911 (d, J = 7.2 Hz, 1 H).
1-phenylpropyl-2-(2-dimethylaminovinyl)benz[c,d]indolium iodide, (10)
Yield 86 %, mp 75-76 °C 1H NMR (400 MHz, DMSO-d6) δ 2.26 (m, 2 H), 2.28 (t, J = 7 Hz, 2 H), 4.00 (s, 3 H), 4.70 (t, J = 7 Hz, 2 H), 7.14 (m, 5 H), 7.96 (t, J = 7.5 Hz, 1 H), 8.10 (t, J = 7.5 Hz, 1 H), 8.41 (m, 2 H), 8.48 (d, J = 8 Hz, 1 H), 8.92 (d, J = 8 Hz, 1 H).
General Synthetic Strategy for Final Dye Compounds 11-13
A mixture of the individual salts 8-10 (0.5 mmol), triethylformate (0.25 mmol), anhydrous sodium acetate (30 mg) and 4 ml of acetic anhydride was boiled for 1 h. The dye was suction filtered, washed with ethyl ether and dried.
1-ethyl-2-[3-(1-ethylbenz[c,d]indol-2(1H)-ylidene)-1-propen-1-yl]-iodide (11)
Yield 75 %; 1H NMR (400 MHz, DMSO-d6) δ 1.40 (t, J = 7 Hz, 3H), 4.36 (d, J = 7 Hz, 2H), 6.90 (d, J = 13 Hz, 1H), 7.20 (m, 1H), 7.57 (d, J = 7.5 Hz, 1H), 7.66 (t, J = 7.5 Hz, 1H), 7.80 (d, J = 8.10 Hz, 1H), 7.97 (t, J = 7.5 Hz, 1H), 8.27 (d, J = 8.10 Hz, 1 H), 8.78 (t, J = 13 Hz, 1 H), 8.94 (d, J = 7.5 Hz, 1H). TOF HRMS m/z (M+) calculated for C29H25N2 401.2018, ound 401.2000. λmax/em = 758/774 nm in methanol.
1-butyl-2-[3-(1-butyllbenz[c,d]indol-2(1H)-ylidene)-1-propen-1-yl]- iodide (12)
Yield 60 %, 1H NMR (300 MHz, DMSO-d6) δ 0.966 (t, J = 7.2 Hz, 6H), 1.434-1.508 (m, 4H), 1.856 (p, J = 6.9 Hz, 4H), 4.404 (t, J = 6.6 Hz, 4H), 7.230 (d, J = 13.2 Hz, 4H), 7.727-7.770(m, 4H), 7.868-7.908 (m, 4H), 8.062 (t, J = 7.5 Hz, 2H), 8.365 (d, J = 8.1 Hz, 1H), 8.661 (d, J = 7.2 Hz, 2H), 9.207 (t, J = 13.2 Hz, 1H). TOF HRMS m/z (M+) calculated for C33H33N2 457.2644, found 457.2624. λmax/em = 758/774 nm in methanol.
1-phenylpropyl-2-[3-(1-phenylpropylbenz[c,d]indol-2(1H)-ylidene)-1-propen-1-yl]iodide (13)
Yield 48 %, 1H NMR (400 MHz, DMSO-d6) δ 2.06 (m, 2H), 2.75 (t, J = 7 Hz, 2H), 4.27 (t, J = 7 Hz, 2H), 6.75 (d, J = 13.6 Hz, 1H), 6.97 (m, 1H), 7.24 (m, 5H), 7.46 (d, J = 7.5 Hz, 1H), 7.63 (t, J = 8 Hz, 1H), 7.76 (d, J = 8 Hz, 1H), 7.94 (t, J = 7.5 Hz, 1H), 8.25 (d, J = 8 Hz, 1H), 8.73 (t, J = 13 Hz, lH), 8.87 (d, J =8 Hz,1H).TOF HRMS m/z (M+)calculated for C43H37N2 581.2957, found 581.2934. λmax/em = 758/774 nm in methanol.
General Synthesis of Indolium and Benz[e]indolium Salts 16-21
A mixture of indolenine 14 or 15 (31.4 mmol) and alkyl halide (94.2 mmol) were refluxed in acetonitrile (25 mL) for 24 hr. The solvent was concentrated and removed in vacuo to afford an oil residue. The reaction mixture was cooled in an ice bath and solid was obtained upon addition of ether and acetone. The resulting crystals were filtered and dried to yield red to light pink solid which was used without further purification in subsequent reactions.
1-ethyl-2,3,3-trimethyl-3H-indol-1-ium iodide (16)
This compound was obtained in 82% yield; mp 217-221 °C; (reported: mp 165-166 °C) (Simmons et al., 2011).
1-butyl-2,3,3-trimethyl-3H-indol-1-ium iodide (17)
This compound was obtained in 80% yield; mp 116-119 °C; (reported: yield 83%, mp 102 °C) (Lee and Hu et al., 2003).
2,3,3-trimethyl-1-(3-phenylpropyl)-3H-indol-1-ium bromide (18)
81% yield; mp 169-171°C; 1H NMR (400 MHz, MeOD-d4) δ 1.587 (s, 6H), 2.32 (p, J = 7.6 Hz, 2H), 2.906 (t, J = 7.6 Hz, 2H), 4.531 (t, J = 8 Hz, 2H), 7.231-7.269 (m, 1H), 7.323-7.340 (m, 4H), 7.633-7.661 (m, 2H), 7.725-7.768 (m, 2H).
3-ethyl-1,1,2-trimethyl-1H-benzo[e]indol-3-ium iodide (19)
This compound was obtained in 81% yield; mp 225-227 °C; (reported: yield 73%, mp 228 °C) (Kuster et al., 2010).
3-butyl-1,1,2-trimethyl-1H-benzo[e]indol-3-ium iodide (20)
This compound was obtained in 87% yield; mp 142-145 °C; (reported: yield 90%, mp 127-129) (Kvach et al., 2008).
1,1,2-trimethyl-3-(3-phenylpropyl)-1H-benzo[e]indol-3-ium iodide (21)
Yield 81%, mp 169-171°C, 1H NMR (400 MHz, MeOD-d4) δ 1.587 (s, 6H), 2.32 (p, J = 7.6 Hz, 2H), 2.906 (t, J = 7.6 Hz, 2H), 4.531 (t, J = 8 Hz, 2H), 7.231-7.269 (m, 1H), 7.323-7.340 (m, 4H), 7.633-7.661 (m, 2H), 7.725-7.768 (m, 2H).
General Synthesis of 22-27
Quaternized ammonium salts 16-21 were heated to 80 °C in acetic anhydride. Triethyl orthoformate was added to the reaction mixture causing an instant color change to deep purple. The reaction mixture was monitored by UV/Vis spectroscopy in methanol and TLC analysis using DCM as the mobile phase. After 2 hr the reaction was stopped and the flask was cooled in a freezer. After 5-10 hr, the product precipitated was filtered and dried. Initial product purification was performed by recrystallization using minimum amounts of methanol, reduced temperature, and titration with ether. Compounds 24, 26 and 27 were additionally purified using silica gel and 2% MeOH in dichloromethane as eluting solvent.
1-ethyl-2-((1E,3E-3-(1-ethyl-3,3-dimethylindolin-2-ylidene)prop-1-en-1-yl)-3,3-dimethyl- 3H-indolium iodide (22)
Yield 77%, 1H NMR (400 MHz, MeOD-d4) δ 1.43 (t, J = 8 Hz, 6H), 1.77 (s, 12H), 4.22 (q, J = 8 Hz, 4H), 6.53 (d, J = 12 Hz, 2H), 7.31 (t, J = 8 Hz, 2H), 7.36 (d, J = 8 H z,2H),7.45 (t, J = 8 Hz, 2H),7.55 (d, J = 8 Hz, 2H), 8.56 (t, J = 12 Hz, 1H); 13C NMR (100 MHz, MeOD-d4) δ 12.69, 28.25, 40.35, 103.44, 112.23, 123.60, 126.77, 130.04, 142.34, 142.95, 152.32, 175.63; TOF HRMS m/z (M+) calculated for C27H33N2 385.2644, found 385.2647. λmax/em = 546/561 nm in methanol.
1-butyl-2-((1E,3E-3-(1-butyl-3,3-dimethylindolin-2-ylidene)prop-1-en-1-yl)-3,3-dimethyl- 3H-indol-1-ium (23)
Yield 74%, mp 172-175 °C, 1H NMR (400 MHz, DMSO) δ 0.95 (t, J = 8 Hz, 6H), 1.41-1.54 (m, 4H), 1.71 (s, 12H), 1.68-1.80 (m, 4H), 4.14 (t, J= 8 Hz, 4H), 6.52 (d, J = 16 Hz, 2H), 7.31 (t, J = 8 Hz, 2H), 7.46 (q, J = 8 Hz, 4H), 7.65 (d, J = 16 Hz, 2H), 8.37 (t, J = 12 Hz, 1H); 13C NMR (MeOD-d4) δ 13.81, 19.57, 27.46, 29.21, 43.70, 48.90, 102.50, 111.56, 122.50, 125.20, 128.64, 140.61, 141.87, 173.84; TOF HRMS m/z (M+)calculated for C31H41N2 441.3270, found 441.3279. λmax/em = 546/561 nm in methanol.
1-phenylpropyl-2-((1E,3E-3-(1-butyl-3,3-dimethylindolin-2-ylidene)prop-1-en-1-yl)-3,3- dimethyl-3H-indol-1-ium (24)
Yield 67%, mp 255-258 °C,1H NMR (400 MHz, MeOD-d4) δ 1.743 (s, 12H), 2.189 (p, J = 7.2 Hz, 4H), 2.856 (t, J = 7.5 Hz, 4H), 4.181 (t, J= 7.8 Hz, 4H), 6.215 (d, J = 13.3 Hz, 2H), 7.45 (t, J = 8 Hz, 2H), 7.55 (d, J = 8 Hz, 2H), 8.56 (t, J = 12 Hz, 1H); 13C NMR (100 MHz, MeOD-d4) δ 24.64, 28.30, 29.97, 33.74, 44.65, 103.69, 112.37, 123.56, 126.82, 127.45, 129.61, 129.71,129.98, 142.08, 142.21, 143.27, 175.96; TOF HRMS m/z (M+) calculated for C27H33N2 385.2644, found 385.2647. λmax/em = 546/561 nm in methanol.
3-ethyl-2-((1E,3E-3-(3-ethyl-1,1-dimethyl-1H-benzo[e]indol-2(3H)-ylidene)prop-1-en-1-yl)- 1,1-dimethyl-1H-benzo[e]indol-3-ium iodide (25)
Yield 66%, mp >260 °C, 1H NMR (400 MHz, MeOD-d4) δ 1.50 (t, J = 8 Hz, 6H), 2.10 (s, 12H), 4.34 (q, J = 8 Hz, 4H), 6.53 (d, J = 16 Hz, 2H), 7.53 (t, J = 8 Hz, 2H), 7.65 (d, J = 8 Hz, 2H), 7.67 (d, J = 8 Hz, 2H), 8.03 (d, J = 8 Hz, 2H), 8.065 (d, J = 12 Hz, 2H), 8.295 (d, J = 8 Hz, 2H), 8.79 (t, J = 8 Hz, 1H); 13C NMR (100 MHz, MeOD-d4) δ 13.13, 28.07, 40.68, 52.59, 102.96, 112.14, 123.49, 126.49, 129.07, 129.55, 131.34, 132.16, 133.82, 140.59; TOF HRMS m/z (M+) calculated for C35H37N2 485.2957, found 485.2960. λmax/em = 586/605 nm in methanol.
3-butyl-2-((1E,3E-3-(3-butyl-1,1-dimethyl-1H-benzo[e]indol-2(3H)-ylidene)prop-1-en-1-yl)-1,1-dimethyl-1H-benzo[e]indol-3-ium iodide (26)
Yield 63%, 1H NMR (400 MHz, MeOD- d4) δ 1.06 (t, J = 8 Hz, 6H), 1.55-1.61 (m, 4H), 1.88-1.94 (m, 4H), 2.11(s, 12H), 4.20 (t, J = 8 Hz, 4H), 6.565 (d, J = 12 Hz, 2H), 7.53 (t, J = 8 Hz, 2H), 7.64 (d, J = 8 Hz, 2H), 7.69 (t, J = 12 Hz, 2H), 8.03 (d, J = 8 Hz, 2H), 8.06 (d, J = 8 Hz, 2H), 8.30 (d, J = 8Hz, 2H), 8.79 (t, J = 12 Hz, 1H); 13C NMR (100 MHz, MeOD-d4): δ 14.41, 21.11, 21.36, 28.18, 31.13, 52.56, 103.27, 112.37, 123.50, 126.52, 129.09, 129.48, 131.34, 132.10, 133.80, 135.10, 141.02; TOF HRMS m/z (M +) calculated for C39H45N2 541.3583, found 541.3582. λmax/em = 586/605 nm in methanol.
2-((1E,3E-3-(1,1-dimethyl-3-(3-phenylpropyl)-1H-benzo[e]indol-2(3H)-ylidene)prop-1-en-1-yl)-1,1-dimethyl-3-(3-phenylpropyl)-1H-benzo[e]indol-3-ium iodide (27)
Yield 55%,1H NMR (400 MHz, MeOD-d4) δ 2.01 (s, 12H), 2.12 (p, J = 8 Hz, 4H), 2.82 (t, J = 8 Hz, 4H), 4.31 (t, J = 8 Hz, 4H), 6.395 (d, J = 12 Hz, 2H), 7.21-7.33 (m, 12H), 7.55 (t, 8 Hz, 2H), 7.740 (t, 8 Hz,2 H), .5 (d, 8 Hz, 2H), 8.11 (t, 8 H z, 4H), 8.30 (d, 8 Hz, 2 H), 8.58 (t, 12 Hz, 1H); 13C NMR(100 MHz, MeOD-d4) δ 26.60, 28.76, 32.32, 101.59, 110.65, 121.92, 125.01, 126.08, 127.55, 127.90, 127.99, 128.07, 128.21, 128.34, 129.78, 130.55, 132.27, 139.29, 140.62. TOF HRMS m/z (M+) calculated for C49H49N2 665.3896, found 665.3893. λmax/em = 586/605 nm in methanol.
Protein Expression and Purification
PRMT1-pET28b plasmid was transformed into Escherichia coli BL21 (DE3) using heat shock method. Protein expression was induced with 0.3-mM IPTG at 16°C for 20 h. After protein expression, cells were pelleted by centrifuge followed by suspension in the lysis buffer (25 mM HEPES pH 8.0, 150 mM NaCl, 1 mM PMSF, 1 mM MgSO4, 5% glycerol, 5% ethylene glycol) and lysed by French Press. The protein supernatant was purified on the Ni-charged His-tag binding resin (Novagen). The beads were equilibrated with column buffer (25 mM Na-HEPES pH 8.0, 500 mM NaCl, 1 mM PMSF, and 30 mM imidazole, 10% glycerol, 1 mM PMSF) followed by protein loading. After that the beads were washed thoroughly with the column buffer, and then the protein was eluted with the elution buffer (25 mM Na-HEPES, pH 7.0, 300 mM NaCl, 1 mM PMSF, 100mM EDTA and 200 mM imidazole, 5% glycerol). Different elution fractions were checked on SDS–PAGE. Then, protein fractions were combined and dialyzed against the storage buffer (25 mM Na-HEPES, pH 7.0, 500 mM NaCl, 1 mM EDTA, 10 mM DTT and 10% glycerol) at 4 °C. After dialysis, the protein solution was concentrated using Millipore centrifugal filters. His6x-tagged PRMT3 was expressed from the pReceiver vector. GST-mCARM1 was expressed from the pGEX-4T1 vector. His6x-tagged PRMT5 and PRMT6 were expressed from the pET28a vector. All of the proteins were expressed in Escherichia coli BL21(DE3). All of the His6x-tagged proteins were purified on Ni-NTA beads. Protein concentrations were determined using Bradford assay.
Radioactive Methyltransferase Assay
The inhibitory activities of small molecule compounds were examined by using 14C-labeled radioactive methylation assays. The assays were carried out in 0.6 mL plastic tubes at 30°C in a reaction volume of 30 μL. The reaction buffer contained 50 mM HEPES (pH 8.0), 0.5 mM dithiothreitol (DTT), 1 mM EDTA, and 50 mM NaCl. In a typical procedure, peptide substrate, [14C]- S-Adenosyl-L-methionine and inhibitor were preincubated in the reaction buffer for 5 min prior to the initiation by the addition of PRMT1 (0.05 μM final). After it was incubated for 8 min, the reaction was quenched by spreading the reaction mixture onto P81 filter paper discs (Whatman). The paper disk was washed with 50 mM NaHCO3, and dried in air for 2 h. The amount of methylated products was quantified by liquid scintillation. To determine the IC50 value of those inhibitors similar experiment was performed with H4-(1-20) (2 μM), [14C]- S-Adenosyl-L-methionine (5 μM) and DMSO (2%) and varied concentration of inhibitors. The IC50 value is the concentration of inhibitor at which half of the maximal activity is reached. The inhibition patterns of 11 were determined by measuring initial velocities of PRMT1 at a range of varied concentrations of one substrate, a fixed concentration of the other substrate, and selected concentrations of the inhibitors. The data were displayed in double reciprocal formats and fitted to competitive or noncompetitive kinetic equations.
Time-Dependent Inhibition Assay
Depending upon the IC50 value of the compound, different concentrations of the compound were incubated with 0.05 μM enzyme so that in the final reaction mixture 11 was present at 0, 1.5, 2.5, 4.0 μM. Incubations were conducted for 0, 2, 4, 6, 8, 10, 15, 20, 25, 30 min at 30 °C After each incubation, the reaction was quenched by spreading 20 μL reaction mixture onto P81 filter paper discs (Whatman).
Dialysis Experiment
For the dialysis assay, 1 μM PRMT1 was incubated with 20 μM 11 in a total reaction volume of 520 μL for 1 h at 30 °C to achieve complete inactivation. Enzyme without any inhibitor was used in experimental controls. The reaction mixtures were then transferred to dialysis cassettes (molecular weight cut off of 10 000Da) and dialyzed for 3 days at 4°C in the 300 ml reaction buffer contained 50 mM HEPES (pH 8.0), 0.5 mM dithiothreitol (DTT), 1 mM EDTA, 50 mM NaCl and 10% DMSO. The buffer was changed six times during this period. After finishing dialysis, residual activity assays were performed on pre- and post-dialysis samples. The activity was measured in the presence of 2 μM peptide substrate, 5 μM [14C]- S-Adenosyl-L-methionine and dialyzed samples were preincubated in the reaction buffer for 5 min prior to the initiation by the addition of PRMT1 (0.05 μM final). After it was incubated for 8 min, the reaction was quenched by spreading the reaction mixture onto P81 filter paper discs (Whatman). The paper discs were washed with 50 mM NaHCO3 and dried in air for 2 h. The amount of methylated products was quantified by liquid scintillation. The amount of product formed in the control reaction was set as 100% and used to determine the percent activity remaining in the inhibited reactions.
Bright Field and Fluorescent Microscopic Imaging
HeLa cells were seeded on coverglass overnight and treated without or with different concentrations of 11 for 6 hours. Cells were then fixed by 4% paraformaldehyde for 10 min at room temperature. Then the optical images were taken under visible light by CoolSNAP CF camera attached to a Nikon Ti-80 microscope. The NIR fluorescent images were taken by an LSM META NLO confocal microscope (Creighton university Integrated biomedical imaging Facility) using a 633-nm laser and a 715 to 785-nm Long Pass filter.
Antiproliferative Assay of HeLa Cells
Hela cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (Cellgro) supplemented with 10% fetal bovine serum (Cellgro) and 1% penicillin-streptomycin in 5% CO2 at 37°C. Cells were washed with PBS and harvested using trypsin. Individual wells of a 96-well tissue culture plate were inoculated with 100 μL of complete medium containing 9000 cells and incubated overnight for attachment at 37 °C in a 5% CO2 incubator. The following day, the media were removed and the cells were treated with different concentrations of 11. The final concentration of DMSO in the culture medium was maintained at 0.2%. Control cells (without any compound) were treated under the same conditions. After 48 hr of incubation period, cell survival was evaluated using an MTT assay: 10 μL of a freshly prepared solution of MTT (5 mg/mL in PBS) was added to each well, and after 4 h of incubation, the medium was removed; after addition of 100 μL of DMSO to each well, the optical density values were detected at 530 nm. Cytotoxicity data were expressed as IC50 values. Data were expressed as mean values of three individual experiments conducted in triplicate.
Cell Cycle Assay
For flow cytometric analysis of DNA content, 1 × 106 cells in exponential growth were treated with different concentrations of 11 at different concentrations for 24 and 48 hr. After the incubation, the cells were collected, centrifuged and fixed with ice-cold ethanol (70%) and stored at 4°C until use. Subsequently, cells were rinsed with PBS and stained with PBS containing 40 μg/ml propidium iodide and followed by the addition of RNase A. Samples were analyzed on a BD-LRFortessa flow cytometer (BD Biosciences). For cell cycle analysis, DNA histograms were analyzed using Flow Jo software.
Supplementary Material
Acknowledgments
This work is supported by Georgia Cancer Coalition Distinguished Cancer Scholar Award and NIH Grant GM086717 to YGZ. YF is supported by an American Heart Association Pre-doctoral Fellowship and a University Doctoral Fellowship at GSU under the instruction of YGZ.
References
- Bedford MT, Richard S. Arginine Methylation: An Emerging Regulator of Protein Function. Mol Cell. 2005;18:263–272. doi: 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Beltowski J, Kedra A. Asymmetric dimethylarginine (ADMA) as a target for pharmacotherapy. Pharmacol Rep. 2006;58:159–78. [PubMed] [Google Scholar]
- Bissinger EM, Heinke R, Spannhoff A, Eberlin A, Metzger E, Cura V, Hassenboehler P, Cavarelli J, Schüle R, Bedford MT, Sippl W, Jung M. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg Med Chem. 2011;19:3717–3731. doi: 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- Bissinger EM, Heinke R, Spannhoff A, Eberlin A, Metzger E, Cura V, Hassenboehler P, Cavarelli J, Schule R, Bedford MT, Sippl W, Jung M. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Bioorg Med Chem. 2011;19:3717–31. doi: 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- Cheng D, Yadav N, King RW, Swanson MS, Weinstein EJ, Bedford MT. Small molecule regulators of protein arginine methyltransferases. J Biol Chem. 2004;279:23892–9. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- Cheng D, Valente S, Castellano S, Sbardella G, Di Santo R, Costi R, Bedford MT, Mai A. Novel 3,5-bis(bromohydroxybenzylidene)piperidin-4-ones as coactivator-associated arginine methyltransferase 1 inhibitors: enzyme selectivity and cellular activity. J Med Chem. 2011a;54:4928–32. doi: 10.1021/jm200453n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng D, Bedford MT. Xenoestrogens regulate the activity of arginine methyltransferases. ChemBioChem. 2011b;12:323–9. doi: 10.1002/cbic.201000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–15. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- Deligeorgiev TG, Gadjev NI. Styryl dyes containing the benz[c,d]indolium heterocycle. Dyes Pigm. 1991;15:215–23. [Google Scholar]
- Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS lett. 2011;585:2024–31. doi: 10.1016/j.febslet.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowden J, Pike RA, Parry RV, Hong W, Muhsen UA, Ward SG. Small molecule inhibitors that discriminate between protein arginine N-methyltransferases PRMT1 and CARM1. Org Biomol Chem. 2011;9:7814–21. doi: 10.1039/c1ob06100c. [DOI] [PubMed] [Google Scholar]
- Feng Y, Li M, Wang B, Zheng YG. Discovery and mechanistic study of a class of protein arginine methylation inhibitors. J Med Chem. 2010;53:6028–39. doi: 10.1021/jm100416n. [DOI] [PubMed] [Google Scholar]
- Flanagan JH1, Khan S, Menchen S, Soper SA, Hammer RP. Functionalized tricarbocyanine dyes as near-infrared fluorescent probes for biomolecules. Bioconjug Chem. 1997;8:751–756. doi: 10.1021/bc970113g. [DOI] [PubMed] [Google Scholar]
- Ficken GE, Kendall JD. The reactivity of the alkylthio group in nitrogen ring compounds. III 2-Methylthiobenz [c,d]indole and its methiodide. J Chem Soc. 1960:1537–41. [Google Scholar]
- Goulet I, Gauvin G, Boisvenue S, Cote J. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J Biol Chem. 2007;282:33009–21. doi: 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- Heinke R, Spannhoff A, Meier R, Trojer P, Bauer I, Jung M, Sippl W. Virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors. ChemMedChem. 2009;4:69–77. doi: 10.1002/cmdc.200800301. [DOI] [PubMed] [Google Scholar]
- Koh SS, Chen D, Lee YH, Stallcup MR. Synergistic enhancement of nuclear receptor function by p160 coactivators and two coactivators with protein methyltransferase activities. J Biol Chem. 2001;276:1089–98. doi: 10.1074/jbc.M004228200. [DOI] [PubMed] [Google Scholar]
- Krause CD, Yang ZH, Kim YS, Lee JH, Cook JR, Pestka S. Protein arginine methyl transferases: evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther. 2007;113:50–87. doi: 10.1016/j.pharmthera.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Kuster S, Sauvage F, Nazeeruddin MK, Grätzel M, Nüesch FA, Geiger T. Unsymmetrical squaraine dimer with an extended π-electron framework: An approach in harvesting near infra-red photons for energy conversion. Dyes Pigm. 2010;87:30–38. [Google Scholar]
- Kvach MV, Ustinov AV, Stepanova IA, Malakhov AD, Skorobogatyi MV, Shmanai VV, Korshun VA. A Convenient Synthesis of Cyanine Dyes: Reagents for the Labeling of Biomolecules. Eur J Org Chem. 2008;12:2107–2117. [Google Scholar]
- Lakshmikantham MV, Chen Wha, Cava Michael P. Thioanhydrides. 3 Synthesis, properties, and Diels-Alder reactions of sulfur analogues of 1,8-naphthalic anhydride. J Org Chem. 1989;54:4746–4750. [Google Scholar]
- Lee CC, Hu AT. Synthesis and optical recording properties of some novel styryl dyes for DVD-R. Dyes Pigm. 2003;59:63–69. [Google Scholar]
- Li KK, Luo C, Wang D, Jiang H, Zheng YG. Chemical and biochemical approaches in the study of histone methylation and demethylation. Med Res Rev. 2010 doi: 10.1002/mrr.20228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losytskyy MY, Volkova KD, Kovalska VB, Makovenko IE, Slominskii Yu L, Tolmachev OI, Yarmoluk SM. Fluorescent Properties of Pentamethine Cyanine Dyes with Cyclopentene and Cyclohexene Group in Presence of Biological Markers. J Fluoresc. 2005;15:849–857. doi: 10.1007/s10895-005-0002-7. [DOI] [PubMed] [Google Scholar]
- Maas R. Pharmacotherapies and their influence on asymmetric dimethylargine (ADMA) Vasc Med. 2005;10(Suppl 1):S49–57. doi: 10.1191/1358863x05vm605oa. [DOI] [PubMed] [Google Scholar]
- Mai A, Cheng D, Bedford MT, Valente S, Nebbioso A, Perrone A, Brosch G, Sbardella G, De Bellis F, Miceli M, Altucci L. Epigenetic multiple ligands: mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (Sirtuin) inhibitors. J Med Chem. 2008;51:2279–2290. doi: 10.1021/jm701595q. [DOI] [PubMed] [Google Scholar]
- Mathioudaki K, Scorilas A, Ardavanis A, Lymberi P, Tsiambas E, Devetzi M, Apostolaki A, Talieri M. Clinical evaluation of PRMT1 gene expression in breast cancer. Tumour Biol. 2011;32:575–82. doi: 10.1007/s13277-010-0153-2. [DOI] [PubMed] [Google Scholar]
- Mishra A, Behera RK, Behera PK, Mishra BK, Behera GB. Cyanines during the 1990s: A Review. Chem Rev. 2000;100:1973–2012. doi: 10.1021/cr990402t. [DOI] [PubMed] [Google Scholar]
- Mojzych M, Henary M. Synthesis of cyanine dyes. In: Strekowski L, editor. Heterocyclic Polymethine Dyes. 2008. pp. 1–9. [Google Scholar]
- Narayanan N, Patonay G. A new method for the synthesis of heptamethine cyanine dyes: synthesis of new near-infrared fluorescent labels. J Org Chem. 1995;60:2391–2395. [Google Scholar]
- Pal S, Sif S. Interplay between chromatin remodelers and protein arginine methyltransferases. J Cell Physiol. 2007;213:306–15. doi: 10.1002/jcp.21180. [DOI] [PubMed] [Google Scholar]
- Papadokostopoulou A, Mathioudaki K, Scorilas A, Xynopoulos D, Ardavanis A, Kouroumalis E, Talieri M. Colon cancer and protein arginine methyltransferase 1 gene expression. Anticancer Res. 2009;29:1361–6. [PubMed] [Google Scholar]
- Ragno R, Simeoni S, Castellano S, Vicidomini C, Mai A, Caroli A, Tramontano A, Bonaccini C, Trojer P, Bauer I, Brosch G, Sbardella G. Small molecule inhibitors of histone arginine methyltransferases: Homology modeling, molecular docking, binding mode analysis, and biological evaluations. J Med Chem. 2007;50:1241–1253. doi: 10.1021/jm061213n. [DOI] [PubMed] [Google Scholar]
- Simmons RL, Yu RT, Myers AG. Storable Arylpalladium(II) Reagents for Alkene Labeling in Aqueous Media. J Am Chem Soc. 2011;133:15870–15873. doi: 10.1021/ja206339s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spannhoff A, Hauser AT, Heinke R, Sippl W, Jung M. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. ChemMedChem. 2009;4:1568–82. doi: 10.1002/cmdc.200900301. [DOI] [PubMed] [Google Scholar]
- Spannhoff A, Heinke R, Bauer I, Trojer P, Metzger E, Gust R, Schüle R, Brosch G, Sippl W, Jung M. Target-Based Approach to Inhibitors of Histone Arginine Methyltransferases. J Med Chem. 2007a;50:2319–2325. doi: 10.1021/jm061250e. [DOI] [PubMed] [Google Scholar]
- Spannhoff A, Machmur R, Heinke R, Trojer P, Bauer I, Brosch G, Schüle R, Hanefeld W, Sippl W, Jung MA. Novel arginine methyltransferase inhibitor with cellular activity. Bioorg Med Chem Lett. 2007b;17:4150–4153. doi: 10.1016/j.bmcl.2007.05.088. [DOI] [PubMed] [Google Scholar]
- Spannhoff A, Machmur R, Heinke R, Trojer P, Bauer I, Brosch G, Schule R, Hanefeld W, Sippl W, Jung MA. Novel arginine methyltransferase inhibitor with cellular activity. Bioorg Med Chem Lett. 2007c;17:4150–3. doi: 10.1016/j.bmcl.2007.05.088. [DOI] [PubMed] [Google Scholar]
- Spannhoff A, Heinke R, Bauer I, Trojer P, Metzger E, Gust R, Schule R, Brosch G, Sippl W, Jung M. Target-based approach to inhibitors of histone arginine methyltransferases. J Med Chem. 2007d;50:2319–25. doi: 10.1021/jm061250e. [DOI] [PubMed] [Google Scholar]
- Strekowski L, Mason CJ, Lee H, Gupta R, Sowell J, Patonay G. Synthesis of water-soluble near-infrared cyanine dyes functionalized with [(succinimido)oxy]carbyonyl group. J Heterocycl Chem. 2003;40:913–916. [Google Scholar]
- Vasilenko NP, Mikhailenko FA, Rozhinskii Yu I. 2-Methylbenz[c,d]indole and its derivatives. Dyes Pigm. 1981;2:231–7. [Google Scholar]
- Wagner S, Weber S, Kleinschmidt MA, Nagata K, Bauer UM. SET-mediated promoter hypoacetylation is a prerequisite for coactivation of the estrogen-responsive pS2 gene by PRMT1. J Biol Chem. 2006;281:27242–50. doi: 10.1074/jbc.M605172200. [DOI] [PubMed] [Google Scholar]
- Wolf SS. The protein arginine methyltransferase family: an update about function, new perspectives and the physiological role in humans. Cell Mol Life Sci. 2009;66:2109–21. doi: 10.1007/s00018-009-0010-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakrzewicz D, Eickelberg O. From arginine methylation to ADMA: a novel mechanism with therapeutic potential in chronic lung diseases. BMC Pulm Med. 2009;9:5. doi: 10.1186/1471-2466-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YG, Wu J, Chen Z, Goodman M. Chemical regulation of epigenetic modifications: Opportunities for new cancer therapy. Med Res Rev. 2008;28:645–687. doi: 10.1002/med.20120. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.