

Abstract
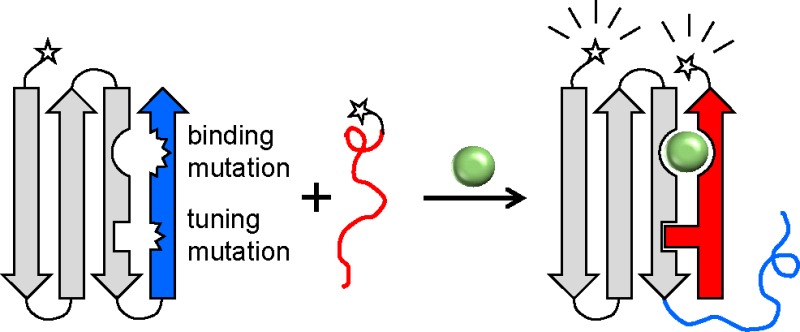
Biosensors can be used in applications ranging from identifying disease biomarkers to detecting spatial and temporal distributions of specific molecules in living cells. A major challenge facing biosensor development is how to functionally couple a biological recognition domain to an output module so that the binding event can be transduced to a visible and quantifiable signal [e.g., Förster resonance energy transfer (FRET)]. Most designs achieve coupling by means of a binding protein that changes conformation upon interacting with its target. This approach is limited by the fact that few proteins possess such natural allosteric mechanisms, and for those that do, the conformational change is frequently not extensive enough to produce a large change in distance between FRET donor and acceptor groups. Here, we introduce protein fragment exchange (FREX) to address both problems. FREX employs two components: a folded binding protein and a fragment duplicated from it, the latter of which can be chosen from many possible fragments. The system is rationally tuned so that addition of ligand induces a conformational change in which the fragment exchanges positions with the corresponding segment of the binding protein. Placing fluorescent donor and acceptor groups on the binding protein and fragment reduces the background level of FRET of the unbound sensor, resulting in a ratiometric FRET response that is expected to be strong and reproducible from protein to protein. FREX is demonstrated using fibronectin III, a monobody binding scaffold that has been tailored to recognize multiple targets. Sensors labeled with Alexa FRET pairs exhibit ratiometric FRET changes of up to 8.6-fold and perform equally well in buffer and serum. A genetically encoded variant of this sensor is shown to be functional in cell lysates and in mammalian cell cultures.
The development of biosensor technology in recent years has been motivated by a growing need to detect, quantify, and monitor biomolecule levels in biological, industrial, and medical fields. Although a number of successful technologies have been introduced, for example, surface plasmon resonance and continuous blood glucose monitors, the development of simpler and more general designs remains a pressing need.
Most biosensors are composed of a biological recognition module (input) and a transducing element with which to convert binding to a visible signal (output). Binding domains of proteins and Förster resonance energy transfer (FRET) have emerged as one of the most potent and widely used combinations of input and output, respectively. Protein interaction domains excel in the former role because they bind their cognate ligands with high affinity and specificity, and some can be engineered to recognize new targets. FRET is powerful because the output response is typically ratiometric and donor/acceptor groups can be genetically encoded in the form of fluorescent proteins. The general approach is to attach donor and acceptor fluorophores to locations on the protein where they report on a distance change induced by ligand binding.
There are two interrelated challenges with constructing biosensors based on the design described above. The first is to develop general mechanisms for introducing ligand-dependent conformational changes into ordinary binding proteins, because the great majority of proteins do not exhibit these changes naturally. Several strategies have been developed for this purpose. One is to take advantage of the natural coupling between binding and folding using proteins that are either intrinsically disordered in the absence of ligand1 or mutated to be so.2 Another is to engineer a binding-dependent fold shift from the native structure to a circularly permuted fold (alternate frame folding).3−5 A third approach is to tether binding domains together such that interaction with ligand triggers open-to-closed rigid body movement, as in SNAP-tag-based semisynthetic fluorescent sensor6,7 and affinity clamp8 technologies. All of these examples are single-component sensors in which distance- or environment-sensitive fluorescent groups are attached to the same molecule.
Although single-component sensors offer numerous advantages, their weakness is exposed by the second challenge of designing a FRET biosensor. For maximal output, the change in donor–acceptor distance should not only be substantial but also span the Förster radius (R0, the distance at which energy transfer is 50% efficient). For the purpose of illustration, for the FRET efficiency to increase from 10 to 90% the donor–acceptor distance must decrease from 1.44R0 to 0.68R0, or from 72 to 34 Å for a typical R0 value of 50 Å. It is difficult to achieve this condition by placing the groups on the same molecule and relying on conformational change (e.g., hinge movement) in the native protein. Even the process of folding, which is arguably the most dramatic transformation that a protein can undergo, results in surprisingly small distance changes. The radius of gyration of an unfolded protein increases roughly linearly from 30 to 70 Å for molecules 100–400 amino acids in length.9,10,28 It is therefore possible to attain large FRET changes upon folding but only for larger (or elongated) proteins and with ideal arrangement of fluorophores in native and unfolded states. For most proteins, the donor–acceptor distance is expected to be less than the R0 in both conformations. Perhaps for this reason, sensors based on binding-induced folding and alternate frame folding have employed short-range, nonratiometric means of detection (e.g., fluorescence quenching or excimer formation) rather than FRET. With their largerscale domain movements, affinity clamp and SNAP-tag sensors produce ratiometric FRET changes of up to 4.9-fold.6−8,11
Here, we develop the protein fragment exchange (FREX) switching mechanism to address both challenges. FREX can in principle be applied to many binding proteins to generate a consistent ligand-dependent conformational change. FREX speaks to the FRET distance problem by placing donor and acceptor groups on separate components that interact only in the presence of the target. The first component is the full-length native protein, and the second component is one of any number of fragments duplicated from that protein. The system is controlled by introducing two point mutations into the full-length protein: one to abolish its ability to bind ligand (binding mutation) and one to decrease its thermodynamic stability (packing mutation). Wild-type (WT) residues are retained in the equivalent positions of the duplicated fragment. In the ligand-free state, the full-length protein is of sufficient stability to assume its native conformation. Addition of a target induces a conformational change in which the duplicated fragment exchanges position with its counterpart peptide on the full-length protein. Ternary complex formation is thus driven by gains in binding energy and stability afforded by the restoration of WT side chain interactions at the remodeled binding and packing interfaces, respectively. This mechanism is analogous to that of alternate frame folding except fragment exchange in FREX occurs in trans and the resulting structure is not circularly permuted. The improvement in the signal-to-noise ratio versus those of single-component sensors is attained by reducing the background level of FRET of the unbound state.
As a proof of principle, we designed a FREX sensor for the detection of biologically relevant targets in vitro and in vivo. For the binding protein, we chose the 10th human fibronectin type III domain (FN3; 100 amino acids), a minimal Ig-like binding scaffold. Also known as a monobody, FN3 has been adapted to recognize the c-Abl SH2 domain (resulting in the FN3-HA4 variant used in this study)12 as well as more than two dozen other targets, including tumor necrosis factor α, hSUMO4, and epidermal growth factor receptor.13−16 FN3 is a versatile platform with which to build sensors because binding variants can be readily generated by modifying its three substrate interacting loops using high-throughput screening. Like Ig domains, FN3 does not undergo a change in conformation upon binding its target. Our purpose is to introduce the core FREX switching mechanism into FN3-HA4.
Experimental Procedures
Gene Construction and Protein Purification
FN3-HA4 and c-Abl SH2 plasmids were gifts from S. Koide (University of Chicago, Chicago, IL). We deleted all purification tags from the original genes and changed the first four residues of FN3-HA4 from GSSV to YGGG as described in the text. For the FN3 variants used in Alexa fluorescence experiments, we either introduced a Cys codon after the codon for Met1 or mutated the Ser48 codon to a Cys codon. P48, P60, and P69 were constructed by ligating the coding sequences of residues 48–100, 60–100, and 69–100, respectively, to the 5′-end of the maltose binding protein (MBP) gene using a linker that translates to GGCGG. The genetically encoded P48 sensor was created by fusing the CyPet gene to the 5′-end of the FN3BN+I75A gene using a linker that translates to GGSGG. For the peptide construct, the YPet gene was inserted between the 3′- and 5′-ends of the MBP and P48 genes, respectively.
A modified pCMV bicistronic vector was constructed for co-expression of mCherry-P48 and EGFP-FN3BN+I75A in mammalian cell cultures. EGFP-FN3BN+I75A and mCherry-P48 genes were constructed as described above. The mCherry-P48 and EGFP-FN3BN+I75A genes were then inserted upstream and downstream of the internal ribosome entry sequence, respectively.
Escherichia coli BL21(DE3) cells were transformed with the plasmids described above, and cultures were grown in Luria-Bertani medium at 37 °C. After induction, cells were allowed to express for 18 h at 20 °C and then centrifuged. FN3BN was purified from lysis supernatants, under native conditions (pH 7.0), using a Q-Sepharose column. FN3BN packing mutants were expressed mostly in lysis pellets. Pellets were solubilized in 6 M urea, and the proteins were purified as described above except in the presence of 6 M urea. Proteins were then refolded by extensive dialysis against 10 mM sodium phosphate (pH 7.0). SH2 was purified by the same native-condition protocol described above, except an SP-Sepharose column was used. We purified P48, P60, and P69 by passing the lysis supernatants (pH 7.5) through an amylose column and eluting with 10 mM maltose. The proteins were then passed through a Superdex-75 size exclusion column. Samples were dialyzed against 10 mM Tris (pH 7.5). All proteins were judged to be >95% pure by sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Size Exclusion Chromatography
Samples were prepared in 20 mM sodium phosphate (pH 7.0), 0.15 M NaCl, 2 mM ethylenediaminetetraacetic acid, and 10 mM β-mercaptoethanol using final concentrations of 5 μM FN3 variants, 5 μM P48, P60, or P69, and 20 μM SH2. Samples were incubated for >16 h at 4 °C to ensure that equilibrium had been reached and then injected onto a Zenix SEC-300 7.8 mm × 300 mm column Sepax Technologies using a Bio-Rad DuoFlow chromatography system.
Alexa Labeling
Samples were reduced with 1 mM tris(2-carboxyethyl)phosphine (TCEP). TCEP was then removed when the sample was passed through a 10 DG desalting column. A 2-fold excess of Alexa488 C5-maleimide or Alexa594 C5-maleimide was immediately added, and the reaction was allowed to proceed for 3 h (pH 7.0). Excess dye was removed by desalting as described above. Final protein concentrations and labeling efficiencies were calculated on the basis of the molar absorptivities and correction factors (CFs) of Alexa488 (ε488 = 72000 M–1 cm–1; CF280 = 0.11) and Alexa594 (ε594 = 96000 M–1 cm–1; CF280 = 0.56).
In Vitro FRET Experiments
All fluorescence data were recorded at 20 °C. Experiments conducted in buffer included 20 mM sodium phosphate (pH 7.0), and those performed with serum included 10% (by volume) fetal bovine serum. For cell lysate experiments, cultures were grown as described above, lysed by sonication, and centrifuged to obtain the soluble fraction. Equilibrium binding studies were performed by adding various amounts of SH2 to a fixed final concentration of FN3 and P48, P60, or P69 (2 μM each, except for cell lysate experiments). Samples were incubated for 3 h prior to data collection. Fluorescence data were recorded on a Horiba Fluoromax-4 fluorometer with excitation at 488 nm (1.5 and 2 nm excitation and emission bandpasses, respectively) for Alexa samples and excitation at 414 nm (1 and 2 nm excitation and emission bandpasses, respectively) for CyPet and YPet samples. The FRET ratio is reported as donor emission divided by acceptor emission (519 and 617 nm for Alexa samples and 468 and 527 nm for CyPet and YPet samples).
In-Cell FRET Experiments
Cos-7 cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum and an antibiotic/antimycotic solution; 105 cells were plated into a 35 mm glass bottom dish at 37 °C with 5% CO2 the day before transfection. Transient transfections were performed using JetPEI transfection reagent with 3 μg of total sensor DNA and 6 μL of JetPEI in each dish. Positive control dishes were cotransfected with 1 μg of FLAG epitope-tagged SH2, while negative control dishes were transfected with sensor DNA only. After 18–20 h, cells were fixed in a 4% paraformaldehyde/phosphate-buffered saline (PBS) mixture at room temperature for 15 min.
Cells were imaged in PBS using a PerkinElmer UltraView VoX spinning disk confocal system mounted on a Nikon Eclipse Ti microscope equipped with a 60×, 1.49 NA APO TIRF objective, a Hamamatsu C9100-50 EMCCD camera, and an environmental chamber to maintain cells at 37 °C. Five images from EGFP (488 nm laser excitation line, 527/55W emission filter) and mCherry (561 nm laser excitation line, EM445/60W-615/70W emission filter) channels were captured before and after mCherry had been photobleached using 25 passes of the 561 nm laser at full power. EGFP images acquired before and after photobleaching were merged to ensure that all pixels were aligned in each image prior to proceeding to subsequent FRET calculations. Following background subtraction, the FRET efficiency for individual pixels was calculated using ImageJ (National Institutes of Health, Bethesda, MD) essentially following the protocol of Deakin et al.17 using the following formula to calculate FRET efficiency: FRETeff = 1 – Dpre/Dpost, where Dpre is the donor intensity before bleaching and Dpost the donor intensity after bleaching. Processed FRET efficiency images were smoothed using the ImageJ smooth function, which replaces each pixel with the average intensity value for the surrounding 3 × 3 pixel region, and displayed on the color intensity scale shown in Figure 5. The fold change in FRET efficiency was calculated by measuring the mean EGFP intensity within the bleached rectangle and then dividing that figure by the mean EGFP intensity in the same size rectangle placed in three to five locations in the cell that were unbleached.
Figure 5.

Performance of the P48 FREX sensor in mammalian cell cultures. Representative raw images of Cos-7 cells transfected with (A) EGFP-FN3BN+I75A and mCherry-P48 and (B) EGFP-FN3BN+I75A, mCherry-P48, and SH2. The top and middle rows show fluorescence before and after, respectively, the mCherry signal in the boxed area had been bleached. The FRET efficiency is plotted in the bottom row. The scale bar is 10 μm.
Results
FREX Mechanism and Simulations
FREX, illustrated schematically in Figure 1A, can be modeled by the following coupled equilibria:
| 1 |
| 2 |
| 3 |
where N is an arbitrary binding protein, U is the unfolded form of that protein, L is its cognate ligand, and P is a peptide duplicated from either terminus of N. FRET donor and acceptor groups are placed on N and P. N*P is the binary complex in which P has displaced the corresponding segment from N, causing it to extend as a tail (Figure 1A). N* indicates that the structure of the protein is identical to that of N except at the point of exchange. L can bind to only N*P, not to N, because N contains a binding knockout mutation that is replaced in N*P by the WT binding residue (which came from P). The binding mutation thus guarantees that ligand binding will exclusively produce the high-FRET ternary complex.
Figure 1.
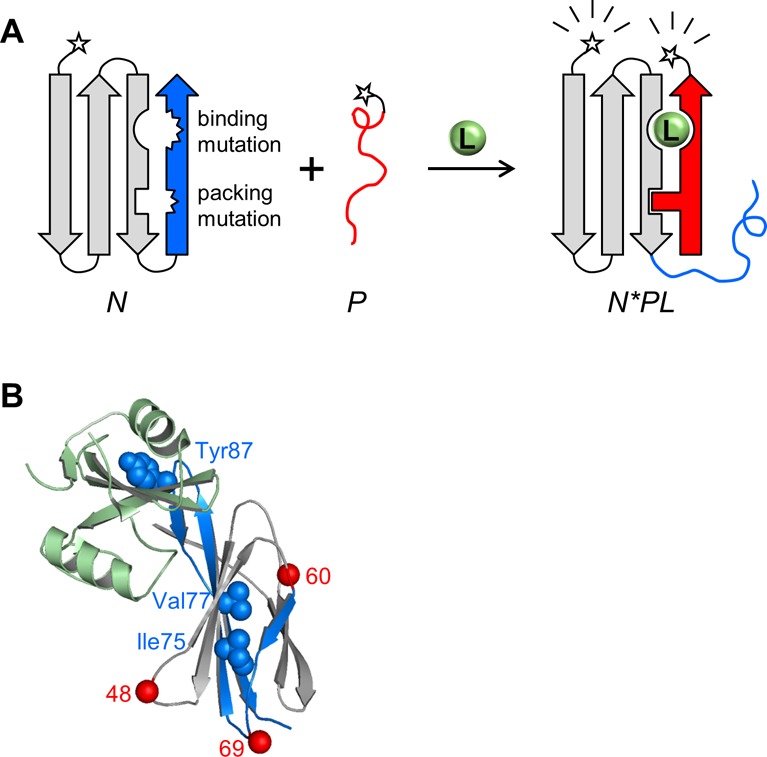
Schematic of FREX and X-ray structure of the FN3-HA4/SH2 complex. (A) An N- or C-terminal segment (blue) of an arbitrary binding protein N (gray) is chosen such that it contains at least one critical ligand binding residue. The blue segment is duplicated to generate peptide P (red). FRET donor and acceptor groups (stars) are attached to N and P at either terminus. The ligand binding residue is mutated in the blue sequence, along with a residue at the packing interface between the blue and gray regions. The resulting protein (N) is destabilized but still folded; consequently, the binary complex of N and P (N*P) does not form to a significant extent. Only in the presence of a ligand (L) do the blue and red segments exchange to generate the ternary complex (N*PL). Formation of N*PL is driven by the restoration of binding and packing interactions, supplied by the WT residues at those positions in P. (B) FN3-HA4 is shown with the starting positions of P48, P60, and P69 indicated by red spheres. Blue/gray color coding is the same as that in panel A; blue denotes the P60 segment. Side chains of binding (Tyr87) and packing (Ile75/Val77) residues are represented by blue spheres. SH2 is colored light green.
A protein need not globally unfold to undergo fragment exchange. U is included to link the thermodynamic stability of N to the probability of forming N*P and N*PL. To illustrate, mixing a protein with a fragment of itself will typically not result in formation of the binary complex, even if the affinity of the protein for the peptide is high. The local concentration of the covalently attached fragment (that would be displaced from N by binding of P) will almost always be greater than the bulk concentration of P. In addition, an entropic cost must be paid for intermolecular folding if the equivalent structure can be produced by intramolecular folding. If these penalties are sufficiently high, then ligand binding energy alone cannot generate the ternary complex necessary for FRET detection. Accordingly, we allow fragment exchange by preferentially destabilizing N relative to N*P and N*PL. Destabilization can be achieved by introducing a packing mutation into the hydrophobic core of N. The role of the packing mutation, which is selected to be distant from the binding site, is to decrease the stability of N (i.e., lower Kunf) without compromising the intrinsic affinity (Ka) or specificity for the ligand. Formation of N*PL is thus driven by the restoration of WT binding and packing interactions provided by the respective WT side chains in the fragment.
The key result of the design described above is that exchange is controlled by the severity of the packing mutation. Adjusting the extent of destabilization allows one to populate the high-FRET N*PL complex in the presence of ligand while minimizing the false-positive N*P state in the absence of ligand. As a demonstration, we simulated FREX in three representative stability regimes of FN3: that of the WT protein (Kunf = 5 × 10–5), a destabilized yet folded mutant (Kunf = 0.02), and an unfolded variant (Kunf = 10). We fixed Kex to 105 M–1, Ka to the value measured for the interaction between WT FN3 and SH2 (108 M–1),12 and protein concentrations to those used in our experiments ([N] = [P] = 2 μM). Panels A and B of Figure S1 of the Supporting Information plot the fractions of N*PL and N*P as a function of ligand concentration. SH2 does not bind appreciably to WT FN3 (Figure S1A of the Supporting Information) because the energy cost to unfold is too great. The unfolded FN3 mutant lacks this barrier and therefore binds SH2 most tightly; the apparent association constant (Ka,app, obtained by fitting the data to the one-site binding equation) is 9.7 × 105 M–1. The trade-off is that a significant fraction of FN3 (14%) exists as the binary complex in the absence of SH2 (Figure S1B of the Supporting Information). The destabilized-yet-folded mutant offers the best combination of high affinity [Ka,app = 2.2 × 105 M–1 (Figure S1A of the Supporting Information)] and a small population of N*P [0.39% (Figure S1B of the Supporting Information)]. Assuming that FRET efficiencies of N*PL and N*P are identical, the theoretical maximal signal-to-noise ratio of the sensor can be calculated by dividing [N*PL] at saturating [L] by [N*P] in the absence of ligand. These values are 257 and 7.4 for the destabilized and unfolded mutants of FN3, respectively.
FN3 Structure and Choice of Mutants
For the binding mutation, we focused on the largest of the three surface loops (residues 79–90 in FN3-HA4) with which FN3 monobodies contact their targets. Tyr87 of FN3-HA4 makes cation−π and polar interactions with SH2 (Figure 1B).12 The same study reported that the Y87A mutant abolishes detectable ligand binding. As insurance, we also changed the adjacent amino acid Met88, which also contacts SH2, to Ala to create the Y87A/M88A binding-null mutant (FN3BN).
The main choice to be made in the FREX methodology involves the identity of the fragment: whether to start at the N- or C-terminus of the parent protein and at what position to end. The fragment must encompass Tyr87 and Met88 and should terminate at a surface loop. We elected to generate three peptides, beginning at residues 48 (P48), 60 (P60), and 69 (P69) and ending at the C-terminus (Figure 1B). To aid in purification, we expressed the peptides with an N-terminal MBP tag.
For the packing sites, we selected the hydrophobic core residues Ile75 and Val77 of FN3BN (Figure 1B). We introduced underpacking mutations in which aliphatic side chains were progressively truncated (I75V, I75A, I75G, V77A, and V77G). All variants unfold cooperatively when they are denatured by guanidine hydrochloride (GdnHCl) (Figure S2 of the Supporting Information), with the exception of I75G, which failed to express. Fitting the data to the two-state equation ΔG = ΔGH2O – m[GdnHCl] finds that all of the mutations destabilize FN3BN as judged by both ΔGH2O and Cm, the midpoint of denaturation (Table S1 of the Supporting Information). Comparison of Cm values yields a rank order of stability commensurate with the number of carbons removed. We singled out the I75A mutant (FN3BN+I75A) for further study because it is the least stable by the Cm criterion and because its Kunf value of 0.021 is close to the value found to be reasonable in the simulations.
For labeling FN3BN with Alexa dyes, we created two mutants in which Cys was inserted at the N-terminus or in place of Ser48 in a surface loop. These Cys variants were used in FRET studies only; the Cys-free version was employed in all other experiments. Peptide constructs contain Cys in the middle of the five-amino acid peptide used to link MBP to P48, P60, and P69. The genetically encoded version of the P48 sensor was constructed by placing the CyPet and YPet fluorescent proteins18 at positions equivalent to those of the Cys residues, i.e., at the N-terminus of FN3BN+I75A and between MBP and P48, respectively.
Of note, we found that WT FN3 dimerizes extensively in solution. Size exclusion chromatography (SEC) showed approximately 50% dimer formation at a protein concentration of ∼30 μM (Figure S3 of the Supporting Information). The X-ray structure suggests a possible dimer interface in which the first four residues (GSSV) of one FN3 form a short β-strand that then adds to the β-sheet of the second molecule in a reciprocal fashion. We changed GSSV to YGGG to disrupt this putative interaction. The YGGG mutant eluted exclusively as a monomer in SEC experiments (Figure S3 of the Supporting Information) and was significantly more stable than WT FN3 (not shown). We therefore incorporated the YGGG mutation into all constructs used in this study.
Binding Tests Conducted via SEC
We first performed negative controls in which SEC was used to determine whether binary complexes form in the absence of the third species. FN3BN+I75A, peptide, and SH2 were mixed pairwise at concentrations of 5, 5, and 20 μM, respectively. Figure 2A confirms that FN3BN+I75A does not form a binary complex with P48, P60, P69, or SH2. FN3BN+I75A (11.0 kDa) and SH2 (13.7 kDa) elute close to the same volume, and the two peaks are not resolved; the peptides (∼46 kDa) are the largest species present because of their MBP purification tags. Likewise, P48, P60, and P69 do not bind SH2 (Figure 2B).
Figure 2.
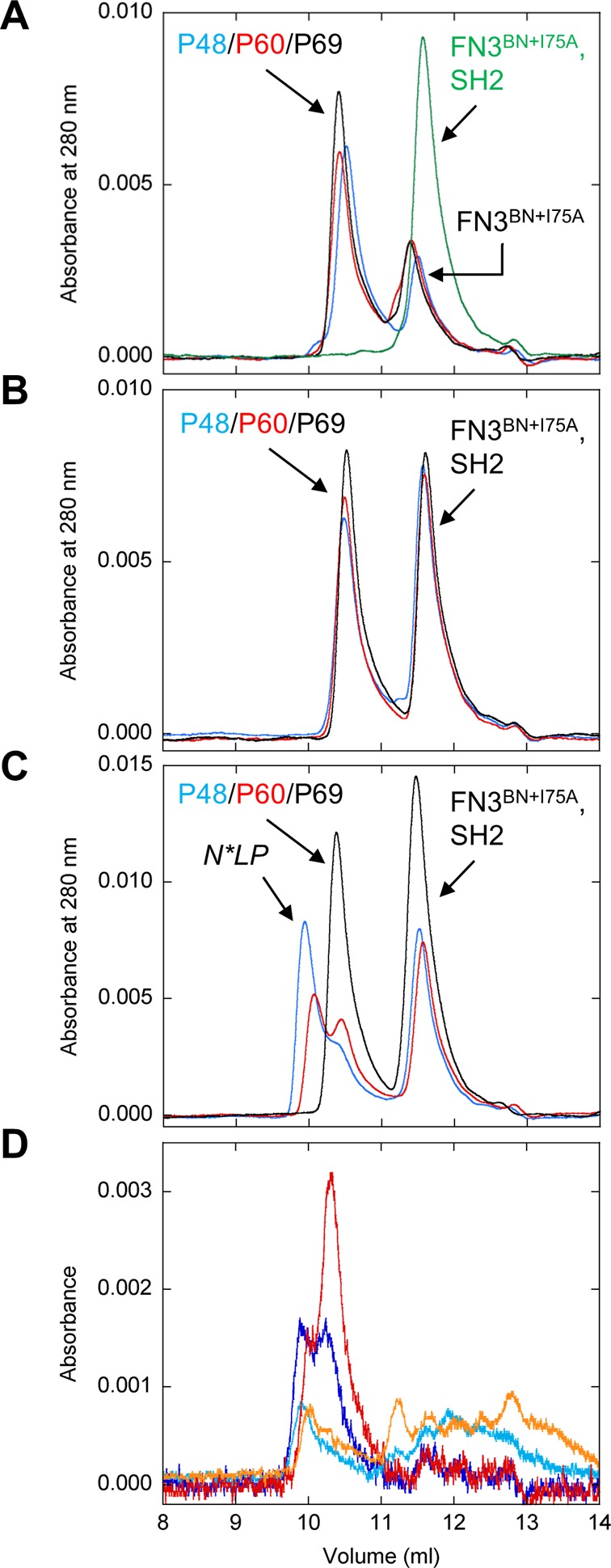
Binding tests conducted via SEC. (A) The first set of binary complex controls consisted of mixing FN3BN+I75A (5 μM) with P48 (blue), P60 (red), P69 (black), and SH2 (green). Peptide and SH2 concentrations are 5 and 20 μM, respectively. (B) The second set of binary complex controls consisted of mixing SH2 with P48 (blue), P60 (red), and P69 (black). (C) Ternary complex formation was tested by mixing FN3BN+I75A, SH2, and P48 (blue), P60 (red), or P69 (black). (D) Components of the N*PL complexes were identified by repeating the experiment in panel C using FN3BN+I75A labeled with Alexa594 and P48/P60 labeled with Alexa488. The chromatogram of the P48-containing sample is shown with absorbance detection at 488 nm (dark blue) and 594 nm (cyan). The chromatogram of the P60-containing sample is shown with absorbance detection at 488 nm (red) and 594 nm (orange).
We next tested for formation of the ternary complex. Mixing FN3BN+I75A, P48, and SH2 results in a higher-molecular mass species (estimated to be 78–88 kDa) consistent with N*PL (Figure 2C). Binding appears to be almost saturated as evidenced by the near disappearance of the free P48 peak (the free SH2 peak remains because it is present in 4-fold excess over P48). To determine whether the new peak is the expected N*PL complex or a nonspecific aggregate, we identified the components in that peak by labeling FN3BN+I75A with Alexa594 and P48 with Alexa488, the same dyes used in the FRET studies described below. The SEC experiment was then repeated using absorbance at 594 and 488 nm to detect FN3BN+I75A and P48, respectively. Figure 2D confirms that the N*PL peak contains both Alexa594 and Alexa488 whereas the P48 peak contains only Alexa488. Mixing P60 with FN3BN+I75A and SH2 also generates the ternary complex (Figure 2C). The decreased peak ratio of N*PL to free P60 suggests that the P60 sensor binds SH2 less tightly than the P48 sensor does. These conclusions are supported by SEC chromatograms of the Alexa-labeled proteins (Figure 2D). In contrast to P60 and P48, P69 does not form a ternary complex (Figure 2C).
The FREX mechanism stipulates that ligand binding is controlled by adjusting the severity of the packing mutation (eq 1). To test that hypothesis, we repeated the SEC experiments using P48, SH2, and FN3BN (Kunf = 4.6 × 10–5) or FN3BN+I75V (Kunf = 8.6 × 10–4) (Table S1 of the Supporting Information). N*PL is not detected in either case (Figure S4 of the Supporting Information), suggesting that binding is too weak to be detected by SEC.
FREX Biosensors
FREX biosensors were created by attaching the Alexa488 donor to the N-terminus of FN3BN+I75A and the Alexa594 acceptor to the N-termini of P48, P60, and P69. Using P48, we observed a ratiometric change in fluorescence intensity, with Alexa488 emission decreasing at 519 nm and Alexa594 emission increasing at 617 nm (Figure 3A). All spectra converge at an isosbestic wavelength of 584 nm. The binding data fit well to the one-site binding equation with a Ka,app of (2.52 ± 0.2) × 105 M–1 and a 3.0-fold change in the FRET ratio (Figure 3B and Table 1). In agreement with SEC data, the SH2 affinity of the P60 sensor [Ka,app = (7.20 ± 1) × 104 M–1] is lower than that of the P48 sensor, but the FRET response of the P60 sensor (8.4-fold change) is substantially larger (Figure S5 of the Supporting Information and Table 1). It is likely that poor labeling efficiency negatively affected the signal change of both sensors: despite repeated trials, we were able to achieve only 26–27% donor labeling (Table 1). Very little SH2 binding is observed for P69 as presaged by the SEC results.
Figure 3.

SH2 binding to FREX sensors monitored by FRET. FN3BN+I75A is labeled with the donor at the N-terminus in panels A–C and F. FN3BN+I75A is labeled with the donor at Cys48 in panels D and E. (A) Unprocessed spectra of donor-labeled FN3BN+I75A (2 μM) and acceptor-labeled P48 (2 μM) are overlaid to show ratiometric changes in fluorescence intensity as a function of increasing SH2 concentration, from 0 (black) to 50 μM SH2 (dark red). (B) Dependence of FRET ratio on SH2 concentration plotted for the P48 (blue circles), P60 (red squares), and P69 (black triangles) sensors. Lines are best fits of the data to the one-site binding equation. Error bars are standard deviations of triplicate experiments. (C) Donor emission at 519 nm (filled green squares) and acceptor emission at 617 nm (filled purple circles), obtained from spectra in panel A, plotted as a function of SH2 concentration. When acceptor-labeled P48 is mixed with unlabeled FN3BN+I75A, the acceptor fluorescence does not increase with SH2 concentration (empty purple circles). Similarly, when donor-labeled FN3BN+I75A is mixed with unlabeled P48, the donor emission does not decrease with SH2 concentration (empty green squares). (D) Unprocessed spectra of donor-labeled FN3BN+I75A and acceptor-labeled P48 are superimposed to show the increase in FRET efficiency resulting from moving the donor to position 48 of FN3BN+I75A from the N-terminus (cf. panel A). SH2 concentrations are identical to those in panel A. (E) The ratiometric output of the P48 sensor (calculated from spectra in panel D) improves when the donor is moved to position 48 of FN3BN+I75A from the N-terminus (cf. panel B). (F) The performance of the P48 sensor in 10% (v/v) fetal bovine serum (●) is comparable to that in buffer (○). Error bars are standard deviations of triplicate measurements.
Table 1. Binding Parameters of FREX Sensorsa.
| sensor variant | donor/acceptor type, location | donor, acceptor labeling efficiency (%) | Ka,app (M–1) | ratiometric response (x-fold change) |
|---|---|---|---|---|
| P48 | Alexa488/Alexa594b | 26, 109 | (2.41 ± 0.3) × 105 | 3.0 ± 0.2 |
| P60 | Alexa488/Alexa594b | 27, 106 | (7.30 ± 1) × 104 | 8.4 ± 0.9 |
| P69 | Alexa488/Alexa594b | 26, 106 | not detected | not detected |
| P48 | Alexa488 (Cys48)/Alexa594 (N-terminus) | 83, 107 | (3.15 ± 0.4) × 105 | 8.5 ± 0.9 |
| P60 | Alexa488 (Cys48)/Alexa594 (N-terminus) | 83, 106 | (5.62 ± 0.7) × 104 | 5.9 ± 0.3 |
| P48 (10% fetal bovine serum) | Alexa488/Alexa594b | 26, 109 | (1.22 ± 0.1) × 105 | 2.8 ± 0.1 |
| P48 (2:1 lysate ratio)c | CyPet/YPetb | not applicable | 1.7 × 105 | 1.3 |
| P48 (1:1 lysate ratio) | CyPet/YPetb | not applicable | 2.8 × 105 | 1.6 |
| P48 (1:2 lysate ratio) | CyPet/YPetb | not applicable | 2.6 × 105 | 1.8 |
Errors are standard deviations of three independent experiments.
The donor and acceptor are at N-termini.
Ratios reflect approximate molar ratios of CyPet-FN3BN+I75A to YPet-P48. Cell lysate volume ratios (CyPet-FN3BN+I75A:YPet-P48) are 1:1, 1:2, and 1:4 for the 2:1, 1:1, and 1:2 molar ratio samples, respectively.
We next considered the possibility that the ratiometric fluorescence changes in panels A and B of Figure 3 might be caused by quenching or another artifact rather than FRET. We repeated the binding assay in Figure 3B except in one experiment we added SH2 to unlabeled P48 (and donor-labeled FN3BN+I75A) and in the other we added SH2 to unlabeled FN3BN+I75A (and acceptor-labeled P48). The donor and acceptor fluorescence values do not change (Figure 3C). By contrast, when both FN3BN+I75A and P48 are labeled, the donor fluorescence decreases and the acceptor fluorescence increases in an SH2-dependent manner. These results signify that the ratiometric changes observed in Figure 3 arise from resonant energy transfer.
To test the effect of fluorophore placement on FRET response, we shifted the position of the donor from the N-terminus of FN3BN+I75A to position 48. Because P48 is labeled with the acceptor at almost the same position, this arrangement might bring the fluorophores closer in the bound state of the P48 sensor. The signal change of the P48 sensor improves to 8.5-fold, but this appears to be due to a higher donor labeling efficiency (83%), as evidenced by the increase in the donor:acceptor emission ratio of the free sensor (Figure 3E and Table 1). The FRET response of the P60 sensor decreases from 8.4- to 5.9-fold despite the improvement in donor labeling efficiency. This result suggests that the donor–acceptor distance is longer when the donor is at position 48 of FN3BN+I75A compared to the N-terminus. As expected, donor placement does not significantly change the affinity of the sensor for SH2 (Table 1).
Kinetics of Switching
To assess the temporal response of the sensors, test for reversibility, and gain insight into the mechanism of switching, we measured on and off rates by monitoring time-dependent changes in FRET. For P48, association and dissociation data fit adequately to single-exponential functions with a kon of (5.68 ± 0.6) × 10–4 s–1 and a koff of (3.58 ± 0.7) × 10–5 s–1 (Figure S6A of the Supporting Information). The FRET ratio returns to the theoretical limit (90% of the original value), signifying that the switch is fully reversible. For the P60 sensor, its kon [(5.38 ± 0.7) × 10–4 s–1] is identical within error to that of the P48 sensor (Figure S6B of the Supporting Information). The off rate of P60 [(6.62 ± 0.4) × 10–5 s–1] is 1.8-fold faster, in agreement with the lower affinity of the P60 switch. The FRET ratio of the P60 switch returns to only ∼2/3 of its original value. One explanation may be that the ternary complex exhibits a slightly greater affinity for donor-labeled FN3BN+I75A than it does for unlabeled FN3BN+I75A.
Association rates were found to be independent of SH2 concentration (Figure S6C of the Supporting Information). Equation 1, equation 2, or a combination of both is therefore rate-limiting. This result is not surprising because the pseudo-first-order association rate for eq 3 (∼500 s–1 at 50 μM SH2, calculated using a diffusion-limited on rate of 107 M–1 s–1) is orders of magnitude faster than the observed kon values. Observed on rates do not change significantly when the concentrations of FN3BN+I75A and P48 are lowered from 2 μM (Figure S6A of the Supporting Information) to 0.5 μM (Figure S6C of the Supporting Information), suggesting that the overall rate-limiting step for formation of the ternary complex is at least partial unfolding of FN3BN+I75A.
Sensor Performance under Real-World Conditions and Genetic Encoding
A major challenge facing any new biosensor design is that it must work in dirty environments rife with off-target binding decoys, proteases, quenchers, and other contaminants. To test the FREX performance under such conditions, we repeated the FRET binding experiment using FN3BN+I75A, P48, and SH2 in the presence of 10% fetal bovine serum. The resulting binding curve is very similar to that obtained in buffer (Figure 3F and Table 1). Thus, the response of the FN3 FREX sensor appears to be robust and resistant to large amounts of contaminants.
We created a genetically encoded variant of the P48 sensor by fusing the CyPet and YPet genes to the 5′-ends of the FN3BN+I75A and P48 genes, respectively. To assess sensor performance in unpurified cell lysates and to delineate the effect of differential expression of the components, two E. coli cultures were transformed separately, grown, and lysed, and the lysates were combined in several ratios. The level of expression of YPet-P48 was roughly twice that of CyPet-FN3BN+I75A as judged by the fluorescence emission of the lysates (not shown). We mixed CyPet-FN3BN+I75A and YPet-P48 lysates at volume ratios of 1:1, 2:1, and 4:1 to simulate up to 2-fold excesses of donor or acceptor that might be encountered upon co-expression of the components in vivo. Purified SH2 was then added to each mixture. Figure 4 shows fluorescence scans and fitted binding curves. Ka,app values are similar for the three samples (Table 1) and are in good agreement with those of the Alexa-labeled sensor obtained in buffer and in 10% serum, indicating that CyPet and YPet do not interfere with ligand binding. The FRET output improves slightly with an increasing acceptor:donor ratio, presumably because the response is optimal when every donor sees an acceptor. The ratiometric response of the genetically encoded P48 sensor in the crude lysate is weaker than that of its purified Alexa-labeled counterpart in buffer (Table 1). The reason is not clear, although it may be due to incomplete maturation of CyPet and YPet chromophores [∼50% (data not shown)]. The 1.3–1.8-fold FRET changes shown in Figure 4 are comparable to those of affinity-tag8 and SNAP-tag6,7 sensors.
Figure 4.

Performance of the genetically encoded P48 sensor in unpurified E. coli lysate. (A) Spectra of CyPet-FN3BN+I75A and YPet-P48 as a function of SH2 concentration. Approximate molar ratios of CyPet-FN3BN+I75A to YPet-P48 are indicated in each figure. Colors and SH2 concentrations are the same as in Figure 3A. Spectra are normalized to the donor emission peak for the sake of clarity. (B) Binding curves are shown below each figure in panel A. Lines are the best fits of the data to the one-site binding equation; fitted parameters are listed in Table 1.
We next tested sensor performance in mammalian cell cultures. We replaced CyPet and YPet with EGFP and mCherry, respectively, to be compatible with the optics of our fluorescence microscope. To help ensure that both sensor components were present in every transfected cell, the mCherry-P48 and EGFP-FN3BN+I75A genes were cloned, in that order, into the coding region of a bicistronic expression plasmid. The SH2 gene was placed on a second plasmid for cotransfection. In-cell FRET experiments were performed by first acquiring fluorescent images of transfected and fixed Cos-7 cells in EGFP and mCherry channels (Figure 5, top row of images). The lone criterion for choosing a cell for FRET imaging was that it exhibit moderate fluorescence in each channel at this stage, prior to the acceptor bleaching step by which FRET efficiency was determined. This selection method was intended to eliminate untransfected cells as well as those that expressed very high levels of one or both fluorescent proteins, which are known to produce FRET artifacts.19 Once a cell was judged to meet this criterion, it was bleached and imaged; no data were discarded thereafter. The FRET efficiency was estimated by bleaching the mCherry signal in a rectangular area within each cell (Figure 5, middle row of cells) and measuring the extent to which EGFP-FN3BN+I75A emission in that rectangular area increased after the bleach (Figure 5, bottom row of cells). Bleaching a defined region allows us to directly compare the EGFP intensity change inside and outside the rectangle in the same cell, thereby reducing false FRET caused by stage movement, pixel misalignment, etc.
Figure S7 of the Supporting Information shows images of 12 representative cells transfected with EGFP-FN3BN+I75A/mCherry-P48 alone (panel A; n = 20) and in combination with SH2 (panel B; n = 28). For cells that were not transfected with SH2, EGFP intensities inside and outside the bleached rectangle are similar, indicating low FRET efficiency (Figure S7A of the Supporting Information). The fold change in FRET efficiency inside versus outside the rectangle is relatively constant at 1.6 ± 0.3. This background FRET signal may result from random collisions between the donor and the acceptor fluorescent proteins. By contrast, most cells cotransfected with SH2 display noticeably brighter EGFP fluorescence inside the rectangle (Figure S7B of the Supporting Information). Of the 28 imaged cells, 25% show high FRET efficiency (>3-fold change; red border around images), 39% show moderate FRET efficiency (2–3-fold change; blue border), and 36% show background levels of FRET (<2-fold change; purple border). Control transfections with the SH2 plasmid alone revealed that the transfection efficiency was ∼70%. It is therefore possible that the 36% population of low-FRET cells represents those that were not successfully transfected with SH2.
Discussion
To the best of our knowledge, FREX is the first example of a biosensor based on a variation of protein fragment complementation. FREX may appear to resemble protein–fragment complementation assays (PCAs) such as split GFP,20 split luciferase,21 split ubiquitin,22 and split DHFR;23 however, the two technologies are fundamentally different as are their intended applications. In PCA, the split protein serves as a reporter only. One piece is fused to a “bait” protein and the other to a “prey” protein, and fragment complementation reports on whether the bait and prey interact. In FREX, the fragment exchange reaction itself serves as the basis for molecular recognition.
It is noteworthy that the FREX methodology employs fragment exchange rather than fragment complementation. It is theoretically possible to build a sensor based on simple complementation by bisecting a binding protein and labeling each piece with a fluorescent reporter. The problem is that it would be difficult to tune the thermodynamics of this system to make the switch respond to the ligand. When proteins are bisected, some fragments exhibit tight binding (always on) while others fail to complement altogether (always off). What would be needed is a pair of fragments that associate with just the right Kd such that they do not interact unless driven to do so by reasonable concentrations of ligand. Tuning in this case would consist of experimentally finding such fragments, because complementation affinity cannot currently be predicted from cleavage sites.
By contrast, FREX employs two copies of the same fragment. Tuning can therefore be achieved by rationally mutating one copy and not the other, using structural and thermodynamic principles that are both well-established and readily quantifiable. As a result, target affinity can be modulated gradually and predictably to match the needs of the application (Figure S1 of the Supporting Information). Although Ka,app depends on both Kex and Kunf, in practice it is usually advisable to choose the fragments with the largest Kex (because our data suggest that even fragments with very high affinity for each other will not exchange in the absence of ligand) and adjust Ka,app by varying the severity of the destabilizing mutation. Importantly, target specificity is not likely to change because the packing position is chosen to be distant from the active site. A secondary advantage of FREX compared to simple complementation is that only one of the components of FREX is a fragment, with the other being a full-length, native protein. This is expected to reduce the extent of potential aggregation and degradation problems associated with fragments and unfolded proteins.
Nonetheless, FREX combines aspects of the binding-induced folding and fragment complementation mechanisms, and as such, it is subject to some of the same limitations. Chief among them are reduced target affinity compared to that of the parent binding protein and the inability to predict Kex, respectively. Ligand interaction energy is used to drive folding in binding-induced folding and to facilitate fragment exchange in FREX. Some reduction in target affinity is therefore inevitable with both mechanisms. For example, the Ka,app values of Kohn and Plaxco’s unfolded SH3 sensor2 and our FREX sensors are both ≈100-fold lower than the Ka values of WT SH3 and WT FN3, respectively. In the case of FREX, it is possible to increase Ka,app by further destabilizing the full-length protein (Figure S1 of the Supporting Information). Koide and colleagues addressed the inability to predict complementation affinity by bisecting a related FN3 (FNfn10) at six sites corresponding to positions 16, 28, 48, 58, 69, and 91 in FN3-HA4.24 Cleavage at position 48 resulted in the tightest fragment complementation, although the fragments lacked residual structure in isolation and aggregation was prominent. Complementation was strong at positions 28 and 58, moderate at position 69, and too weak to be detected at positions 16 and 91. Perhaps not surprisingly, the rank order of complementation strength correlates with Ka,app of our sensors. PCA screening methods such as the yeast two-hybrid system have been employed to identify fragments that complement with high affinity.24 It may be useful to apply these techniques to generate viable fragments for FREX, although structural inspection proved to be sufficient in this case.
It should be noted that the FN3 sensors respond to changes in target concentration more slowly than do some other existing designs. The reason is that the rate-limiting step in FN3 switching appears to be an unfolding–dissociation event (displacement of the duplicate segment in FN3BN) rather than a binding or folding reaction as in most cases. The response time may be shortened by accelerating the rate of unfolding (e.g., by increasing the temperature or by employing a more destabilizing packing mutation). Nevertheless, the kon half-time of ∼30 min is sufficient for many in vitro applications and for monitoring cellular processes that occur on a time scale of minutes to hours, and especially when one needs to detect a scarce analyte, in which case a slow off rate is desirable for signal integration.25
FRET biosensors are expected to perform best when the levels of donor and acceptor are approximately equal. FREX can easily achieve this condition when the application is to detect targets outside of the cell. For in vivo uses, single-component sensors are convenient because the FRET pair can be encoded in the same gene. Two-component sensors like FREX can increase the dynamic range by reducing the background level of FRET in the off state. In this study, we co-expressed the two FREX components from a single promoter using an internal ribosome entry sequence. It is known that the level of expression of the downstream gene (EGFP-FN3BN+I75A in this case) is typically much lower than that of the upstream gene (mCherry-P48).26 In qualitative agreement, we found mCherry fluorescence to be greater than EGFP fluorescence for all cells examined (not shown). It may be feasible to improve FRET output by equalizing donor and acceptor expression, e.g., by placing the components on separate plasmids (we avoided this strategy here because of the need to introduce the target ligand using a second plasmid).
Conclusions
We have introduced the FREX switching mechanism into the FN3-HA4 binding scaffold. When FN3 monobodies are modified to recognize different targets, their cores are conserved, with structural differences largely limited to the three substrate recognition loops.12,27 Single mutations have been shown to disrupt binding of other engineered FN3s to their ligands, and these residues lie in the same loop that harbors the Tyr87 and Met88 binding mutations in FN3-HA4. Thus, it is reasonable to speculate that the FREX mechanism can be transferred to other FN3 monobodies by using the same peptides and packing mutations employed in this study, with minimal additional optimization. Future studies will reveal whether the FREX methodology can be applied to unrelated binding proteins to convert them into ligand-driven molecular switches.
Acknowledgments
We thank S. Koide (University of Chicago) for the FN3-HA4 and SH2 genes, P. Daugherty (University of California, Santa Barbara, CA) for the CyPet and YPet genes, B. Knox for the EGFP and mCherry genes, P. Calvert for the modified pCMV IRES bicistronic vector, N. Walker-Kopp for assistance with gene construction of WT FN3, c-Abl SH2, CyPet-FN3BN+I75A, and YPet-P48, N. Deakin and P. Calvert for advice on in-cell FRET, V. Sirotkin for technical assistance with confocal microscopy, and J.-H. Ha, D. Mitrea, and M. Stratton for review of the manuscript.
Supporting Information Available
One table and seven figures characterizing the stability, binding, and in vitro and in-cell FRET of sensor variants. This material is available free of charge via the Internet at http://pubs.acs.org.
This work was supported by National Institutes of Health Grant GM069755 to S.N.L.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Cissell K. A.; Shrestha S.; Purdie J.; Kroodsma D.; Deo S. K. (2008) Molecular biosensing system based on intrinsically disordered proteins. Anal. Bioanal. Chem. 391, 1721–1729. [DOI] [PubMed] [Google Scholar]
- Kohn J. E.; Plaxco K. W. (2005) Engineering a signal transduction mechanism for protein-based biosensors. Proc. Natl. Acad. Sci. U.S.A. 102, 10841–10845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M. M.; Mitrea D. M.; Loh S. N. (2008) A Ca2+-sensing molecular switch based on alternate frame protein folding. ACS Chem. Biol. 3, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton M. M.; Loh S. N. (2010) On the mechanism of protein fold-switching by a molecular sensor. Proteins: Struct., Funct., Bioinf. 78, 3260–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J. H.; Shinsky S. A.; Loh S. N. (2013) Stepwise conversion of a binding protein to a fluorescent switch: Application to Thermoanaerobacter tengcongensis ribose binding protein. Biochemistry 52, 600–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun M. A.; Tan K. T.; Nakata E.; Hinner M. J.; Johnsson K. (2009) Semisynthetic fluorescent sensor proteins based on self-labeling protein tags. J. Am. Chem. Soc. 131, 5873–5884. [DOI] [PubMed] [Google Scholar]
- Brun M. A.; Tan K. T.; Griss R.; Kielkowska A.; Reymond L.; Johnsson K. (2012) A semisynthetic fluorescent sensor protein for glutamate. J. Am. Chem. Soc. 134, 7676–7678. [DOI] [PubMed] [Google Scholar]
- Huang J.; Koide S. (2010) Rational conversion of affinity reagents into label-free sensors for peptide motifs by designed allostery. ACS Chem. Biol. 5, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millett I. S.; Doniach S.; Plaxco K. W. (2002) Toward a taxonomy of the denatured state: Small angle scattering studies of unfolded proteins. Adv. Protein Chem. 62, 241–262. [DOI] [PubMed] [Google Scholar]
- Fleming P. J., and Rose G. D. (2005) Conformational Properties of Unfolded Proteins, Protein Folding Handbook, Wiley-VCH, Weinheim, Germany. [Google Scholar]
- Kohn J. E.; Millett I. S.; Jacob J.; Zagrovic B.; Dillon T. M.; Cingel N.; Dothager R. S.; Seifert S.; Thiyagarajan P.; Sosnick T. R.; Hasan M. Z.; Pande V. S.; Ruczinski I.; Doniach S.; Plaxco K. W. (2004) Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. U.S.A. 101, 12491–12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun M. A.; Griss R.; Reymond L.; Tan K. T.; Piguet J.; Peters R. J.; Vogel H.; Johnsson K. (2011) Semisynthesis of fluorescent metabolite sensors on cell surfaces. J. Am. Chem. Soc. 133, 16235–16242. [DOI] [PubMed] [Google Scholar]
- Wojcik J.; Hantschel O.; Grebien F.; Kaupe I.; Bennett K. L.; Barkinge J.; Jones R. B.; Koide A.; Superti-Furga G.; Koide S. (2010) A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol. 17, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom L.; Calabro V. (2009) FN3: A new protein scaffold reaches the clinic. Drug Discovery Today 14, 949–955. [DOI] [PubMed] [Google Scholar]
- Jacobs S., and O’Neil K. (2012) in FN3 Domain Engineering (Kaumaya P., Ed.) InTech, Morn Hill, U.K. [Google Scholar]
- Koide A.; Wojcik J.; Gilbreth R. N.; Hoey R. J.; Koide S. (2012) Teaching an old scaffold new tricks: Monobodies constructed using alternative surfaces of the FN3 scaffold. J. Mol. Biol. 415, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide S.; Koide A.; Lipovsek D. (2012) Target-binding proteins based on the 10th human fibronectin type III domain (10Fn3). Methods Enzymol. 503, 135–156. [DOI] [PubMed] [Google Scholar]
- Deakin N. O.; Bass M. D.; Warwood S.; Schoelermann J.; Mostafavi-Pour Z.; Knight D.; Ballestrem C.; Humphries M. J. (2009) An integrin-α4-14-3-3ζ-paxillin ternary complex mediates localised Cdc42 activity and accelerates cell migration. J. Cell Sci. 122, 1654–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen A. W.; Daugherty P. S. (2005) Evolutionary optimization of fluorescent proteins for intracellular FRET. Nat. Biotechnol. 23, 355–360. [DOI] [PubMed] [Google Scholar]
- Karpova T., and McNally J. G. (2006) Detecting protein-protein interactions with CFP-YFP FRET by acceptor photobleaching. Current Protocols in Cytometry, Chapter 12, Unit 12, 17, Wiley, New York. [DOI] [PubMed] [Google Scholar]
- Cabantous S.; Waldo G. S. (2006) In vivo and in vitro protein solubility assays using split GFP. Nat. Methods 3, 845–854. [DOI] [PubMed] [Google Scholar]
- Ozawa T.; Kaihara A.; Sato M.; Tachihara K.; Umezawa Y. (2001) Split luciferase as an optical probe for detecting protein-protein interactions in mammalian cells based on protein splicing. Anal. Chem. 73, 2516–2521. [DOI] [PubMed] [Google Scholar]
- Johnsson N.; Varshavsky A. (1994) Split ubiquitin as a sensor of protein interactions in vivo. Proc. Natl. Acad. Sci. U.S.A. 91, 10340–10344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy I.; Campbell-Valois F. X.; Michnick S. W. (2007) Detection of protein-protein interactions using a simple survival protein-fragment complementation assay based on the enzyme dihydrofolate reductase. Nat. Protoc. 2, 2120–2125. [DOI] [PubMed] [Google Scholar]
- Dutta S.; Batori V.; Koide A.; Koide S. (2005) High-affinity fragment complementation of a fibronectin type III domain and its application to stability enhancement. Protein Sci. 14, 2838–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L.; Hires S. A.; Mao T.; Huber D.; Chiappe M. E.; Chalasani S. H.; Petreanu L.; Akerboom J.; McKinney S. A.; Schreiter E. R.; Bargmann C. I.; Jayaraman V.; Svoboda K.; Looger L. L. (2009) Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J.; van Weeren L.; Adjobo-Hermans M. J.; Elzenaar I.; Hink M. A.; Gadella T. W. Jr. (2011) Quantitative co-expression of proteins at the single cell level: Application to a multimeric FRET sensor. PLoS One 6, e27321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyani A.; Vitriol E.; Allen R.; Wu J.; Gremyachinskiy D.; Lewis S.; Dewar B.; Graves L. M.; Kay B. K.; Kuhlman B.; Elston T.; Hahn K. M. (2011) A biosensor generated via high-throughput screening quantifies cell edge Src dynamics. Nat. Chem. Biol. 7, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.