

Abstract
Background
Milk production is an economically important sector of global agriculture. Much attention has been paid to the identification of quantitative trait loci (QTL) associated with milk, fat, and protein yield and the genetic and molecular mechanisms underlying them. Copy number variation (CNV) is an emerging class of variants which may be associated with complex traits.
Results
In this study, we performed a genome-wide association between CNVs and milk production traits in 26,362 Holstein bulls and cows. A total of 99 candidate CNVs were identified using Illumina BovineSNP50 array data, and association tests for each production trait were performed using a linear regression analysis with PCA correlation. A total of 34 CNVs on 22 chromosomes were significantly associated with at least one milk production trait after false discovery rate (FDR) correction. Some of those CNVs were located within or near known QTL for milk production traits. We further investigated the relationship between associated CNVs with neighboring SNPs. For all 82 combinations of traits and CNVs (less than 400 kb in length), we found 17 cases where CNVs directly overlapped with tag SNPs and 40 cases where CNVs were adjacent to tag SNPs. In 5 cases, CNVs located were in strong linkage disequilibrium with tag SNPs, either within or adjacent to the same haplotype block. There were an additional 20 cases where CNVs did not have a significant association with SNPs, suggesting that the effects of those CNVs were probably not captured by tag SNPs.
Conclusion
We conclude that combining CNV with SNP analyses reveals more genetic variations underlying milk production traits than those revealed by SNPs alone.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-683) contains supplementary material, which is available to authorized users.
Keywords: Copy number variation (CNV), dPTA, Association, Milk production traits
Background
Milk production is an economically important sector of global agriculture and much attention has been paid to improve milk performance-related traits in cattle. Various methods have been employed to identify significant genetic markers for milk production. These methods include quantitative trait loci (QTL) mapping with different mapping designs and genome-wide association studies with a variety of statistical tests. Many QTL related to milk production traits have been reported using different populations and DNA markers, such as microsatellite [1–4] and SNPs [5–11]. The identification of QTL and investigation of genetic and molecular mechanisms underlying those QTL may result in more efficient animal selection and increased rates of genetic progress. However, most of these cattle QTL studies did not identify the casual variant, which is useful information for breeding applications to avoid losses in accuracy because of recombination between associated QTL markers and the actual quantitative trait nucleotide (QTN). Only a few casual mutations within genes of known function, such as DGAT [3, 12, 13], ABCG [4] and GRH [14, 15], have been identified with their large effects on milk production validated. Additionally, most of those GWAS studies concluded that SNP may only explain a small portion of genetic variance. Alternative frameworks to explain the missing heritability of complex traits were proposed [16].
Genomic structural variants are comprised mainly of copy number variations (CNVs) in the form of large-scale insertions and deletions, as well as inversions and translocations [17]. Compared to SNPs, CNVs involve more genomic sequence and have potentially greater effects, including changing gene structure and dosage, alternating gene regulation and exposing recessive alleles [18]. Human and mouse studies have found that CNVs capture 18 to 30% of the genetic variation in gene expression [19, 20]. Those CNVs were shown to be important in both normal phenotypic variability and disease susceptibility. In livestock, most CNV studies have used limited CNV detection methods, including CGH arrays, SNP arrays, and next generation sequencing [21–31]. Our previous studies have indicated that some CNVs could be associated with resistance or susceptibility to gastrointestinal nematodes in Angus cattle [32] and residual feed intake in Holstein cows [33]. Moreover, Glick et al. identified a CNV associated with fertility in Israeli Holsteins [34]. A recent study reported a 660 kb deletion with antagonistic effects on fertility and milk production in Nordic Red cattle [35]. Kadri et al. previously reported linkage disequilibrium (LD) between one deletion and its neighboring SNPs in Holsteins cattle [36]. However, no study has reported about genome wide CNV association directly with milk production traits. Furthermore, no systematic study of the relationship between CNVs and SNPs in the bovine genome has been published.
In this study, we reported a systematic CNV association analysis with milk production traits in 26,362 US Holsteins. Thirty-four CNVs have been identified as significantly associated with milk production traits using an association test, and most of them overlap known QTL. Haplotype analysis for associated CNVs and neighboring SNPs produced further evidence that CNVs provide additional information that is not captured by SNPs alone. Therefore, CNVs could be utilized as additional molecular markers for use in genetic improvement programs.
Methods
Samples
Holstein bulls and cows (26,362 samples) were genotyped using the Illumina BovineSNP50 array version 1 (Illumina Inc., San Diego, CA). Genotypes of those animals have been included in the routine genomic evaluation program for the United States and Canada since 2009 [37, 38]. The main source of extracted DNA for bulls was semen from the Cooperative Dairy DNA Repository and from the National Center for Genetic Resources Preservation, ARS, USDA (Fort Collins, CO). The research did not involve any experiment on animals and for this reason no ethics approval was necessary.
Phenotypic and dPTA values
Traditional predicted transmitting abilities (PTAs) for five production traits, including milk yield (MY), fat yield (FY), protein yield (PY), fat percentage (FP), and protein percentage (PP), were calculated by USDA ARS AIPL (Beltsville, MD). Those PTA are predicted additive genetic effects after removing fixed non-genetic effects, and the reliabilities of the PTA were used to quantify the amount of information available for different individuals [39, 40]. De-regressed PTAs (dPTA) were computed as in Garrick et al. [40] by dividing PTA by their squared reliability [dPTA = PTA/(reliability)2]. The dPTA were used as the phenotypes for genome-wide association studies.
CNV segmentation and genotyping
The intensity data of 56,947 SNP probes were generated by Illumina BovineSNP50 arrays. We imported Log R Ratios (LRR) from the GenomeStudio software into Golden Helix SNP & Variation Suite (SVS) 7.7 (Golden Helix Inc., Bozeman, MT, USA) using its DSF Export Plug-In 4.1.
A total of 48,669 SNPs were mapped onto the Bos taurus genome assembly UMD 3.1 (https://ccb.jhu.edu/bos_taurus_assembly.shtml) within 29 autosomes.
To normalize the LRR, we used the default GC correlation file (GC Reference bos_taurus_UMD3.1.gc_digest.dsf) to correct for the waviness contributed by GC content. We then utilized the copy number analysis module (CNAM) under the multivariate option to segment chromosomes with a maximum of 20 segments per window, a minimum of 3 markers per segment, and a significance level of p = 0.01 for pairwise permutations (n = 1,000) as described previously [41].
PCA-corrected association testing
In Golden Helix SVS 7.7, a linear regression under the additive genetic model was employed to identify CNVs associated with each production trait with the option of Full Scan Permutations (10,000 permutations). We used the principal component analysis (PCA) option to correct batch effects/stratification of the test input data. Significant CNVs were counted at the significance level of (p-value < 0.05) after FDR correction.
Association tests based on SNPs
Association tests were carried out using the linear model plugin of SVS 7.7 software with PCA correction. Significant SNPs were detected when their adjusted p-values passed the threshold of p < 1 × 10-8 after FDR correction.
Haplotype block analysis
To investigate linkage disequilibrium (LD) patterns in the regions containing associated CNVs, pairwise LD statistics D’ and r2 were calculated using Haploview (Version 4.2) [42] and the LD blocks were defined by the criteria of Gabriel et al. [43]. Qanbari et al. [44] presented a linkage disequilibrium (LD) map for Holsteins based on the same Bovine SNP50 array. Based on their maximum haplotype block length (1.26 Mb), the regions considered for LD analysis were extended 25 SNPs on the both upstream and downstream directions of each CNV (i.e., 1.26 Mb/average marker spacing of 50 kb).
Relationship between significantly associated CNVs and significantly associated SNPs
We classified all 82 significant combinations of traits and CNVs (less than 400 kb in length and p values < 0.05 after FDR correction as shown in bold and italic in Table 1) in the following way. First, when a significant SNP directly overlapped with a CNV by genomic coordinate, we called the situation as “IN”. If a SNP was directly adjacent to a CNV, we called it “IN*”. When a SNP was the second SNP next to a CNV, we called it “IN**”. Any SNPs beyond the neighboring 2 positions of a CNV were not considered in this comparison. We then evaluated the linkage relationship between CNVs and neighboring SNPs. When a CNV was in the same haploblock with at least one significant SNP, we called it “LD”. When a CNV was adjacent to a haploblock which contains at least one significant SNP, we called it “LD*”. In contrast, when a CNV was in a haploblock which contained no significant SNP, we called it “LD NS”. When a CNV was adjacent to a haploblock which contained no significant SNP, we called it “LD* NS”. For complicated situation where both “IN” and “LD” existed, “IN” was considered first and used instead of “LD” as the category. Finally, when a CNV did not overlap with any SNPs and was not in a haploblock, we called it “NN”.
Table 1.
Relationship classification between trait associated CNVs and their neighboring SNPs for 82 combinations of trait and CNVs
| CNV# | P value after FDR correction | Tagged by SNPs | Breakpoint | PennCNV | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MY | FY | PY | FP | PP | MY | FY | PY | FP | PP | Support | ||
| 1 | 2.56E-06 | 2.12E-08 | 0.0246 | 0.0657 | 3.96E-21 | IN* | IN* | IN* | NN | |||
| 2 | 8.53E-07 | 1.32E-07 | 0.0020 | 0.8913 | 1.46E-09 | IN | IN | IN | NN | Yes | ||
| 3 | 0.0810 | 0.1375 | 0.9468 | 0.6499 | 1.32E-32 | IN* | ||||||
| 4 | 1.26E-09 | 1.99E-08 | 4.06E-09 | 0.1334 | 0.0756 | IN** | IN** | IN** | Yes | |||
| 5 | 0.2506 | 0.0473 | 0.2410 | 0.0758 | 0.0008 | IN* | IN* | Yes | Yes | |||
| 6 | 0.0008 | 1.91E-06 | 4.95E-05 | 0.0004 | 6.95E-22 | NN | NN | IN | NN | NN | Yes | |
| 7 | 0.0020 | 0.0001 | 0.0418 | 0.2851 | 1.38E-05 | IN** | IN** | NN | IN** | Yes | ||
| 8 | 9.03E-10 | 5.38E-10 | 1.61E-06 | 1.0000 | 9.35E-05 | IN* | IN* | IN* | IN* | Yes | ||
| 9 | 0.0005 | 2.50E-06 | 0.2701 | 0.0049 | 9.47E-18 | IN** | IN** | NN | IN | |||
| 10 | 9.29E-12 | 8.38E-12 | 2.23E-07 | 1.0000 | 0.0997 | IN | IN | IN* | ||||
| 11 | 3.81E-05 | 6.52E-05 | 0.0345 | 0.7610 | 0.0037 | IN* | IN* | IN* | IN | Yes | Yes | |
| 12 | 0.0006 | 0.1336 | 0.5275 | 3.70E-09 | 1.52E-22 | IN* | IN* | NN | Yes | |||
| 14 | 0.1484 | 0.5341 | 0.7490 | 0.0700 | 0.0371 | IN* | Yes | |||||
| 16 | 0.2552 | 0.8739 | 0.0352 | 0.0033 | 0.2416 | NN | LD* | Yes | ||||
| 17 | 6.96E-07 | 2.87E-06 | 2.99E-05 | 0.4144 | 0.9401 | IN* | IN** | IN* | Yes | |||
| 18 | 0.0890 | 2.02E-05 | 0.0062 | 1.79E-13 | 6.21E-15 | LD | LD | IN* + LD | IN* | Yes | Yes | |
| 19 | 0.0032 | 3.53E-05 | 0.2698 | 0.0061 | 0.0035 | IN* | IN* | IN* | IN | Yes | ||
| 20 | 0.9468 | 0.8614 | 0.6384 | 1.0000 | 0.0286 | NN | ||||||
| 21 | 0.0798 | 0.1692 | 0.5656 | 0.3360 | 0.0433 | IN + LD | Yes | |||||
| 22 | 0.6780 | 0.4652 | 0.3753 | 0.9048 | 0.0005 | NN | Yes | Yes | ||||
| 23 | 0.0065 | 0.0209 | 0.0008 | 0.5674 | 7.60E-05 | NN | IN** | IN** | NN | |||
| 24 | 0.0616 | 0.0844 | 0.6569 | 0.8919 | 6.98E-09 | NN | Yes | Yes | ||||
| 25 | 4.43E-08 | 1.58E-08 | 1.94E-05 | 0.9206 | 0.4082 | IN | IN** | IN** | ||||
| 26 | 0.8385 | 0.7831 | 0.8926 | 0.9465 | 2.51E-16 | IN + LD | Yes | |||||
| 28 | 0.0016 | 1.70E-08 | 0.0008 | 2.21E-13 | 0.0002 | LD* NS | LD* NS | LD* NS | IN* + LD | LD* | Yes | Yes |
| 29 | 8.09E-07 | 2.03E-07 | 0.0007 | 0.7631 | 3.02E-05 | IN + LD | IN + LD | IN + LD | NN | Yes | ||
| 31 | 0.1197 | 0.0159 | 0.1152 | 0.0773 | 3.15E-05 | IN | IN** | Yes | Yes | |||
| 32 | 9.08E-09 | 3.95E-08 | 0.0001 | 0.5293 | 4.20E-07 | LD NS | NN | LD | IN + LD | |||
CNV27, CNV33 to CNV37 were not considered due to their large sizes (> 400 kb). For CNVs less than 400 kb in length, their p values < 0.05 after FDR correction were shown in bold and italic. For the meanings of IN*, IN**, LD*, please refer to the main text.
Results and discussion
Trait properties and correlations
We selected five traits related to milk production for an association analysis: milk yield (MY), fat yield (FY), protein yield (PY), fat percentage (FP), protein percentage (PP). The descriptive statistics of PTA, including reliability and heritability, are given in Additional file 1: Figure S1 and Additional file 2: Figure S2, respectively. Pearson correlation coefficients for all pairs of traits are provided in Table 2. As expected, the three yield traits (MY, FY and PY) are strongly and positively correlated. The two percentage traits (FP and PP) are positively correlated with each other, but are negatively correlated with the yield traits.
Table 2.
Pairwise Pearson correlation coefficients for all pairs of traits
| Milk Yield | Fat Yield | Protein Yield | Fat Percentage | Protein Percentage | |
|---|---|---|---|---|---|
| Milk Yield | - | 0.7181 | 0.9040 | -0.3285 | -0.3596 |
| Fat Yield | 0.7181 | - | 0.7938 | 0.4206 | 0.0581 |
| Protein Yield | 0.9040 | 0.7938 | - | -0.1012 | 0.0723 |
| Fat Percentage | -0.3285 | 0.4206 | -0.1012 | - | 0.5470 |
| Protein Percentage | -0.3596 | 0.0581 | 0.0723 | 0.5470 | - |
CNV segmentation and genotyping
In contrast to conventional CNV discovery studies, which try to detect as many variable regions as possible, this CNV-based GWAS is intended to identify the common CNVs shared among samples in order to detect associations with common diseases or traits. Using the multivariate method of CNAM in SVS, a total of 2,626,669 distinct segments were detected in the 26,362 samples. After merging across samples, 99 nonredundant CNVs were left for subsequent association test (Additional file 3: Table S1). Within these 99 segments, each sample was genotyped (i.e., called as loss, neutral or gain event) according to a three-state model with strict threshold levels of marker mean ± 0.5. Since the multivariate CNAM method was developed to identify common CNVs, only those segments with frequencies above 0.4% were retained for further analysis in order to filter away false positive calls. A total of 39 CNVs ranging in size from 45,109 bp to 7.16 Mb were retained (frequency > 0.04). These 39 CNVs have an estimated average size and SNP count of 962.71 kb and 18.4, respectively.
CNV association analyses
A total of 26,362 Holstein cattle were employed to test associations between CNVs and dPTA data. Using a linear regression, we identified a total of 34 CNVs that were significantly associated with at less one trait (Figure 1 and Additional file 4: Table S2). Among those 34 associated CNVs, the CNV with the highest frequency (96.03%) was found at chr14:11,250,157-11,307,423, while the CNV with the lowest frequency (2.09%) was localized at chr29:46,099,425-51,502,868 (Additional file 4: Table S2). After we removed 6 CNVs larger than 400 kb (CNV27, CNV33 to CNV37), we compared the 28 remaining CNVs with 5 previously published results [23, 26, 27, 45, 46]. We found 23 out of these 28 CNVs (82.1%) were also previously reported, and the 5 non-overlapping CNVs (CNV4, 5, 8, 9 and 23) had high frequencies (ranging from 30 to 70%) and large marker mean changes (ranging from -0.56 to -1.31, deviated from 0), suggesting they are probably real CNVs. Three CNVs: CNV6 (chr13:70,496,054-70,623,303), CNV28 (chr7:42,700,425-42,788,788), and CNV33 (chr17:73,055,503-75,058,715) had P values < 0.05 after FDR correction for all five traits. Their frequencies were 66.88%, 16.95% and 7.55%, respectively (Additional file 4: Table S2). Another 12 CNVs were significantly associated with four traits. The overlapping relationship of these associated CNVs among 5 traits was shown in a Venn diagram (Additional file 5: Figure S3).
Figure 1.
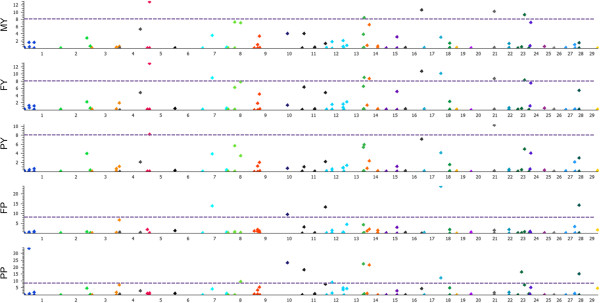
Manhattan plots of associated CNVs for five milk production traits (Milk Yield, Fat Yield, Protein Yield, Fat Percentage and Protein Percentage) using linear regression model. Negative log10-transformed P values from a genome-wide scan are plotted against genomic coordinates on 29 autosomal chromosomes.
We then overlapped these 34 associated CNVs with the reported cattle QTL as reported by the Animal QTL database at http://www.animalgenome.org/QTLdb/cattle.html [47]. When considered together, 21 of the 34 significant CNVs overlapped with at least one of the known QTL for milk production. Among these 34 CNVs, we found 15, 15, 14, 7 and 6 CNVs overlapping with QTL for MY, FY, PY, FP and PP traits, respectively (Additional file 4: Table S2). Based on the UMD 3.1 assembly, Additional file 4: Table S2 summarizes these 34 CNV regions, bovine RefSeq gene annotations within the CNV regions (1X), and flanking regions (3X: extended regions by one CNV length in both downstream and upstream directions).
CNVs significantly associated with milk traits
Of the 34 CNVs, 19, 23 and 18 were significantly associated with MY, FY and PY, respectively (Additional file 4: Table S2). Taken together, 15 CNVs were significantly associated with all three yield traits. One top-ranked CNV, CNV10 (chr5:9,756,491-9,837,147), reached significance values of 9.66 × 10-12, 8.38 × 10-12 and 2.23 × 10-7 for MY, FY, and PY, respectively. In the proximity of this region, two QTL effect peaks were reported at the 1 to 3 cM region of chr5 for milk traits in Holstein cattle using SNP data [11]. On chr14, we identified CNV1 (chr14:11,250,157-11,307,423) starting from SNP Hapmap29947-BTC-070181, near the previously reported milk production QTL regions. This CNV had the highest frequency (96.03%) and is 57,267 bp long. Approximately 6 Mb upstream of this CNV segment, the NIBP gene has been reported to also have highly significant effect for milk yield [8, 48]. Several important genes located further upstream, including DGAT1 and VPS28, have been validated to have highly significant effects for milk production traits [12, 49]. Within the gene cluster including DGAT1 and NIBP, our previous SNP-based study using U.S. Holstein cows also identified some SNPs, which are significantly associated with effects on milk production trait [8].
We also identified 11 and 29 CNVs that were significantly associated with FP and PP, respectively. The most significant CNV associated with FP was CNV33 (chr17:73,055,503-75,058,715) with a p-value of 7.00 × 10-23). We also found other CNVs like CNV12, CNV18, CNV28 and CNV34. However, CNV28 overlaps with only one QTL previously reported on chr7 [50]. Using PP, we identified the largest number (23) of significantly associated CNVs. Eleven of them were also significantly associated with all three yield traits (Table 1 and Additional file 5: Figure S3). The top five CNVs were CNV3, CNV6, CNV1, CNV12 and CNV9 based on their p values of the association test. QTL evidence from previous studies were also found in CNV1 [50–52], CNV24 [53], CNV26 [53, 54] CNV36 [55] and CNV37 [56–58].
Relationship between associated CNVs and associated SNPs
To further explore the relationship between SNPs and CNVs, we carried out association tests based on SNPs using the same 26,362 Holstein cattle SNP array data. Additional file 6: Table S3 contains SNP information near CNV regions. For all 82 possible combinations of traits and CNVs (less than 400 kb in length), we found 17 cases where significant SNPs directly overlapped with CNVs (e.g. IN for MY and CNV2, Figure 2A), 26 cases where significant SNPs were directly adjacent to CNVs (IN* for MY and CNV8, Figure 2B), and 14 cases where significant SNPs are the second SNPs next to CNVs (IN** for PP and CNV31, Figure 2C). To determine if there is any linkage between CNVs and neighboring SNPs, we performed LD analysis by computing pairwise D’ around CNV regions which included 25 SNPs both downstream and upstream of associated CNVs. Our results showed three cases (LD for FY and CNV18, Figure 3A) where CNVs were enclosed in the same haplotype block with at least one significant SNP, two cases where CNVs were directly adjacent to a haploblock which contains at least one significant SNP (LD* for FP and CNV16, Figure 3B), which suggested these CNVs could be represented by tagged SNPs within the same haploblock.
Figure 2.
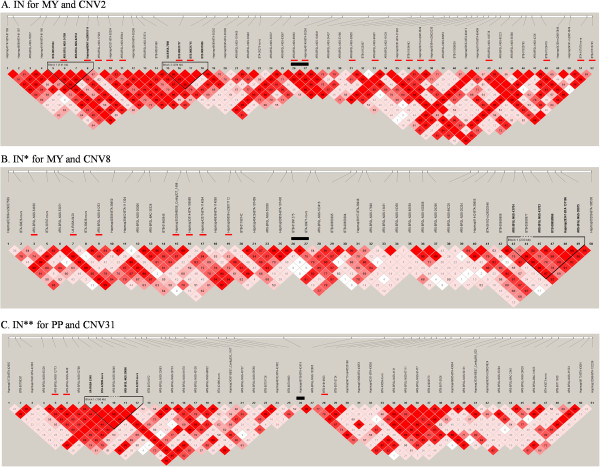
Haplotype analysis of (A) IN for MY, (B) IN* for MY and CNV8 and (C) IN** for PP and CNV31. Black bar represents CNV and red bars represent significant tag SNPs.
Figure 3.

Haplotype analysis of (A) LD for FY and CNV18 and (B) LD* for FP and CNV16. Black bar represents CNV and red bars represent significant tag SNPs.
Some cases were complex situations involving both IN and LD. We generally assigned them to the IN, IN* and IN** instead of LD or LD* classes. However, no matter which class was used, CNVs in both the IN and LD classes were apparently well-captured by neighboring tag SNPs. It is interesting to note that although 9 out of 31 CNVs were located within breakpoints of haploblocks (Table 1 and Additional file 7: Figure S4), 7 of them were still tagged by neighboring SNPs. For example, we found one haploblock from Hapmap55972-rs29011387 to Hapmap50751-BTA-64830 in CNV28 with a length of 385 kb. We observed one SNP ARS-BFGL-NGS-63581 which was significantly associated with MY, FY, and PY (Additional file 7: Figure S4). We obtained one haploblock from Hapmap54599-rs29019617 to Hapmap48210-BTA-120730 in CNV21 and one SNPs Hapmap48210-BTA-120730 were detected to associate with MY, FY, PY and PP. For CNV32, one haploblock from ARS-BFGL-NGS-109612 to ARS-BFGL-NGS-26195 that contained a PY-associated SNP INRA-655 was identified. For CNV26, one large region containing three adjacent haploblocks (161 kb, 286 kb, 85 kb), which ranged from ARS-BFGL-BAC-28908 to ARS-BFGL-NGS-501, was found. These regions contained many SNPs associated with production traits. Additionally, two adjacent haploblocks were detected near CNV4, from ARS-BFGL-NGS-234 to ARS-BFGL-NGS-35131. This region contains one SNP ARS-BFGL-NGS-102090, which was significantly associated with MY, FY, and PY. Moreover, three adjacent haploblocks were found near CNV 28 (from ARS-BFGL-NGS-100845 to ARS-BFGL-NGS-13798), several significant associated SNPs were also found in this region.
Finally, we also found 20 cases where CNVs were not related (overlapping, neighboring or LD) with significantly associated SNPs (Table 3), suggesting that the impacts of those CNVs were probably not captured (e.g., NN for PP and CNV6, Figure 4. For more examples, please see Additional file 7: Figure S4). The possibility for this observation is that CNVs are likely to work as independent variants besides SNP. For example, we indentified a haploblock embedded in CNV16, which spanned 363 kb on chr27. However, no significant associated SNPs were found in this haploblock region for PY.
Table 3.
Summary of relationship between CNVs and SNPs
| MY | FY | PY | FP | PP | All | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Count | % | Count | % | Count | % | Count | % | Count | % | Count | % | |
| NN | 2 | 11.76% | 2 | 10.53% | 2 | 12.50% | 2 | 28.57% | 8 | 34.78% | 16 | 19.51% |
| LD NS | 2 | 11.76% | 1 | 5.26% | 1 | 6.25% | 0 | 0.00% | 0 | 0.00% | 4 | 4.88% |
| Not tagged | 4 | 23.53% | 3 | 15.79% | 3 | 18.75% | 2 | 28.57% | 8 | 34.78% | 20 | 24.39% |
| LD | 0 | 0.00% | 1 | 5.26% | 2 | 12.50% | 0 | 0.00% | 0 | 0.00% | 3 | 3.66% |
| LD* | 0 | 0.00% | 0 | 0.00% | 0 | 0.00% | 1 | 14.29% | 1 | 4.35% | 2 | 2.44% |
| IN | 4 | 23.53% | 4 | 21.05% | 3 | 18.75% | 0 | 0.00% | 6 | 26.09% | 17 | 20.73% |
| IN* | 6 | 35.29% | 5 | 26.32% | 5 | 31.25% | 4 | 57.14% | 6 | 26.09% | 26 | 31.71% |
| IN** | 3 | 17.65% | 6 | 31.58% | 3 | 18.75% | 0 | 0.00% | 2 | 8.70% | 14 | 17.07% |
| Tagged | 13 | 76.47% | 16 | 84.21% | 13 | 81.25% | 5 | 71.43% | 15 | 65.22% | 62 | 75.61% |
| Total | 17 | 19 | 16 | 7 | 23 | 82 | ||||||
For the meanings of IN*, IN**, LD*, please refer to the main text.
Figure 4.

Haplotype analysis of NN for PP and CNV6. Black bar represents CNV and red bars represent significant tag SNPs.
Conclusions
Previous and current genome wide association studies have been investigated to identify significant genes or linked markers based on SNPs. In this CNV-based study, our results indicate that CNV are associated with, and likely contribute to, differences in milk production. Our study provides a systematic estimate that approximately one-quarter of CNVs are not captured by LD with nearby SNPs. This provides an alternative framework to explain the missing heritability of complex traits. This study helps to fill gaps left by current SNP-based genome wide association and selection studies. Therefore, we conclude that combining CNV with SNP analyses reveals more genetic variations underlying milk production traits than can be revealed by SNPs alone. Interrogation of the genome for both CNVs and SNPs, including common and rare variations, could be an effective way to identify the causes of complex diseases and traits [16, 59]. A more comprehensive appreciation of the full spectrum of genetic variation may unravel the genetic basis for milk production.
Availability of supporting data
Supporting information is available in the additional files and further supporting data is available from the authors on request.
Electronic supplementary material
Additional file 1: Figure S1: Boxplot of reliability of five production traits. (PDF 76 KB)
Additional file 2: Figure S2: Heritability of five production traits. (PDF 75 KB)
Additional file 3: Table S1: List of CNV segments. (XLSX 25 KB)
Additional file 4: Table S2: Cattle QTL and RefSeq genes of 34 CNVs based on the UMD 3.1 assembly. (XLSX 20 KB)
Additional file 5: Figure S3: Overlapping relationships of 34 significantly associated CNV segments among five milk production traits. (PDF 107 KB)
Additional file 6: Table S3: SNPs near CNV regions. (XLSX 210 KB)
Additional file 7: Figure S4: Haplotype block views. (PDF 1 MB)
Acknowledgements
We thank Reuben Anderson and Alexandre Dimitriv for technical assistance. This work was supported in part by AFRI grant No. 2011-67015-30183 from USDA NIFA. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture. The USDA is an equal opportunity provider and employer.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
GEL and LX conceived and designed the experiments. LX, YH, DMB, JS and GEL performed in silico prediction and computational analyses. JBC, PMV, TSS and CPVT collected the phenotypic and genotypic data. GEL, LX, and JBC wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Lingyang Xu, Email: xulingyang2008@gmail.com.
John B Cole, Email: John.Cole@ars.usda.gov.
Derek M Bickhart, Email: Derek.Bickhart@ars.usda.gov.
Yali Hou, Email: houyali1210@gmail.com.
Jiuzhou Song, Email: songj88@umd.edu.
Paul M VanRaden, Email: Paul.Vanraden@ars.usda.gov.
Tad S Sonstegard, Email: Tad.Sonstegard@ars.usda.gov.
Curtis P Van Tassell, Email: Curt.Vantassell@ars.usda.gov.
George E Liu, Email: George.Liu@ars.usda.gov.
References
- 1.Schnabel RD, Kim JJ, Ashwell MS, Sonstegard TS, Van Tassell CP, Connor EE, Taylor JF. Fine-mapping milk production quantitative trait loci on BTA6: analysis of the bovine osteopontin gene. Proc Natl Acad Sci U S A. 2005;102:6896–6901. doi: 10.1073/pnas.0502398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gautier M, Barcelona RR, Fritz S, Grohs C, Druet T, Boichard D, Eggen A, Meuwissen THE. Fine mapping and physical characterization of two linked quantitative trait loci affecting milk fat yield in dairy cattle on BTA26. Genetics. 2006;172:425–436. doi: 10.1534/genetics.105.046169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P, Cambisano N, Mni M, Reid S, Simon P, Spelman R, Georges M, Snell R. Positional candidate cloning of a QTL in dairy cattle: Identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 2002;12:222–231. doi: 10.1101/gr.224202. [DOI] [PubMed] [Google Scholar]
- 4.Cohen-Zinder M, Seroussi E, Larkin DM, Loor JJ, Everts-van der Wind A, Lee JH, Drackley JK, Band MR, Hernandez AG, Shani M, Lewin HA, Weller JI, Ron M. Identification of a missense mutation in the bovine ABCG2 gene with a major effect on the QTL on chromosome 6 affecting milk yield and composition in Holstein cattle. Genome Res. 2005;15:936–944. doi: 10.1101/gr.3806705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang L, Liu JF, Sun DX, Ma PP, Ding XD, Yu Y, Zhang Q. Genome Wide Association Studies for Milk Production Traits in Chinese Holstein Population. Plos One. 2010;5:e13661. doi: 10.1371/journal.pone.0013661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meredith BK, Kearney FJ, Finlay EK, Bradley DG, Fahey AG, Berry DP, Lynn DJ. Genome-wide associations for milk production and somatic cell score in Holstein-Friesian cattle in Ireland. BMC Genet. 2012;13:21. doi: 10.1186/1471-2156-13-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maxa J, Neuditschko M, Russ I, Forster M, Medugorac I. Genome-wide association mapping of milk production traits in Braunvieh cattle. J Dairy Sci. 2012;95:5357–5364. doi: 10.3168/jds.2011-4673. [DOI] [PubMed] [Google Scholar]
- 8.Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ, Jr, Crooker BA, Van Tassell CP, Yang J, Wang S, Matukumalli LK, Da Y. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011;12:408. doi: 10.1186/1471-2164-12-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang XL, Wurmser C, Pausch H, Jung S, Reinhardt F, Tetens J, Thaller G, Fries R. Plos One. 2012. Identification and Dissection of Four Major QTL Affecting Milk Fat Content in the German Holstein-Friesian Population. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kolbehdari D, Wang Z, Grant JR, Murdoch B, Prasad A, Xiu Z, Marques E, Stothard P, Moore SS. A whole genome scan to map QTL for milk production traits and somatic cell score in Canadian Holstein bulls. J Anim Breed Genet. 2009;126:216–227. doi: 10.1111/j.1439-0388.2008.00793.x. [DOI] [PubMed] [Google Scholar]
- 11.Daetwyler HD, Schenkel FS, Sargolzaei M, Robinson JAB. A genome scan to detect quantitative trait loci for economically important traits in Holstein cattle using two methods and a dense single nucleotide polymorphism map. J Dairy Sci. 2008;91:3225–3236. doi: 10.3168/jds.2007-0333. [DOI] [PubMed] [Google Scholar]
- 12.Winter A, Kramer W, Werner FAO, Kollers S, Kata S, Durstewitz G, Buitkamp J, Womack JE, Thaller G, Fries R. Association of a lysine-232/alanine polymorphism in a bovine gene encoding acyl-CoA : diacylglycerol acyltransferase (DGAT1) with variation at a quantitative trait locus for milk fat content. Proc Natl Acad Sci U S A. 2002;99:9300–9305. doi: 10.1073/pnas.142293799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grisart B, Farnir F, Karim L, Cambisano N, Kim JJ, Kvasz A, Mni M, Simon P, Frere JM, Coppieters W, Georges M. Genetic and functional confirmation of the causality of the DGAT1 K232A quantitative trait nucleotide in affecting milk yield and composition. Proc Natl Acad Sci U S A. 2004;101:2398–2403. doi: 10.1073/pnas.0308518100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Viitala S, Szyda J, Blott S, Schulman N, Lidauer M, Maki-Tanila A, George M, Vilkki J. The role of the bovine growth hormone receptor and prolactin receptor genes in milk, fat and protein production in Finnish Ayrshire dairy cattle. Genetics. 2006;173:2151–2164. doi: 10.1534/genetics.105.046730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blott S, Kim JJ, Moisio S, Schmidt-Kuntzel A, Cornet A, Berzi P, Cambisano N, Ford C, Grisart B, Johnson D, Karim L, Simon P, Snell R, Spelman R, Wong J, Vilkki J, Georges M, Farnir F, Coppieters W. Molecular dissection of a quantitative trait locus: A phenylalanine-to-tyrosine substitution in the transmembrane domain of the bovine growth hormone receptor is associated with a major effect on milk yield and composition. Genetics. 2003;163:253–266. doi: 10.1093/genetics/163.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scherer SW, Lee C, Birney E, Altshuler DM, Eichler EE, Carter NP, Hurles ME, Feuk L. Challenges and standards in integrating surveys of structural variation. Nat Genet. 2007;39:S7–15. doi: 10.1038/ng2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang F, Gu W, Hurles ME, Lupski JR. Copy number variation in human health, disease, and evolution. Annu Rev Genomics Hum Genet. 2009;10:451–481. doi: 10.1146/annurev.genom.9.081307.164217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stranger BE, Forrest MS, Dunning M, Ingle CE, Beazley C, Thorne N, Redon R, Bird CP, de Grassi A, Lee C, Tyler-Smith C, Carter N, Scherer SW, Tavare S, Deloukas P, Hurles ME, Dermitzakis ET. Relative impact of nucleotide and copy number variation on gene expression phenotypes. Science. 2007;315:848–853. doi: 10.1126/science.1136678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henrichsen CN, Vinckenbosch N, Zollner S, Chaignat E, Pradervand S, Schutz F, Ruedi M, Kaessmann H, Reymond A. Segmental copy number variation shapes tissue transcriptomes. Nat Genet. 2009;41:424–429. doi: 10.1038/ng.345. [DOI] [PubMed] [Google Scholar]
- 21.Liu GE, Hou Y, Zhu B, Cardone MF, Jiang L, Cellamare A, Mitra A, Alexander LJ, Coutinho LL, Dell'Aquila ME, Gasbarre LC, Lacalandra G, Li RW, Matukumalli LK, Nonneman D, Regitano LC, Smith TP, Song J, Sonstegard TS, Van Tassell CP, Ventura M, Eichler EE, McDaneld TG, Keele JW. Analysis of copy number variations among diverse cattle breeds. Genome Res. 2010;20:693–703. doi: 10.1101/gr.105403.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bae JS, Cheong HS, Kim LH, NamGung S, Park TJ, Chun JY, Kim JY, Pasaje CFA, Lee JS, Shin HD. Identification of copy number variations and common deletion polymorphisms in cattle. BMC Genomics. 2010;11:232. doi: 10.1186/1471-2164-11-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hou Y, Liu GE, Bickhart DM, Cardone MF, Wang K, Kim ES, Matukumalli LK, Ventura M, Song J, VanRaden PM, Sonstegard TS, Van Tassell CP. Genomic characteristics of cattle copy number variations. BMC Genomics. 2011;12:127. doi: 10.1186/1471-2164-12-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi JW, Lee KT, Liao XP, Stothard P, An HS, Ahn S, Lee S, Lee SY, Moore SS, Kim TH. Genome-wide copy number variation in Hanwoo, Black Angus, and Holstein cattle. Mamm Genome. 2013;24:151–163. doi: 10.1007/s00335-013-9449-z. [DOI] [PubMed] [Google Scholar]
- 25.Cicconardi F, Chillemi G, Tramontano A, Marchitelli C, Valentini A, Ajmone-Marsan P, Nardone A. Massive screening of copy number population-scale variation in Bos taurus genome. BMC Genomics. 2013;14:124. doi: 10.1186/1471-2164-14-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hou Y, Bickhart DM, Hvinden ML, Li C, Song J, Boichard DA, Fritz S, Eggen A, DeNise S, Wiggans GR, Sonstegard TS, Van Tassell CP, Liu GE. Fine mapping of copy number variations on two cattle genome assemblies using high density SNP array. BMC Genomics. 2012;13:376. doi: 10.1186/1471-2164-13-376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang L, Jiang JC, Yang J, Liu X, Wang JY, Wang HF, Ding XD, Liu JF, Zhang Q. Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genomics. 2013;14:131. doi: 10.1186/1471-2164-14-131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bickhart DM, Hou Y, Schroeder SG, Alkan C, Cardone MF, Matukumalli LK, Song J, Schnabel RD, Ventura M, Taylor JF, Garcia JF, Van Tassell CP, Sonstegard TS, Eichler EE, Liu GE. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012;22:778–790. doi: 10.1101/gr.133967.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fadista J, Thomsen B, Holm LE, Bendixen C. Copy number variation in the bovine genome. BMC Genomics. 2010;11:284. doi: 10.1186/1471-2164-11-284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seroussi E, Glick G, Shirak A, Yakobson E, Weller JI, Ezra E, Zeron Y. Analysis of copy loss and gain variations in Holstein cattle autosomes using BeadChip SNPs. BMC Genomics. 2010;11:673. doi: 10.1186/1471-2164-11-673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang L, Jia S, Yang M, Xu Y, Li C, Sun J, Huang Y, Lan X, Lei C, Zhou Y, Zhang C, Zhao X, Chen H. Detection of copy number variations and their effects in Chinese bulls. BMC Genomics. 2014;15:480. doi: 10.1186/1471-2164-15-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hou Y, Liu GE, Bickhart DM, Matukumalli LK, Li C, Song J, Gasbarre LC, Van Tassell CP, Sonstegard TS. Genomic regions showing copy number variations associate with resistance or susceptibility to gastrointestinal nematodes in Angus cattle. Funct Integr Genomics. 2012;12:81–92. doi: 10.1007/s10142-011-0252-1. [DOI] [PubMed] [Google Scholar]
- 33.Hou Y, Bickhart DM, Chung H, Hutchison JL, Norman HD, Connor EE, Liu GE. Analysis of copy number variations in Holstein cows identify potential mechanisms contributing to differences in residual feed intake. Funct Integr Genomics. 2012;12:717–723. doi: 10.1007/s10142-012-0295-y. [DOI] [PubMed] [Google Scholar]
- 34.Glick G, Shirak A, Seroussi E, Zeron Y, Ezra E, Weller JI, Ron M. Fine Mapping of a QTL for Fertility on BTA7 and Its Association With a CNV in the Israeli Holsteins. G3 (Bethesda) 2011;1:65–74. doi: 10.1534/g3.111.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kadri NK, Sahana G, Charlier C, Iso-Touru T, Guldbrandtsen B, Karim L, Nielsen US, Panitz F, Aamand GP, Schulman N, Georges M, Vilkki J, Lund MS, Druet T. A 660-kb deletion with antagonistic effects on fertility and milk production segregates at high frequency in nordic red cattle: additional evidence for the common occurrence of balancing selection in livestock. PLoS Genet. 2014;10:e1004049. doi: 10.1371/journal.pgen.1004049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadri NK, Koks PD, Meuwissen TH. Prediction of a deletion copy number variant by a dense SNP panel. Genet Sel Evol. 2012;44:7. doi: 10.1186/1297-9686-44-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.VanRaden PM, Van Tassell CP, Wiggans GR, Sonstegard TS, Schnabel RD, Taylor JF, Schenkel FS. Invited review: Reliability of genomic predictions for North American Holstein bulls. J Dairy Sci. 2009;92:16–24. doi: 10.3168/jds.2008-1514. [DOI] [PubMed] [Google Scholar]
- 38.Wiggans GR, Sonstegard TS, Vanraden PM, Matukumalli LK, Schnabel RD, Taylor JF, Schenkel FS, Van Tassell CP. Selection of single-nucleotide polymorphisms and quality of genotypes used in genomic evaluation of dairy cattle in the United States and Canada. J Dairy Sci. 2009;92:3431–3436. doi: 10.3168/jds.2008-1758. [DOI] [PubMed] [Google Scholar]
- 39.Vanraden PM, Wiggans GR. Derivation, Calculation, and Use of National Animal-Model Information. J Dairy Sci. 1991;74:2737–2746. doi: 10.3168/jds.S0022-0302(91)78453-1. [DOI] [PubMed] [Google Scholar]
- 40.Garrick DJ, Taylor JF, Fernando RL. Deregressing estimated breeding values and weighting information for genomic regression analyses. Genet Sel Evol. 2009;41:1. doi: 10.1186/1297-9686-41-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu L, Hou Y, Bickhart DM, Song J, Van Tassell CP, Sonstegard TS, Liu GE. A genome-wide survey reveals a deletion polymorphism associated with resistance to gastrointestinal nematodes in Angus cattle. Funct Integr Genomics. 2014;14:333–339. doi: 10.1007/s10142-014-0371-6. [DOI] [PubMed] [Google Scholar]
- 42.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 43.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, Higgins J, DeFelice M, Lochner A, Faggart M, Liu-Cordero SN, Rotimi C, Adeyemo A, Cooper R, Ward R, Lander ES, Daly MJ, Altshuler D. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 44.Qanbari S, Pimentel EC, Tetens J, Thaller G, Lichtner P, Sharifi AR, Simianer H. The pattern of linkage disequilibrium in German Holstein cattle. Anim Genet. 2010;41:346–356. doi: 10.1111/j.1365-2052.2009.02011.x. [DOI] [PubMed] [Google Scholar]
- 45.Zhan B, Fadista J, Thomsen B, Hedegaard J, Panitz F, Bendixen C. Global assessment of genomic variation in cattle by genome resequencing and high-throughput genotyping. BMC Genomics. 2011;12:557. doi: 10.1186/1471-2164-12-557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jiang L, Jiang JC, Wang JY, Ding XD, Liu JF, Zhang Q. Genome-Wide Identification of Copy Number Variations in Chinese Holstein. Plos One. 2012;7:e48732. doi: 10.1371/journal.pone.0048732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu ZL, Park CA, Wu XL, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41:D871–D879. doi: 10.1093/nar/gks1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma L, Wiggans GR, Wang S, Sonstegard TS, Yang J, Crooker BA, Cole JB, Van Tassell CP, Lawlor TJ, Da Y. Effect of sample stratification on dairy GWAS results. BMC Genomics. 2012;13:536. doi: 10.1186/1471-2164-13-536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thaller G, Kramer W, Winter A, Kaupe B, Erhardt G, Fries R. Effects of DGAT1 variants on milk production traits in German cattle breeds. J Anim Sci. 2003;81:1911–1918. doi: 10.2527/2003.8181911x. [DOI] [PubMed] [Google Scholar]
- 50.Boichard D, Grohs C, Bourgeois F, Cerqueira F, Faugeras R, Neau A, Rupp R, Amigues Y, Boscher MY, Leveziel H. Detection of genes influencing economic traits in three French dairy cattle breeds. Genet Sel Evol. 2003;35:77–101. doi: 10.1186/1297-9686-35-1-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaupe B, Brandt H, Prinzenberg EM, Erhardt G. Joint analysis of the influence of CYP11B1 and DGAT1 genetic variation on milk production, somatic cell score, conformation, reproduction, and productive lifespan in German Holstein cattle. J Anim Sci. 2007;85:11–21. doi: 10.2527/jas.2005-753. [DOI] [PubMed] [Google Scholar]
- 52.Bennewitz J, Reinsch N, Grohs C, Leveziel H, Malafosse A, Thomsen H, Xu N, Looft C, Kuhn C, Brockmann GA, Schwerin M, Weimann C, Hiendleder S, Erhardt G, Medjugorac I, Russ I, Forster M, Brenig B, Reinhardt F, Reents R, Averdunk G, Blumel J, Boichard D, Kalm E. Combined analysis of data from two granddaughter designs: A simple strategy for QTL confirmation and increasing experimental power in dairy cattle. Genet Sel Evol. 2003;35:319–338. doi: 10.1186/1297-9686-35-3-319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viitala SM, Schulman NF, de Koning DJ, Elo K, Kinos R, Virta A, Virta J, Maki-Tanila A, Vilkki JH. Quantitative trait loci affecting milk production traits in Finnish Ayrshire dairy cattle. J Dairy Sci. 2003;86:1828–1836. doi: 10.3168/jds.S0022-0302(03)73769-2. [DOI] [PubMed] [Google Scholar]
- 54.Elo KT, Vilkki J, de Koning DJ, Velmala RJ, Maki-Tanila AV. A quantitative trait locus for live weight maps to bovine chromosome 23. Mamm Genome. 1999;10:831–835. doi: 10.1007/s003359901098. [DOI] [PubMed] [Google Scholar]
- 55.Bagnato A, Schiavini F, Rossoni A, Maltecca C, Dolezal M, Medugorac I, Solkner J, Russo V, Fontanesi L, Friedmann A, Soller M, Lipkin E. Quantitative trait loci affecting milk yield and protein percentage in a three-country Brown Swiss population. J Dairy Sci. 2008;91:767–783. doi: 10.3168/jds.2007-0507. [DOI] [PubMed] [Google Scholar]
- 56.Schopen GC, Visker MH, Koks PD, Mullaart E, van Arendonk JA, Bovenhuis H. Whole-genome association study for milk protein composition in dairy cattle. J Dairy Sci. 2011;94:3148–3158. doi: 10.3168/jds.2010-4030. [DOI] [PubMed] [Google Scholar]
- 57.Magee DA, Sikora KM, Berkowicz EW, Berry DP, Howard DJ, Mullen MP, Evans RD, Spillane C, Machugh DE. DNA sequence polymorphisms in a panel of eight candidate bovine imprinted genes and their association with performance traits in Irish Holstein-Friesian cattle. BMC Genet. 2010;11:93. doi: 10.1186/1471-2156-11-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bagnicka E, Siadkowska E, Strzalkowska N, Zelazowska B, Flisikowski K, Krzyzewski J, Zwierzchowski L. Association of polymorphisms in exons 2 and 10 of the insulin-like growth factor 2 ( IGF2) gene with milk production traits in Polish Holstein-Friesian cattle. J Dairy Res. 2010;77:37–42. doi: 10.1017/S0022029909990197. [DOI] [PubMed] [Google Scholar]
- 59.McCarroll SA. Extending genome-wide association studies to copy-number variation. Hum Mol Genet. 2008;17:R135–R142. doi: 10.1093/hmg/ddn282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1: Boxplot of reliability of five production traits. (PDF 76 KB)
Additional file 2: Figure S2: Heritability of five production traits. (PDF 75 KB)
Additional file 3: Table S1: List of CNV segments. (XLSX 25 KB)
Additional file 4: Table S2: Cattle QTL and RefSeq genes of 34 CNVs based on the UMD 3.1 assembly. (XLSX 20 KB)
Additional file 5: Figure S3: Overlapping relationships of 34 significantly associated CNV segments among five milk production traits. (PDF 107 KB)
Additional file 6: Table S3: SNPs near CNV regions. (XLSX 210 KB)
Additional file 7: Figure S4: Haplotype block views. (PDF 1 MB)