

Summary
The human kidneys produce approximately 160–170 L of ultrafiltrate per day. The proximal tubule contributes to fluid, electrolyte, and nutrient homeostasis by reabsorbing approximately 60%–70% of the water and NaCl, a greater proportion of the NaHCO3, and nearly all of the nutrients in the ultrafiltrate. The proximal tubule is also the site of active solute secretion, hormone production, and many of the metabolic functions of the kidney. This review discusses the transport of NaCl, NaHCO3, glucose, amino acids, and two clinically important anions, citrate and phosphate. NaCl and the accompanying water are reabsorbed in an isotonic fashion. The energy that drives this process is generated largely by the basolateral Na+/K+-ATPase, which creates an inward negative membrane potential and Na+-gradient. Various Na+-dependent countertransporters and cotransporters use the energy of this gradient to promote the uptake of HCO3− and various solutes, respectively. A Na+-dependent cotransporter mediates the movement of HCO3− across the basolateral membrane, whereas various Na+-independent passive transporters accomplish the export of various other solutes. To illustrate its homeostatic feat, the proximal tubule alters its metabolism and transport properties in response to metabolic acidosis. The uptake and catabolism of glutamine and citrate are increased during acidosis, whereas the recovery of phosphate from the ultrafiltrate is decreased. The increased catabolism of glutamine results in increased ammoniagenesis and gluconeogenesis. Excretion of the resulting ammonium ions facilitates the excretion of acid, whereas the combined pathways accomplish the net production of HCO3− ions that are added to the plasma to partially restore acid-base balance.
Introduction
The extracellular fluid (ECF) space provides a constant environment for the cells of a multicellular organism and prevents wide fluctuations in the ambient environment. This enables the cells to devote their gene products to more productive functions. The kidney is the principal organ that maintains the amount and composition of the ECF by executing functions of excretion, metabolism, and provision of endocrine substances. Most of these kidney functions occur in the proximal tubule, which is an ancient segment in mammalian nephron evolution.
In terms of excretion, the proximal tubule maintains an array of secretory mechanisms inherited from the more archaic secretory nephrons, which are ancestors of mammalian nephrons. The proximal tubule is also a tour de force of reabsorption of the glomerular filtrate. The filtration-reabsorption scheme is critical because, as metabolic rates escalated during mammalian evolution, GFR had to increase accordingly. The high GFR mandates a corresponding increase in reabsorption to prevent loss of valuable solutes and water. The proximal tubule fulfills most of the reabsorptive role for NaCl and NaHCO3, leaving the fine-tuning to the distal nephron. The proximal tubule also completes the reabsorption of glucose, amino acids, and important anions, including phosphate and citrate, because it is the sole site of transport of these filtered solutes.
In addition to solute reabsorption and secretion, the proximal tubule is also a metabolic organ. For example, within the proximal tubule, 25-hydroxy-vitamin D is converted to 1,25-dihydroxy-vitamin D, a hormone that increases blood Ca2+ levels. The proximal tubule is also the site of the 24-hydroxylase reaction that converts 25-hydroxy-vitamin D and 1,25-dihydroxy-vitamin D to their inactive forms (1). In addition, the proximal tubule is an important site of gluconeogenesis that parallels the liver (2). As an endocrine organ, the kidney also releases erythropoietin, renin, and Klotho into the systemic circulation and produces a plethora of locally active paracrine/autocrine and intracrine hormones, such as dopamine, endothelin, PGs, renin, angiotensin II, and so forth (1,3–5).
Space constraints do not permit a comprehensive account of proximal tubule function in this article. Thus, we will highlight NaCl and NaHCO3 handling as examples of reclamation of filtrate that are critical in preventing shock and fatal acidosis and where the proximal tubule accomplishes the bulk uptake, leaving the completion to the distal nephron. We will also briefly cover the reabsorption of glucose, amino acids, phosphate, and one organic anion, citrate, where the entire regulatory and absorptive function is confined to the proximal tubule. Whereas glucose and phosphate are primarily returned to the circulation, citrate represents one substrate that is partially metabolized in the proximal tubule. Another organic substrate that is absorbed and metabolized is the amino acid glutamine. This process provides the nitrogen and carbon skeleton necessary to support renal gluconeogenesis and ammoniagenesis. Finally, the proximal tubule constantly adjusts its functions in response to needs, which is the hallmark of a stringent homeostatic system (6). Metabolic acidosis represents a state where there is concerted adaptations in multiple proximal tubule transport and metabolic functions aimed at minimization of the effect of the excess acid on the organism and rectification of the disturbance.
NaCl and NaHCO3 Transport
Na+ is the primary cation that maintains the ECF volume (ECFV). Because Cl− is four times more abundant than HCO3− as an ECFV anion, NaCl balance has become synonymous with ECFV regulation. NaHCO3 is also a major ECFV solute, second only to NaCl, but it is the principal intracellular and extracellular buffer for H+. Thus, NaHCO3 is better known for its role in acid-base balance than ECFV maintenance. There is limited regulation of gastrointestinal Na+, Cl−, or HCO3− absorption so the kidney is the primary organ that regulates external electrolyte balance. The high GFR in humans (160–170 L/d) mandates reabsorption of the valuable filtered solutes. Otherwise, approximately 24,000 mmol of filtered Na+ and approximately 4000 mmol of filtered HCO3− would be lost per day with disastrous consequences. The proximal tubule is the first nephron segment after the glomerulus where reabsorption commences. It is important to note that proximal solute and water reabsorption proceeds primarily in an isotonic fashion with very small changes in luminal osmolarity. Figure 1A shows the profile of changes in selected solutes along the length of the proximal tubule. Figure 1B shows a generic cell model of how transepithelial transport is achieved. Transporters can broadly be viewed from a thermodynamic standpoint as being driven primarily by changes in enthalpy or entropy. Enthalpy-based or active transporters are directly coupled to ATP hydrolysis. They use energy released from the hydrolysis of phosphoanhydride bonds to move solutes uphill; hence, such transporters are by nature ATPases. Entropy-based or secondary active transporters dissipate existing electrochemical gradients to move a solute against a concentration gradient. Thus, they use the downhill free energy change of one solute to energize the uphill movement of another solute.
Figure 1.
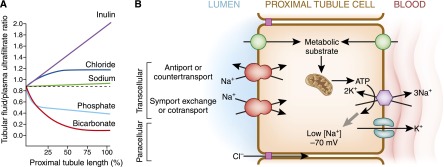
General considerations of proximal tubule transport. (A) Profile of the tubular fluid to plasma ultrafiltrate ratio (TF/PUF). Selected solutes are shown along the length of the proximal tubule. PUF is a surrogate for the proto-urine in Bowman’s space. Inulin represents a filtered molecule that is neither secreted nor reabsorbed and the rise in its TF/PUF solely reflects reabsorption of water, which concentrates luminal inulin. Sodium reabsorption is near isotonic with water, which results in a very small increase in TF/PUF by the end of the proximal tubule. HCO3− absorption in the early proximal convoluted tubule is particularly avid leading to rapid fall in TF/PUF. The fall in luminal [HCO3−] is accompanied by a reciprocal rise in luminal [Cl−] as reabsorption remains by-and-large isotonic. Inorganic phosphate (Pi) reabsorption is more avid in the earlier parts of the proximal tubule. (B) Generic scheme of the proximal tubule cell. The primary energy currency is organic metabolic substrates that enter the proximal tubule and are catabolized to produce ATP, which serves as the secondary energy currency. Some transporters are directly coupled to ATP hydrolysis (enthalpic transport), such as the H+-ATPase and Na+/K+-ATPase. The latter represents the main workhorse of the proximal tubule responsible for the majority of the cellular ATP consumption. The Na+/K+-ATPase converts the energy stored in ATP into low cellular [Na+] and high cellular [K+]. The presence of K+ conductance allows the [K+] gradient to increase the negative interior potential. The low cell [Na+] and negative voltage serve as the tertiary energy currencies that drive multiple secondary active apical transporters to achieve uphill movement of solutes coupled to downhill movement of Na+ (entropic transport). The transported solutes move in the same (symport or cotransport) or opposite (antiport, exchanger, or countertransport) direction as Na+. Movement of solute can also proceed via paracellular routes driven by electrochemical forces.
Transepithelial transport can occur via the paracellular or transcellular route, both of which are driven by electrochemical forces. The energy for solute movement is derived ultimately from high energy bonds in organic substrates taken up from the blood whose catabolism converts the energy into ATP (Figure 1B). Although there are multiple active transport systems directly coupled to ATP hydrolysis, the basolateral Na+-K+-ATPase is the principal consumer of ATP in the proximal tubule. It creates a low cellular [Na+] and negative voltage, which provides the ultimate energy to transport a multitude of solutes across the proximal tubule (Figure 1B). Apical secondary active solute entry can proceed through Na+-dependent cotransporters (symporters), exchangers (antiporters), parallel transporter systems, or other Na+-independent facilitated transporters (6).
NaCl Transport
Approximately 60%–70% of the filtered NaCl and the accompanying water are reabsorbed by the proximal tubule in a near isotonic fashion. This process is vital to the preservation of ECFV in the face of a high GFR. The foremost driving force is provided by the basolateral Na+-K+-ATPase, which sets the electrochemical gradient to drive a number of apical transporters that mediate Na+ and Cl− entry (Figure 2) as bipartite-coupled parallel exchangers or tripartite-coupled parallel exchangers that all eventuate in net NaCl entry into the cell. Basolateral Cl− exit is mediated by Cl−-carrying exchangers and cotransporters (Figure 2) (7–9). Na+-coupled transport is a general mechanism in the apical membrane so not all of the Na+ ions that enter the cell are devoted to NaCl transport (6). For example, a significant amount of glucose enters the apical membrane via Na+-glucose cotransport (10). This electrogenic process (net positive charge moving into the cell) contributes to a slight negative luminal potential. In addition, the avid absorption of HCO3− in the early proximal tubule and the isotonic nature of the transport elevate the luminal [Cl−] to above that of plasma (11). This combined electrochemical driving force in coalition with paracellular Cl− permeability results in Cl− movement from urine to blood, which is tantamount to essentially “Na+-glucose-Cl− absorption” (Figure 2). Alternatively, Na+ can leak back from the paracellular space into the lumen, providing a recycling system for Na+-coupled transport of glucose.
Figure 2.

Proximal tubule NaCl reabsorption. Unlike the thick ascending limb or distal convoluted tubule, there are no dedicated “NaCl” transporters in the proximal tubule. The proximal tubule uses parallel arrays of symporters and antiporters to affect NaCl entry. (A) Parallel Na+/H+ exchange (NHE3) and Cl−/base− exchange (CFEX, SLC26A6) with recycling of the conjugate acid HX resulting in net NaCl entry. (B) A triple coupling mechanism where Na-sulfate cotransport (NaS-1) runs in parallel with two anion exchangers to achieve NaCl cotransport. (C) Na+-coupled organic solute transport with concurrent paracellular Cl− transport driven by the lumen-to-interstitial space downhill chemical gradient of [Cl−]. Basolateral Na+ exit is mediated by the Na+/K+-ATPase but some Na+ also exit with HCO3− (see Figure 3). Cl− exit is less well defined utilizing a variety of transporters and possibly a Cl− channel.
The regulation of proximal tubule NaCl reabsorption facilitates the maintenance of ECFV. Hormones that maintain the ECFV and the integrity of the circulation through antinatriuresis stimulate proximal NaCl absorption. These include angiotensin II (12), endothelin (13), and α-adrenergic stimuli (14). Conversely, natriuretic hormones, such as dopamine, inhibit proximal tubule NaCl reabsorption (15). Hormonal factors regulate largely transcellular NaCl flux via modulation of the Na+ transporters (16). Because NHE3, the apical Na+/H+ exchanger, serves both NaCl and NaHCO3 reabsorption, it is not entirely clear how modulation of these two modes of transport can be dissociated when NHE3 is the prime target of regulation (17). In the proximal tubule, there is also a unique set of regulators that are largely physical in nature and involve coordinate coupling to regulation of GFR. These regulators are unlikely to operate in other nephron segments. When effective arterial blood volume is reduced, GFR is maintained by changes in arteriolar resistances that increase the filtration fraction despite lower renal plasma flow. This increases the protein concentration and oncotic pressure in the postglomerular blood, which along with the lower hydrostatic pressure jointly promote proximal fluid to move into the peritubular capillary.
NaHCO3 Transport
Whereas NaHCO3 contributes to the maintenance of ECFV, HCO3− is one of the major buffers that protect an organism from constant and pervasive acidification. A 70-kg human contains a free [H+] of 40 nM in about 42 L of water. Consumption of a high-protein Western diet results in a net production of 50–70 mEq of H+ per day. Thus, in the absence of an appropriate buffer, the daily production of H+ will decrease the body pH to <3 within an hour, which is clearly not compatible with life. HCO3− ions provide the major buffer system that prevents the rapid acidification of the ECF. The kidney is the primary organ that controls plasma [HCO3−]. The burden of renal transport is to excrete an amount of acid equivalent to the daily net H+ production plus the amount of filtered HCO3−, which is equivalent to the addition of acid if not reclaimed. HCO3− reclamation is achieved not so much by HCO3− reabsorption but, rather, by H+ secretion. The approximately 4000 mEq/d of HCO3− reclaimed is much higher than any daily dietary acid or base burden. Whether the organism is consuming a net acid diet of 50 mEq H+ equivalent per day versus a net alkaline diet of 50 mEq OH− equivalent per day, the H+ that needs to be secreted into the lumen is 4050 mEq/d versus 3950 mEq/d, respectively. Therefore, the proximal tubular epithelium is perpetually engaged in an H+-secreting mode regardless of the dietary load.
The proximal tubule plays a pivotal role in many aspects of acid-base homeostasis, which extends beyond HCO3− reclamation and H+ excretion (18). Because the urine cannot possibly maintain a pH low enough to hold 50–70 mEq of free H+, urinary buffers carry the majority of the H+. The proximal tubule synthesizes the most important open buffer (NH3/NH4+) and regulates the most abundant closed buffer (HPO4−/H2PO42−), which will be discussed in more detail below. Finally, the most abundant base equivalent in urine is citrate2−/3−, whose urinary excretion is also exclusively regulated by the proximal tubule (19).
The proximal tubule fulfills the roles of reclamation of HCO3− and secretion of H+ through the uphill transport of H+ into the lumen, a process collectively referred to as renal acidification. Classic disequilibrium pH experiments have detected very little direct HCO3− reabsorption (20). In the proximal tubule, H+ is secreted into the lumen mostly by electroneutral Na+/H+ exchange. The active transport of H+ by the H+-ATPase (V-ATPase) also contributes, but to a lesser extent (Figure 3) (21,22). The main Na+/H+ exchanger isoform is NHE3 but NHE8 also participates, particularly in the neonate where NHE3 is not fully expressed (23,24). In addition to H+, NHE3 also mediates the secretion of NH4+ formed in the cell by titration of NH3, which is tantamount to net H+ secretion (25). The H+ exported into the lumen has multiple fates (Figure 3A). It combines with the filtered HCO3− and, under the action of luminal carbonic anhydrase (CA-IV), generates CO2, which diffuses into the cell and reconstitutes H+ and HCO3−. The combined reaction accomplishes the reclamation of filtered HCO3− (Figure 3A). The secreted H+ also titrates citrate from its trivalent form into its divalent form, which is the preferred substrate for uptake by the Na+ dicarboxylate cotransporter, NaDC-1 (Figure 3B) (26). The reabsorption of citrate2−/3− is equivalent to reabsorption of alkali (27). Finally, H+ secretion also titrates divalent HPO42− to monovalent H2PO4− that is not transported by NaPi-2a and NaPi-2c (Figure 3B), leading to phosphaturia and increased titratable acid or nonammonia urinary buffer carrying H+ ions in the urine. The HCO3− generated intracellularly by apical H+ secretion or ammoniagenesis exits the basolateral membrane via the family of Na+-bicarbonate cotransporters (28). Notably, the highly electrogenic NBCe1A (NBC1, SLC4A4), which is a splice variant of the electrogenic family of NBCe1, is responsible for basolateral HCO3− exit (29).
Figure 3.
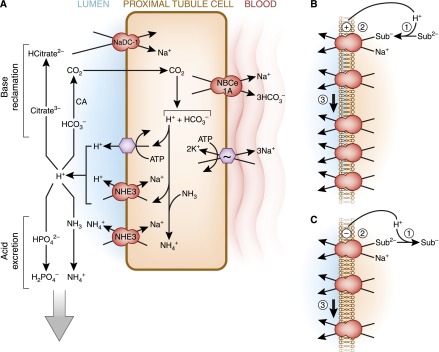
Proximal tubule HCO3− reabsorption and H+ secretion. Unlike NaCl reabsorption, luminal acidification is mediated by dedicated acid-base transporters. (A) Luminal H+ secretion is mediated by direct coupling to ATP hydrolysis via the H+-ATPase, but a higher proportion of luminal H+ secretion occurs by the Na+/H+ exchanger NHE3. In the neonate before maturational expression of NHE3, NHE8 is the more important NHE isoform. In addition to H+, NHE3 also directly transports NH4+ formed in the cell from NH3 and H+ into the lumen. The H+ secreted into the lumen has several fates. Reclamation of filtered base is shown on the top left. The titration of filtered HCO3− leads to formation of CO2 under the influence of carbonic anhydrase (CA). The CO2 enters the cell and is tantamount to HCO3− reabsorption. The titration of trivalent citrate to its bivalent form facilitates citrate reabsorption. Citrate is the most important and abundant organic urinary base equivalent. The bottom left depicts acid secretion. In addition to direct Na+/NH4+ exchange by NHE3, luminal H+ can combine with NH3 to form de novo NH4+ in the lumen. Titration of divalent to monovalent phosphate reduces phosphate absorption and allows the titrated phosphate to function as a H+ carrier in the urine. CO2 in the cell is hydrated to H2CO3, which dissociates to form a H+ and HCO3−. The metabolism of citrate2−/3− also consumes H+ in the cell, a reaction equivalent to generating HCO3−. The generated HCO3− exits the cell via Na+-coupled transport. There are three generic mechanisms by which changes in luminal and cell pH can alter apical transporters. (B and C) Stimulation is shown in B and inhibition is shown in C. (1) Luminal pH can alter the substrate concentration by titration and regulate transport kinetically. (2) Direct regulatory gating of the transporters by pH. (3) Changes in the number of transporters on the apical membrane by changes in trafficking, protein synthesis, and transcript levels. Sub, substrate.
Approximately 70%–90% of the filtered HCO3− is reabsorbed by the proximal tubule. Thus, this segment plays a pivotal role in the reclamation of HCO3−. In terms of net H+ excretion, all of the NH3/NH4+ in the final urine is synthesized in the proximal tubule. The luminal pH at the end of the proximal straight tubule is approximately 6.7–6.8, which means virtually all of the NH3 is titrated to NH4+ (pK = 9.3). Likewise, half of the phosphate is already in its monovalent form. Therefore, the proximal tubule plays an essential role in net acid excretion because a large fraction of the urinary buffers is already titrated by the end of the proximal tubule.
Other Solute Transport
Another primary function of the proximal tubule is the recovery of metabolites from the glomerular filtrate. Approximately 180 g of glucose (1000 mEq) and 50 g of amino acids (400 mEq) are filtered by the human kidney per day. The process of transepithelial transport normally accomplishes the recovery of 99.8% of these metabolites from the luminal fluid of the proximal tubule. As mentioned previously, this process requires the asymmetric association of distinct transporters in the apical and basolateral membranes. Typically, a secondary active Na+-dependent transporter in the brush border membrane uses the Na+ gradient to accomplish the initial uptake of solutes. By contrast, the subsequent transport of the solutes across the basolateral membrane frequently utilizes a Na+-independent passive transporter. Important examples of transepithelial transport are those involved in the recovery of glucose, glutamine, citrate, and phosphate.
Glucose Transport
Two distinct Na+-dependent transporters mediate the uptake of glucose from the lumen of the proximal tubule (30). SGLT2 (SLC5A2) is a moderate affinity glucose transporter that mediates the cotransport of glucose and one Na+ ion. The SGLT2 transporter is localized to the brush border membrane of the S1 and S2 segments of the proximal tubule where it extracts the bulk of the filtered glucose. By contrast, SGLT1 (SLC5A1) has a slightly greater affinity, cotransports two Na+ ions per glucose, and is preferentially expressed in the apical membrane of the S3 segment. The cotransport of two Na+ ions makes the uptake of glucose energetically more favorable and thereby increases the concentrative power of the SGLT1. Thus, the sequential positioning of the two transporters that favor capacity and affinity in the early and late tubule, respectively, creates an effective mechanism to ensure that <1% of the filtered glucose exits the proximal tubule. Selective knockout studies indicate that SGLT2 normally accounts for 97% of the net glucose reabsorption, whereas SGLT1 removes the residual glucose and provides reserve capacity (31). Selective inhibition of SGLT2 has been proposed as a potential therapy for treatment of diabetes that may ameliorate glycemia and reduce the associated hyperfiltration (32,33). The basolateral membranes of the early and late segments of the proximal tubule contain the Na+-independent glucose transporters, GLUT2 and GLUT1, respectively. Both transporters facilitate the passive movement of glucose from the proximal tubular cells to the interstitial space. The proximal tubule metabolizes little or no glucose, which is compatible with the very low hexokinase in this segment (34). In normal acid-base balance, the arterial-venous difference for glucose across the kidney is zero or slightly positive, which reflects substantial proximal tubule gluconeogenesis counterbalanced by glucose consumption by the rest of the nephron segments (35).
Amino Acid Transport
The renal transport of amino acids is a complex process due to the range of structures and ionic properties of the free amino acids in the plasma (36,37). However, >80% of the filtered amino acids are neutral amino acids, all of which are recovered by the apical BoAT1 (SLC6A19) transporter. BoAT1 is a broad specificity Na+-dependent cotransporter that is expressed in the early portion of the proximal tubule and that binds various neutral amino acids, including glutamine, with relatively low affinities (38). Previous micropuncture studies established that the filtered glutamine is nearly quantitatively reabsorbed from the lumen of the early proximal tubule (39). Mutations in the BoAT1 transporter result in Hartnup disorder, which is characterized by a pronounced urinary loss of neutral amino acids (40,41). A separate Na+ and H+-dependent cotransporter (SLC1A1) recovers the acidic amino acids (42), whereas an antiporter (SLC7A9) mediates the uptake of basic amino acids and cysteine in exchange for a neutral amino acid (37). The transporters that mediate the export of amino acids across the basolateral membrane are less well characterized. However, it is thought that in normal acid-base balance, LAT2-4F2hc (SLC7A8), a heterodimeric obligatory neutral amino acid exchanger, mediates the efflux of glutamine from the proximal tubule (36,43). A related heterodimeric exchanger, y+LAT1-4F2hc (SLC7A7), promotes the export of basic amino acids in exchange for a neutral amino acid (44). The two antiporters contribute to the maintenance of normal intracellular levels of amino acids. However, net efflux requires the participation of a uniporter that facilitates the passive export of neutral amino acids. The TAT1 transporter (SLC16A10) may accomplish the latter process (37).
Organic Cation and Anion Transport
The proximal tubule handles a very broad range of organic cations and anions that utilizes a variety of transporters operating in absorptive and secretory modes (45); however, due to space limitations, we will focus our discussion on citrate. The reabsorption of filtered citrate occurs in the proximal tubule apical membrane by NaDC-1 (SLC13A2), a Na+-dependent dicarboxylic acid cotransporter (46). The preferred substrates are dicarboxylates, such as citrate, succincate, fumarate, and α-ketoglutarate. In the proximal tubular lumen, citrate exists in equilibrium between its divalent and trivalent forms but citrate2− is the transported species. Once it is absorbed from the lumen, citrate can be metabolized by cytoplasmic ATP citrate lyase to oxaloacetate and acetyl-CoA or shuttled into the mitochondria where it enters the citric acid cycle (47). When citrate2−/3− is converted to CO2 and H2O, 2 or 3 H+ ions are consumed. Therefore, each milliequivalent of citrate excreted in the urine is tantamount to 2 or 3 OH− lost. In the normal day-to-day setting in a human on a Western diet, there is a negligible amount of HCO3− in the urine. Citrate is the only organic anion in millimolar quantities in the urine and represents the main mode of base excretion under normal circumstances. In the face of large alkali loads, bicarbonaturia becomes the main mechanism of base excretion.
Citrate serves a dual purpose in the urine. As indicated above, it is a urinary base. In addition to base excretion, the 1:1 Ca2+:citrate3− complex has a very high association constant and solubility. These properties render citrate the most effective chelator of calcium in the urine, which prevents its precipitation with phosphate and oxalate (48,49). Hypocitraturia is a major underlying cause of human kidney stones and, thus, citrate is the most important urinary anion for clinicians to understand (49).
Phosphate Transport
Phosphate homeostasis at the whole-organism level involves coordinated fluxes in the intestine, bone, and kidney, and endocrine cross-talk constituting a complex multiorgan network (50). Although both the intestine and bone are critical organs for phosphate homeostasis, this manuscript will only discuss the renal component. It is important to state that intestinal phosphate absorption involves significant paracellular uptake that is poorly regulated (51). Thus, the kidney is the paramount regulator of external balance. Unlike Na+ reabsorption where the finishing fine-tunings are achieved by more distal segments, phosphate reabsorption is accomplished almost entirely by the proximal tubule (52). A small contribution from the distal tubule has been proposed, but is still disputed (53). Plasma levels reflect total body phosphate status but are very insensitive; spot urinary concentrations are confounded by water excretion rate; and excretion rates of phosphate are affected by ingestion. Thus, all of these parameters are less than ideal to evaluate renal tubular phosphate handling. The parameters listed in Figure 4A are better suited to probe the proximal tubular handling independent of the excreted or filtered load.
Figure 4.
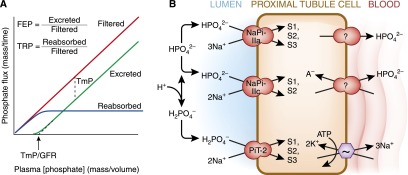
Proximal tubule phosphate transport. (A) Concepts of renal inorganic phosphate (Pi) homeostasis by the proximal tubule. The flux of filtered and reabsorbed Pi is plotted against plasma phosphate concentration; the difference between the two yields the rate of excretion of Pi. There are a number of terms used to quantify the proximal tubule’s Pi reabsorption at the whole organism level. Fractional excretion of Pi (FEP) and tubular reabsorption of Pi (TRP) sum to unity (FEP=1−TRP). The maximal tubular reabsorptive capacity of Pi (TmP in units of mass/time) refers to the saturating transepithelial flux of Pi that the tubule can mount and is equal to the difference between filtered and absorbed phosphate when the filtered load is higher than TmP. The plasma concentration threshold at which Pi starts to appear in the urine is TmP/GFR (in units of mass/volume). (B) Cell model of proximal tubule Pi transport. Three apical transporters mediate Pi entry with different preferred valence of Pi, stoichiometry of Na+, electrogenicity, and pH gating. The affinities for Na+ are all approximately 30–50 mM but are much higher for phosphate (0.1, 0.07, and 0.025 mM for NaPi-Ila, NaPi-Ilc, and PiT-2, respectively). Distribution in the proximal tubule segments (S1, S2, S3). Basolateral Pi exit occurs via unknown mechanisms. Apical Na-coupled Pi transport is inhibited in acidosis by alteration in luminal substrate, directly gating of the transporter by pH, and decreased apical NaPi transporters as described in Figure 3B.
The cellular model for proximal tubule phosphate transport is shown in Figure 4B. The primary driving force is the Na+-K+-ATPase generating an electrochemical gradient for apical phosphate entry. The current model predicts three transporters for apical entry; NaPi-2a (Npt2a, SLC34A1), NaPi-2c (Npt2c, SLC34A2), and Pit-2 (Npt3, SLC20A2, Ram-1). The disparate properties of the three transporters were reviewed in great detail by Virkki and colleagues (54). At present, the basolateral mechanism of phosphate exit still remains enigmatic. It is possible that one or more of the plethora of anion exchange mechanisms may mediate phosphate exit. Thus far, there is little evidence for regulation of basolateral phosphate exit.
Regulation of phosphate transport at the proximal tubule apical membrane is precise because this is the only and final site of determination of extracellular phosphate balance by the kidney. Phosphate uptake is affected by incoming signals, such as parathyroid hormone (55), dopamine (56), fibroblast growth factor-23 (57), and Klotho (58), which inhibit phosphate transport and induce phosphaturia. One of the most potent regulators of phosphaturia is dietary phosphate intake itself, which may involve a variety of hormones, including unknown intestinal enterokines (59), and direct sensing by the proximal tubule (60). This is one of the most important, yet least known, areas in phosphate homeostasis. The modulation of proximal phosphate transport is achieved largely by trafficking of the transporters in and out of the apical membrane (52) with the exception of Klotho, which can directly affect phosphate transport activity (58).
Response to Acidosis
The catabolism of acidic and sulfur-containing amino acids results in the net production of acids. As a result, a high-protein diet leads to a mild chronic metabolic acidosis that is usually well compensated. The common clinical condition of metabolic acidosis is characterized by a more significant decrease in plasma pH and bicarbonate concentration. This disturbance in acid-base balance can be caused by genetic or acquired alterations in metabolism, in renal handling of bicarbonate, and in the excretion of acid. In addition, patients with cachexia, trauma, uremia, ESRD, and HIV infection frequently develop acidosis as a secondary complication that adversely affects their outcome. Chronic acidosis also causes impaired growth, bone loss, muscle wasting, nephrocalcinosis, and urolithiasis. In acute situations with massive acid production that overwhelms the renal capacity, the kidney’s response is not relevant; however, in more chronic conditions, renal compensation is crucial.
An essential renal compensatory response to metabolic acidosis is initiated by increased extraction and catabolism of plasma glutamine that occur predominately in the proximal convoluted tubule. The resulting increases in renal ammoniagenesis and transport into the urine accomplish the excretion of acid, whereas the increased bicarbonate synthesis and transport into the blood partially correct the systemic acidosis. These adaptations occur rapidly after acute onset of acidosis and are subsequently sustained by more gradual changes in gene expression.
During normal acid-base balance, the kidneys extract and metabolize very little of the plasma glutamine. Although approximately 20% of the plasma glutamine is filtered, the measured rat renal arterial-venous difference is <3% of the arterial concentration of glutamine (61), and only 7% of the plasma glutamine is extracted by the human kidneys even after an overnight fast (62). Therefore, renal utilization is significantly less than the fraction of plasma glutamine filtered by the glomeruli. To account for the effective reabsorption of glutamine, either the activity of the mitochondrial glutamine transporter or the glutaminase must be largely inhibited or inactivated in vivo during normal acid-base balance.
Acute onset of metabolic acidosis produces rapid changes in the interorgan metabolism of glutamine (63) that support a rapid and pronounced increase in renal catabolism of glutamine. Within 1–3 hours, the arterial plasma glutamine concentration is increased 2-fold (64) due primarily to an increased release of glutamine from muscle (65). Significant renal extraction of glutamine becomes evident as the arterial plasma concentration is increased. Net extraction by the kidney reaches 35% of the plasma glutamine, a level that exceeds the proportion (20%) filtered by the glomeruli. Thus, the normal direction of the basolateral glutamine exchange transporter, LAT2-4F2hc, is reversed in order for the proximal convoluted tubule cells to extract glutamine from both the glomerular filtrate and the venous blood (Figure 5). In addition, the transport of glutamine into the mitochondria may be acutely activated (66). Additional responses include a prompt acidification of the urine that results from translocation, as evidenced in OKP cells (67), and by acute activation of NHE3 (68). This process facilitates the rapid removal of cellular ammonium ions (69) and ensures that the bulk of the ammonium ions generated from the amide and amine nitrogens of glutamine are excreted in the urine. Finally, the cellular concentrations of glutamate and α-ketoglutarate are significantly decreased within the rat renal cortex (70). The latter compounds are products and inhibitors of the glutaminase and glutamate dehydrogenase reactions, respectively. The decrease in concentrations of the two regulatory metabolites may result from a pH-induced activation of α-ketoglutarate dehydrogenase (69). Thus, the acute increase in renal ammoniagenesis may result from a rapid activation of key transport processes, an increased availability of glutamine, and a decrease in product inhibition of the enzymes of ammoniagenesis.
Figure 5.
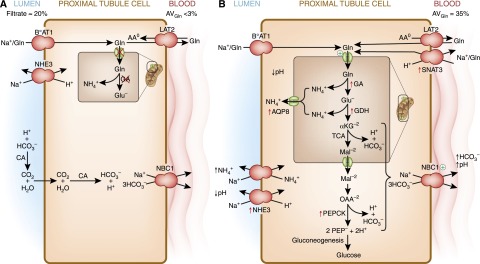
Renal proximal tubular catabolism of glutamine. (A) During normal acid-base balance, the glutamine filtered by the glomeruli is nearly quantitatively extracted from the lumen of the proximal convoluted tubule and largely returned to the blood. The transepithelial transport utilizes BoAT1, a Na+-dependent neutral amino acid cotransporter in the apical membrane, and LAT2, a neutral amino acid antiporter in the basolateral membrane. To accomplish this movement, either the mitochondrial glutamine transporter or the mitochondrial glutaminase (GA) must be inhibited (red X). The apical Na+/H+ exchanger functions to slightly acidify the lumen to facilitate the recovery of HCO3− ions. (B) During chronic acidosis, approximately one third of the plasma glutamine is extracted and catabolized within the early portion of the proximal tubule. BoAT1 continues to mediate the extraction of glutamine from the lumen. Uptake of glutamine through the basolateral membrane occurs by reversal of the neutral amino acid exchanger, LAT2, and through increased expression of SNAT3. Increased renal catabolism of glutamine is facilitated by increased expression (red arrows) of the genes that encode glutaminase (GA), glutamate dehydrogenase (GDH), phosphoenolpyruvate carboxykinase (PEPCK), the mitochondrial aquaporin-8 (AQP8), the apical Na+/H+ exchanger (NHE3), and the basolateral glutamine transporter (SNAT3). In addition, the activities of the mitochondrial glutamine transporter and the basolateral Na+/3HCO3− are increased (+). Increased expression of NHE3 contributes to the transport of ammonium ions and the acidification of the luminal fluid. The combined increases in renal ammonium ion excretion and gluconeogenesis result in a net synthesis of HCO3− ions that are transported across the basolateral membrane by the Na+/3HCO3− cotransporter (NBC1). CA, carbonic anhydrase; αKG, α-ketoglutarate; Mal, malate; OAA, oxaloacetate; PEP, phosphoenolpyruvate.
During chronic acidosis, many of the acute responses are reversed and the arterial plasma concentration is decreased to a new steady state that is 70% of normal. However, more than one third of the plasma glutamine is still extracted in a single pass through the kidneys. The increased renal catabolism of glutamine in the proximal convoluted tubule is now sustained by increased expression of genes that encode key transporters and enzymes of glutamine metabolism (Figure 5). A comprehensive survey of the adaptive response of known amino acid transporters in mouse kidney demonstrated that only the basolateral SNAT3 (SLC38A3) transporter exhibits a rapid and pronounced increase during onset of acidosis (71). The SNAT3 transporter has a high affinity for glutamine (72). Under physiologic conditions, it catalyzes a reversible Na+-dependent uptake of glutamine that is coupled to the efflux of H+ ions (73). SNAT3 is normally localized solely to the basolateral membrane of the S3 segment of the proximal tubule (74). This segment expresses high levels of glutamine synthetase (75). Thus, the SNAT3 transporter may normally facilitate the pH-dependent release of glutamine. However, during chronic acidosis, increased expression of the SNAT3 transporter occurs primarily in the basolateral membranes of the S1 and S2 segments of proximal tubule, the site of increased glutamine catabolism (76). Given the sustained increase in H+ ion concentration within these cells, the increased expression of the SNAT3 transporter may now facilitate the basolateral uptake of glutamine and contribute to its sustained extraction during chronic acidosis. Within 8–24 hours after onset of acidosis, a pronounced increase in phosphoenolpyruvate carboxykinase (77) also occurs only in the proximal convoluted tubule. More gradual increases in levels of mitochondrial glutaminase (78,79) and glutamate dehydrogenase (80) that require 4–7 days also occur solely within the proximal convoluted tubule. In addition, the level of aquaporin-8, a potential mitochondrial ammonia transporter, is increased (81).
Glutamine uptake in mitochondria from normal rats is mediated through two glutamine antiporters, whereas a highly active glutamine uniporter is evident only in mitochondria prepared from acidotic rats (82). Therefore, acidosis leads to increased expression or activation of a unique, but unidentified, mitochondrial glutamine transporter. Acute activation and subsequent increase in expression of NHE3 (83–85) acidifies the fluid in the tubular lumen and contributes to the active transport of ammonium ions as a direct substrate of NHE3 (69). The adaptation in NHE3 likely reflects the increased demand for ammonium ion secretion although this is difficult to prove. The filtered HCO3− load is certainly decreased in metabolic acidosis, so increased HCO3− absorptive capacity is not required. As a result, increased renal ammoniagenesis continues to provide an expendable cation that facilitates excretion of strong acids while conserving sodium and potassium ions. In rats (86,87) and humans (35), the α-ketoglutarate generated from renal catabolism of glutamine is primarily converted to glucose. This process requires phosphoenolpyruvate carboxykinase to divert oxaloacetate, derived from intermediates of the tricarboxylic acid cycle, into the pathway of gluconeogenesis. The combined pathways of ammoniagenesis and gluconeogenesis result in a net production of 2 NH4+ and 2 HCO3− ions per glutamine. Activation of NBCe1A (83), the basolateral Na+/3HCO3− cotransporter, facilitates the translocation of reabsorbed and de novo–synthesized HCO3− ions into the renal venous blood. Thus, the combined adaptations also create a net renal release of HCO3− ions that partially restores acid-base balance.
The adaptative increase in the NaDC-1 transporter contributes to increased reabsorption and metabolism of citrate within the proximal tubule. This reduces the excretion of a valuable base in the urine. This adaptation occurs at multiple levels. Acidification of luminal pH titrates citrate3− to citrate2−, which is the preferred substrate, and low pH also directly activates NaDC1 to increase transport independent of [citrate2−] (Figure 3B) (26,88). In addition to transport, increased cellular metabolism also drives citrate reabsorption. After cellular uptake, citrate is metabolized through one of two pathways: a cytoplasmic pathway involving citrate lyase or a mitochondrial pathway involving the citric acid cycle (47). During acidosis, the activities of cytoplasmic citrate lyase and mitochondrial aconitase are also increased (89). Because both pathways generate HCO3−, the increased reabsorption of citrate is equivalent to a decrease in base excretion (90). Enhanced catabolism of citrate also produces substrates that support the increased gluconeogenesis.
Acid-base disturbances are a major regulator of proximal tubule phosphate handling. Metabolic and respiratory acidosis increase phosphate excretion and metabolic and respiratory alkalosis decrease phosphate excretion. The mechanism of phosphaturia in acidosis is complex and mediated by many factors, including increased release of phosphate from bone (91), titration of luminal phosphate to monovalent form, direct gating of NaPi-IIa and NaPi-IIc by pH (54), decrease in apical membrane protein (92), and transcripts (93) of the transporters, although disparate results on protein levels have been described (Figure 3B) (94). The acid-induced phosphaturia serves to increase urinary H+ buffer but also accommodates the phosphate released from bone associated with acid loading.
The gradual increases in glutaminase (95–97) and glutamate dehydrogenase (98) result from selective stabilization of their mRNAs. By contrast, the rapid increase in phosphoenolpyruvate carboxykinase results from enhanced transcription of the PCK-1 gene (99), whereas mRNA stabilization contributes to the sustained increase (100,101). The presence of a pH-response element (pH-RE) that regulates the turnover of the glutaminase mRNA was initially demonstrated by stable expression of a chimeric β-globin reporter mRNA, which includes a 956-bp segment of the 3′-untranslated region of glutaminase mRNA (95). RNA gel shift analyses of various deletion constructs mapped the pH-RE to two 8-nt AU-sequences within the 3′-untranslated region. Mutation of the pH-RE within the β-globin reporter mRNA blocked the pH-responsive stabilization. In addition, insertion of only a 29-bp segment containing the pH-RE was sufficient to produce both rapid degradation and pH-responsive stabilization (102). Therefore, the identified pH-RE contributes to the rapid turnover of the glutaminase mRNA and is both necessary and sufficient to mediate its pH-responsive stabilization.
Proteomic analysis of isolated rat renal proximal convoluted tubules identified an additional 60 proteins that are increased during acute and chronic acidosis (103–105). More than 50% of the mRNAs that encode the induced proteins contain an AU-sequence that has >85% identity with the pH-response element found in the glutaminase mRNA. This finding strongly suggests that mRNA stabilization is a primary mechanism by which protein expression is increased in response to onset of metabolic acidosis.
The maintenance of the composition and content of the extracellular fluid volume is critical to health, as evidenced by the multiple organ failure seen in kidney disease. The mammalian proximal tubule uses an array of polarized membrane transporters to accomplish vectoral solute transport. By lowering the luminal content of many abundant solutes to a level that is within the lower reabsorptive capacity of the distal nephron segments, it enables the absorption of these solutes to be fine-tuned downstream. Without the large proximal reabsorption capacity, it would be impossible for the kidney to maintain the high GFR that is necessary to sustain the high metabolic rates characteristic of terrestrial vertebrates. For many solutes with lower plasma concentrations and, hence, lower filtered load, proximal reabsorption is the final arbitrator of urinary excretion. The kidney has enormous capacity to cope with a wide range of physiologic challenges. Whereas volatile acids can be excreted in a gaseous phase in the lung, the kidney is the only organ where nonvolatile acids can be excreted and where discomposed body buffers, such as bicarbonate, can be regenerated. Metabolic acid loading and metabolic acidosis have undesirable acute and chronic consequences and they trigger a coordinated multiorgan network of adaptive responses that partially rectify the disturbance. Although intuitive in principle, the actual mechanisms are actually extremely complex and the proximal tubule is in the center stage spotlight. With a built-in cast of players inherent to this epithelium, the proximal tubule elicits a complex set of mechanisms that includes intrinsic pH sensing and adaptations in membrane transporters and metabolic enzymes to take a neutral molecule, partition it into an acid and a base, and transfer the acid into urine and the base into the blood. Although the short account in this article summarizes significant advances made over decades, the understanding of this system is just beginning. With the current upsurge of powerful investigative methods, the delineation of mechanisms of adaptation to acidosis will emerge with greater rapidity and clarity.
Disclosures
N.P.C. is a consultant for Calithera and O.W.M. has received research support from Genzyme.
Acknowledgments
This study was supported in part by grants from the National Institutes of Health (DK037124 to N.P.C. and AI041612, DK078596, and DK091392 to O.W.M.). O.W.M. also received support from the O’Brien Kidney Research Center (DK-079328) and Genzyme.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Jones G, Prosser DE, Kaufmann M: 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch Biochem Biophys 523: 9–18, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Meyer C, Dostou JM, Gerich JE: Role of the human kidney in glucose counterregulation. Diabetes 48: 943–948, 1999 [DOI] [PubMed] [Google Scholar]
- 3.De Vito E, Cabrera RR, Fasciolo JC: Renin production and release by rat kidney slices. Am J Physiol 219: 1042–1045, 1970 [DOI] [PubMed] [Google Scholar]
- 4.Koury ST, Bondurant MC, Koury MJ: Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood 71: 524–527, 1988 [PubMed] [Google Scholar]
- 5.Moe OW, Ujiie K, Star RA, Miller RT, Widell J, Alpern RJ, Henrich WL: Renin expression in renal proximal tubule. J Clin Invest 91: 774–779, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moe OW, Giebisch G, Seldin DW: The logic of the kidney. In: Genetic Diseases of the Kidney, edited by Lifton R, Somlo S, Seldin DW, Giebisch G, Amsterdam, Elsevier, 2009, pp 29–76 [Google Scholar]
- 7.Aronson PS: Ion exchangers mediating NaCl transport in the renal proximal tubule. Cell Biochem Biophys 36: 147–153, 2002 [DOI] [PubMed] [Google Scholar]
- 8.Moe OW, Preisig PA, Alpern RJ: Cellular model of proximal tubule NaCl and NaHCO3 absorption. Kidney Int 38: 605–611, 1990 [DOI] [PubMed] [Google Scholar]
- 9.Preisig PA, Rector FC, Jr: Role of Na+-H+ antiport in rat proximal tubule NaCl absorption. Am J Physiol 255: F461–F465, 1988 [DOI] [PubMed] [Google Scholar]
- 10.Aronson PS, Sacktor B: The Na+ gradient-dependent transport of D-glucose in renal brush border membranes. J Biol Chem 250: 6032–6039, 1975 [PubMed] [Google Scholar]
- 11.Rector FC, Jr: Sodium, bicarbonate, and chloride absorption by the proximal tubule. Am J Physiol 244: F461–F471, 1983 [DOI] [PubMed] [Google Scholar]
- 12.Harris PJ, Young JA: Dose-dependent stimulation and inhibition of proximal tubular sodium reabsorption by angiotensin II in the rat kidney. Pflugers Arch 367: 295–297, 1977 [DOI] [PubMed] [Google Scholar]
- 13.Garcia NH, Garvin JL: Endothelin’s biphasic effect on fluid absorption in the proximal straight tubule and its inhibitory cascade. J Clin Invest 93: 2572–2577, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gill JR, Jr, Casper AG: Effect of renal alpha-adrenergic stimulation on proximal tubular sodium reabsorption. Am J Physiol 223: 1201–1205, 1972 [DOI] [PubMed] [Google Scholar]
- 15.Baum M, Quigley R: Inhibition of proximal convoluted tubule transport by dopamine. Kidney Int 54: 1593–1600, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moe OW, Tejedor A, Levi M, Seldin DW, Preisig PA, Alpern RJ: Dietary NaCl modulates Na(+)-H+ antiporter activity in renal cortical apical membrane vesicles. Am J Physiol 260: F130–F137, 1991 [DOI] [PubMed] [Google Scholar]
- 17.Bobulescu IA, Moe OW: Luminal Na(+)/H (+) exchange in the proximal tubule. Pflugers Arch 458: 5–21, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bobulescu IA, Moe OW: Na+/H+ exchangers in renal regulation of acid-base balance. Semin Nephrol 26: 334–344, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamm LL: Renal handling of citrate. Kidney Int 38: 728–735, 1990 [DOI] [PubMed] [Google Scholar]
- 20.DuBose TD, Jr, Pucacco LR, Seldin DW, Carter NW, Kokko JP: Microelectrode determination of pH and PCO2 in rat proximal tubule after benzolamide: Evidence for hydrogen ion secretion. Kidney Int 15: 624–629, 1979 [DOI] [PubMed] [Google Scholar]
- 21.Chantrelle B, Cogan MG, Rector FC, Jr: Evidence for coupled sodium/hydrogen exchange in the rat superficial proximal convoluted tubule. Pflugers Arch 395: 186–189, 1982 [DOI] [PubMed] [Google Scholar]
- 22.Preisig PA, Ives HE, Cragoe EJ, Jr, Alpern RJ, Rector FC, Jr: Role of the Na+/H+ antiporter in rat proximal tubule bicarbonate absorption. J Clin Invest 80: 970–978, 1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amemiya M, Loffing J, Lötscher M, Kaissling B, Alpern RJ, Moe OW: Expression of NHE-3 in the apical membrane of rat renal proximal tubule and thick ascending limb. Kidney Int 48: 1206–1215, 1995 [DOI] [PubMed] [Google Scholar]
- 24.Becker AM, Zhang J, Goyal S, Dwarakanath V, Aronson PS, Moe OW, Baum M: Ontogeny of NHE8 in the rat proximal tubule. Am J Physiol Renal Physiol 293: F255–F261, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nagami GT: Luminal secretion of ammonia in the mouse proximal tubule perfused in vitro. J Clin Invest 81: 159–164, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brennan S, Hering-Smith K, Hamm LL: Effect of pH on citrate reabsorption in the proximal convoluted tubule. Am J Physiol 255: F301–F306, 1988 [DOI] [PubMed] [Google Scholar]
- 27.Hamm LL, Simon EE: Roles and mechanisms of urinary buffer excretion. Am J Physiol 253: F595–F605, 1987 [DOI] [PubMed] [Google Scholar]
- 28.Boron WF, Boulpaep EL: Intracellular pH regulation in the renal proximal tubule of the salamander. Basolateral HCO3- transport. J Gen Physiol 81: 53–94, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmitt BM, Biemesderfer D, Romero MF, Boulpaep EL, Boron WF: Immunolocalization of the electrogenic Na+-HCO-3 cotransporter in mammalian and amphibian kidney. Am J Physiol 276: F27–F38, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Hummel CS, Lu C, Loo DD, Hirayama BA, Voss AA, Wright EM: Glucose transport by human renal Na+/D-glucose cotransporters SGLT1 and SGLT2. Am J Physiol Cell Physiol 300: C14–C21, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vallon V, Platt KA, Cunard R, Schroth J, Whaley J, Thomson SC, Koepsell H, Rieg T: SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol 22: 104–112, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bakris GL, Fonseca VA, Sharma K, Wright EM: Renal sodium-glucose transport: Role in diabetes mellitus and potential clinical implications. Kidney Int 75: 1272–1277, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Vallon V, Rose M, Gerasimova M, Satriano J, Platt KA, Koepsell H, Cunard R, Sharma K, Thomson SC, Rieg T: Knockout of Na-glucose transporter SGLT2 attenuates hyperglycemia and glomerular hyperfiltration but not kidney growth or injury in diabetes mellitus. Am J Physiol Renal Physiol 304: F156–F167, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vandewalle A, Wirthensohn G, Heidrich HG, Guder WG: Distribution of hexokinase and phosphoenolpyruvate carboxykinase along the rabbit nephron. Am J Physiol 240: F492–F500, 1981 [DOI] [PubMed] [Google Scholar]
- 35.Gerich JE, Meyer C, Woerle HJ, Stumvoll M: Renal gluconeogenesis: Its importance in human glucose homeostasis. Diabetes Care 24: 382–391, 2001 [DOI] [PubMed] [Google Scholar]
- 36.McGivan JD, Bungard CI: The transport of glutamine into mammalian cells. Front Biosci 12: 874–882, 2007 [DOI] [PubMed] [Google Scholar]
- 37.Verrey F, Singer D, Ramadan T, Vuille-dit-Bille RN, Mariotta L, Camargo SM: Kidney amino acid transport. Pflugers Arch 458: 53–60, 2009 [DOI] [PubMed] [Google Scholar]
- 38.Romeo E, Dave MH, Bacic D, Ristic Z, Camargo SM, Loffing J, Wagner CA, Verrey F: Luminal kidney and intestine SLC6 amino acid transporters of B0AT-cluster and their tissue distribution in Mus musculus. Am J Physiol Renal Physiol 290: F376–F383, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Silbernagl S: Tubular reabsorption of L-glutamine studied by free-flow micropuncture and microperfusion of rat kidney. Int J Biochem 12: 9–16, 1980 [DOI] [PubMed] [Google Scholar]
- 40.Kleta R, Romeo E, Ristic Z, Ohura T, Stuart C, Arcos-Burgos M, Dave MH, Wagner CA, Camargo SR, Inoue S, Matsuura N, Helip-Wooley A, Bockenhauer D, Warth R, Bernardini I, Visser G, Eggermann T, Lee P, Chairoungdua A, Jutabha P, Babu E, Nilwarangkoon S, Anzai N, Kanai Y, Verrey F, Gahl WA, Koizumi A: Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat Genet 36: 999–1002, 2004 [DOI] [PubMed] [Google Scholar]
- 41.Seow HF, Bröer S, Bröer A, Bailey CG, Potter SJ, Cavanaugh JA, Rasko JE: Hartnup disorder is caused by mutations in the gene encoding the neutral amino acid transporter SLC6A19. Nat Genet 36: 1003–1007, 2004 [DOI] [PubMed] [Google Scholar]
- 42.Shayakul C, Kanai Y, Lee WS, Brown D, Rothstein JD, Hediger MA: Localization of the high-affinity glutamate transporter EAAC1 in rat kidney. Am J Physiol 273: F1023–F1029, 1997 [DOI] [PubMed] [Google Scholar]
- 43.Rossier G, Meier C, Bauch C, Summa V, Sordat B, Verrey F, Kühn LC: LAT2, a new basolateral 4F2hc/CD98-associated amino acid transporter of kidney and intestine. J Biol Chem 274: 34948–34954, 1999 [DOI] [PubMed] [Google Scholar]
- 44.Torrents D, Estévez R, Pineda M, Fernández E, Lloberas J, Shi YB, Zorzano A, Palacín M: Identification and characterization of a membrane protein (y+L amino acid transporter-1) that associates with 4F2hc to encode the amino acid transport activity y+L. A candidate gene for lysinuric protein intolerance. J Biol Chem 273: 32437–32445, 1998 [DOI] [PubMed] [Google Scholar]
- 45.Moe OW, Palacin M, Wright S: Renal Organic Solute Transport, Philadelphia, Saunders Elsevier, 2007 [Google Scholar]
- 46.Pajor AM: Sequence and functional characterization of a renal sodium/dicarboxylate cotransporter. J Biol Chem 270: 5779–5785, 1995 [DOI] [PubMed] [Google Scholar]
- 47.Simpson DP: Citrate excretion: A window on renal metabolism. Am J Physiol 244: F223–F234, 1983 [DOI] [PubMed] [Google Scholar]
- 48.Moe OW, Preisig PA: Dual role of citrate in mammalian urine. Curr Opin Nephrol Hypertens 15: 419–424, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Moe OW: Kidney stones: Pathophysiology and medical management. Lancet 367: 333–344, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Hu MC, Shiizaki K, Kuro-o M, Moe OW: Fibroblast growth factor 23 and Klotho: Physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol 75: 503–533, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marks J, Debnam ES, Unwin RJ: Phosphate homeostasis and the renal-gastrointestinal axis. Am J Physiol Renal Physiol 299: F285–F296, 2010 [DOI] [PubMed] [Google Scholar]
- 52.Biber J, Hernando N, Forster I, Murer H: Regulation of phosphate transport in proximal tubules. Pflugers Arch 458: 39–52, 2009 [DOI] [PubMed] [Google Scholar]
- 53.Peraino RA, Suki WN: Phosphate transport by isolated rabbit cortical collecting tubule. Am J Physiol 238: F358–F362, 1980 [DOI] [PubMed] [Google Scholar]
- 54.Virkki LV, Biber J, Murer H, Forster IC: Phosphate transporters: A tale of two solute carrier families. Am J Physiol Renal Physiol 293: F643–F654, 2007 [DOI] [PubMed] [Google Scholar]
- 55.Haramati A, Haas JA, Knox FG: Nephron heterogeneity of phosphate reabsorption: Effect of parathyroid hormone. Am J Physiol 246: F155–F158, 1984 [DOI] [PubMed] [Google Scholar]
- 56.Glahn RP, Onsgard MJ, Tyce GM, Chinnow SL, Knox FG, Dousa TP: Autocrine/paracrine regulation of renal Na(+)-phosphate cotransport by dopamine. Am J Physiol 264: F618–F622, 1993 [DOI] [PubMed] [Google Scholar]
- 57.Gattineni J, Bates C, Twombley K, Dwarakanath V, Robinson ML, Goetz R, Mohammadi M, Baum M: FGF23 decreases renal NaPi-2a and NaPi-2c expression and induces hypophosphatemia in vivo predominantly via FGF receptor 1. Am J Physiol Renal Physiol 297: F282–F291, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu MC, Shi M, Zhang J, Pastor J, Nakatani T, Lanske B, Razzaque MS, Rosenblatt KP, Baum MG, Kuro-o M, Moe OW: Klotho: A novel phosphaturic substance acting as an autocrine enzyme in the renal proximal tubule. FASEB J 24: 3438–3450, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berndt T, Thomas LF, Craig TA, Sommer S, Li X, Bergstralh EJ, Kumar R: Evidence for a signaling axis by which intestinal phosphate rapidly modulates renal phosphate reabsorption. Proc Natl Acad Sci U S A 104: 11085–11090, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pfister MF, Hilfiker H, Forgo J, Lederer E, Biber J, Murer H: Cellular mechanisms involved in the acute adaptation of OK cell Na/Pi-cotransport to high- or low-Pi medium. Pflugers Arch 435: 713–719, 1998 [DOI] [PubMed] [Google Scholar]
- 61.Squires EJ, Hall DE, Brosnan JT: Arteriovenous differences for amino acids and lactate across kidneys of normal and acidotic rats. Biochem J 160: 125–128, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meyer C, Stumvoll M, Dostou J, Welle S, Haymond M, Gerich J: Renal substrate exchange and gluconeogenesis in normal postabsorptive humans. Am J Physiol Endocrinol Metab 282: E428–E434, 2002 [DOI] [PubMed] [Google Scholar]
- 63.Tamarappoo BK, Joshi S, Welbourne TC: Interorgan glutamine flow regulation in metabolic acidosis. Miner Electrolyte Metab 16: 322–330, 1990 [PubMed] [Google Scholar]
- 64.Hughey RP, Rankin BB, Curthoys NP: Acute acidosis and renal arteriovenous differences of glutamine in normal and adrenalectomized rats. Am J Physiol 238: F199–F204, 1980 [DOI] [PubMed] [Google Scholar]
- 65.Schröck H, Cha CJ, Goldstein L: Glutamine release from hindlimb and uptake by kidney in the acutely acidotic rat. Biochem J 188: 557–560, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sastrasinh M, Sastrasinh S: Effect of acute pH change on mitochondrial glutamine transport. Am J Physiol 259: F863–F866, 1990 [DOI] [PubMed] [Google Scholar]
- 67.Yang X, Amemiya M, Peng Y, Moe OW, Preisig PA, Alpern RJ: Acid incubation causes exocytic insertion of NHE3 in OKP cells. Am J Physiol Cell Physiol 279: C410–C419, 2000 [DOI] [PubMed] [Google Scholar]
- 68.Horie S, Moe O, Tejedor A, Alpern RJ: Preincubation in acid medium increases Na/H antiporter activity in cultured renal proximal tubule cells. Proc Natl Acad Sci U S A 87: 4742–4745, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tannen RL, Ross BD: Ammoniagenesis by the isolated perfused rat kidney: The critical role of urinary acidification. Clin Sci (Lond) 56: 353–364, 1979 [DOI] [PubMed] [Google Scholar]
- 70.Lowry M, Ross BD: Activation of oxoglutarate dehydrogenase in the kidney in response to acute acidosis. Biochem J 190: 771–780, 1980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moret C, Dave MH, Schulz N, Jiang JX, Verrey F, Wagner CA: Regulation of renal amino acid transporters during metabolic acidosis. Am J Physiol Renal Physiol 292: F555–F566, 2007 [DOI] [PubMed] [Google Scholar]
- 72.Christensen HN: Role of amino acid transport and countertransport in nutrition and metabolism. Physiol Rev 70: 43–77, 1990 [DOI] [PubMed] [Google Scholar]
- 73.Chaudhry FA, Reimer RJ, Edwards RH: The glutamine commute: Take the N line and transfer to the A. J Cell Biol 157: 349–355, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Karinch AM, Lin CM, Wolfgang CL, Pan M, Souba WW: Regulation of expression of the SN1 transporter during renal adaptation to chronic metabolic acidosis in rats. Am J Physiol Renal Physiol 283: F1011–F1019, 2002 [DOI] [PubMed] [Google Scholar]
- 75.Taylor L, Curthoys NP: Glutamine metabolism: Role in acid-base balance. Biochem Mol Biol Educ 32: 291–304, 2004 [DOI] [PubMed] [Google Scholar]
- 76.Solbu TT, Boulland JL, Zahid W, Lyamouri Bredahl MK, Amiry-Moghaddam M, Storm-Mathisen J, Roberg BA, Chaudhry FA: Induction and targeting of the glutamine transporter SN1 to the basolateral membranes of cortical kidney tubule cells during chronic metabolic acidosis suggest a role in pH regulation. J Am Soc Nephrol 16: 869–877, 2005 [DOI] [PubMed] [Google Scholar]
- 77.Schoolwerth AC, deBoer PA, Moorman AF, Lamers WH: Changes in mRNAs for enzymes of glutamine metabolism in kidney and liver during ammonium chloride acidosis. Am J Physiol 267: F400–F406, 1994 [DOI] [PubMed] [Google Scholar]
- 78.Curthoys NP, Lowry OH: The distribution of glutaminase isoenzymes in the various structures of the nephron in normal, acidotic, and alkalotic rat kidney. J Biol Chem 248: 162–168, 1973 [PubMed] [Google Scholar]
- 79.Wright PA, Knepper MA: Phosphate-dependent glutaminase activity in rat renal cortical and medullary tubule segments. Am J Physiol 259: F961–F970, 1990 [DOI] [PubMed] [Google Scholar]
- 80.Wright PA, Knepper MA: Glutamate dehydrogenase activities in microdissected rat nephron segments: Effects of acid-base loading. Am J Physiol 259: F53–F59, 1990 [DOI] [PubMed] [Google Scholar]
- 81.Molinas SM, Trumper L, Marinelli RA: Mitochondrial aquaporin-8 in renal proximal tubule cells: Evidence for a role in the response to metabolic acidosis. Am J Physiol Renal Physiol 303: F458–F466, 2012 [DOI] [PubMed] [Google Scholar]
- 82.Atlante A, Passarella S, Minervini GM, Quagliariello E: Glutamine transport in normal and acidotic rat kidney mitochondria. Arch Biochem Biophys 315: 369–381, 1994 [DOI] [PubMed] [Google Scholar]
- 83.Preisig PA, Alpern RJ: Chronic metabolic acidosis causes an adaptation in the apical membrane Na/H antiporter and basolateral membrane Na(HCO3)3 symporter in the rat proximal convoluted tubule. J Clin Invest 82: 1445–1453, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ambühl PM, Amemiya M, Danczkay M, Lötscher M, Kaissling B, Moe OW, Preisig PA, Alpern RJ: Chronic metabolic acidosis increases NHE3 protein abundance in rat kidney. Am J Physiol 271: F917–F925, 1996 [DOI] [PubMed] [Google Scholar]
- 85.Wu MS, Biemesderfer D, Giebisch G, Aronson PS: Role of NHE3 in mediating renal brush border Na+-H+ exchange. Adaptation to metabolic acidosis. J Biol Chem 271: 32749–32752, 1996 [DOI] [PubMed] [Google Scholar]
- 86.Pagliara AS, Goodman AD: Relation of renal cortical gluconeogenesis, glutamate content, and production of ammonia. J Clin Invest 49: 1967–1974, 1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roobol A, Alleyne GA: Control of renal cortex ammoniagenesis and its relationship to renal cortex gluconeogenesis. Biochim Biophys Acta 362: 83–91, 1974 [DOI] [PubMed] [Google Scholar]
- 88.Wright SH, Kippen I, Wright EM: Effect of pH on the transport of Krebs cycle intermediates in renal brush border membranes. Biochim Biophys Acta 684: 287–290, 1982 [DOI] [PubMed] [Google Scholar]
- 89.Aruga S, Wehrli S, Kaissling B, Moe OW, Preisig PA, Pajor AM, Alpern RJ: Chronic metabolic acidosis increases NaDC-1 mRNA and protein abundance in rat kidney. Kidney Int 58: 206–215, 2000 [DOI] [PubMed] [Google Scholar]
- 90.Preisig PA: The acid-activated signaling pathway: Starting with Pyk2 and ending with increased NHE3 activity. Kidney Int 72: 1324–1329, 2007 [DOI] [PubMed] [Google Scholar]
- 91.Lemann J, Jr, Bushinsky DA, Hamm LL: Bone buffering of acid and base in humans. Am J Physiol Renal Physiol 285: F811–F832, 2003 [DOI] [PubMed] [Google Scholar]
- 92.Ambühl PM, Zajicek HK, Wang H, Puttaparthi K, Levi M: Regulation of renal phosphate transport by acute and chronic metabolic acidosis in the rat. Kidney Int 53: 1288–1298, 1998 [DOI] [PubMed] [Google Scholar]
- 93.Jehle AW, Hilfiker H, Pfister MF, Biber J, Lederer E, Krapf R, Murer H: Type II Na-Pi cotransport is regulated transcriptionally by ambient bicarbonate/carbon dioxide tension in OK cells. Am J Physiol 276: F46–F53, 1999 [DOI] [PubMed] [Google Scholar]
- 94.Nowik M, Picard N, Stange G, Capuano P, Tenenhouse HS, Biber J, Murer H, Wagner CA: Renal phosphaturia during metabolic acidosis revisited: Molecular mechanisms for decreased renal phosphate reabsorption. Pflugers Arch 457: 539–549, 2008 [DOI] [PubMed] [Google Scholar]
- 95.Hansen WR, Barsic-Tress N, Taylor L, Curthoys NP: The 3′-nontranslated region of rat renal glutaminase mRNA contains a pH-responsive stability element. Am J Physiol 271: F126–F131, 1996 [DOI] [PubMed] [Google Scholar]
- 96.Laterza OF, Hansen WR, Taylor L, Curthoys NP: Identification of an mRNA-binding protein and the specific elements that may mediate the pH-responsive induction of renal glutaminase mRNA. J Biol Chem 272: 22481–22488, 1997 [DOI] [PubMed] [Google Scholar]
- 97.Laterza OF, Curthoys NP: Specificity and functional analysis of the pH-responsive element within renal glutaminase mRNA. Am J Physiol Renal Physiol 278: F970–F977, 2000 [DOI] [PubMed] [Google Scholar]
- 98.Schroeder JM, Liu W, Curthoys NP: pH-responsive stabilization of glutamate dehydrogenase mRNA in LLC-PK1-F+ cells. Am J Physiol Renal Physiol 285: F258–F265, 2003 [DOI] [PubMed] [Google Scholar]
- 99.Hanson RW, Reshef L: Regulation of phosphoenolpyruvate carboxykinase (GTP) gene expression. Annu Rev Biochem 66: 581–611, 1997 [DOI] [PubMed] [Google Scholar]
- 100.Hwang JJ, Perera S, Shapiro RA, Curthoys NP: Mechanism of altered renal glutaminase gene expression in response to chronic acidosis. Biochemistry 30: 7522–7526, 1991 [DOI] [PubMed] [Google Scholar]
- 101.Mufti J, Hajarnis S, Shepardson K, Gummadi L, Taylor L, Curthoys NP: Role of AUF1 and HuR in the pH-responsive stabilization of phosphoenolpyruvate carboxykinase mRNA in LLC-PK₁-F⁺ cells. Am J Physiol Renal Physiol 301: F1066–F1077, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Schroeder JM, Ibrahim H, Taylor L, Curthoys NP: Role of deadenylation and AUF1 binding in the pH-responsive stabilization of glutaminase mRNA. Am J Physiol Renal Physiol 290: F733–F740, 2006 [DOI] [PubMed] [Google Scholar]
- 103.Curthoys NP, Taylor L, Hoffert JD, Knepper MA: Proteomic analysis of the adaptive response of rat renal proximal tubules to metabolic acidosis. Am J Physiol Renal Physiol 292: F140–F147, 2007 [DOI] [PubMed] [Google Scholar]
- 104.Freund DM, Prenni JE, Curthoys NP: Response of the mitochondrial proteome of rat renal proximal convoluted tubules to chronic metabolic acidosis. Am J Physiol Renal Physiol 304: F145–F155, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Walmsley SJ, Freund DM, Curthoys NP: Proteomic profiling of the effect of metabolic acidosis on the apical membrane of the proximal convoluted tubule. Am J Physiol Renal Physiol 302: F1465–F1477, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]