

Abstract
Purpose
Osteogenesis imperfecta (OI) is a group of inherited disorders characterized by bone fragility. Ocular findings include blue sclera, low ocular rigidity, and thin corneal thickness. However, there are no documented cases linking OI and primary open angle glaucoma (POAG). In this report, we describe three individuals, one isolated case and two from a multiplex family, with OI type I and POAG.
Methods
Available family members with OI and POAG had a complete eye examination, including visual acuity, intraocular pressure (IOP), pachymetry, slit-lamp exam, dilated fundus exam, and visual fields. DNA from blood samples was sequenced and screened for mutations in COL1A1/2 and myocilin (MYOC).
Results
All subjects had OI type I. Findings of POAG included elevated IOP, normal gonioscopy, and glaucomatous optic disc cupping and visual field loss. POAG cosegregated with OI in the multiplex family. The multiplex family had a single nucleotide insertion (c.540_541insC) in COL1A1 resulting in a frameshift mutation and a premature termination codon. The sporadic case had a COL1A1 splice acceptor site mutation (c.2452–2A>T or IVS36–2A>T) predicted to result in a premature termination codon due to intron inclusion or a cryptic splice site. None of the glaucoma cases had mutations or sequence changes in MYOC.
Conclusions
We identified two novel mutations in COL1A1 in individuals with OI type I and POAG. Thus, some mutations in COL1A1 may be causative for OI and POAG. Alternatively, susceptibility genes may interact with mutations in COL1A1 to cause POAG.
Introduction
Osteogenesis imperfecta (OI) is a group of inherited disorders characterized by bone fragility and is usually inherited as an autosomal dominant trait. There are multiple forms of OI, ranging in severity from perinatal lethal to a mild phenotype with increased fracture frequency. OI type I is the most common adult form of the disease and is caused by mutations in the collagen type 1 alpha 1 gene (COL1A1, OMIM 120150). COL1A1 is located on chromosome 17, and it encodes the pro〈1(I) chains of type I procollagen [1]. OI results from mutations that produce premature termination codons and mRNA instability in the COL1A1 transcript. Type I collagen is the major structural protein in bone and many other tissues. Mutations in COL1A1 result in production of approximately half the usual amount of type I collagen. OI type I is characterized by normal height, bone fragility, ocular findings, and progressive hearing loss.
Ocular findings associated with OI include blue sclerae, low ocular rigidity, and thin corneas [2]. Various other reported ocular abnormalities include arcus senilis, small corneal diameter, small globe length, myopia, secondary glaucoma, optic atrophy, retinal detachment, subhyaloid hemorrhage, vitreous hyperplasia, congenital absence of Bowman’s layer, megalocornea, corneal opacities, and keratoconus. Although secondary forms of glaucoma have been reported in conjunction with surgery for retinal detachment or other interventions, there is no documented association between OI and primary open angle glaucoma (POAG). Herein, we report three individuals, one who is the only affected family member and two members of a multiplex family, with OI type I and POAG.
Methods
Informed consent was obtained from all participating individuals. This research adhered to the ARVO statement on human subjects as well as the tenets of the Declaration of Helsinki and was reviewed and approved by the Institutional Review Board of Duke University Medical Center (Durham, NC). Three individuals with OI type I, two members of one family (Figure 1) and an isolated case from a second family, were seen and evaluated at the Duke Eye Center Glaucoma Service. Subjects received a complete eye examination including visual acuity, intraocular pressure (IOP), pachymetry, slit-lamp exam, dilated fundus exam, and visual fields.
Figure 1.

Pedigree of the multiplex family. Subjects with OI and the genotypes of tested subjects are indicated. Filled circles indicate the presence of POAG. Subject 1 is the proband (0001), and Subject 2 is 107.
Blood samples were obtained, and genomic DNA was extracted using a salting out procedure from nucleated cells from the venous blood samples from all subjects. Type I collagen genes (COL1A1 and COL1A2, OMIM 120160) were sequenced. Coding exons and flanking intron sequences were amplified with PCR using 17 primer pairs for the COL1A1 gene and 23 primer pairs for the COL1A2 gene. The primer sequences and amplification conditions are available upon request. The identified mutations were confirmed by sequencing a new amplification product. To analyze the outcome of the splice-site mutation, cDNA was synthesized from RNA made from cultured fibroblasts after cyclohexamide incubation to inhibit translation-dependent mRNA degradation. The cDNA fragment containing the site of interest was amplified using a sense PCR primer located in exon 31 and an antisense primer in exon 47 and examined with polyacrylamide gel electrophoresis (PAGE). Fragments that contained additional bands compared to those seen in the control were excised from the gel, reamplified with the same primers, and sequenced with primers located in exon 32 and exon 41. Primers flanking the entire coding sequence of myocilin (MYOC, OMIM 601652) were designed with Primer3 software [3]. The targeted region including all three exons included at least 80 base pairs into flanking introns to screen for exon splicing variants.
Results
Subject 1
This patient is a 74-year-old woman with a history of multiple fractures and the clinical diagnosis of OI type I. POAG was diagnosed at age 50. The maximum reported IOP was 29 mmHg oculus uterque or both eyes (OU). The patient had three siblings with OI, and one of them was deceased (Figure 1). All family members with OI also had documented POAG. There were no family members with glaucoma other than those with OI. On examination, visual acuity was 20/20 OU with a refractive error of +1.75–0.50 X 076 oculus dexter (OD) and +2.25–0.50 X 026 oculus sinister (OS). IOP was 12 OD and 13 OS on treatment with latanoprost OU. Central corneal thickness (CCT) was 377 OD and 374 OS. Slit-lamp examination revealed sclera with a blue appearance. Gonioscopy was normal with an angle that was open to the ciliary body band OU. Dilated fundus examination revealed bilateral glaucomatous cupping (Figure 2). Optical coherence tomography revealed diffuse loss of the retinal nerve fiber layer OU (Figure 3). Humphrey visual fields demonstrated early visual field loss (Figure 4). The remainder of the ocular exam was normal.
Figure 2.

Optic nerve photographs of Subject 1 demonstrate characteristic glaucomatous optic nerve cupping.
Figure 3.
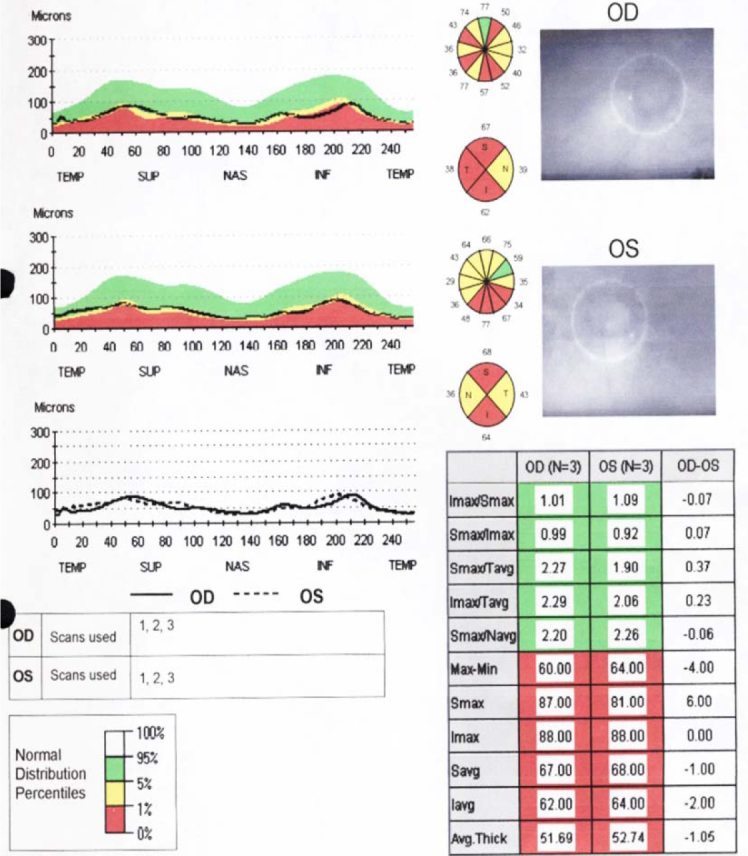
Optic coherence tomography of the retinal nerve fiber layers reveals diffuse nerve fiber layer thinning in both eyes of Subject 1.
Figure 4.

Humphrey visual fields (24–2 SITA-Standard) demonstrate early nasal field defects in both eyes of Subject 1.
Genomic DNA sequencing of COL1A1 and COL1A2 revealed a single nucleotide insertion in exon 6 of COL1A1 (c.540_541insC). This mutation causes a shift in the reading frame that would produce a premature termination codon in exon 8. This mutation is predicted to cause mRNA instability.
Subject 2
This subject was the 70-year-old sister of Subject 1. The patient had a history of remote femur and ankle fractures. On eye exam, visual acuity was 20/20 OU with refractive errors of +0.50–0.50 X 054 OD and +0.75–0.25 X 154 OS. CCTs were 426 OD and 432 OS. IOP was 13 mmHg OU. IOP was controlled with latanoprost, timolol, and dorzolamide OU. On slit-lamp examination, the sclerae were noted to be thin and bluish. Gonioscopy was normal with angles open to the ciliary body band. Dilated fundus examination revealed bilateral glaucomatous cupping (Figure 5). Humphrey visual fields demonstrated glaucomatous visual field loss consistent with the optic nerve exam (Figure 6). Due to medication intolerance, the patient had selective laser trabeculoplasty (SLT) performed OU. Her pressure had remained well controlled off topical therapy to date.
Figure 5.

Optic nerve photographs of Subject 2 demonstrate glaucomatous optic nerve cupping in both eyes.
Figure 6.

Humphrey visual fields (24–2 SITA-Standard) of Subject 2 demonstrate a superior altitudinal defect involving fixation in the left eye and early visual field changes in the right eye.
Directed DNA sequencing of COL1A1 revealed an insertion in exon 6 of COL1A1 (c.540_541insC), the same mutation as her sibling. Due to age and illness, the other family members affected by OI were either not able or unwilling to undergo ocular examination and blood sampling.
Subject 3
The patient is a 49-year-old woman with established diagnoses of OI type I and POAG. There was no known family history of OI or POAG. On ocular examination, visual acuity was 20/60 OD and 20/30 OS. The refractive error was −10.75–1.50 X 020 OD and −5.75–1.50 X 169 OS. IOP by applanation was 34 OD and 13 OS. Pachymetry revealed CCTs measuring 452 microns OU. Gonioscopy was normal with the angle open to the ciliary body band OU. She had glaucomatous cupping of both optic nerves. Humphrey visual fields demonstrated advanced visual field loss OU (Figure 7). The patient required placement of a Baerveldt 350 glaucoma implant surgery in the right eye for IOP control.
Figure 7.

Humphrey visual fields (24–2 SITA-Standard) of Subject 3 indicate advanced visual field loss in the right eye and an inferior arcuate and superior nasal step in the left eye.
Genomic DNA sequencing of COL1A1 and COL1A2 identified a COL1A1 acceptor splice site mutation (c.2452–2A>T or IVS36–2A>T) in intron 36. Analysis of the effect of the mutation on splice outcome in cultured dermal fibroblasts identified three splicing products, two of which were unstable and resulted in a small amount of stable mRNA. The first transcript used a cryptic acceptor 58–56 nt upstream from the constitutive site. This caused inclusion of the last 55 nt of intron 36 in the mRNA, producing a frameshift and premature termination codon in exon 37. The second transcript used a cryptic acceptor site in exon 37 that resulted in deletion of 28 nt from the mRNA. This product was also unstable. Unexpectedly, the third transcript used a cryptic donor site in the upstream exon 36 that deleted 44 nt from this exon, spliced to the cryptic acceptor site in exon 37, and deleted another 28 nt. The resulting product from the 72 nt deletion was small. Although it was stable, none was seen upon screening in cultured fibroblasts.
All glaucoma cases were screened for MYOC. No mutations or sequence changes were found in any sample.
Discussion
We identified two mutations in COL1A1 in two families affected by OI type I and POAG. The two mutations (c.540_541insC and c.2452–2A>T) result in a reading frame shift, create premature termination codons downstream, and produce nonsense-mediated mRNA decay of transcripts. These mutations result in the production of about half the amount of normal type I procollagen, the characteristic genetic defect in OI type I. The mutation identified in the multiplex family (subjects 1 and 2), c.540_541insC, has not been previously reported. The three affected family members all had typical findings of OI and POAG. The mutation in the isolated case, Subject 3, c.2452–2A>T (IVS36–2A>T), has been described in two individuals with OI type I; however, neither study completed RNA processing studies [4,5]. Although there are known ocular associations with OI, the relationship between OI and POAG has not been well described. Manschot et al. described a post-mortem histological study of eyes from an infant with OI congenita (likely OI type II) with blue sclerae, markedly thin corneas, open angles, and deep excavation of the optic disc that was hypothesized to be due to “insufficient development of the lamina cribrosa.” However, this change was not thought to result from increased IOP [6]. Berggren et al. described five patients with OI with eyes that had blue sclerae, normal scleral rigidity, low IOP (range: 10–14 mmHg), and high facility of outflow. There was no description of glaucomatous cupping or visual field loss in any of these individuals [7]. Beighton et al. described an autosomal recessive form of OI marked by blindness from hyperplasia of the vitreous, corneal opacities, and secondary glaucoma, possibly after penetrating keratoplasty [8]. This is now recognized as a distinct entity known as osteoporosis-pseudoglioma syndrome that results from mutations in LRP5 [9,10]. Superti-Furga et al. described a family with OI (likely type IV) due to a defect in COL1A2 in which one affected family member had glaucoma in one eye, but no further details of the ocular history, optic nerve examination, or visual field were provided [11]. POAG is a common ocular disorder, and this finding of POAG in patients with OI may have occurred by chance. Thus, to rule out known glaucoma-associated genetic variants we sequenced MYOC, the only known glaucoma-associated gene that would behave as an autosomal dominant condition and produce glaucoma with highly elevated IOP [12]. We found no sequence changes or mutations in these glaucoma cases that might explain our observation.
Type I collagen is found in many ocular tissues that play an important role in normal aqueous humor dynamics and optic nerve function. These include the trabecular meshwork, the uveoscleral pathway, and the optic nerve. Decreased amounts of type I collagen within the trabecular meshwork could potentially increase aqueous outflow resistance, which would produce a corresponding increase in IOP. Similarly, changes in the uveoscleral pathway could affect aqueous humor outflow. In addition, collagen abnormalities within the optic nerve head and lamina cribrosa could render the optic nerve more susceptible to damage from elevated IOP.
Dimasi et al. analyzed the role of mutations in type I collagen genes in CCT. They measured CCT in a cohort of 28 patients with OI type I and found that the mean CCT was significantly lower than that of a normal population; this was observed in our patients. The authors also reported that common single nucleotide polymorphisms in COL1A1 and COL1A2 are associated with CCT variation in a normal population [13]. Lower CCT is a known risk factor for POAG, and mutations in other collagen genes, including SNPs in collagen type 5 alpha 1 (COL5A1, OMIM 120215), collagen type 8 alpha 2 (COL8A2, OMIM 120252) and collagen type 8 alpha 1 (COL8A1, OMIM 120251) have also been linked to decreased CCT in Caucasian and Asian populations [14-17]. Mutations in other collagen genes, COL15A1 and COL18A1, which are expressed in the ciliary body, astrocytes in the optic nerve, and ganglion cell layer, have also been shown to affect the age of onset of POAG [18]. Based on these observations, further study of the role of variants in collagen genes in POAG appears warranted.
Interestingly, studies by Aihara et al. in a transgenic mouse model lend support to the association between type I collagen variants and glaucoma. In this report, a transgenic mouse model with a targeted mutation in COL1A1 developed ocular hypertension [19]. In a follow-up report, Mabuchi and colleagues noted the loss of optic nerve axons in addition to ocular hypertension, consistent with glaucomatous optic neuropathy [20]. Outflow facility is reduced by approximately 25% until 35 weeks of age in this mouse model. After this time period, outflow facility increases, and IOP subsequently normalizes [21]. Unlike the OI cases in which collagen production decreases, this mouse model proposes an increase in type I collagen deposition as the cause of ocular hypertension and glaucoma. The interactions between various extracellular matrix molecules are complex; therefore, it is difficult to predict the effect of decreased or increased synthesis of type I collagen in a specific connective tissue in humans. However, these studies and our report suggest that abnormalities in the synthesis of COL1A1 are associated with glaucoma.
Here, we reported three individuals in two families with OI type I and associated POAG. In one of these families with multiple affected members, POAG appeared to cosegregate with the OI trait. Another genetic factor or factors working separately or in concert with mutations in COL1A1 might be associated with the development of glaucoma in these individuals. However, viewed in light of other studies that suggest the role of collagen type I in POAG, it is not unreasonable to suggest that some mutations for OI may increase lifetime risk for POAG. These mutations may produce collapse of the trabecular meshwork or other adverse physiologic effects that could produce elevated IOP with resultant glaucoma. Additional effects on the lamina cribrosa and optic nerve might increase the risk of glaucomatous optic neuropathy. Further genetic evaluation of families with OI, as well as the role of other inherited collagen variants, may provide insight into mechanisms that lead to the development of POAG.
Acknowledgments
Kathleen Yang B.S., Melanie Pepin M.S., and Peter Byers M.D. at the Collagen Diagnostic Laboratory at the University of Washington performed DNA sequencing of COL1A1 and COL1A2. Research is supported by a grant from Research to Prevent Blindness, NEI R01EY015543 (RRA)
References
- 1.Byers PH, Cole WG. Osteogenesis imperfecta. In: Royce PM, Steinmann B, editors. Connective Tissue and its Heritable Disorders. Molecular, Genetic, and Medical Aspects. New York:Wiley-Liss; 2002. p. 385–430. [Google Scholar]
- 2.Kaiser-Kupfer MI, McCain L, Shapiro JR, Podgor MJ, Kupfer C, Rowe D. Low ocular rigidity in patients with osteogenesis imperfecta. Invest Ophthalmol Vis Sci. 1981;20:807–9. [PubMed] [Google Scholar]
- 3.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 4.Lee KS, Song HR, Cho TJ, Kim HJ, Lee TM, Jin HS, Park HY, Kang S, Jung SC, Koo SK. Mutational spectrum of type I collagen genes in Korean patients with osteogenesis imperfecta. Hum Mutat. 2006;27:599. doi: 10.1002/humu.9423. [DOI] [PubMed] [Google Scholar]
- 5.Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, Hyland JC, Korkko J, Prockop DJ, De Paepe A, Coucke P, Symoens S, Glorieux FH, Roughley PJ, Lund AM, Kuurila-Svahn K, Hartikka H, Cohn DH, Krakow D, Mottes M, Schwarze U, Chen D, Yang K, Kuslich C, Troendle J, Dalgeish R, Byers PH. Consortium for osteogenesis imperfecta mutations in the helical domain of type I collagen: regions rich in lethal mutations align with collagen binding sites for integrins and proteoglycans. Hum Mutat. 2007;28:209–21. doi: 10.1002/humu.20429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manschot WA. Ocular Anomalies in osteogenesis imperfecta. Ophthalmologica. 1965;149:241–5. doi: 10.1159/000304777. [DOI] [PubMed] [Google Scholar]
- 7.Berggren L, Wessler E, Wennerström J. Intraocular pressure and excretion of mucopolysaccharides in osteogenesis imperfecta. Acta Ophthalmol (Copenh) 1969;47:122–8. doi: 10.1111/j.1755-3768.1969.tb05616.x. [DOI] [PubMed] [Google Scholar]
- 8.Beighton P, Winship I, Behari D. The ocular form of osteogenesis imperfecta: a new autosomal recessive syndrome. Clin Genet. 1985;28:69–75. doi: 10.1111/j.1399-0004.1985.tb01220.x. [DOI] [PubMed] [Google Scholar]
- 9.Gong Y, Vikkula M, Boon L, Liu J, Beighton P, Ramesar R, Peltonen L, Somer H, Hirose T, Dallapiccola B, De Paepe A, Swoboda W, Zabel B, Superti-Furga A, Steinmann B, Brunner HG, Jans A, Boles RG, Adkins W, van den Boogaard MJ, Olsen R, Warman ML. Osteoporosis-pseudoglioma syndrome, a disorder affecting skeletal strength and vision, is assigned to chromosome region11q12–13. Am J Hum Genet. 1996;59:146–51. [PMC free article] [PubMed] [Google Scholar]
- 10.Gong Y, Slee RB, Fukai N, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Juppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML. Osteoporosis- Pseudoglioma Syndrome Collaborative Group. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 11.Superti-Furga A, Pistone F, Romano C, Steinmann B. Clinical variability of osteogenesis imperfecta linked to COL1A2 and associated with a structural defect in the type I collagen molecule. J Med Genet. 1989;26:358–62. doi: 10.1136/jmg.26.6.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stone EM, Fingert JH, Alward WL. Identification of a gene that causes primary open angle glaucoma. Science. 1997;275:668–70. doi: 10.1126/science.275.5300.668. [DOI] [PubMed] [Google Scholar]
- 13.Dimasi DP, Chen JY, Hewitt AW, Klebe S, Davey R, Stirling J, Thompson E, Forbes R, Tan TY, Savarirayan R, Mackey DA, Healey PR, Mitchell P, Burdon KP, Craig JE. Novel quantitative trait loci for central corneal thickness identified by candidate gene analysis of osteogenesis imperfecta genes. Hum Genet. 2010;127:33–44. doi: 10.1007/s00439-009-0729-3. [DOI] [PubMed] [Google Scholar]
- 14.Hoehn R, Zeller T, Verhoeven VJ, Grus F, Adler M, Wolfs RC, Uitterlinden AG, Castagne R, Schillert A, Klaver CC, Pfeiffer N, Mirshahi A. Population-based meta-analysis in Caucasians confirms association with COL5A1 and ZNF469 but not COL8A2 wiht central corneal thickness. Hum Genet. 2012;131:1783–93. doi: 10.1007/s00439-012-1201-3. [DOI] [PubMed] [Google Scholar]
- 15.Vithana EN, Aung T, Khor CC, Cornes BK, Tay WT, Sim X, Lavanya R, Wu R, Zheng Y, Hibberd ML, Chia KS, Seielstad M, Goh LK, Saw SM, Tai ES, Wong TY. Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum Mol Genet. 2011;20:649–58. doi: 10.1093/hmg/ddq511. [DOI] [PubMed] [Google Scholar]
- 16.Desronvil T, Logan-Wyatt D, Abdrabou W, Triana M, Jones R, Taheri S, Del Bono E, Pasquale LR, Olivier M, Haines JL, Fan BJ, Wiggs JL. Distribution of COL8A2 and COL8A1 gene variants in Caucasian primary open angle glaucoma patients with thin central corneal thickness. Mol Vis. 2010;16:2185–91. [PMC free article] [PubMed] [Google Scholar]
- 17.Vitart V, Bencic G, Hayward C, Skunca Herman J, Huffman J, Campbell S, Bućan K, Navarro P, Gunjaca G, Marin J, Zgaga L, Kolcić I, Polasek O, Kirin M, Hastie ND, Wilson JF, Rudan I, Campbell H, Vatavuk Z, Fleck B, Wright A. New loci associated with central cornea thickness include COL5A1, AKAP13, and AVGR8. Hum Mol Genet. 2010;19:4304–11. doi: 10.1093/hmg/ddq349. [DOI] [PubMed] [Google Scholar]
- 18.Wiggs JL, Howell GR, Linkroum K, Abdrabou W, Hodges E, Braine CE, Pasquale LR, Hannon GJ, Haines JL, John SW. Variations in COL15A1 and COL18A1 influence age of onset of primary open angle glaucoma. Clin Genet. 2013;84:167–74. doi: 10.1111/cge.12176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aihara M, Lindsey JD, Weinreb RN. Ocular hypertension in mice with a targeted type I collagen mutation. Invest Ophthalmol Vis Sci. 2003;44:1581–5. doi: 10.1167/iovs.02-0759. [DOI] [PubMed] [Google Scholar]
- 20.Mabuchi F, Lindsey JD, Aihara M, Mackey MR, Weinreb RN. Optic nerve damage in mice with a targeted type I collagen mutation. Invest Ophthalmol Vis Sci. 2004;45:1841–5. doi: 10.1167/iovs.03-1008. [DOI] [PubMed] [Google Scholar]
- 21.Dai Y, Lindsey JD, Duong-Polk X, Nguyen D, Hofer A, Weinreb RN. Outflow facility in mice with a targeted type I collagen mutation. Invest Ophthalmol Vis Sci. 2009;50:5749–53. doi: 10.1167/iovs.08-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]