

Abstract
Background
Supernumerary teeth are often observed in patients suffering from cleidocranial dysplasia due to a mutation in Runx2 that results in haploinsufficiencty. However, the underlying molecular mechanisms are poorly defined. In this study, we assessed the roles of Runx2 and its functional antagonist Twist1 in regulating fibroblast growth factor (FGF) signaling using in vitro biochemical approaches.
Results
We showed that Twist1 stimulated Fgfr2 and Fgf10 expression in a mesenchymal cell line and that it formed heterodimers with ubiquitously expressed E12 (together with E47 encoded by E2A gene) and upregulated Fgfr2 and Fgf10 promoter activities in a dental mesenchyme-derived cell line. We further demonstrated that the bHLH domain of Twist1 was essential for its synergistic activation of Fgfr2 promoter with E12 and that the binding of E12 stabilized Twist1 by preventing it from undergoing lysosomal degradation. Although Runx2 had no apparent effects on Fgfr2 and Fgf10 promoter activities, it inhibited the stimulatory activity of Twist1 on Fgfr2 promoter.
Conclusions
These findings suggest that Runx2 haploinsufficiency might result in excessive unbound Twist1 that can freely bind to E12 and enhance FGF signaling, thereby promoting the formation of extra teeth.
Keywords: Runx2, Twist1, E12, FGF10, Fgfr2, tooth morphogenesis
INTRODUCTION
The patterning of mammalian dentition involves the highly coordinated processes of cell migration, growth, selective proliferation and programmed cell death. This results in a critical mass of cells that are shaped, sized and positioned for terminal differentiation and the formation of bone, dentin, cementum and enamel. Without morphogenesis, cell differentiation and matrix formation cannot proceed normally. Hence, understanding the molecular mechanisms responsible for restraining the differentiation and mineralization potential of cells while patterning morphogenesis is in progress remains central questions for bone and tooth biologists today. Human cleidocranial dysplasia (CCD) is a skeletal disorder that offers an excellent model to study how patterning and cell differentiation are regulated. Mutations in the transcription factor, Runx2, cause CCD that is characterized by dysplastic clavicles, patent sutures and multiple supernumerary teeth (Jensen and Kreiborg, 1990; Lee et al., 1997; Bergwitz et al., 2001). Over the past decade, multiple studies have demonstrated the key roles of Runx2 in the transcription of genes that mark the terminal differentiation of osteoblasts, odontoblasts and hypertrophic chondrocytes (Ducy et al., 1997; Komori et al., 1997; Otto et al., 1997; Hinoi et al., 2006; Galler et al., 2007) . Other studies have also elucidated how Runx2’s functions are regulated at the levels of gene expression and protein-protein interactions (Bialek et al., 2004). Despite these advances, there is relatively little known about the precise molecular role of Runx2 in controlling the patterning of dentition.
Runx2 homozygous null mice die shortly after birth due to respiratory failure caused by the lack of bone formation (Komori et al., 1997; Otto et al., 1997). Our phenotypic analyses revealed that in the absence of Runx2, molar and incisor organs fail to develop beyond the late bud and bell stages of development, respectively (D’Souza et al., 1999; Aberg et al., 2004a). The presence of accessory epithelial buds on the lingual aspect of mutant upper molars further suggests an inhibitory role for Runx2 in successional tooth formation (Aberg et al., 2004a; Wang et al., 2005). While an extensive analysis of potential downstream targets of Runx2 revealed that Runx2 might regulate fibroblast growth factor (FGF) signaling (Aberg et al., 2004b), definitive studies are needed to better understand this molecular relationship.
The basic helix-loop-helix (bHLH) transcription factor, Twist1 is of particular interest as a regulatory protein partner for Runx2. Mutations in Twist1 result in Saethre-Chotzen Syndrome, an autosomal dominant condition that is characterized by craniosynostoses, limb anomalies and tooth agenesis (Reardon and Winter, 1994; el Ghouzzi et al., 1997; El Ghouzzi et al., 1999; El Ghouzzi et al., 2000). Twist1-heterozygous mice also present a Saethre-Chotzen syndrome-like phenotype (Bourgeois et al., 1998). Previous studies have suggested that the early fusion of cranial sutures is caused by a deregulation in FGF receptor 2 (Fgfr2) mediated signaling and the untimely (premature) differentiation of osteoblasts (Rice et al., 2000; Guenou et al., 2005; Connerney et al., 2006; Connerney et al., 2008). It is well established that Twist1 shows functional cooperativity with E12 and E47, two bHLH – containing transcription factors that result from the alternative splicing of E2A pre-mRNA (Sun and Baltimore, 1991). Interestingly, Twist1 also interacts with the DNA-binding Runt domain of Runx2 and it is the relief of this inhibition that leads to the onset of osteoblast differentiation (Bialek et al., 2004). In light of the observations that Runx2 and Twist1 are co-expressed in dental mesenchyme (D’Souza et al., 1999; Yamashiro et al., 2002; Rice et al., 2005), it is highly likely that Runx2 and Twist1 share a molecular relationship with FGF signaling during early tooth morphogenesis.
FGF signaling plays essential roles in mediating the reciprocal interactions between dental epithelial and mesenchymal cells throughout tooth development. The epithelial-derived Fgf4, Fgf8 and Fgf9 regulate cell proliferation mainly in the dental mesenchyme through activating Fgfr1IIIc and Fgfr2IIIc receptors, whereas mesenchymal Fgf3 and Fgf10 stimulate cell proliferation only in the dental epithelium through activating Fgfr1IIIb and Fgfr2IIIb receptors (Ornitz et al., 1996; Kettunen et al., 1998; Kettunen et al., 2000; Ohuchi et al., 2000). This FGF-mediated reciprocal signaling is critical for tooth development not only in regulating the size and shape of teeth formed but also in controlling the position and number of teeth formed (Ohuchi et al., 2000; Klein et al., 2006; Milunsky et al., 2006; Tekin et al., 2007; Wang et al., 2007; Charles et al., 2009; Charles et al., 2011).
In this study, we assessed the molecular relationship of Runx2 and Twist1 together with E12 in regulating FGF signaling, using their effects on Fgfr2 and/or Fgf10 promoter activities as readouts. Our data indicated that Twist1 formed heterodimers with E12 and synergistically stimulated Fgfr2 and Fgf10 expression. While Runx2 showed no apparent effects on the Fgfr2 and Fgf10 promoter activities, it antagonized the stimulatory effects of Twist1 on Fgfr2 promoter. The antagonistic action of Runx2 on Twist1 function suggests a novel mechanism that explains the supernumerary teeth formed in human CCD patients.
RESULTS
Twist1 and E12 synergistically regulate Fgfr2 and Fgf10 expression
C3H10T1/2 cells are pluripotent stem cells that were able to differentiate into odontoblast-like cells (Narayanan et al., 2001). To determine whether Twist1 was able to stimulate endogenous Fgfr2 and Fgf10 expression, we transfected C3H10T1/2 cells with Twist1 expression construct. We found that the forced expression of Twist1 resulted in about 2-fold increase in endogenous Fgfr2 (Fig. 1A) and Fgf10 (Fig. 1B) transcript levels. To further determine the underlying molecular mechanisms, we generated a 4.9 kb Fgfr2 promoter-luciferase construct and a 5.9 kb Fgf10 promoter-luciferase construct (Fig. 1C) and examined the effects of Twist1 and its binding partner E12 in MDPC-23 odontoblast-like cells. Co-transfection with the Twist1 expression vector alone slightly stimulated Fgfr2 promoter activity in MDPC-23 cells and the E12 expression vector had no effect, but co-transfection with both the Twist1 and E12 expression vectors produced a 3-fold increase in Fgfr2 promoter activities (Fig. 1D). Furthermore, Twist1 and E12 also activated Fgfr2 promoter activity in a dose-dependent manner (Fig. 1E). Similarly, co-transfection with either the Twist1 or E12 expression vector only slightly increased Fgf10 promoter activity, while co-transfection of both vectors stimulated a 4-fold increase in Fgf10 promoter activity (Fig. 1F). These findings suggest that Twist1 and E12 synergistically upregulate Fgfr2 and Fgf10 expression.
Fig. 1. Effects of Twist1 and E12 on Fgfr2 and Fgf10 expression.
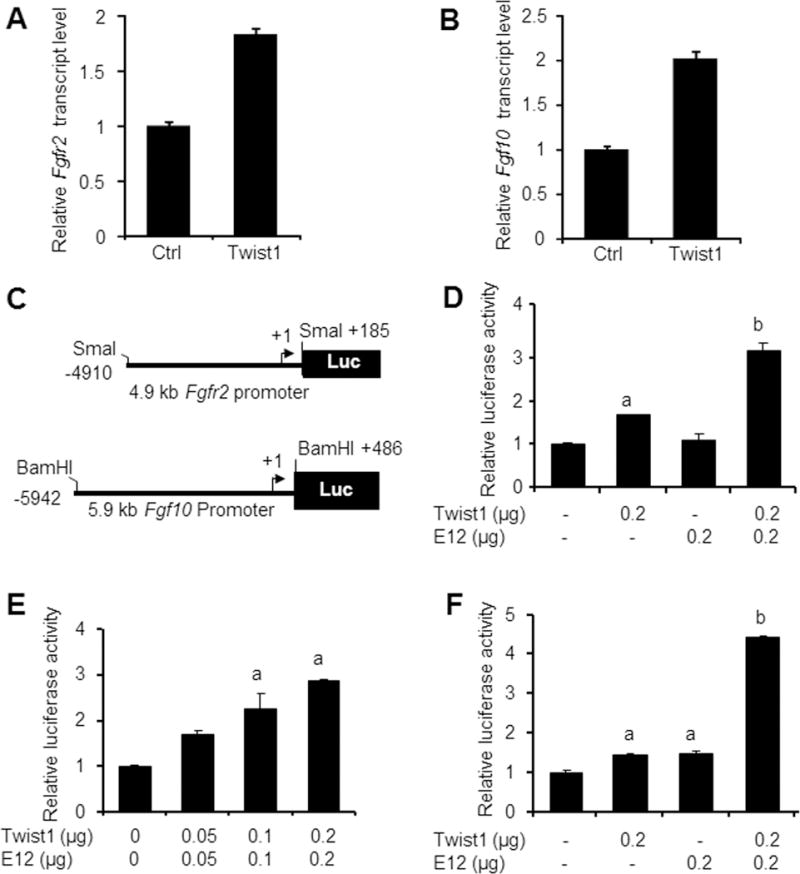
A. and B. C3H10T1/2 cells were transfected with 2 μg of either empty expression vector (Ctrl) or expression vector for Twist1 and total RNA was isolated 48 hours after transfection. The expression of Fgfr2 (A) and Fgf10 (B) was determined by quantitative real-time PCR. Data were presented as mean ± SEM of triplicate samples. C. Diagrams of 4.9 kb Fgfr2 promoter luciferase construct (pFgfr2-luc) and 5.9 kb Fgf10 promoter luciferase construct (pFgf10-luc). D. MDPC-23 cells were co-transfected with pFgfr2-luc construct and with the indicated expression vectors along with pRL-TK construct. Twist1 and E12 synergistically stimulate Fgfr2 promoter over 3 folds. E. MDPC-23 cells were co-transfected with the pFgfr2-luc construct and the indicated expression vectors along with pRL-TK construct. Twist1 and E12 activated Fgfr2 promoter activity in a dose-dependent manner. F. MDPC-23 cells were co-transfected with the pFgf10-luc construct and the indicated expression vectors along with pRL-TK construct. Twist1 and E12 synergistically stimulate Fgf10 promoter over 4 folds. Note: “a” indicates significant difference from the control (p<0.05); “b” means significant difference from all other groups (p<0.05).
The Twist1 bHLH domain is necessary for its synergistic action with E12
To determine whether Twist1 and E12 form heterodimers to activate Fgfr2 and Fgf10 promoters, 293FT cells were transfected with the expression vector for E12 together with expression vectors for full-length Twist1 or three Twist1 mutants, including NTwist1, NHTwist1 and NCTwist1 (Fig. 2A). All three Twist1 mutants contained the N-terminus of Twist1 (NTwist1), a domain that contains the nuclear localization signal (Hamamori et al., 1999) but has no apparent effect on osteoblast gene expression (Bialek et al., 2004). NHTwist1 had the bHLH domain that mediates the DNA binding and dimerization (Murre et al., 1994), whereas NCTwist1 comprised of the C-terminal Twist box, a domain that binds to and inhibits Runx2 transactivation function (Bialek et al., 2004). Coimmunoprecipitation demonstrated that only the full-length Twist1 was able to bind to E12, while Twist1 mutants failed to interact with E12 (Fig. 2B). However, the failure to detect the NHTwist1 interaction with E12 might be partially due to its less abundance in the input (Fig. 2B), which could be attributed to its rapid degradation after synthesis (El Ghouzzi et al., 2000). Indeed, not only was the full-length Twist1 able to stimulate Fgfr2 promoter activity alone and synergistically with E12, but also the NHTwist1 mutant had the ability to do so in the presence of E12 (Fig. 2C). These data highlighted that the bHLH domain is essential for the synergistic action of Twist1 with E12.
Fig. 2. Interaction between Twist1 and E12.
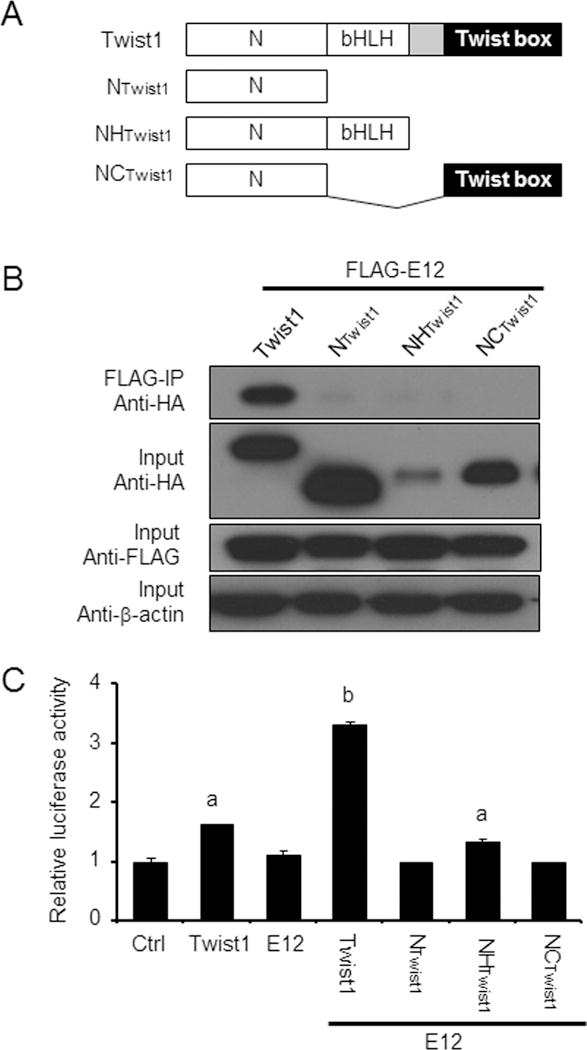
A. Schematic representation of constructs containing Twist1 and its various mutants, full-length Twist1, NTwist1, NHTwist1 and NCTwist1. B. Coimmunoprecipitation assay was used to determine the interactions of E12 with Twist1 as well as its domains. Western-blotting showed that only full-length HA-Twist1 interacted with Flag-E12, but not Twist1 mutants; However, There was remarkably less HA-NHTwist1 protein when the same amounts of proteins (20 μg) were loaded in each lane, suggesting that HA-NHTwist1 was unstable; C. MDPC-23 cells were transiently co-transfected with pFgfr2-luc construct and with the indicated expression vectors along with pRL-TK construct as an internal control. Again, the full-length Twist1 showed the stimulation of Fgfr2 promoter activity alone and synergistically with E12. Although it is weaker, the NHTwist1 mutant still significantly activated Fgfr2 promoter together with E12. However, NTwist1 and NCTwist1 failed to do so. Data are relative luciferase activity of one representative experiment performed in triplicate, and are expressed as mean ± SEM. n=3 in each group. “a” indicates significant difference from the control (p<0.05); “b” means significant difference from all other groups (p<0.05).
E12 prevents Twist1 from lysosomal degradation
To further understand the molecular mechanisms underlying the synergistic action of Twist1 and E12 on the Fgfr2 and Fgf10 promoters, 293FT cells were transfected with the vectors expressing HA-Twist1 or Flag-E12 either alone or together. Western-blot analyses showed that the level of Twist1 protein was lower in the cells transfected with only Twist1 vector, compared with the cells transfected with both the Twist1 and E12 vectors at 24 hours after transfection (Fig. 3A). By 48 hours, Twist1 proteins were accumulated in the cells transfected with both vectors, but in the cells transfected with the Twist1 vector alone, this protein was dramatically reduced (Fig. 3A). However, we did not notice any apparent difference in the level of E12 in the presence or absence of Twist1 (Fig. 3A). These data suggest that binding of E12 to Twist1 stabilizes the Twist1 proteins. Intracellular proteins are degraded by either a proteasomal or a lysosomal degradation pathway. To determine which pathway is responsible for Twist1 degradation, 293FT cells transfected with vector expressing HA-Twist1 were treated with or without the proteasomal inhibitor (MG-132) or the lysosomal inhibitor (ammonium chloride (NH4Cl)) for 6 hours. Western-blot analyses showed that Twist1 degradation was slowed down by NH4Cl, but not by MG132 (Fig. 3B). These data imply that Twist1 is degraded by the lysosomal degradation pathway.
Fig. 3. E12 prevents Twist1 from undergoing lysosomal degradation.
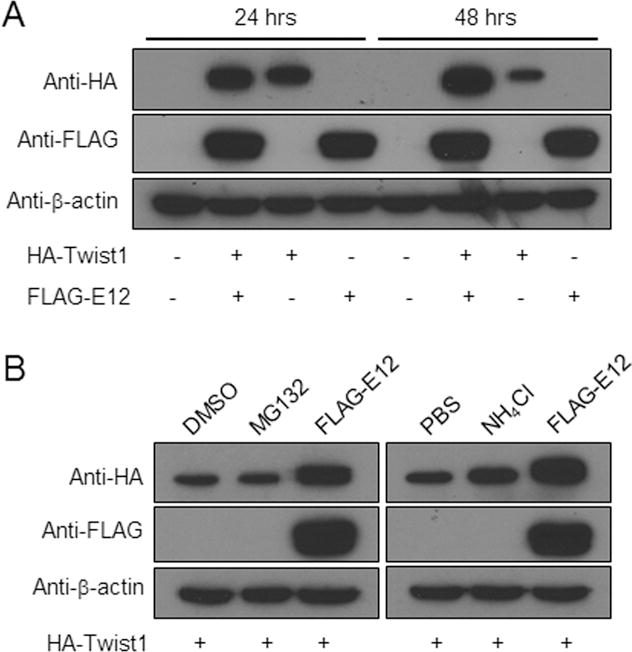
A. E12 stabilizes Twist1. 293FT cells were transiently transfected with the indicated expression vectors. Total proteins were extracted 24 and 48 hours after transfection and were subjected to western blotting analysis and immunoblotted with anti-HA, anti-Flag and anti-β-actin antibodies. Note that HA-Twist1 was stabilized when 293FT cells were co-transfected with Flag-E12 expression construct. B. Twist1 degradation was inhibited by lysosomal inhibitor. 293FT cells transfected with a vector expressing HA-Twist1 were treated with a proteasomal inhibitor (MG132) or DMSO as a control, or a lysosomal inhibitor (NH4Cl) or 1xPBS as a control for 6 hours. Cells cotransfected with vectors expressing both HA-Twist1 and Flag-E12 were used as controls. Total proteins were extracted 48 hours after transfection (or 6 hours after the addition of inhibitors), and immunoblotted with anti-HA, anti-Flag and anti-β-actin antibodies. Note that Twist1 degradation was suppressed by NH4Cl, but not by MG132.
Differential effects of Runx2 and Twist1 on Fgfr2 and Fgf10 promoter activities
Runx2 is another binding partner of Twist1 and the binding of Twist1 inhibits the transactivation function of Runx2 (Bialek et al., 2004). Runx2, like Twist1, is also induced in dental mesenchyme by FGFs (D’Souza et al., 1999; Aberg et al., 2004b; Rice et al., 2005). To determine whether Runx2 plays a similar function as Twist1 in regulating FGF signaling, we first examined the effects of Runx2 on the activities of both Fgfr2 and Fgf10 promoter-luciferase constructs in MDPC-23 odontoblast-like cells. As shown in Figs. 4A and 4C, co-transfection with the Runx2 expression vector alone had no apparent effects on the activities of Fgfr2 or Fgf10 promoter, respectively. Next, we examined whether Runx2 would affect the function of Twist1 since they bind to each other (Bialek et al., 2004). We found that Runx2 inhibited the stimulatory effects of Twist1 on the Fgfr2 promoter activity in a dose-dependent manner (Fig. 4B), but it had no influence on the effects of Twist1 in stimulating the Fgf10 promoter activity (Fig. 4D). These findings suggest that Runx2 might indirectly inhibit FGF signaling through antagonizing the function of Twist1.
Fig. 4. Antagonistic action of Runx2 on Twist1 function.
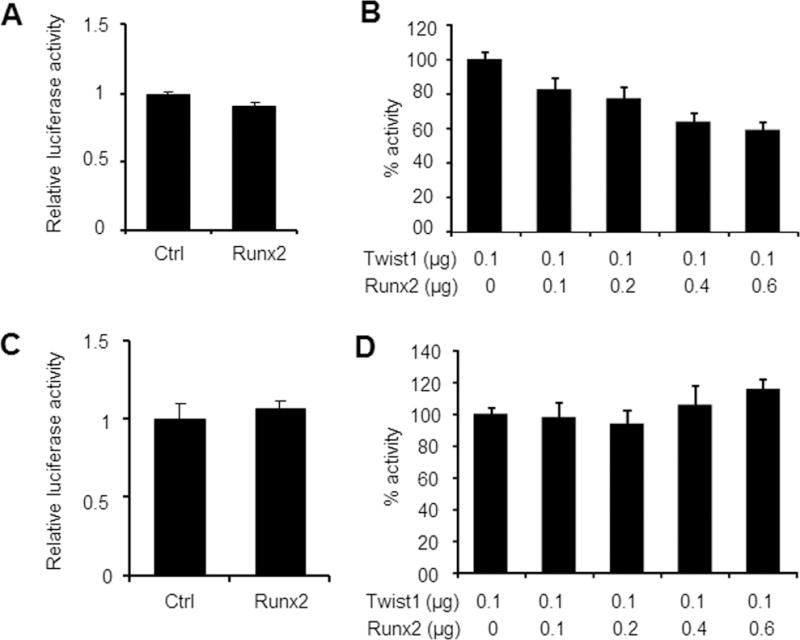
A. MDPC-23 cells were co-transfected with pFgfr2-luc construct and the indicated empty expression vector (Ctrl) or expression vector for Runx2 along with the pRL-TK construct as an internal control. Runx2 had no apparent effects on Fgfr2 promoter activity. B. MDPC-23 cells were co-transfected with pFgfr2-luc construct and the indicated expression vectors along with the pRL-TK construct. Fold activity was set at 100% in the absence of Runx2. Runx2 inhibited the stimulatory effects of Twist1 on Fgfr2 promoter activity in a dose-dependent manner. C. MDPC-23 cells were co-transfected with the pFgf10-luc construct and the indicated empty expression vector (Ctrl) or expression vector for Runx2 along with pRL-TK construct. Runx2 had no apparent effects on Fgf10 promoter activity. D. MDPC-23 cells were transfected with pFgf10-luc construct and the indicated expression vectors along with the pRL-TK construct. Fold activity was set at 100% in the absence of Runx2. Runx2 did not interfere with the effects of Twist1 on Fgf10 promoter.
DISCUSSION
By using organ culture and mouse genetic studies, we and other groups have previously shown that both Runx2 and its binding partner Twist1 are expressed and induced in dental mesenchyme by FGFs during tooth morphogenesis (D’Souza et al., 1999; Aberg et al., 2004b; Rice et al., 2005). However, the way in which these two transcription factors regulate FGF signaling is largely unknown. In this study, we showed that Twist1 and E12 stimulated endogenous Fgfr2 and Fgf10 expression and that Runx2 antagonized the stimulatory effects of Twist1 on Fgfr2 promoter activities. These findings suggest that the antagonistic action of Runx2 and Twist1 in controlling FGF signaling may be essential for normal tooth development.
In addition to teeth, Twist1 has also been shown to regulate FGF signaling in developing cranial sutures. Haploinsufficiency for Twist1 results in the altered Fgfr2 expression in the cranial sutures (Rice et al., 2000). In addition, Guenou et al (2005) found that FGFR2 expression is reduced in human cranial osteoblasts derived from patients with Saethre-Chotzen syndrome due to Twist1 haploinsufficiency, and that Twist1 overexpression restores FGFR2 expression. They further showed that Twist1 binds to a specific region in the FGFR2 promoter in cultured human calvarial osteoblasts, suggesting that Twist1 directly regulates FGFR2 expression (Guenou et al., 2005). Furthermore, Connerney et al (2006) demonstrated that Twist1 homodimers or heterodimers with E proteins differentially regulate the expression of target genes including Fgfr2 gene. Taken together, these studies and our current findings support the essential roles of Twist1 homodimers and/or heterodimers with E proteins in regulating FGF signaling in different organ systems.
Mouse genetic studies have documented the essential roles of FGF signaling in controlling the number and position of teeth formed. While molars undergo complete development in Fgf3 or Fgf10 single knockout embryos, they are arrested prior to the bud stage in the double knockout embryos (Ohuchi et al., 2000; Harada et al., 2002; Wang et al., 2007), suggesting that ablation of mesenchymal Fgfs could result in tooth agenesis. Consistently, when their cognate receptor, the epithelium isoform of Fgfr2 (Fgfr2IIIb) is deleted, tooth development is also arrested at the bud stage (De Moerlooze et al., 2000). On the contrary, excessive FGF signaling often results in the formation of supernumerary teeth. Sprouty genes encode general intracelllular antagonists of receptor tyrosine kinase signaling, including FGF signaling (Hacohen et al., 1998; Kramer et al., 1999; Reich et al., 1999). Deletions of sprouty genes lead to the formation of diastema teeth and supernumerary incisors in mice, and these extra teeth can be prevented by lowering the dosage of the relevant FGF or FGF receptor genes (Klein et al., 2006; Charles et al., 2011).
Our current findings are in contrast to the previous study showing the reduced FGF signaling in Runx2-deficient embryos (Aberg et al., 2004b). The underlying reason for this discrepancy is unknown. However, several lines of evidence suggest that the roles of Runx2 in the development of successional teeth might be different from those in the development of primary teeth. First, Runx2 haploinsufficiency in humans affects permanent dentition but not primary dentition (Jensen and Kreiborg, 1990; Bergwitz et al., 2001); Second, decreased levels of Runx2 in mice are also adequate for the formation of one layer of teeth, but the complete lack of Runx2 results in extra tooth buds on the lingual aspect of upper molars (Aberg et al., 2004a; Wang et al., 2005). In this regard, the roles of Runx2 might be different in humans and mice since the extra tooth buds are formed only in the complete loss of Runx2 function in mice. In addition, although Msx1 is essential for the development of mouse normal dentition, it is dispensable for the formation of supernumerary teeth caused by Apc loss-of-function (Wang et al., 2009), which also supports the different molecular mechanisms involved in the development of normal and supernumerary teeth.
Our current and previous studies suggest that Twist1 might be the key molecule in the dental mesenchyme that couples epithelial and mesenchymal FGF signaling. Epithelial FGFs induces Twist1 in dental mesenchyme (Rice et al., 2005), which in turn stimulates Fgfr2 expression; more Fgfr2 proteins on the cell surface subsequently make cell more responsive to epithelial FGFs and produce more Twist1. In addition, Twist1 also upregulates the expression of Fgf10, which acts on epithelial cells. Therefore, Runx2 inhibits the stimulatory effects of Twist1 on Fgfr2 expression, which would indirectly result in the inhibition of Fgf10 expression as well. These findings suggest that the ablation of Runx2 function might lead to a relative abundance of unbound Twist1 and subsequently higher levels of Fgfr and/or Fgf expression in mesenchyme. This will most likely result in elevated FGF signaling as well, subsequently an increase in the proliferation of the overlying dental lamina and the formation of extra-lingual epithelial buds in mice and morphologically normal supernumerary teeth in humans.
EXPERIMENTAL PROCEDURES
Cells and Constructs
C3H10T1/2 cells were pluripotent mesenchymal stem cells obtained from the American Type Culture Collection (ATCC). MDPC-23 cells are mouse dental mesenchymal cells with the potential to differentiate into odontoblast-like cells and to form multilayered nodules in the presence of ascorbic acid and β-glycerophosphate (Hanks et al., 1998; Gaikwad et al., 2001). 293FT cells were obtained from Invitrogen, and were fast-growing and highly transferable cells. All three cell lines were grown in DMEM supplemented with 10% FBS in a humidified incubator with 5% CO2 at a temperature of 37°C. A 4.9 kb Fgfr2 promoter fragment was released by the restriction enzyme SmaI from a 7 kb genomic DNA fragment containing Fgfr2 upstream sequence, initially isolated from a mouse genomic library (Ali et al., 1995). A 5.9 kb Fgf10 promoter fragment was isolated by restriction enzyme BamHI from a mouse bacterial artificial chromosome (BAC) clone containing the Fgf10 gene (Source BioScience LifeSciences). Both promoter fragments were separately subcloned into pGL3-basic vector (Promega). The pCMV-Flag-E12 construct was generated by cloning human E12 cDNA into the BamHI and XhoI sites of pCMV-Tag 2B vector (Stratagene). Constructs for HA-tagged full-length Twist1 (carrying an N-terminal domain, a bHLH domain and a C-terminal Twist box) and its various mutants, NTwist1 (containing the N-terminal domain only), NHTwist1 (comprising of N-terminal plus the bHLH) and NCTwist1 (consisting of N-terminal plus the C-terminal region) were obtained from Dr. Gerard Karsenty (Bialek et al., 2004).
Quantitative real-time PCR
To determine whether Twist1 was able to induce endogenous Fgfr2 and Fgf10 expression, we transfected C3H10T1/2 cells with 2 μg of either an empty vector (pCDNA3) or a construct expressing Twist1 with Fugene 6 transfection reagent (Roche Applied Science). Forty-eight hours after transfection, total RNA was isolated from the transfected C3H10T1/2 cells with Trizol® reagent (Invitrogen Corporation) and reverse transcribed into cDNA using the QuantiTect® Reverse Transcription Kit (Qiagen). Quantitative real-time PCR was carried out on a Bio-Rad CFX96 system (Bio-Rad) using GoTaq qPCR Master Mix (Promega), with an initial denaturation at 95°C for 2 min followed by 40 cycles of 95°C for 30 s and 60°C for 1 min. PCR product accumulation was monitored by measuring the increase in fluorescence intensity resulting from the binding of the fluorescent dye to double stranded DNA. The relative expression of target genes was determined using the comparative threshold cycle (CT) method (CT number is the number of amplification cycles required to reach a threshold fluorescence level) and Gapdh as a reference gene. The gene expression data were expressed as fold changes relative to the experimental controls. The primer sequences for the Fgfr2 gene were forward primer 5′- gtgcttggcgggtaattcta −3′ and reverse primer 5′- gatgactgtcaccaccatgc −3′; The primer sequences for the Fgf10 gene were forward primer 5′- ctggaaagcacttgggtcat −3′ and reverse primer 5′- ggagacagaatgcacaagca −3′; The primer sequences for Gapdh gene were forward primer 5′- gaagcccatcaccatcttc −3′ and reverse primer 5′-gttcacacccatcacaaaca −3′.
Luciferase assay
For the promoter-luciferase assay, subconfluent MDPC-23 cells in 24-well plates were transiently transfected with 0.1 μg of pFgfr2-luc or pFgf10-luc construct and the designated expression vectors with Fugene 6 transfection reagent (Roche Applied Science). The pRL-TK construct, which expressed renilla luciferase, was used as an internal control to monitor transfection efficiency. Total amounts of transfected DNA were balanced by the addition of empty vector (pcDNA3). Transfected cells were harvested using passive lysis buffer 48 hours after transfection. The firefly and renilla luciferase activities were assayed using a Dual-luciferase Reporter Assay System (Promega). Promoter activities were measured by the firefly luciferase activities normalized to renilla luciferase activities and expressed as luciferase activities relative to the activities from the control. All luciferase assays were performed at least three times in triplicate, and representative data are presented.
Twist1 degradation assays
For analysis of Twist1 stability, 293FT cells in 6-well plates were co-transfected with expression vectors for Twist1 or E12 alone or both with Fugene 6 transfection reagent (Roche Applied Science), proteins were then extracted at 24 and 48 hours after transfection and assayed by western-blotting with antibodies against HA (for Twist1) or against Flag (for E12). To further determine the pathways that account for Twist1 degradation, the 293FT cells transfected with expression vector for Twist1 were treated with or without proteasomal inhibitor, 25 μM MG-132 (Sigma) or lysosomal inhibitor, 25 μM NH4Cl (Sigma) for 6 hours. Twist1 protein levels were then analyzed by western-blotting with antibodies against HA (for Twist1) as described below.
SDS-PAGE and Western Blot Analysis
Transfected 293FT cell lysates were electrophoresed using 10% SDS-polyacrylamide gel, and transferred onto nitrocellulose membranes. Blots were then immunoblotted with rabbit polyclonal anti-HA antibody (Santa Cruz Biotechnology; 1:1000) or mouse monoclonal anti-Flag antibody (Stratagene, 1:20, 000) followed by secondary antibody with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology; 1:1000) or HRP-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology; 1:1000). β-actin was immunoblotted with mouse monoclonal anti-β-actin-peroxidase antibody (Sigma; 1:20,000). The immunostained bands were detected with ECL™ Chemiluminescent Detection reagents (Amersham Biosciences) and imaged using a CL-XPosure film (Pierce Biotechnology Inc.).
Immunoprecipitations
The protein-protein interaction of E12 and Twist1 as well as its various mutants were assayed by co-immunoprecipitation (Co-IP). Briefly, 293FT cells transfected with expression vectors for E12 and Twist1 or various Twist1 mutants were lysed with cell lysis buffer (100 mM NaCl, 50 mM Tris HCL, pH 7.5, 0.5% Triton X-100). Two hundred micrograms of protein extracts were then incubated with mouse monoclonal anti-Flag antibodies for E12 overnight, followed by incubation with protein G beads (Sigma-Aldrich) in 100 μl of cell lysis buffer for 3 hours with rotation at 4°C. After beads were washed with cell lysis buffer, eluates were subjected to SDS-PAGE and immunoblotted using rabbit polyclonal anti-HA antibodies for Twist1, as described above.
Statistical Analysis
Student t-test was performed for two group comparison. One-way ANOVA was conducted for multiple group comparison followed by Bonferroni method to determine which groups were significantly different from others. The quantified data are expressed as the mean ± standard error of the mean (SEM). P<0.05 was considered statistically significant.
Key findings.
Twist1 forms heterodimers with ubiquitously expressed E12 and synergistically stimulates Fgfr2 and Fgf10 promoter activities
E12 prevents Twist1 from undergoing lysosomal degradation
Runx2 has no direct effects on Fgfr2 and Fgf10 promoter activities, but it inhibited the stimulatory effects of Twist1 on Fgfr2 promoter
A relative abundance of unbound Twist1, due to Runx2 haploinsufficiency, may lead to elevated FGF signaling, and subsequently the formation of supernumerary teeth in human CCD
Acknowledgments
This study was supported by NIDCR/NIH Grants, DE013368 and DE16472 to RDS, and DE021773 to YL.
References
- Aberg T, Cavender A, Gaikwad JS, Bronckers AL, Wang X, Waltimo-Siren J, Thesleff I, D’Souza RN. Phenotypic changes in dentition of Runx2 homozygote-null mutant mice. J Histochem Cytochem. 2004a;52:131–139. doi: 10.1177/002215540405200113. [DOI] [PubMed] [Google Scholar]
- Aberg T, Wang XP, Kim JH, Yamashiro T, Bei M, Rice R, Ryoo HM, Thesleff I. Runx2 mediates FGF signaling from epithelium to mesenchyme during tooth morphogenesis. Dev Biol. 2004b;270:76–93. doi: 10.1016/j.ydbio.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Ali J, Mansukhani A, Basilico C. Fibroblast growth factor receptors 1 and 2 are differentially regulated in murine embryonal carcinoma cells and in response to fibroblast growth factor-4. J Cell Physiol. 1995;165:438–448. doi: 10.1002/jcp.1041650225. [DOI] [PubMed] [Google Scholar]
- Bergwitz C, Prochnau A, Mayr B, Kramer FJ, Rittierodt M, Berten HL, Hausamen JE, Brabant G. Identification of novel CBFA1/RUNX2 mutations causing cleidocranial dysplasia. J Inherit Metab Dis. 2001;24:648–656. doi: 10.1023/a:1012758925617. [DOI] [PubMed] [Google Scholar]
- Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, Justice MJ, Karsenty G. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–435. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- Bourgeois P, Bolcato-Bellemin AL, Danse JM, Bloch-Zupan A, Yoshiba K, Stoetzel C, Perrin-Schmitt F. The variable expressivity and incomplete penetrance of the twist-null heterozygous mouse phenotype resemble those of human Saethre-Chotzen syndrome. Hum Mol Genet. 1998;7:945–957. doi: 10.1093/hmg/7.6.945. [DOI] [PubMed] [Google Scholar]
- Charles C, Hovorakova M, Ahn Y, Lyons DB, Marangoni P, Churava S, Biehs B, Jheon A, Lesot H, Balooch G, Krumlauf R, Viriot L, Peterkova R, Klein OD. Regulation of tooth number by fine-tuning levels of receptor-tyrosine kinase signaling. Development. 2011;138:4063–4073. doi: 10.1242/dev.069195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles C, Lazzari V, Tafforeau P, Schimmang T, Tekin M, Klein O, Viriot L. Modulation of Fgf3 dosage in mouse and men mirrors evolution of mammalian dentition. Proc Natl Acad Sci U S A. 2009;106:22364–22368. doi: 10.1073/pnas.0910086106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connerney J, Andreeva V, Leshem Y, Mercado MA, Dowell K, Yang X, Lindner V, Friesel RE, Spicer DB. Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev Biol. 2008;318:323–334. doi: 10.1016/j.ydbio.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connerney J, Andreeva V, Leshem Y, Muentener C, Mercado MA, Spicer DB. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev Dyn. 2006;235:1345–1357. doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- D’Souza RN, Aberg T, Gaikwad J, Cavender A, Owen M, Karsenty G, Thesleff I. Cbfa1 is required for epithelial-mesenchymal interactions regulating tooth development in mice. Development. 1999;126:2911–2920. doi: 10.1242/dev.126.13.2911. [DOI] [PubMed] [Google Scholar]
- De Moerlooze L, Spencer-Dene B, Revest JM, Hajihosseini M, Rosewell I, Dickson C. An important role for the IIIb isoform of fibroblast growth factor receptor 2 (FGFR2) in mesenchymal-epithelial signalling during mouse organogenesis. Development. 2000;127:483–492. doi: 10.1242/dev.127.3.483. [DOI] [PubMed] [Google Scholar]
- Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89:747–754. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- El Ghouzzi V, Lajeunie E, Le Merrer M, Cormier-Daire V, Renier D, Munnich A, Bonaventure J. Mutations within or upstream of the basic helix-loop-helix domain of the TWIST gene are specific to Saethre-Chotzen syndrome. Eur J Hum Genet. 1999;7:27–33. doi: 10.1038/sj.ejhg.5200240. [DOI] [PubMed] [Google Scholar]
- el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin AL, Munnich A, Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- El Ghouzzi V, Legeai-Mallet L, Aresta S, Benoist C, Munnich A, de Gunzburg J, Bonaventure J. Saethre-Chotzen mutations cause TWIST protein degradation or impaired nuclear location. Hum Mol Genet. 2000;9:813–819. doi: 10.1093/hmg/9.5.813. [DOI] [PubMed] [Google Scholar]
- Gaikwad JS, Hoffmann M, Cavender A, Bronckers AL, D’Souza RN. Molecular insights into the lineage-specific determination of odontoblasts: the role of Cbfa1. Adv Dent Res. 2001;15:19–24. doi: 10.1177/08959374010150010501. [DOI] [PubMed] [Google Scholar]
- Galler KM, Yasue A, Cavender AC, Bialek P, Karsenty G, D’Souza RN. A novel role for Twist-1 in pulp homeostasis. J Dent Res. 2007;86:951–955. doi: 10.1177/154405910708601007. [DOI] [PubMed] [Google Scholar]
- Guenou H, Kaabeche K, Mee SL, Marie PJ. A role for fibroblast growth factor receptor-2 in the altered osteoblast phenotype induced by Twist haploinsufficiency in the Saethre-Chotzen syndrome. Hum Mol Genet. 2005;14:1429–1439. doi: 10.1093/hmg/ddi152. [DOI] [PubMed] [Google Scholar]
- Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92:253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- Hamamori Y, Sartorelli V, Ogryzko V, Puri PL, Wu HY, Wang JY, Nakatani Y, Kedes L. Regulation of histone acetyltransferases p300 and PCAF by the bHLH protein twist and adenoviral oncoprotein E1A. Cell. 1999;96:405–413. doi: 10.1016/s0092-8674(00)80553-x. [DOI] [PubMed] [Google Scholar]
- Hanks CT, Fang D, Sun Z, Edwards CA, Butler WT. Dentin-specific proteins in MDPC-23 cell line. Eur J Oral Sci. 1998;106(Suppl 1):260–266. doi: 10.1111/j.1600-0722.1998.tb02185.x. [DOI] [PubMed] [Google Scholar]
- Harada H, Toyono T, Toyoshima K, Yamasaki M, Itoh N, Kato S, Sekine K, Ohuchi H. FGF10 maintains stem cell compartment in developing mouse incisors. Development. 2002;129:1533–1541. doi: 10.1242/dev.129.6.1533. [DOI] [PubMed] [Google Scholar]
- Hinoi E, Bialek P, Chen YT, Rached MT, Groner Y, Behringer RR, Ornitz DM, Karsenty G. Runx2 inhibits chondrocyte proliferation and hypertrophy through its expression in the perichondrium. Genes Dev. 2006;20:2937–2942. doi: 10.1101/gad.1482906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BL, Kreiborg S. Development of the dentition in cleidocranial dysplasia. J Oral Pathol Med. 1990;19:89–93. doi: 10.1111/j.1600-0714.1990.tb00803.x. [DOI] [PubMed] [Google Scholar]
- Kettunen P, Karavanova I, Thesleff I. Responsiveness of developing dental tissues to fibroblast growth factors: expression of splicing alternatives of FGFR1, −2, −3, and of FGFR4; and stimulation of cell proliferation by FGF-2, −4, −8, and −9. Dev Genet. 1998;22:374–385. doi: 10.1002/(SICI)1520-6408(1998)22:4<374::AID-DVG7>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Kettunen P, Laurikkala J, Itaranta P, Vainio S, Itoh N, Thesleff I. Associations of FGF-3 and FGF-10 with signaling networks regulating tooth morphogenesis. Dev Dyn. 2000;219:322–332. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1062>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Klein OD, Minowada G, Peterkova R, Kangas A, Yu BD, Lesot H, Peterka M, Jernvall J, Martin GR. Sprouty genes control diastema tooth development via bidirectional antagonism of epithelial-mesenchymal FGF signaling. Dev Cell. 2006;11:181–190. doi: 10.1016/j.devcel.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Kramer S, Okabe M, Hacohen N, Krasnow MA, Hiromi Y. Sprouty: a common antagonist of FGF and EGF signaling pathways in Drosophila. Development. 1999;126:2515–2525. doi: 10.1242/dev.126.11.2515. [DOI] [PubMed] [Google Scholar]
- Lee B, Thirunavukkarasu K, Zhou L, Pastore L, Baldini A, Hecht J, Geoffroy V, Ducy P, Karsenty G. Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet. 1997;16:307–310. doi: 10.1038/ng0797-307. [DOI] [PubMed] [Google Scholar]
- Milunsky JM, Zhao G, Maher TA, Colby R, Everman DB. LADD syndrome is caused by FGF10 mutations. Clin Genet. 2006;69:349–354. doi: 10.1111/j.1399-0004.2006.00597.x. [DOI] [PubMed] [Google Scholar]
- Murre C, Bain G, van Dijk MA, Engel I, Furnari BA, Massari ME, Matthews JR, Quong MW, Rivera RR, Stuiver MH. Structure and function of helix-loop-helix proteins. Biochim Biophys Acta. 1994;1218:129–135. doi: 10.1016/0167-4781(94)90001-9. [DOI] [PubMed] [Google Scholar]
- Narayanan K, Srinivas R, Ramachandran A, Hao J, Quinn B, George A. Differentiation of embryonic mesenchymal cells to odontoblast-like cells by overexpression of dentin matrix protein 1. Proc Natl Acad Sci U S A. 2001;98:4516–4521. doi: 10.1073/pnas.081075198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchi H, Hori Y, Yamasaki M, Harada H, Sekine K, Kato S, Itoh N. FGF10 acts as a major ligand for FGF receptor 2 IIIb in mouse multi-organ development. Biochem Biophys Res Commun. 2000;277:643–649. doi: 10.1006/bbrc.2000.3721. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Reardon W, Winter RM. Saethre-Chotzen syndrome. J Med Genet. 1994;31:393–396. doi: 10.1136/jmg.31.5.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich A, Sapir A, Shilo B. Sprouty is a general inhibitor of receptor tyrosine kinase signaling. Development. 1999;126:4139–4147. doi: 10.1242/dev.126.18.4139. [DOI] [PubMed] [Google Scholar]
- Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, Thesleff I. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- Rice R, Thesleff I, Rice DP. Regulation of Twist, Snail, and Id1 is conserved between the developing murine palate and tooth. Dev Dyn. 2005;234:28–35. doi: 10.1002/dvdy.20501. [DOI] [PubMed] [Google Scholar]
- Sun XH, Baltimore D. An inhibitory domain of E12 transcription factor prevents DNA binding in E12 homodimers but not in E12 heterodimers. Cell. 1991;64:459–470. doi: 10.1016/0092-8674(91)90653-g. [DOI] [PubMed] [Google Scholar]
- Tekin M, Hismi BO, Fitoz S, Ozdag H, Cengiz FB, Sirmaci A, Aslan I, Inceoglu B, Yuksel-Konuk EB, Yilmaz ST, Yasun O, Akar N. Homozygous mutations in fibroblast growth factor 3 are associated with a new form of syndromic deafness characterized by inner ear agenesis, microtia, and microdontia. Am J Hum Genet. 2007;80:338–344. doi: 10.1086/510920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XP, Aberg T, James MJ, Levanon D, Groner Y, Thesleff I. Runx2 (Cbfa1) inhibits Shh signaling in the lower but not upper molars of mouse embryos and prevents the budding of putative successional teeth. J Dent Res. 2005;84:138–143. doi: 10.1177/154405910508400206. [DOI] [PubMed] [Google Scholar]
- Wang XP, O’Connell DJ, Lund JJ, Saadi I, Kuraguchi M, Turbe-Doan A, Cavallesco R, Kim H, Park PJ, Harada H, Kucherlapati R, Maas RL. Apc inhibition of Wnt signaling regulates supernumerary tooth formation during embryogenesis and throughout adulthood. Development. 2009;136:1939–1949. doi: 10.1242/dev.033803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XP, Suomalainen M, Felszeghy S, Zelarayan LC, Alonso MT, Plikus MV, Maas RL, Chuong CM, Schimmang T, Thesleff I. An integrated gene regulatory network controls stem cell proliferation in teeth. PLoS Biol. 2007;5:e159. doi: 10.1371/journal.pbio.0050159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashiro T, Aberg T, Levanon D, Groner Y, Thesleff I. Expression of Runx1, −2 and −3 during tooth, palate and craniofacial bone development. Mech Dev. 2002;119(Suppl 1):S107–110. doi: 10.1016/s0925-4773(03)00101-1. [DOI] [PubMed] [Google Scholar]