

Abstract
Recently, patient mutations that activate PI3K signaling have been linked to a primary antibody deficiency. Here, we used whole-exome sequencing and characterized the molecular defects in 4 patients from 3 unrelated families diagnosed with hypogammaglobulinemia and recurrent infections. We identified 2 different heterozygous splice site mutations that affect the same splice site in PIK3R1, which encodes the p85α subunit of PI3K. The resulting deletion of exon 10 produced a shortened p85α protein that lacks part of the PI3K p110-binding domain. The hypothetical loss of p85α-mediated inhibition of p110 activity was supported by elevated phosphorylation of the known downstream signaling kinase AKT in patient T cell blasts. Analysis of patient blood revealed that naive T and memory B cell counts were low, and T cell blasts displayed enhanced activation-induced cell death, which was corrected by addition of the PI3Kδ inhibitor IC87114. Furthermore, B lymphocytes proliferated weakly in response to activation via the B cell receptor and TLR9, indicating a B cell defect. The phenotype exhibited by patients carrying the PIK3R1 splice site mutation is similar to that of patients carrying gain-of-function mutations in PIK3CD. Our results suggest that PI3K activity is tightly regulated in T and B lymphocytes and that various defects in the PI3K-triggered pathway can cause primary immunodeficiencies.
Introduction
It was shown recently that some primary antibody deficiencies are caused by hyperactivation of the PI3K signaling pathway, as gain-of-function mutations in PIK3CD encoding for the p110δ subunit result in activated PI3Kδ syndrome (APDS) (1, 2). Class IA PI3K molecules are composed of a p110 catalytic subunit (p110α, p110β, or p110δ) and a regulatory subunit (p85α, p55α, p50α, p85β, or p55γ) that regulates p110’s stability, cellular localization, and function. Each of the catalytic subunits can bind to any of the regulatory subunits. Expression of the p110δ catalytic subunit is restricted mainly to leukocytes, whereas p110α and p110β are ubiquitously expressed (3). The function of class IA PI3Ks is to convert phosphatidylinositol 4,5-bisphosphate into phosphatidylinositol 3,4,5-trisphosphate (PIP3), a phospholipid secondary messenger that interacts with intracellular enzymes containing pleckstrin homology domains. The most important of these enzymes is the serine/threonine kinase AKT, whose important role in cell proliferation, growth, survival, and metabolism in many cell types has been conserved throughout evolution (3). PI3K signaling notably has an important role in several distinct stages of B and T cell development, differentiation, and function, and their activation occurs downstream of the antigen receptor (either the B cell receptor [BCR] or TCR) and costimulatory molecules (4). The expression of an active form of p110α was enough to maintain B cell survival in the periphery in the absence of a BCR, highlighting the importance of PI3K for B lymphocytes (5). Furthermore, PI3K signaling has an important role in the antibody maturation process, because it is involved in the regulation of Ig class switch recombination and plasma cell differentiation (6). It was reported recently that a patient presenting with agammaglobulinemia and the absence of B lineages carried a premature stop codon in PIK3R1, resulting in the absence of p85α (7). PIK3R1 encodes p85α and 2 additional isoforms (p55α and p50α), the production of which is regulated by 2 distinct promoters and alternative first exons. The last nine 3′ exons of PIK3R1 are expressed in all isoforms (7).
Results and Discussion
Patients.
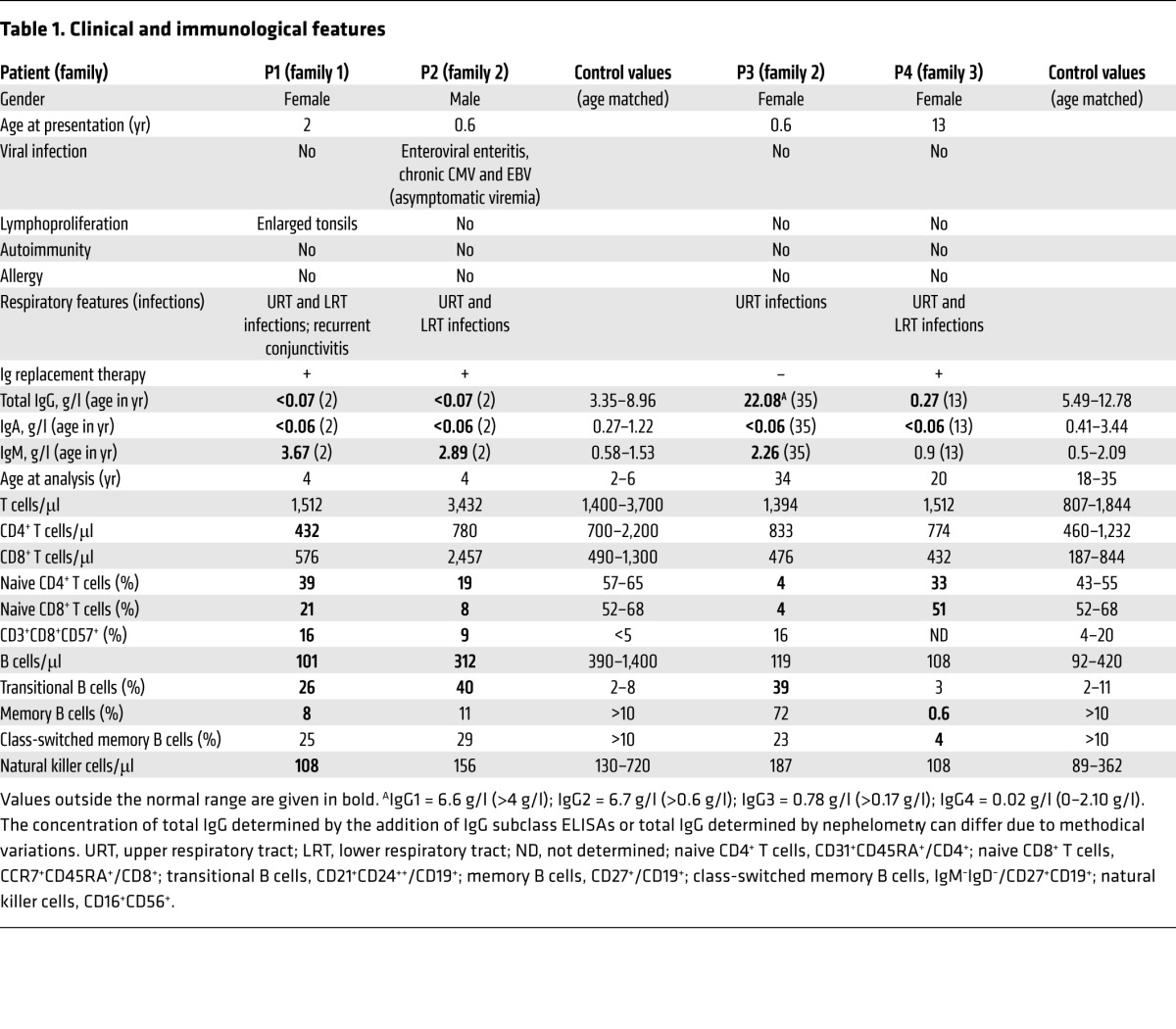
All 4 patients had been suffering from recurrent respiratory bacterial tract infections from soon after birth, although none of them was hospitalized to treat infections (Table 1 and Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI75746DS1). They had no signs of allergy, autoimmunity, splenomegaly, or lymphadenopathy (with the exception of patient 1 [P1], whose tonsils had been removed in 2 separate operations). Although viral infections and opportunistic bacterial or fungal infections were not reported, P2, but not P1 and P4, presented with CMV and EBV viremia, as detected by PCR (CMV, 9,300 copies per ml; EBV, 1,500 copies per ml; reference values, <446 and <182, respectively). Lymphocyte counts were in the normal range, but the frequency of CD31+CD45RA+ naive CD4 and CCR7+CD45RA+ naive CD8 peripheral blood T cells was low in all patients (Table 1). An elevated proportion of CD8 T cells expressing the senescence-associated marker CD57 was observed in the 2 youngest patients (P1 and P2). Two of four patients displayed a memory B cell deficiency. P4 exhibited a nearly complete absence of IgM–IgD–CD27+ switched memory B cells. P1, P2, and P3 had elevated transitional B cell counts (Table 1). Impaired B cell function was suggested by the serum Ig profile: undetectable IgA levels in all patients; very low IgG levels in P1, P2, and P4 (but elevated levels in P3); and elevated IgM levels in 3 patients. All the patients but P3 were receiving Ig replacement therapy. The immunological phenotype of these patients was somewhat heterogeneous and reminiscent of that observed in APDS. The presence of dominant gain-of-function mutations of PIK3CD was ruled out for all 4 patients (data not shown). P2 presented with growth retardation (–2 SD for weight and –2.5 SD for height), without improvement after treatment with Ig replacement therapy.
Table 1.
Clinical and immunological features
Identification of a heterozygous gain-of-function mutation in PIK3R1.
By applying whole-exome sequencing (Supplemental Methods) and then targeted Sanger sequencing, we identified 2 different heterozygous splice site mutations affecting the same splice site in PIK3R1 (Figure 1, A and B) in all 4 patients. A heterozygous G-to-T mutation (position g.67589663) at the +1 position of the splice donor site of PIK3R1 for intron 10 was found in P1. Sanger sequencing confirmed that the heterozygous mutation was present in the patient but not in her healthy siblings or parents, indicating that it was a de novo mutation. The 3 other patients carry a heterozygous G-to-C mutation at the same nucleotide position as P1 (Figure 1, C and D). Neither our in-house variant database nor the Exome Sequencing Project’s Exome Variant Server (http://evs.gs.washington.edu/EVS/) contained a record of a nucleotide variation at this position. The mutation in P2 was inherited from his mother (P3). DNA from the healthy fathers of P2 and P4 was not available for genetic analysis. Sequencing of RT-PCR products from RNA samples from P1 and P3 confirmed that both mutations resulted in the same PIK3R1 mRNA splice variant (i.e., exclusion of exon 10, Figure 1E). The splicing from exon 9 to exon 11 is in-frame and therefore resulted in deletion of amino acid residues 434–475 of p85α (encoded by exon 10); the p55α and p50α isoforms were similarly affected. This peptide sequence is part of one of the α-helices involved in p85α binding to the cognate domain in p110 (ref. 8, Figure 1F, and Supplemental Figure 2). A shortened protein, p85αΔ434_475, was observed by Western blot analysis, although in lower quantity compared with WT p85α (Supplemental Figure 3A). A lower amount of mutant protein p85αΔ434_475 in comparison with that of WT p85α was also observed in transfection experiments in 293T cells, indicating that the mutant protein is less stable (Supplemental Figure 3B). The description of heterozygous mutations, including a series of overlapping in-frame deletions, in exon 10 of the PIK3R1 gene in cancer cells supports the hypothesis whereby this novel splice site mutation is disease causing (9, 10). These mutations do not affect p85α’s ability to stabilize p110 but abrogate inhibitory activity and thus promote AKT activation (10). To test the hypothesis in which p85αΔ434_475 behaves similarly to the mutant p85α proteins found in cancer cells, we examined the abundance of p110δ protein and phosphorylation of AKT at Ser473 in the patients’ T cell blasts. The expression of p110δ was found to be normal. Phosphorylation of AKT at Ser473 and Thr308 was detected in nonstimulated patients’ cells but not in nonstimulated control cells. Following stimulation with an anti-CD3 antibody, the level of phosphorylation of AKT at Ser473 in patients’ cells was comparable to that in control cells when analyzed by immunoblotting but consistently higher in patients’ cells than in control cells when analyzed by flow cytometry (Figure 2, A and B, and Supplemental Figure 4, A–C). Phosphorylation of AKT was also higher in patients’ cells than in control cells following a 30-minute stimulation with anti-CD3 and anti-CD28 antibodies when analyzed by flow cytometry (Supplemental Figure 5A). Phosphorylation of AKT was no longer detected by flow cytometry in patients’ or control cells when the PI3Kδ inhibitor IC87114 was present in a high concentration during the activation (Figure 2B) or added for the last 20 minutes of stimulation (Supplemental Figure 5A). Lower concentrations of the inhibitor strongly but not completely abrogated phosphorylation of AKT at Ser473, arguing for p110δ-independent AKT phosphorylation (Supplemental Figure 5B).
Figure 1. A splice site mutation in PIK3R1 leads to deletion of part of the p110-binding domain.

(A) Exons of the PIK3R1 gene, which encodes p85α and the isoforms p50α (purple) and p55α (orange). (B) Protein structure of p85α and the isoforms p50 and p55α. Phosphorylated tyrosine (Y) and serine (S) residues are indicated. (C) Pedigrees of the families carrying PIK3R1 splice site mutations. (D) A sequencing chromatogram showing the heterozygous splice site mutations. Corresponding protein sequence is shown on the top. (E) RT-PCR covering exon 10 of PIK3R1 from indicated samples. (F) Protein structure of p85αΔ434_475 mutant protein.
Figure 2. Abundance of p110δ protein, phosphorylation of AKT at Ser473, and increased AICD in T cell blasts from patients.

(A) Expression of p110δ, AKT, and GAPDH and phosphorylation of AKT at Ser473 and at Thr308 by Western blotting of cell lysates derived from T cell blasts from P1, P2, and P3 and controls before (Ø) and after (+) stimulation with anti-CD3 antibody. (B) Intracellular staining of phosphorylated AKT at Ser473 in nonstimulated and CD3-activated T cell blasts from patients and controls with or without 10 μM IC87114 inhibitor, measured by flow cytometry. (C) A representative experiment showing the expression of flag-tagged WT p85α, flag-tagged p85αΔ434_475, AKT, and Ku-70 and phosphorylation of AKT at Ser473 by Western blotting of cell lysates derived from NIH3T3 cells 2 days after transfection with the indicated expression vector. (D) Representative AICD experiment with T cell blasts from P1 and the control. The results are expressed as the percentage of apoptotic/dead blasts after OKT3 activation minus the percentage of apoptotic/dead blasts in the absence of stimulation. AICD was analyzed with or without 3 μM IC87114 inhibitor with T cell blasts cultured for 6 days. Similar results were obtained at later time points and for P2 and P3 (data not shown).
Hyperactivated PI3K/AKT signaling in T cell blasts has been reported to result in increased mTORC1 signaling (2). We indeed found elevated phosphorylation of the ribosomal protein S6 at Ser240 and Ser244 in nonstimulated patients’ cells compared with that in nonstimulated control cells, indicating increased mTORC1 signaling (Supplemental Figure 5, C and D).
To confirm that the shortened protein p85αΔ434_475 was responsible for the hyperactivation of AKT, we ectopically expressed the mutant and WT form of p85α in NIH3T3 cells. We found elevated levels of phosphorylated AKT in the cells expressing p85αΔ434_475 in comparison with controls (Figure 2C). These results strongly suggest that the PIK3R1 splice site mutation is a disease-causing mutation that increases PI3K signaling.
Increased cell death following T lymphocyte activation.
Given that mutations in p110δ result in increased PI3K signaling and greater susceptibility of T cell blasts to activation-induced cell death (AICD), we measured the latter phenomenon in T cell blasts from patients carrying the PIK3R1 splice site mutations. After stimulation with anti-CD3, the patients’ T cell blasts displayed higher levels of AICD than control blasts cultured for the same period of time (Figure 2D). Elevated levels of AICD were observed at a lower intensity when the inhibitor IC87114 was added 15 minutes before stimulation (Figure 2D), indicating that the cell death was, to a large part, a consequence of stronger PI3Kδ signaling.
Impaired T and B lymphocyte proliferation in vitro.
To study the effect of the PIK3R1 splice site mutation on B and T lymphocyte function, we examined the in vitro proliferation activity of the patients’ peripheral blood lymphocytes after exposure to various stimuli. As assessed by the loss of CFSE fluorescence intensity after cell stimulation with CD40L and IL-4, the proliferative activity of the patients’ B cells was similar to that of healthy controls. In contrast, low proliferative activity was observed when the patients’ B cells were activated with anti-IgM plus CpG oligonucleotides, indicating that p85αΔ434_475 interferes with signaling through the BCR and/or the TLR9 (Figure 3A). Impaired proliferation activity (relative to controls) was also observed when the patients’ CD8 T cells were stimulated with anti-CD3– and anti-CD28–coated beads (Figure 3B). It remains to be established whether the decreased proliferative activity of CD8 T cells results from more intense PI3K signaling. Indeed composition of peripheral blood cells was biased toward effector T cells, which are known to be less able to proliferate after TCR stimulation in vitro (relative to naive T cells) (11).
Figure 3. Proliferative ability of peripheral blood B and T lymphocytes.

Proliferation profile as determined by dilution of CFSE-labeled (A) CD19+ gated peripheral blood lymphocytes from patients and the control 5 days after the indicated activation and (B) CD4+ or CD8+ gated peripheral blood lymphocytes from the patients and the control 3 days after stimulation with anti-CD3– and anti-CD28–coated beads. The gray peak indicates CFSE labeling of nonstimulated cells.
The phenotype of the patients described herein appears to be a phenocopy of that seen in APDS, including phenotypic heterogeneity that may be related to the patient’s history of infections, environmental factors, and/or the presence of modifier genes. Considering the broad expression of PIK3R1-derived regulatory components and their implication in many signal transduction pathways, a nonlymphocyte component might contribute to the phenotype.
The PI3K signaling product PIP3 is dephosphorylated by the phosphatase and tensin homolog (PTEN) (12). The p85α regulatory protein also binds to PTEN and enhances the latter’s lipid phosphatase activity, making it a dual regulatory protein for both the PI3K p110 catalytic subunit and PTEN. Hence, p85α has a critical regulatory function in maintaining the balance of PI3K/PTEN signaling (13). The PTEN protein abundance was normal in P1’s T cell blasts (Supplemental Figure 6). It remains to be seen to what extent the p85αΔ434_475 mutant protein impairs PTEN activity and thereby strengthens PI3K signaling.
Mutations in PIK3R1, which either impair p85α function or completely abrogate p85α expression, cause primary antibody deficiencies (hypogammaglobulinemia or agammaglobulinemia, respectively). Agammaglobulinemia is the result of severely impaired B cell development, and such a B cell intrinsic effect in patients carrying homozygous loss-of-function mutation results in nearly complete PI3K deficiency (7). In contrast, the variable hypogammaglobulinemia observed in patients carrying heterozygous PIK3R1 splice site mutations is the result of activated p110 and can be explained by (a) an intrinsic B cell defect (impaired B cell development/differentiation and responses to BCR and TLR stimuli); (b) an extrinsic B cell defect, e.g., defective T cell function (increased AICD); or (c) a combination of B cell intrinsic and extrinsic defects.
Given that B cell lymphoma has been reported in APDS, patients carrying a heterozygous PIK3R1 splice site mutation are also at risk of carcinogenesis (1, 2). The spectrum of disorders might be broader, since p85α can also interact with p110α and p110β; this might lead to hyperactivation of PI3K signaling in other cell types. Our present work describes a new genetic defect responsible for an immunodeficiency that is caused by activating mutations in PIK3R1 and increased PI3K signaling. We suggest naming this new immunodeficiency “activated PI3Kδ syndrome 2” (APDS2), since uncontrolled activity of p110δ is (at least to a large part) responsible for the disease. Our results highlight the role of PI3K signaling in the homeostasis of immune responses. Further screening for defects that lead to hyperactivation of PI3K signaling should therefore be performed in other patients with similar primary immunodeficiencies and no known genetic defect.
Methods
Further details are provided as Supplemental Methods.
Statistics.
Analyses were performed with PRISM software (version 4 for Macintosh, GraphPad Inc.). Statistical hypotheses were tested using 2-tailed t test. A P value of less than 0.05 was considered significant.
Study approval.
Peripheral blood samples were obtained from the patients, their relatives, and healthy donors after the provision of informed, written consent. Genetic studies and data collection procedures were approved by the Comité de Protection des Personnes Ile de France II and the French Advisory Committee on Data Processing in Medical Research.
Supplementary Material
Acknowledgments
We thank Sylvain Breton, Erwan Dumontet, Monique Forveille, Stéphanie Ndaga, Nathalie Lambert, Virginie Grandin, Ioana Morel, and Aminata Diabate for technical support and Cécile Masson for help with exome analysis. S. Kracker is a CNRS researcher. This study was supported by INSERM, the Fondation pour la Recherche Médicale (grant ING20130526624), the Ligue Contre le Cancer – Comité de Paris, the EUFP7 European Research Council PIDIMMUNE program (grant 249816, to A. Fischer), and a government grant managed by the French Agence Nationale de la Recherche as part of the “Investments for the Future” program (ANR-10-IAHU-01).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(9):3923–3928. doi:10.1172/JCI75746.
See the related commentary beginning on page 3688.
References
- 1.Angulo I, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342(6160):866–871. doi: 10.1126/science.1243292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lucas CL, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol. 2014;15(1):88–97. doi: 10.1038/ni.2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. 2003;3(4):317–330. doi: 10.1038/nri1056. [DOI] [PubMed] [Google Scholar]
- 4.Deane JA, Fruman DA. Phosphoinositide 3-kinase: diverse roles in immune cell activation. Annu Rev Immunol. 2004;22:563–598. doi: 10.1146/annurev.immunol.22.012703.104721. [DOI] [PubMed] [Google Scholar]
- 5.Srinivasan L, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139(3):573–586. doi: 10.1016/j.cell.2009.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omori SA, et al. Regulation of class-switch recombination and plasma cell differentiation by phosphatidylinositol 3-kinase signaling. Immunity. 2006;25(4):545–557. doi: 10.1016/j.immuni.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 7.Conley ME, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med. 2012;209(3):463–470. doi: 10.1084/jem.20112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mandelker D, et al. A frequent kinase domain mutation that changes the interaction between PI3Kα and the membrane. Proc Natl Acad Sci U S A. 2009;106(40):16996–17001. doi: 10.1073/pnas.0908444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Urick ME, et al. PIK3R1 (p85α) is somatically mutated at high frequency in primary endometrial cancer. Cancer Res. 2011;71(12):4061–4067. doi: 10.1158/0008-5472.CAN-11-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jaiswal BS, et al. Somatic mutations in p85α promote tumorigenesis through class IA PI3K activation. Cancer Cell. 2009;16(6):463–474. doi: 10.1016/j.ccr.2009.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho BK, et al. Functional differences between memory and naive CD8 T cells. Proc Natl Acad Sci U S A. 1999;96(6):2976–2981. doi: 10.1073/pnas.96.6.2976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci U S A. 1999;96(8):4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chagpar RB, et al. Direct positive regulation of PTEN by the p85 subunit of phosphatidylinositol 3-kinase. Proc Natl Acad Sci U S A. 2010;107(12):5471–5476. doi: 10.1073/pnas.0908899107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.