

Abstract
Implantation is an essential process during establishment of pregnancy in mammals. It is initiated with the attachment of the blastocyst to a receptive uterine epithelium followed by its invasion into the stromal tissue. These events are profoundly regulated by the steroid hormones 17β-estradiol and progesterone. During the past several years, mouse models harboring conditional gene knockout mutations have become powerful tools for determining the functional roles of cellular factors involved in various aspects of implantation biology. Studies using these genetic models as well as primary cultures of human endometrial cells have established that the estrogen receptor α, the progesterone receptor, and their downstream target genes critically regulate uterine growth and differentiation, which in turn control embryo-endometrial interactions during early pregnancy. These studies have uncovered a diverse array of molecular cues, which are produced under the influence of estrogen receptor α and progesterone receptor and exchanged between the epithelial and stromal compartments of the uterus during the progressive phases of implantation. These paracrine signals are critical for acquisition of uterine receptivity and functional interactions with the embryo. This review highlights recent work describing paracrine mechanisms that govern steroid-regulated uterine epithelial-stromal dialogue during implantation and their roles in fertility and disease.
In mammals, the principal function of the uterus is to support the growth and development of the fetus during pregnancy. During establishment of pregnancy, the embryo comes into intimate physical contact with the uterine endometrium and implants into this tissue (1–3). Successful implantation requires attachment of the embryo to the uterine luminal epithelium and its subsequent invasion into the underlying stromal bed (4). Embryo invasion triggers a unique transformation of the stromal cells, known as decidualization, which involves sequential proliferation and differentiation of these cells (5, 6). The differentiated stromal tissue supports uterine remodeling and development of an elaborate maternal vasculature that plays a critical role in embryonic growth and survival (7, 8). The events during early pregnancy are profoundly influenced by the ovarian steroid hormones, 17β-estradiol (E) and progesterone (P). The molecular mechanisms via which steroid-regulated pathways control these processes are under intense investigation by many laboratories around the world.
In humans, infertility is one of the most common disturbances of reproductive health. Despite significant advances in assisted reproductive technologies (ART), many couples experience infertility as a result of failed implantation of the fertilized embryos into the uterus and subsequent loss of pregnancy. The implantation rates in ART remain low, even with high-quality embryos, emphasizing the importance of this process as a major cause of pregnancy failure and infertility (9). An understanding of the signaling mechanisms central to implantation has the potential to alleviate many problems associated with infertility and improve the outcome of ART. Because of ethical considerations, research to study the early events during human pregnancy is restricted. However, the mouse has served as an important animal model to investigate the molecular mechanisms underlying female infertility (10). The mouse and human share many common features of implantation: (1) occurrence of implantation at the blastocyst stage, (2) a restricted window of uterine receptivity, (3) extensive stromal differentiation and remodeling to produce decidual tissue, (4) invasion of the embryo into the stromal bed, and (5) establishment of hemochorial placentation. These similarities raise the probability that mechanistic information gleaned from mouse models, when carefully interpreted, is relevant to humans. This review focuses on our emerging understanding of the network of steroid hormone–regulated molecular pathways that regulate endometrial function during implantation in the mouse. We will also describe how this knowledge is currently being extended to the clinical realm through the use of human endometrial specimens and primary cell cultures. Several factors identified to be critical for implantation in the mouse have now been shown to be important for human endometrial proliferation and differentiation. We believe that these conserved pathways are likely to represent the most fundamental mediators of the implantation process.
During early pregnancy, E and P act in an opposing fashion to orchestrate changes in the uterine epithelium that render it competent for embryo attachment. In mice, preovulatory ovarian E stimulates uterine epithelial growth and proliferation on days 1 and 2 of pregnancy (11–13). In the E-dominated proliferative phase, the epithelium displays a distinct columnar phenotype and expresses proteins, such as E-cadherin, which play a key role in the maintenance of the adhesive and polarized phenotype of uterine epithelial cells (14, 15). Starting on day 3 of pregnancy, in response to rising P levels, epithelial cells cease to proliferate and enter a differentiation program. On day 4 of pregnancy, uterine epithelial cells lose their polarity, as indicated by the down-regulation of E-cadherin from junctional complexes, and acquire adhesiveness for the embryo (16). The attachment of the embryo to the uterine epithelium is followed by the P-driven differentiation of subjacent fibroblastic stromal cells into secretory decidual cells. Thus, critical uterine changes during implantation are orchestrated by an intricate interplay of E and P signaling via their cognate receptors (1, 2). Recent studies have revealed that a series of molecular communications between the epithelial and stromal cells, guided by the steroid hormone receptors present in these tissues, is critical for acquisition of uterine receptivity and progressive functional interactions with the embryo (17–19). In this review, we summarize our emerging understanding of the paracrine mechanisms that mediate this cell-cell dialogue.
ESR1 and PGR Control Implantation
The cellular actions of E and P are mediated through their intracellular receptors, estrogen receptor α (ESR1) and progesterone receptor (PGR), respectively. These receptors function as ligand-inducible transcription factors (20). Ligand-occupied ESR1 and PGR bind to specific DNA sequences, known as steroid response elements, to activate or repress the expression of their target genes. Use of chromatin immunoprecipitation coupled to high throughput sequencing (ChIP-Seq) has identified genome-wide binding sites of ESR1 and PGR in the mouse uterus (21, 22). In addition to interacting directly with the hormone response elements, these receptors can also control target gene transcription by tethering to other transcription factors bound to the gene regulatory regions. Recent studies indicate that binding of these receptors can occur at distal enhancer regions, which are situated many thousands of base pairs away from the transcription start site but are able to form complexes with the proximal promoter through chromosomal looping events (23, 24).
A large body of literature indicates that E and P, acting through their receptors, regulate cell proliferation, differentiation, and secretory protein production in the uterus (25, 26). Accordingly, a dynamic pattern of expression of ESR1 and PGR proteins is seen in both epithelial and stromal compartments of the uterus during the reproductive cycle and pregnancy. A mouse model harboring a global knockout of Esr1 established a critical role of this receptor in the regulation of overall uterine function, particularly in E-induced growth of this tissue (27, 28). Female mice lacking Pgr showed that P signaling via its receptor is essential for decidualization and is a prerequisite to successful implantation (29). In addition, several cellular factors, which modulate the transcriptional functions of ESR1 and PGR in the uterine tissue, were shown to control implantation. Studies by Dey and colleagues (30, 31) revealed that immunophilin FKBP52, which serves as a chaperone that maintains the structure and function of PGR in the uterine cells, is critical for implantation. Simmen and colleagues (32) reported that Krüppel-like factor (KLF) 9, a member of the Krüppel-like family of transcription factors, interacts with PGR to control the expression of P-regulated target genes in endometrial epithelial cells. Studies by this group further indicated that the loss of KLF9 in the uterine stromal cells down-regulates PGR expression and creates P resistance, resulting in the failure of the suppression of epithelial proliferation by P (33). However, Simmen et al did not elaborate on the P- and KLF9-regulated pathways that mediate this paracrine effect.
Over the past several years, it has become evident that steroid receptor function in uterine cells is modulated by distinct coregulator factors termed coactivators and corepressors (34, 35). Whereas a coactivator enhances the transcriptional function of the receptor, a corepressor generally represses its activity. Since the pregnant uterus progresses through physiological stages in which steroid receptor functions are alternately switched on or switched off in various tissue compartments, the modulatory roles of these proteins on receptor function are extremely significant. Indeed, recent gene knockout studies by O'Malley and DeMayo and colleagues (36, 37) revealed an essential modulatory role of the steroid receptor coactivator-2 (SRC-2) in PGR function during uterine decidualization. Katzenellenbogen and colleagues (38, 39) have identified a unique corepressor, repressor of estrogen receptor activity (REA), which is a specific regulator of estrogen receptor function. This group generated a uterus-specific knockout of REA and established that it plays a crucial role in modulating ESR1 activity and thereby controls uterine proliferation and differentiation at different stages of the implantation process.
Tissue Recombination Studies: Initial Evidence for Epithelial-Stromal Cross Talk
In a series of elegant studies, Cunha et al (40) used uterine tissue recombinants obtained from Esr1-null and Pgr-null mice to dissect the individual roles of epithelial and stromal steroid receptors in regulating uterine proliferation and differentiation. To examine the role of ESR1 in E-induced epithelial proliferation, tissue recombinants were prepared using epithelium and stroma from wild-type and Esr1-null mice (41). This study reported that the mitogenic effect of E on uterine epithelium is independent of epithelial ESR1 but is dependent on stromal ESR1. This surprising result suggested that uterine epithelial proliferation is a paracrine event dictated by E signaling originating in the stromal compartment. Using a similar approach with Pgr-null mice, Cunha and colleagues (42) demonstrated that stromal PGR is both necessary and sufficient to mediate the antiproliferative effects of P on E-induced epithelial cell proliferation. Collectively, the experiments of Cunha et al suggested that uterine epithelial cell proliferation is controlled by stromal ESR1 and its antagonism is mediated by stromal PGR, indicating that E or P acting on uterine stromal cells initiates paracrine mechanisms that modulate epithelial proliferation. However, because these studies used neonatal mouse tissue, questions were raised whether these results truly reflect adult uterine physiology. Nevertheless, these studies underscored, for the first time, the importance of epithelial-stromal communications in regulating key uterine functions.
Conditional Deletion of Esr1 and Pgr in the Uterus
The advent of the Cre-Lox strategy for generating tissue-specific gene deletion has allowed researchers to make great advances in understanding the cell type–specific roles of ESR1 and PGR in the adult mouse uterus. Recently, Korach and colleagues (43) addressed the epithelial cell–specific role of ESR1 by using the wingless-type MMTV integration site family (Wnt)7a-Cre mouse model, which allows ablation of the Esr1 gene specifically in luminal and glandular epithelia. They reported that the epithelial ESR1 does not control E-mediated epithelial cell proliferation, raising the possibility that stromal ESR1 actually regulates epithelial growth via paracrine mechanisms. This observation is in excellent agreement with the results of the tissue recombination experiments of Cunha and colleagues (41). Pollard and colleagues (44) previously showed that IGF-I is a paracrine effector of stromal origin that mediates E-driven uterine epithelial growth via IGF-I receptors located on the luminal and glandular epithelia. Korach and colleagues (45) showed further that ESR1 induces the expression of IGF-I in the stromal cells. It is pertinent to mention here that our recent findings also support a role of stroma-derived growth factors, although distinct from Igf-1, in mediating E-induced epithelial proliferation (46). It is conceivable that an interplay of multiple E-regulated growth factors of stromal origin controls uterine epithelial cell growth and proliferation.
Uterine compartment–specific conditional knockouts also proved valuable in delineating the role of PGR in epithelial-stromal cross talk. DeMayo and colleagues (47) generated and characterized a uterine epithelium-specific knockout of Pgr. These mice were infertile owing to defects in embryo attachment, stromal cell decidualization, and the inability to suppress E-induced epithelial cell proliferation. This study highlighted some of the mechanisms controlled by uterine epithelial PGR during early pregnancy. Loss of epithelial PGR led to the down-regulation of Indian hedgehog (IHH), a morphogen, whose production by the luminal epithelium is regulated by P (48). IHH acts via a paracrine mechanism to promote stromal cell decidualization. Surprisingly, this study also reported that epithelial PGR suppresses E-induced epithelial proliferation, although no mechanism has been proposed for this effect (47). This finding contradicts an earlier report by Cunha and colleagues (42) demonstrating that stromal PGR is both necessary and sufficient for P-mediated suppression of E-induced epithelial cell proliferation. This contradiction emphasizes the important point that, although the tissue recombinant approach is useful in identifying critical epithelial-stromal interactions, one still needs to verify these mechanisms in adult mice using tissue-specific knockouts.
Downstream Mediators of E and P Function During Implantation
Extensive use of gene expression profiling has uncovered many genes that are regulated by E and P during implantation (49–51). During the past several years, with the advent and continued refinement of gene knockout and knockin strategies, transgenic mice have become powerful tools for determining the functional roles of several of these E- and P-regulated pathways in various aspects of uterine physiology (52, 53). Studies using these animal models have established that these factors critically regulate uterine growth and differentiation, which in turn control embryo-endometrial interactions during early pregnancy. Here we provide a brief description of the physiological relevance of these steroid-regulated genes in the context of epithelial-stromal interactions during implantation and decidualization.
Steroid-Induced Epithelial-Stromal Cross Talk Involving Glandular Epithelium, Luminal Epithelium, and Stroma: Role of LIF, STAT3, and the EGF Family
Leukemia inhibitory factor (LIF) was discovered as an obligatory factor for implantation by Stewart et al (54) more than 20 years ago. It is a member of the IL-6 family and functions through the LIF receptor, which is associated with the signal transducer GP130 (55). At the time of implantation, LIF, produced by the endometrial glands, acts on the LIF receptors, which are primarily located in the luminal epithelium. In female mice lacking LIF, the embryos fail to attach to the luminal epithelium due to a defect in uterine receptivity (54). Accumulated evidence indicates that LIF plays a central role in the development of uterine receptivity by orchestrating a cross talk involving different uterine cell types: glandular epithelial, luminal epithelial, and stromal cells.
In rodent uterus, the expression of LIF is induced specifically in the glandular epithelium in response to a transient surge of E immediately before implantation (56). Our recent studies indicated that the conditional ablation of uterine epithelial Esr1 in mice leads to a loss of LIF production by the glands, confirming the E regulation of this factor (Pawar, S., I. C. Bagchi, and M. K. Bagchi, unpublished results). However, it is not clear whether ESR1 directly controls LIF expression in uterine epithelium. There is strong evidence that the tumor suppressor p53 directly regulates LIF expression in this tissue (57). It was also reported recently that ESR1 directly controls p53 gene transcription in breast cancer cells (58). Based on these findings, it is conceivable that ESR1 controls LIF expression in the uterine glands via an indirect mechanism in which p53 plays a mediator role.
During implantation, LIF, secreted from the glands, binds to its receptors in luminal epithelial cells and activates the Janus tyrosine kinase-Signal transducer and activator of transcription 3 (JAK-STAT3) pathway (59). To understand the molecular mechanisms via which the LIF-STAT3 signaling pathway controls uterine functions during implantation, Pawar et al (60) created a mutant mouse model in which Stat3 is specifically deleted in the uterine epithelium but is retained in the stroma. Using this mouse model, it was revealed that STAT3, upon activation after LIF signaling, alters the molecular organization of epithelial junctional complexes at the time of implantation. STAT3 suppresses the expression of the tight junction proteins claudin-1, -3, and -4 within the implantation window. It also down regulates the expression of α- and β-catenin, which probably contributes to the redistribution of E-cadherin away from the lateral adherens junctional complexes during implantation. The concerted down-regulation of the tight and adherens junction complexes in the luminal epithelium, driven by STAT3 signaling, critically alters the uterine epithelial phenotype by disrupting cell-cell linkages and triggering a loss of epithelial polarization. This presumably leads to proper organization and presentation of cell surface adhesion molecules that allow interaction with the blastocyst trophectoderm, initiating the process of implantation.
STAT3, acting downstream of LIF, also influences stromal function during implantation. Pawar et al (60) reported that the conditional loss of uterine epithelial STAT3 results in impaired decidualization. Specifically, it was noted that, in the absence of uterine epithelial STAT3, the stromal cells exhibit a significantly reduced capacity to undergo proliferation in response to a decidual stimulus. These results suggested a role of epithelial STAT3 in promoting production and/or secretion of a paracrine factor(s) from the uterine epithelium, which in turn induces stromal cell proliferation, as depicted in Figure 1. Indeed, a marked reduction in the levels of a subset of epidermal growth factor (EGF) family growth factors, including heparin-binding epidermal growth factor (Hb-egf), was observed in the Stat3-null uterine epithelium in the period after implantation, indicating that these growth factors are probably downstream paracrine effectors generated by epithelial STAT3 signaling. Consistent with this notion, a marked reduction in the level of active, phosphorylated EGF receptor was observed in the uterine stroma of these mutant mice. Furthermore, administration of exogenous EGF family factors was effective in rescuing the uterine stromal proliferation defect in mice lacking uterine epithelial STAT3. These findings are in agreement with the results reported previously by Dey and colleagues (61), indicating an important role of EGF-like factors downstream of LIF during implantation. Dey et al (62) have also shown that conditional ablation of HB-EGF expression in mouse uterine tissue markedly reduces the rate of implantation.
Figure 1.
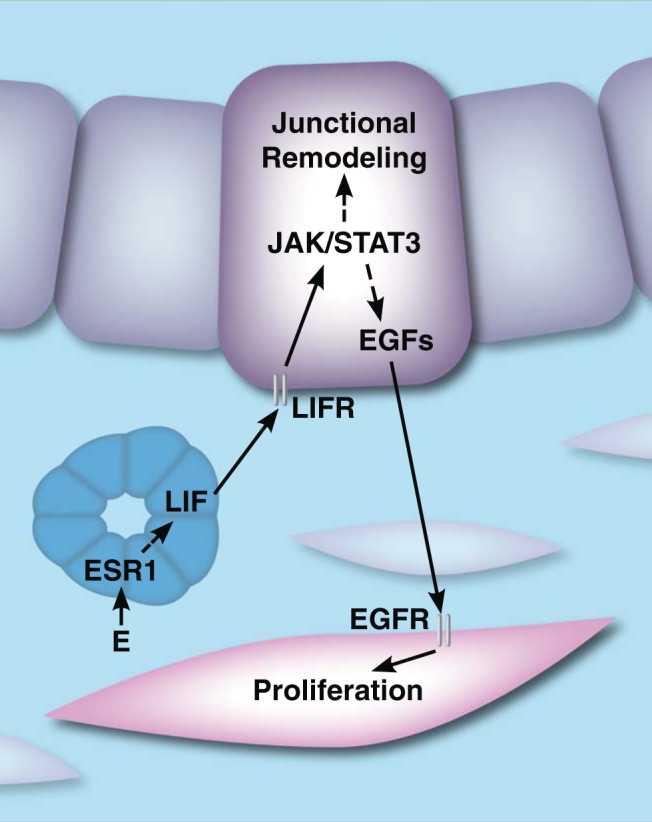
Steroid-mediated glandular epithelial-luminal epithelial-stromal cross talk in the uterus. E induces LIF in the glandular epithelium (blue), which acts in a paracrine manner on the luminal epithelium (purple) to activate the JAK/STAT pathway to mediate a shift in junctional integrity required for implantation. Activation of JAK/STAT also promotes proliferation of the stroma (pink) via expression and secretion of EGF family proteins from the luminal epithelium. Dashed lines represent indirect events. LIFR, LIF receptor; EGFR, EGF receptor.
Recent studies by Jeong and colleagues (63) and Dey and colleagues (64) have indicated a role for uterine stromal STAT3 in controlling the decidualization process. They used conditional mutant mice, developed by crossing floxed Stat3 mice with Pgr-Cre mice, which resulted in Stat3 gene deletion in both the epithelial and stromal compartments of the uterus. Loss of uterine STAT3 led to heightened E responsiveness of the luminal epithelium and decreased Pgr expression and P responsiveness in the stroma (63). Collectively, these studies provided valuable insights into the mechanisms by which LIF-STAT3 signaling allows transition of the uterine epithelium and stroma to proper functional states that permit embryo attachment, invasion, and subsequent decidualization of the underlying stroma.
Although the LIF-STAT3 pathway is essential for implantation in rodents, the role of LIF in human implantation is not as clearly established. In the human, LIF is abundantly expressed in the secretory phase of the menstrual cycle in both luminal and glandular epithelium (65). Immunohistochemical analysis of LIF expression in endometrial samples derived from women who have recurrent implantation failure after in vitro fertilization (IVF) showed reduced LIF in the glandular epithelium during the midsecretory phase compared with that in normal subjects (66). These results indicated that LIF is required for successful implantation in the human. Additional support for the role of LIF was provided by Kang et al (67), who studied polymorphisms in p53, which directly controls LIF expression. They discovered that a single nucleotide polymorphism in amino acid residue 72 of p53 is enriched in women having recurrent implantation failure. This genetic variation in p53 results in reduced LIF expression in cell cultures, presenting the possibility that it affects LIF levels in the endometrium and is therefore responsible for decreased uterine receptivity and implantation rates in these patients. However, conflicting reports also exist regarding the effectiveness of LIF in alleviating human infertility. One study measured serum LIF levels in women undergoing IVF treatment and reported that systemic levels of LIF have no association with the implantation rate or miscarriage rate (68). In addition, a clinical trial wherein recombinant LIF was administered subcutaneously to women with a history of 2 or more unexplained implantation failures after ART showed that LIF supplementation failed to increase pregnancy rates over those in the control group (69). It is important to note, however, that the route of LIF administration may have a profound effect on its efficacy, as the levels reaching the endometrium may be much lower than expected. This is a relevant concern because pharmacokinetic studies have shown rapid absorption and clearance of recombinant LIF after subcutaneous injection (70). A recent experiment used in vitro human endometrial constructs containing stroma and glandular epithelium obtained from uterine biopsy specimens to measure the human embryo attachment rate (71). After exposure to a LIF antagonist, attachment of the embryo to the construct and subsequent hatching were both decreased. These results support the view that a more direct route of administration may be necessary to elicit a functional effect of LIF in the human.
Evidence that STAT3 plays a critical role in the human uterus during pregnancy is also accumulating. Using primary cultures of human endometrial stromal cells (HESCs), Wang et al (72) demonstrated that the expression of STAT3 gene is directly regulated by C/EBPβ, a critical steroid-induced regulator of endometrial stromal proliferation and differentiation in mice and humans (50). Attenuation of STAT3 mRNA expression in HESCs resulted in markedly reduced differentiation of these cells, indicating an important role for STAT3 in human decidualization. Gene expression profiling, using STAT3-deficient HESCs, indicated that STAT3 regulates a variety of genes involved in inflammation mediated by chemokines/cytokines (eg, IL-11 and IL-11 receptorα), Wnt signaling (eg, WNT4), angiogenesis (eg, vascular endothelial growth factor [VEGF]-A and SPHK1 [sphingosine kinase 1]), and the TGFβ signaling pathway (eg, bone morphogenic protein 2 [BMP2]), supporting our view that it is a critical regulator of endometrial differentiation in women.
Steroid-Induced Paracrine Signals Originating in the Epithelium: Role of the IHH-COUP-TFII-BMP2-WNT4 Network
IHH represents a prototype hormone-regulated paracrine factor that links epithelial and stromal functions in the uterus. It is a member of the hedgehog family of morphogens and is induced by P in the uterus (73). Recent studies by DeMayo and colleagues (22), using ChIP-Seq, showed that PGR binds directly to the Ihh promoter. In the endometrium, IHH is expressed in the luminal and glandular epithelium before implantation and its expression decreases thereafter (48, 74). Its receptor patched-1 (PTCH-1) is also up-regulated in a similar temporal fashion in the luminal epithelium and stroma. In the human, IHH is expressed in both epithelial and stromal compartments during the secretory phase of the uterine cycle (75). To study IHH-mediated signaling in the uterus, Lee et al (73) created a conditionally deleted mutant of Ihh using Pgr-Cre mice. The resulting mutant females are infertile, exhibiting implantation defects and failure to respond to an artificial decidual stimulus. Defects in implantation were attributed to heightened ESR1 activity in mutant mice (76). Loss of IHH also resulted in reduced expression of PTCH-1 and the transcription factor chicken ovalbumin upstream promoter–transcription factor II (COUP-TFII, also known as NR2F2) in the subepithelial stroma (73). Taken together, these observations suggested that IHH is a major downstream factor mediating PGR signaling in the uterus. When secreted from the luminal epithelium, it influences stromal function by regulating COUP-TFII, as shown in Figure 2, and thereby mediates the communication between the uterine epithelium and stroma required for embryo implantation.
Figure 2.
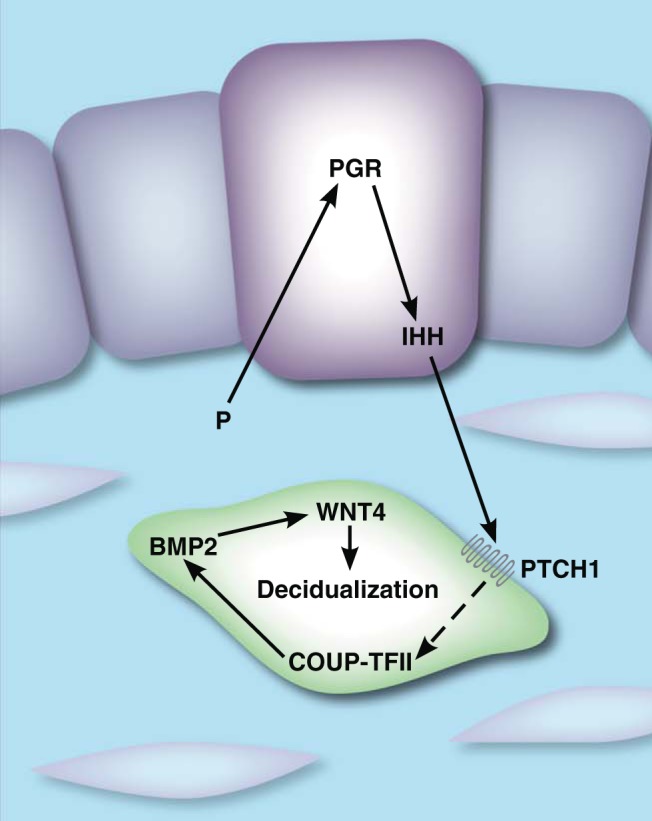
Steroid-mediated luminal epithelial-stromal cross talk in the uterus. P drives IHH expression in the luminal epithelium (purple). IHH acts on the stroma to promote decidualization through activation of COUP-TFII signaling. Decidualization results in morphologically distinct stroma (green) compared with undifferentiated stroma (pale pink). Dashed lines represent indirect events.
COUP-TFII, which is expressed almost exclusively in the uterine stroma, acts downstream of IHH in both mouse and human (73, 77). To assess its uterine function, a conditional COUP-TFII knockout mouse was generated using Pgr-Cre mice. Similar to Ihh-null mice, COUP-TFII–null females are infertile owing to implantation failure (78). It was demonstrated that COUP-TFII–null mice also exhibit enhanced E activity in the epithelium during the implantation window, which inhibits epithelial maturation and thus leads to failure of embryo attachment (79). It was reported that COUP-TFII regulates PGR expression in the uterine stroma (78). Stromal PGR, acting through paracrine mechanisms, down-regulates ESR1 activity in the luminal epithelium to promote a receptive uterus. Thus, suppression of stromal PGR in COUP-TFII–null mice may result in dysregulated E signaling in the epithelium.
The COUP-TFII–null mice also failed to undergo experimentally induced decidualization, a defect linked to decreased expression of BMP2, a factor induced in the uterine stroma in response to P stimulation (78). Previous studies by DeMayo and colleagues (80) and Bagchi and colleagues (81) have shown that BMP2 is an essential regulator of decidualization in mice and humans. Furthermore, these groups demonstrated that the morphogen WNT4 operates downstream of PGR and BMP2 to regulate decidualization of mouse and HESCs (81–83). In addition, COUP-TFII was shown to induce gene networks involved in inflammation, cell adhesion, and angiogenesis in human endometrial stromal decidualization (77). Collectively, these results indicate that P action in the epithelium initiates the IHH-COUP-TFII signaling pathway, which functionally links the epithelial and stromal compartments and then continues on via the PGR-BMP2-WNT4 pathway in the stroma, as indicated in Figure 2, forming an elaborate network of factors that play a central role in the regulation of implantation and decidualization.
Another noteworthy paracrine factor produced in the receptive rat uterus and cycling human endometrium within the implantation window is calcitonin (CT), a peptide hormone that controls calcium homeostasis. In pregnant rat uterus, CT is transiently induced in the glands in response to P at the time of implantation (84). Treatment of uterine epithelial cells with CT triggered a transient rise in intracellular Ca2+ and resulted in the remodeling of the adherens junctions between epithelial cells, indicating that this change in epithelial phenotype might be a critical event during the implantation of the blastocyst (85). Recent studies suggested that administration of exogenous CT promotes blastocyst implantation in mice by up-regulating the expression of integrin β3, LIF, and HB-EGF in endometrial epithelial cells (86). Intriguingly, only low levels of endogenous CT are present in the peri-implantation mouse uterus, compared with that in the rat uterus at the same stage, raising the point that a knockout mouse model may not be appropriate for the study of its in vivo role during implantation.
Role of Small Molecule Paracrine Signals in Steroid-Induced Decidualization
For many years, scientists were intrigued by the observation that endometrial stromal cells can be experimentally induced to differentiate in vivo by mechanically stimulating the luminal epithelium or injecting oil into the uterine lumen of a steroid-primed uterus (87). It was thought that physical perturbation of the uterine epithelium mimics embryo attachment and releases a diffusible signal(s) that acts on the stromal cells to trigger the downstream events leading to decidualization. Many laboratories investigated the nature of this signal(s), and an extensive body of literature supports the concept that cyclooxygenase-derived prostaglandins, produced at the epithelial-stromal interface, are important mediators of decidualization (88, 89). Recent studies revealed that the endometrial epithelial sodium (Na+) channels (ENaC), which are up-regulated in steroid-primed uterus during implantation, undergo activation by mechanical stimulation or embryo-derived proteases, leading to membrane depolarization and increased Ca2+ influx (90). This Ca2+ mobilization facilitates cyclooxygenase-2–dependent production of prostaglandins, including prostaglandin E2, in the epithelial cells. Prostaglandins are believed to act in a paracrine manner via the EP2 or EP4 receptors on stromal cells to promote decidualization. Although this elegant study described the sequence of events leading to the production of prostaglandins at the uterine epithelial-stromal interface in the mouse, further studies are needed to elaborate how the ovarian steroids regulate ENaC and whether ENaC and its downstream pathways are involved in the control of embryo implantation in the human.
Steroid-Induced Epithelial-Stromal Cross Talk Originating in the Stroma: Regulation by HAND2 and FGFs
Although the tissue recombination studies of Cunha and colleagues (42) indicated that stromal PGR plays a pivotal role in mediating the antiproliferative effects of P on the uterine epithelium, the molecular mechanisms by which P achieves this regulation remained unclear until recently. Microarray-based profiling of P-responsive transcripts during the implantation window in the mouse identified the basic helix-loop-helix transcription factor heart and neural crest derivatives expressed protein 2 (Hand2) as a P-regulated gene in the uterine stroma (46, 91). In cultured HESCs, HAND2 is induced by P treatment and inhibited by cotreatment with the PGR antagonist RU486 (92). HAND2 is robustly expressed in mouse uterine stroma by day 3 of gestation but is notably absent in the epithelium. Its stromal expression persists through the peri-implantation phase and is detectable until day 8.5 of pregnancy (93). Recent mouse genome-wide PGR binding studies indicated that Hand2 is a direct target of regulation by this receptor (22).
Studies by Li et al (46) and Hewitt and Korach (94) revealed that HAND2 plays a central role in controlling the paracrine mechanisms that mediate the antiproliferative effects of P in the endometrium. Deletion of Hand2 in the mouse uterus, using Pgr-Cre, resulted in infertility due to failed implantation. Hand2-null mice showed persistent luminal epithelial proliferation on day 4 of pregnancy, indicating that, in the absence of HAND2, pregnant uteri fail to achieve a receptive state. HAND2 was shown to regulate the expression of a subset of stromal fibroblast growth factors (FGFs): FGF1, FGF2, FGF9, and FGF18. Unopposed, stromal FGFs, presumably induced by E via stromal ESR1, act in a paracrine fashion to target receptors on the luminal epithelium and mediate epithelial proliferation through extracellular-signal-related kinase 1/2 (ERK1/2) activation. HAND2 mediates the antiproliferative effects of P by inhibiting the production of FGFs and thus terminating E-driven epithelial proliferation as illustrated in Figure 3. In addition, phosphorylation and activation of epithelial ESR1 was also shown to be persistent in Hand2-null mice. Taken together, these findings show that loss of HAND2 removes the brake by which the uterus shifts from a proliferative state to a differentiated state and creates a tissue unreceptive to the blastocyst. This study provides a plausible mechanism by which epithelial proliferation is controlled in a paracrine manner by signaling molecules produced downstream of steroid action in the stroma.
Figure 3.
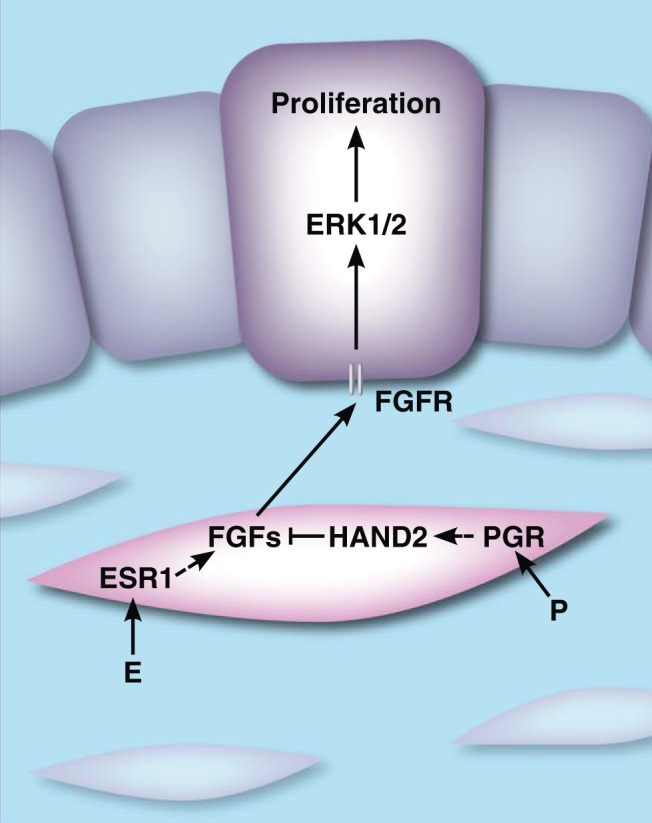
Steroid-mediated epithelial-stromal cross talk in the uterus. Before implantation, E, acting through ESR1 in the stroma (pink), drives epithelial (purple) proliferation through the secretion of paracrine acting FGFs. During the periimplantation period, P, acting on PGR in the stroma, promotes the expression of HAND2. HAND2 inhibits expression of FGFs and blocks E-induced epithelial proliferation. Dashed lines represent indirect events. FGFR, FGF receptor.
Apart from HAND2, two other uterine factors, muscle segment homeobox (MSX) 1 and MSX2, which are only modestly influenced by the ovarian steroids, regulate epithelial proliferation and differentiation by controlling epithelial-stromal cross talk (95, 96). Conditional ablation of both Msx1 and Msx2 in the uterus resulted in female infertility due to a failure in implantation. Nallasamy et al (95) showed that, in the absence of MSX1 and MSX2, increased canonical WNT signaling in the stromal cells activated β-catenin, stimulating the production of FGF1, FGF10, FGF18, and FGF21 in these cells. It was proposed that the FGFs act in a paracrine manner via the FGF receptors in the epithelium to promote epithelial proliferation, thereby preventing differentiation of this tissue and creating a nonreceptive uterus refractory to implantation. Interestingly, it appears that MSX1/MSX2 functions via a mechanism very similar to that operating downstream of HAND2, although it is not clear how these pathways converge, if at all. Daikoku et al (96) proposed a somewhat different mechanism for the role of MSX1/MSX2 in implantation. They reported that the loss of uterine expression of these factors correlates with altered E-cadherin/β-catenin complex formation in the luminal epithelium in response to elevated WNT5A expression in the endometrium. This study also indicated that the MSX factors directly control Wnt5a gene transcription. Clearly, more work needs to be done to determine how HAND2 and MSX1/MSX2 interact with the WNT and FGF signaling pathways to control stromal-epithelial interactions during implantation.
Interestingly, Pollard and colleagues (97, 98) have implicated a third set of factors, KLF4 and KLF15, in the control of uterine epithelial proliferation. According to this group, these factors act in a reciprocal fashion in the epithelium to induce or suppress E-induced uterine epithelial proliferation. E, acting via KLF4, stimulates the expression of the mini-chromosome maintenance (MCM) proteins 2 to 7 in the uterine epithelium. The MCMs play a critical role in the initiation of DNA replication. Interestingly, KLF4 is reported to bind directly to the CCAAT/enhancer-binding protein β (C/ebpβ) promoter to induce its expression in some tissues (99). If this mechanism of activation by KLF4 holds in the uterine epithelium, it will be consistent with our finding that C/EBPβ mediates E-induced proliferation of the epithelial cells (13). Pollard and Ray (98) further showed that administration of P promotes the expression of KLF15, which occupies the Mcm2 promoter and inhibits its E-mediated transcription, resulting in the blockade of epithelial proliferation. These results suggest that the antiproliferative effects of P can be exerted without the involvement of a paracrine mechanism originating in the stroma and therefore argue in favor of a cell autonomous mechanism regulating uterine epithelial proliferation. However, one should note that this study was performed entirely in nonpregnant mice treated exogenously with E or E plus P. It will be important to generate targeted genetic deletions of individual KLF genes to verify their functions in fertility regulation. This would help us understand whether the proposed cell autonomous mechanism has relevance in the physiological context of implantation in which several paracrine mechanisms, such as HAND2-FGF and IGF-I, are reported to mediate the actions of the ovarian steroids.
Dysfunction of Epithelial-Stromal Cross Talk Leads to Reproductive Diseases
During the past several years, the use of conditional knockout mice has allowed us to make great strides in identifying key molecular players important for the implantation process. Many of these factors, of both stromal and epithelial origin, have been well characterized in vitro and in vivo. An emerging concept is that dysregulation of these factors not only affects fertility but is also linked to other reproductive pathological conditions, such as endometriosis and endometrial cancer. Endometriosis, a chronic disease characterized by endometrial epithelium and stroma implanting outside of the uterus, is often associated with inflammation, abdominal pain, and infertility (100, 101). Because endometriotic tissue exhibits deregulation in differentiation gene networks associated with PGR signaling, endometriosis is often considered a P-resistant disease, although the mechanism is not fully understood (102). In addition, several lines of evidence link endometriosis with elevated expression of P450 aromatase and a consequent increase in E signaling in the endometriotic tissue (103).
Bulun et al (103) have shown that specific steroid hormone receptor isoforms are differentially regulated in stromal cells isolated from normal and endometriotic tissue. For example, the PGR-B isoform, but not PGR-A, is down-regulated in endometriotic tissue (104). It was proposed that the stromal dysregulation of PGR contributes to endometriosis through the down-regulation of 17β-hydroxysteroid dehydrogenase type 2, an enzyme that metabolizes E into estrone, a weakly estrogenic metabolite (105). This impaired E metabolism, coupled with the abnormal high expression of P450 aromatase that is seen in endometriotic tissue, leads to excessive local E signaling, which presumably drives the growth of endometriotic lesions. Additional studies have linked impaired expression of other key P-regulated pathways in the stroma to the pathogenesis of endometriosis (106–108). Bulun et al (109) have also reported that the level of estrogen receptor β (ESR2) expression is significantly greater in endometriotic stroma vs normal stroma. They postulated that the increased ESR2 levels observed in endometriosis are a consequence of hypomethylation at a CpG island within the Esr2 promoter, and that ESR2 overexpression leads to the observed suppression of ESR1 and PGR. Taken together, these reports support the concept that aberrant steroid receptor expression in endometriosis, driven in part by epigenetic alterations, results in dysregulation of paracrine signaling between the stroma and the epithelium, leading to the abnormal epithelial proliferation and stromal differentiation observed in this disease. Identification of these steroid-regulated factors would be of importance for understanding the molecular basis of endometriosis.
Recent studies revealed that impaired stromal-epithelial interactions also contribute to endometrial hyperplasia, a precursor to endometrial cancer, which is associated with increased E signaling and decreased P signaling (110). Treatment regimens for this condition often include progestin therapy to inhibit E-mediated proliferation. Remarkably, Jones et al (111) identified HAND2 as the most commonly hypermethylated and silenced gene in endometrial hyperplasia and cancer (111). The degree of HAND2 methylation was positively correlated with the severity of hyperplasia. It was further shown that uterine samples from patients who did not respond to progestin treatment exhibited greater HAND2 methylation than positive responders. Morphological changes similar to complex atypical hyperplasia, including an increased gland/stroma ratio and irregularities in glandular shape compared with those for age-matched control mice, were also observed in uteri of aged Hand2-null mice (Figure 4). Loss of Hand2 also results in down-regulation of the tumor suppressor Pten, an event that often occurs early during endometrial carcinogenesis (111). Collectively, these results indicated that stromal epigenetic modifications of key target genes, such as HAND2, have a profound effect on the protective effects of P on the epithelium and can now be considered to be predictors of progestin responsiveness in endometrial cancer treatment regimens. This novel and important insight, along with future studies, will help develop early detection methods and new therapeutic approaches in treating endometrial cancer.
Figure 4.

Epigenetic silencing of Hand2 through hormonal dysregulation or environmental insults leads to signs of complex atypical uterine hyperplasia.
Unresolved Questions and Future Challenges
This review has highlighted several key steroid-regulated pathways that play important roles in mediating the epithelial-stromal cross talk that controls early pregnancy. Tissue-specific gene knockout models combined with gene expression profiling and powerful bioinformatic approaches have helped us understand how these pathways act in different uterine compartments to synchronize the critical events leading to implantation. Despite these advancements, there are many unanswered questions in the field of implantation biology that need to be addressed to complete our understanding of the mechanisms underlying successful establishment of pregnancy. We list below a few important and unresolved issues that need to be tackled in the future.
A precise understanding of the luminal epithelial factors that mediate direct interactions of this tissue with the embryo is still lacking. Although several candidate cell adhesion molecules, such as αvβ3 integrin and its ligand osteopontin, are reported to be involved in this process (112), a stringent assessment of their roles, using uterine epithelium-specific gene knockout models, is necessary. Similarly, it is critical to identify the factors that control luminal epithelial ion transport, influencing uterine fluid absorption and luminal closure, thereby having an impact on the interactions of the epithelium with the embryo during the peri-implantation period. Previous studies indicated that the ENaC, which regulates epithelial Na+ transport, facilitates uterine receptivity in mice (113). Brosens and colleagues (114) have recently reported that up-regulation of serum- and glucocorticoid-inducible kinase 1 (SGK1) in uterine epithelium, which promotes the expression of ENaCs, is linked to unexplained infertility. These apparently contradictory results indicate that the relationships among these factors are complex. It is possible that the primary function of SGK1 in implantation is not via direct regulation of ENaCs but may involve other aspects of uterine epithelial function, such as regulation of cell proliferation, during establishment of pregnancy. Further experiments are needed to explore these issues.
For implantation to occur successfully, highly coordinated development of the embryo and the maternal tissue is absolutely critical. It is thought that this is achieved by a timely exchange of signals between the growing embryo and the uterus. There is, however, very little understanding of the signals that emanate from the steroid-primed maternal tissue to influence the transformation of the trophectoderm during embryo development. The notion that such factors are critical is supported by the phenomenon of diapause or delayed implantation in which embryonic development beyond the blastocyst stage is arrested until a surge of the maternal steroid hormone E induces embryo attachment to the receptive uterus (115, 116). A recent report by Lee et al (117) suggests that during delayed implantation blastocysts exhibit increased autophagy. Presumably, administration of E, which ends embryonic dormancy and reduces autophagy, induces maternal factors that act directly on the trophoblast. The identity of these factors remains unknown. There is also a paucity of knowledge regarding the nature of embryonic factors that act on the uterine cells to directly influence implantation in the mouse and the human. However, in ruminants and pigs, a large body of work established that the embryo-derived interferon tau is a critical paracrine signal for pregnancy. It functions as a pregnancy recognition signal and acts on the uterine epithelium to block the luteolytic pulsatile release of prostaglandin F2α (118, 119). Additional evidence of an embryonic factor supporting implantation came from Fazleabas et al (120), who used a nonhuman primate model to present evidence that trophoblast-derived chorionic gonadotropin may act directly on the maternal tissue to regulate uterine receptivity.
Besides the epithelial-stromal cross talk, other modes of paracrine interactions within the endometrium are also critical for the establishment and maintenance of pregnancy. There is increasing evidence that paracrine factors secreted by the stromal cells act directly on the endothelial cells to influence the establishment of an elaborate uterine angiogenic network that supports embryonic growth and survival. For example, Laws et al (121) demonstrated that gap junction communication between adjacent mouse or human endometrial stromal cells is mediated by connexin-43 and that it is critical for regulation of signaling events that promote local production of VEGF, which in turn helps expand the endothelial cell population in the uterine stroma. Endometrial stromal cells are also known to secrete cytokines and chemokines that control the recruitment of immune cells, such as the uterine natural killer cells and regulatory T cells, to the implantation sites (122, 123). Interestingly, our recent gene expression profiling data indicate that STAT3 may control the immune response in human endometrial stroma by regulating inflammation-related gene pathways during decidualization (72). To gain further insights into the stromal paracrine mechanisms that promote uterine angiogenesis and suppress maternal immune response to the embryo, it is important to create uterine stromal cell–specific gene knockouts that can be induced at progressive phases of the implantation process. No such model currently exists.
It is increasingly becoming clear that the metabolic status of the uterine cells controls the implantation process. Previous studies have suggested a role of amino acids and glucose provided by the maternal tissue in regulating the onset of trophoblast differentiation (124). It has been reported by Bazer et al (125) that in ruminants and gilts, addition of leucine, arginine, and glutamine, as constituents of uterine secretions or histotrophs, promotes proliferation and motility of cultured trophoblast cells. Whereas these studies collectively suggest an important role played by maternal nutrients in the regulation of trophoblast function, much remains to be learned about the mechanisms by which the uterine cells interact with each other in vivo to provide the nutrients that regulate implantation. Studies by Moley and colleagues (126) showed that uptake of glucose via glucose transporter 1 and its metabolism via the pentose phosphate pathway are important for decidualization of mouse and human endometrial stromal cells, presumably to meet the energy demands to support their high rate of proliferation before differentiation (126). Recent studies by O'Malley and colleagues (127) revealed that SRC-2 plays an obligatory role during decidualization by increasing the glycolytic flux in the uterine stromal cells. However, the impact of these stromal metabolic events on epithelial function and on embryo development, particularly under adaptive conditions, such as hypoxia, remain unknown.
Although in vivo genetic models have provided invaluable information regarding many factors that play important roles during implantation, there is still a critical need for development of effective in vitro models for further analysis of the mechanisms involved. Although some success has been achieved in this direction (128–130), continued efforts are needed to develop 3-dimensional primary cultures and blastocyst-endometrium coculture systems to study embryo attachment and invasion processes. These systems would provide an easier avenue for performing proteomic and metabolomic analyses to assess the functional roles of paracrine factors that mediate various stages of the embryo-uterine dialogue.
Concerted use of genome-based analyses, such as ChIP-Seq, microarray-based mRNA profiling and RNA-sequencing, and proteomic and metabolomics approaches, using endometrial specimens from the human, mouse, and other species, will be necessary to build a comprehensive map of pathways that control implantation and to decipher their mechanisms of action. Knowledge of the precise sequence of interactions and the hierarchical arrangement and convergence of different pathways will emerge from these studies. It is encouraging that the data generated from genome-based studies are already being used to construct customized microarrays to identify the window of endometrial receptivity in the human (131). Continued fine tuning and testing of these arrays is necessary to validate the clinical usefulness of such gene signatures in predicting uterine receptivity and diagnosis of endometrial dysfunctions associated with infertility.
It is also crucial that we continue to gather evidence for the genetic and epigenetic basis of endometrial dysfunctions leading to human infertility. Recent studies by Kang et al (67) provided strong support for the hypothesis that genetic variations at certain genomic loci may have an impact on uterine functions during implantation in the human. It was further observed that selected alleles of single nucleotide polymorphisms in p53, LIF, and other components of the p53 pathway are enriched in patients undergoing IVF, and some of these variants are linked to recurrent implantation failure (67, 132). There is increasing documentation of dynamic epigenetic alterations, such as DNA methylation and histone acetylation or methylation, that occur in the endometrial cells at specific gene loci (133, 134). There is also emerging evidence that certain of these epigenetic changes, accentuated by in vivo hormonal imbalance and exposure to environmental agents, disrupt normal uterine functions, leading to infertility, various reproductive disorders, and cancer.
Acknowledgments
We thank Jason Neff for creating the figures.
S.P., I.C.B., and M.K.B. are supported by the Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health (Cooperative Agreement Grant U54 HD055787 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research and Grant R21 HD078983). A.M.H. is supported by National Institutes of Health Predoctoral Traineeship in Endocrine, Developmental and Reproductive Toxicology T32ES007326.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ART
- assisted reproductive technologies
- BMP2
- bone morphogenic protein 2
- C/EBPβ
- CCAAT/enhancer-binding protein β
- ChIP-Seq
- chromatin immunoprecipitation coupled to high throughput sequencing
- COUP-TFII
- chicken ovalbumin upstream promoter–transcription factor II
- CT
- calcitonin
- E
- 17β-estradiol
- EGF
- epidermal growth factor
- ENaC
- endometrial epithelial sodium (Na+) channel
- ESR1
- estrogen receptor α
- FGF
- fibroblast growth factor
- HAND2
- heart and neural crest derivatives expressed protein 2
- HB-EGF
- heparin-binding epidermal growth factor
- HESC
- human endometrial stromal cell
- IHH
- Indian hedgehog
- IVF
- in vitro fertilization
- JAK
- Janus tyrosine kinase
- KLF
- Krüppel-like factor
- LIF
- leukemia inhibitory factor
- MCM
- mini-chromosome maintenance
- MSX
- muscle segment homeobox
- P
- progesterone
- PGR
- progesterone receptor
- PTCH-1
- patched-1
- REA
- repressor of estrogen receptor activity
- SGK1
- serum- and glucocorticoid-inducible kinase 1
- SRC-2
- steroid receptor coactivator-2
- STAT3
- signal transducer and activator of transcription 3
- VEGF
- vascular endothelial growth factor
- WNT
- wingless-type MMTV integration site family.
References
- 1. Bazer FW, Spencer TE, Johnson GA, Burghardt RC, Wu G. Comparative aspects of implantation. Reproduction. 2009;138:195–209 [DOI] [PubMed] [Google Scholar]
- 2. Cha J, Sun X, Dey SK. Mechanisms of implantation: strategies for successful pregnancy. Nat Med. 2012;18:1754–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vasquez YM, DeMayo FJ. Role of nuclear receptors in blastocyst implantation. Semin Cell Dev Biol. 2013;24:724–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Paria BC, Huet-Hudson YM, Dey SK. Blastocyst's state of activity determines the “window” of implantation in the receptive mouse uterus. Proc Natl Acad Sci USA. 1993;90:10159–10162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Finn C. The implantation reaction. In: Wynn R, ed. Biology of the Uterus: New York, NY: Springer US; 1977:245–308 [Google Scholar]
- 6. Ramathal CY, Bagchi IC, Taylor RN, Bagchi MK. Endometrial decidualization: of mice and men. Semin Reprod Med. 2010;28:17–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chang K, Lubo Z. Review article: steroid hormones and uterine vascular adaptation to pregnancy. Reprod Sci. 2008;15:336–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Douglas NC, Tang H, Gomez R, et al. Vascular endothelial growth factor receptor 2 (VEGFR-2) functions to promote uterine decidual angiogenesis during early pregnancy in the mouse. Endocrinology. 2009;150:3845–3854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sharkey AM, Smith SK. The endometrium as a cause of implantation failure. Best Pract Res Clin Obsteet Gynecol. 2003;17:289–307 [DOI] [PubMed] [Google Scholar]
- 10. Greenhouse S, Rankin T, Dean J. Genetic causes of female infertility: targeted mutagenesis in mice. Am J Hum Genet. 1998;62:1282–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carson DD, Bagchi I, Dey SK, et al. Embryo implantation. Dev Biol. 2000;223:217–237 [DOI] [PubMed] [Google Scholar]
- 12. Dey SK, Lim H, Das SK, et al. Molecular cues to implantation. Endocr Rev. 2004;25:341–373 [DOI] [PubMed] [Google Scholar]
- 13. Ramathal C, Bagchi IC, Bagchi MK. Lack of CCAAT enhancer binding protein β (C/EBPβ) in uterine epithelial cells impairs estrogen-induced DNA replication, induces DNA damage response pathways, and promotes apoptosis. Mol Cell Biol. 2010;30:1607–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thie M, Fuchs P, Denker HW. Epithelial cell polarity and embryo implantation in mammals. Int J Dev Biol. 1996;40:389–393 [PubMed] [Google Scholar]
- 15. Paria BC, Zhao X, Das SK, Dey SK, Yoshinaga K. Zonula occludens-1 and E-cadherin are coordinately expressed in the mouse uterus with the initiation of implantation and decidualization. Dev Biol. 1999;208:488–501 [DOI] [PubMed] [Google Scholar]
- 16. Singh H, Aplin JD. Adhesion molecules in endometrial epithelium: tissue integrity and embryo implantation. J Anat. 2009;215:3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simon L, Spiewak KA, Ekman GC, et al. Stromal progesterone receptors mediate induction of Indian hedgehog (IHH) in uterine epithelium and its downstream targets in uterine stroma. Endocrinology. 2009;150:3871–3876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rubel CA, Jeong JW, Tsai SY, Lydon JP, Demayo FJ. Epithelial-stromal interaction and progesterone receptors in the mouse uterus. Semin Reprod Med. 2010;28:27–35 [DOI] [PubMed] [Google Scholar]
- 19. Wetendorf M, DeMayo FJ. The progesterone receptor regulates implantation, decidualization, and glandular development via a complex paracrine signaling network. Mol Cell Endocrinol. 2012;357:108–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tsai MJ, O'Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem. 1994;63:451–486 [DOI] [PubMed] [Google Scholar]
- 21. Hewitt SC, Li L, Grimm SA, et al. Research resource: Whole-genome estrogen receptor α binding in mouse uterine tissue revealed by ChIP-seq. Mol Endocrinol. 2012;26:887–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rubel CA, Lanz RB, Kommagani R, Franco HL, Lydon JP, DeMayo FJ. Research resource: Genome-wide profiling of progesterone receptor binding in the mouse uterus. Mol Endocrinol. 2012;26:1428–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pan YF, Wansa KD, Liu MH, et al. Regulation of estrogen receptor-mediated long range transcription via evolutionarily conserved distal response elements. J Biol Chem. 2008;283:32977–32988 [DOI] [PubMed] [Google Scholar]
- 24. Li W, Notani D, Ma Q, et al. Functional roles of enhancer RNAs for oestrogen-dependent transcriptional activation. Nature. 2013;498:516–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Spencer TE, Bazer FW. Biology of progesterone action during pregnancy recognition and maintenance of pregnancy. Front Biosci. 2002;7:d1879–d1898 [DOI] [PubMed] [Google Scholar]
- 26. Critchley HO, Saunders PT. Hormone receptor dynamics in a receptive human endometrium. Reprod Sci. 2009;16:191–199 [DOI] [PubMed] [Google Scholar]
- 27. Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? Endocr Rev. 1999;20:358–417 [DOI] [PubMed] [Google Scholar]
- 28. Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development. 2000;127:4277–4291 [DOI] [PubMed] [Google Scholar]
- 29. Lydon JP, DeMayo FJ, Funk CR, et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev. 1995;9:2266–2278 [DOI] [PubMed] [Google Scholar]
- 30. Tranguch S, Wang H, Daikoku T, Xie H, Smith DF, Dey SK. FKBP52 deficiency-conferred uterine progesterone resistance is genetic background and pregnancy stage specific. J Clin Invest. 2007;117:1824–1834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hirota Y, Acar N, Tranguch S, et al. Uterine FK506-binding protein 52 (FKBP52)-peroxiredoxin-6 (PRDX6) signaling protects pregnancy from overt oxidative stress. Proc Natl Acad Sci USA. 2010;107:15577–15582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang D, Zhang XL, Michel FJ, Blum JL, Simmen FA, Simmen RC. Direct interaction of the Krüppel-like family (KLF) member, BTEB1, and PR mediates progesterone-responsive gene expression in endometrial epithelial cells. Endocrinology. 2002;143:62–73 [DOI] [PubMed] [Google Scholar]
- 33. Velarde MC, Geng Y, Eason RR, Simmen FA, Simmen RC. Null mutation of Kruppel-like factor9/basic transcription element binding protein-1 alters peri-implantation uterine development in mice. Biol Reprod. 2005;73:472–481 [DOI] [PubMed] [Google Scholar]
- 34. Collingwood TN, Urnov FD, Wolffe AP. Nuclear receptors: coactivators, corepressors and chromatin remodeling in the control of transcription. J Mol Endocrinol. 1999;23:255–275 [DOI] [PubMed] [Google Scholar]
- 35. McKenna NJ, Lanz RB, O'Malley BW. Nuclear receptor coregulators: cellular and molecular biology. Endocr Rev. 1999;20:321–344 [DOI] [PubMed] [Google Scholar]
- 36. Mukherjee A, Soyal SM, Fernandez-Valdivia R, et al. Steroid receptor coactivator 2 is critical for progesterone-dependent uterine function and mammary morphogenesis in the mouse. Mol Cell Biol. 2006;26:6571–6583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jeong JW, Lee KY, Han SJ, et al. The p160 steroid receptor coactivator 2, SRC-2, regulates murine endometrial function and regulates progesterone-independent and -dependent gene expression. Endocrinology. 2007;148:4238–4250 [DOI] [PubMed] [Google Scholar]
- 38. Park S, Yoon S, Zhao Y, et al. Uterine development and fertility are dependent on gene dosage of the nuclear receptor coregulator REA. Endocrinology. 2012;153:3982–3994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao Y, Park S, Bagchi MK, Taylor RN, Katzenellenbogen BS. The coregulator, repressor of estrogen receptor activity (REA), is a crucial regulator of the timing and magnitude of uterine decidualization. Endocrinology. 2013;154:1349–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cunha GR, Cooke PS, Kurita T. Role of stromal-epithelial interactions in hormonal responses. Arch Histol Cytol. 2004;67:417–434 [DOI] [PubMed] [Google Scholar]
- 41. Cooke PS, Buchanan DL, Young P, et al. Stromal estrogen receptors mediate mitogenic effects of estradiol on uterine epithelium. Proc Natl Acad Sci USA. 1997;94:6535–6540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kurita T, Young P, Brody JR, Lydon JP, O'Malley BW, Cunha GR. Stromal progesterone receptors mediate the inhibitory effects of progesterone on estrogen-induced uterine epithelial cell deoxyribonucleic acid synthesis. Endocrinology. 1998;139:4708–4713 [DOI] [PubMed] [Google Scholar]
- 43. Winuthayanon W, Hewitt SC, Orvis GD, Behringer RR, Korach KS. Uterine epithelial estrogen receptor α is dispensable for proliferation but essential for complete biological and biochemical responses. Proc Natl Acad Sci USA. 2010;107:19272–19277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu L, Pollard JW. Estradiol-17β regulates mouse uterine epithelial cell proliferation through insulin-like growth factor 1 signaling. Proc Natl Acad Sci USA. 2007;104:15847–15851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of Igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem. 2010;285:2676–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li Q, Kannan A, DeMayo FJ, et al. The antiproliferative action of progesterone in uterine epithelium is mediated by Hand2. Science 2011;331:912–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Franco HL, Rubel CA, Large MJ, et al. Epithelial progesterone receptor exhibits pleiotropic roles in uterine development and function. FASEB J. 2012;26:1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Takamoto N, Zhao B, Tsai SY, DeMayo FJ. Identification of Indian hedgehog as a progesterone-responsive gene in the murine uterus. Mol Endocrinol. 2002;16:2338–2348 [DOI] [PubMed] [Google Scholar]
- 49. Cheon YP, Li Q, Xu X, DeMayo FJ, Bagchi IC, Bagchi MK. A genomic approach to identify novel progesterone receptor regulated pathways in the uterus during implantation. Mol Endocrinol. 2002;16:2853–2871 [DOI] [PubMed] [Google Scholar]
- 50. Mantena SR, Kannan A, Cheon YP, et al. C/EBPβ is a critical mediator of steroid hormone-regulated cell proliferation and differentiation in the uterine epithelium and stroma. Proc Natl Acad Sci USA. 2006;103:1870–1875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Reese J, Das SK, Paria BC, et al. Global gene expression analysis to identify molecular markers of uterine receptivity and embryo implantation. J Biol Chem. 2001;276:44137–44145 [DOI] [PubMed] [Google Scholar]
- 52. Korach KS. Estrogen receptor knock-out mice: molecular and endocrine phenotypes. J Soc Gynecol Investig. 2000;7(1 suppl):S16–S17 [DOI] [PubMed] [Google Scholar]
- 53. Mulac-Jericevic B, Conneely OM. Reproductive tissue-selective actions of progesterone receptors. Ernst Schering Res Found Workshop. 2005;51:19–37 [DOI] [PubMed] [Google Scholar]
- 54. Stewart CL, Kaspar P, Brunet LJ, et al. Blastocyst implantation depends on maternal expression of leukaemia inhibitory factor. Nature. 1992;359:76–79 [DOI] [PubMed] [Google Scholar]
- 55. Fujio Y, Maeda M, Mohri T, et al. Glycoprotein 130 cytokine signal as a therapeutic target against cardiovascular diseases. J Pharmacol Sci. 2011;117:213–222 [DOI] [PubMed] [Google Scholar]
- 56. Yang ZM, Le SP, Chen DB, et al. Leukemia inhibitory factor, LIF receptor, and gp130 in the mouse uterus during early pregnancy. Mol Reprod Dev. 1995;42:407–414 [DOI] [PubMed] [Google Scholar]
- 57. Hu W, Feng Z, Teresky AK, Levine AJ. p53 regulates maternal reproduction through LIF. Nature. 2007;450:721–724 [DOI] [PubMed] [Google Scholar]
- 58. Berger CE, Qian Y, Liu G, Chen H, Chen X. p53, a target of estrogen receptor (ER) α, modulates DNA damage-induced growth suppression in ER-positive breast cancer cells. J Biol Chem. 2012;287:30117–30127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cheng JG, Chen JR, Hernandez L, Alvord WG, Stewart CL. Dual control of LIF expression and LIF receptor function regulate Stat3 activation at the onset of uterine receptivity and embryo implantation. Proc Natl Acad Sci USA. 2001;98:8680–8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pawar S, Starosvetsky E, Orvis GD, Behringer RR, Bagchi IC, Bagchi MK. STAT3 regulates uterine epithelial remodeling and epithelial-stromal crosstalk during implantation. Mol Endocrinol. 2013;27:1996–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Song H, Lim H, Das SK, Paria BC, Dey SK. Dysregulation of EGF family of growth factors and COX-2 in the uterus during the preattachment and attachment reactions of the blastocyst with the luminal epithelium correlates with implantation failure in LIF-deficient mice. Mol Endocrinol. 2000;14:1147–1161 [DOI] [PubMed] [Google Scholar]
- 62. Xie H, Wang H, Tranguch S, et al. Maternal heparin-binding-EGF deficiency limits pregnancy success in mice. Proc Natl Acad Sci USA. 2007;104:18315–18320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee JH, Kim TH, Oh SJ, et al. Signal transducer and activator of transcription-3 (Stat3) plays a critical role in implantation via progesterone receptor in uterus. FASEB J. 2013;27:2553–2563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun X, Bartos A, Whitsett JA, Dey SK. Uterine deletion of Gp130 or Stat3 shows implantation failure with increased estrogenic responses. Mol Endocrinol. 2013;27:1492–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cullinan EB, Abbondanzo SJ, Anderson PS, Pollard JW, Lessey BA, Stewart CL. Leukemia inhibitory factor (LIF) and LIF receptor expression in human endometrium suggests a potential autocrine/paracrine function in regulating embryo implantation. Proc Natl Acad Sci USA. 1996;93:3115–3120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mariee N, Li TC, Laird SM. Expression of leukaemia inhibitory factor and interleukin 15 in endometrium of women with recurrent implantation failure after IVF; correlation with the number of endometrial natural killer cells. Hum Reprod. 2012;27:1946–1954 [DOI] [PubMed] [Google Scholar]
- 67. Kang HJ, Feng Z, Sun Y, et al. Single-nucleotide polymorphisms in the p53 pathway regulate fertility in humans. Proc Natl Acad Sci USA. 2009;106:9761–9766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Thum MY, Abdalla HI, Bhaskaran S, et al. The effect of serum concentration of leukaemia inhibitory factor on in vitro fertilization treatment outcome. Am J Reprod Immunol. 2006;55:76–80 [DOI] [PubMed] [Google Scholar]
- 69. Brinsden PR, Alam V, de Moustier B, Engrand P. Recombinant human leukemia inhibitory factor does not improve implantation and pregnancy outcomes after assisted reproductive techniques in women with recurrent unexplained implantation failure. Fertil Steril. 2009;91(4 suppl):1445–1447 [DOI] [PubMed] [Google Scholar]
- 70. Goggin T, Nguyen QT, Munafo A. Population pharmacokinetic modelling of Emfilermin (recombinant human leukaemia inhibitory factor, r-hLIF) in healthy postmenopausal women and in infertile patients undergoing in vitro fertilization and embryo transfer. Br J Clin Pharmacol. 2004;57:576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Lalitkumar S, Boggavarapu NR, Menezes J, et al. Polyethylene glycated leukemia inhibitory factor antagonist inhibits human blastocyst implantation and triggers apoptosis by down-regulating embryonic AKT. Fertil Steril. 2013;100:1160–1169 [DOI] [PubMed] [Google Scholar]
- 72. Wang W, Taylor RN, Bagchi IC, Bagchi MK. Regulation of human endometrial stromal proliferation and differentiation by C/EBPβ involves cyclin E-cdk2 and STAT3. Mol Endocrinol. 2012;26:2016–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Lee K, Jeong J, Kwak I, et al. Indian hedgehog is a major mediator of progesterone signaling in the mouse uterus. Nat Genet. 2006;38:1204–1209 [DOI] [PubMed] [Google Scholar]
- 74. Matsumoto H, Zhao X, Das SK, Hogan BL, Dey SK. Indian hedgehog as a progesterone-responsive factor mediating epithelial-mesenchymal interactions in the mouse uterus. Dev Biol. 2002;245:280–290 [DOI] [PubMed] [Google Scholar]
- 75. Wei Q, Levens ED, Stefansson L, Nieman LK. Indian hedgehog and its targets in human endometrium: menstrual cycle expression and response to CDB-2914. J Clin Endocrinol Metab. 2010;95:5330–5337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Franco HL, Lee KY, Broaddus RR, et al. Ablation of Indian hedgehog in the murine uterus results in decreased cell cycle progression, aberrant epidermal growth factor signaling, and increased estrogen signaling. Biol Reprod. 2010;82:783–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Li X, Large MJ, Creighton CJ, et al. COUP-TFII regulates human endometrial stromal genes involved in inflammation. Mol Endocrinol. 2013;27:2041–2054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kurihara I, Lee DK, Petit FG, et al. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007;3:e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Lee DK, Kurihara I, Jeong JW, et al. Suppression of ERα activity by COUP-TFII is essential for successful implantation and decidualization. Mol Endocrinol. 2010;24:930–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Lee KY, Jeong JW, Wang J, et al. Bmp2 is critical for the murine uterine decidual response. Mol Cell Biol. 2007;27:5468–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li Q, Kannan A, Wang W, et al. Bone morphogenetic protein 2 functions via a conserved signaling pathway involving Wnt4 to regulate uterine decidualization in the mouse and the human. J Biol Chem. 2007;282:31725–31732 [DOI] [PubMed] [Google Scholar]
- 82. Franco HL, Dai D, Lee KY, et al. WNT4 is a key regulator of normal postnatal uterine development and progesterone signaling during embryo implantation and decidualization in the mouse. FASEB J. 2011;25:1176–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Li Q, Kannan A, Das A, et al. WNT4 acts downstream of BMP2 and functions via β-catenin signaling pathway to regulate human endometrial stromal cell differentiation. Endocrinology. 2013;154:446–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ding YQ, Zhu LJ, Bagchi MK, Bagchi IC. Progesterone stimulates calcitonin gene expression in the uterus during implantation. Endocrinology. 1994;135:2265–2274 [DOI] [PubMed] [Google Scholar]
- 85. Li Q, Wang J, Armant DR, Bagchi MK, Bagchi IC. Calcitonin down-regulates E-cadherin expression in rodent uterine epithelium during implantation. J Biol Chem. 2002;277:46447–46455 [DOI] [PubMed] [Google Scholar]
- 86. Xiong T, Zhao Y, Hu D, et al. Administration of calcitonin promotes blastocyst implantation in mice by up-regulating integrin β3 expression in endometrial epithelial cells. Hum Reprod. 2012;27:3540–3551 [DOI] [PubMed] [Google Scholar]
- 87. Finn CA, Keen PM. The induction of deciduomata in the rat. J Embryol Exp Morphol. 1963;11:673–682 [PubMed] [Google Scholar]
- 88. Bonventre JV, Huang Z, Taheri MR, et al. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature. 1997;390:622–625 [DOI] [PubMed] [Google Scholar]
- 89. Lim H, Paria BC, Das SK, et al. Multiple female reproductive failures in cyclooxygenase 2-deficient mice. Cell. 1997;91:197–208 [DOI] [PubMed] [Google Scholar]
- 90. Ruan YC, Guo JH, Liu X, et al. Activation of the epithelial Na+ channel triggers prostaglandin E2 release and production required for embryo implantation. Nat Med. 2012;18:1112–1117 [DOI] [PubMed] [Google Scholar]
- 91. Bagchi IC, Li Q, Cheon YP, Mantena SR, Kannan A, Bagchi MK. Use of the progesterone receptor antagonist RU 486 to identify novel progesterone receptor-regulated pathways in implantation. Semin Reprod Med. 2005;23:38–45 [DOI] [PubMed] [Google Scholar]
- 92. Cho H, Okada H, Tsuzuki T, Nishigaki A, Yasuda K, Kanzaki H. Progestin-induced heart and neural crest derivatives expressed transcript 2 is associated with fibulin-1 expression in human endometrial stromal cells. Fertil Steril. 2013;99:248–255 [DOI] [PubMed] [Google Scholar]
- 93. Huyen DV, Bany BM. Evidence for a conserved function of heart and neural crest derivatives expressed transcript 2 in mouse and human decidualization. Reproduction. 2011;142:353–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Hewitt SC, Korach KS. Cell biology. A hand to support the implantation window. Science. 2011;331:863–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Nallasamy S, Li Q, Bagchi MK, Bagchi IC. Msx homeobox genes critically regulate embryo implantation by controlling paracrine signaling between uterine stroma and epithelium. PLoS Genet. 2012;8:e1002500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Daikoku T, Cha J, Sun X, et al. Conditional deletion of Msx homeobox genes in the uterus inhibits blastocyst implantation by altering uterine receptivity. Dev Cell. 2011;21:1014–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Pan H, Deng Y, Pollard JW. Progesterone blocks estrogen-induced DNA synthesis through the inhibition of replication licensing. Proc Natl Acad Sci USA. 2006;103:14021–14026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Ray S, Pollard JW. KLF15 negatively regulates estrogen-induced epithelial cell proliferation by inhibition of DNA replication licensing. Proc Natl Acad Sci USA. 2012;109:E1334–E1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Birsoy K, Chen Z, Friedman J. Transcriptional regulation of adipogenesis by KLF4. Cell Metab. 2008;7:339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ryan IP, Taylor RN. Endometriosis and infertility: new concepts. Obstet Gynecol Surv. 1997;52:365–371 [DOI] [PubMed] [Google Scholar]
- 101. Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364:1789–1799 [DOI] [PubMed] [Google Scholar]
- 102. Kao LC, Germeyer A, Tulac S, et al. Expression profiling of endometrium from women with endometriosis reveals candidate genes for disease-based implantation failure and infertility. Endocrinology. 2003;144:2870–2881 [DOI] [PubMed] [Google Scholar]
- 103. Bulun SE, Cheng YH, Pavone ME, et al. Estrogen receptor-β, estrogen receptor-α, and progesterone resistance in endometriosis. Semin Reprod Med. 2010;28:36–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Attia GR, Zeitoun K, Edwards D, Johns A, Carr BR, Bulun SE. Progesterone receptor isoform A but not B is expressed in endometriosis. J Clin Endocrinol Metab. 2000;85:2897–2902 [DOI] [PubMed] [Google Scholar]
- 105. Zeitoun K, Takayama K, Sasano H, et al. Deficient 17β-hydroxysteroid dehydrogenase type 2 expression in endometriosis: failure to metabolize 17β-estradiol. J Clin Endocrinol Metab. 1998;83:4474–4480 [DOI] [PubMed] [Google Scholar]
- 106. Pabona JM, Simmen FA, Nikiforov MA, et al. Krüppel-like factor 9 and progesterone receptor coregulation of decidualizing endometrial stromal cells: implications for the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2012;97:E376–E392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yang H, Zhou Y, Edelshain B, Schatz F, Lockwood CJ, Taylor HS. FKBP4 is regulated by HOXA10 during decidualization and in endometriosis. Reproduction. 2012;143:531–538 [DOI] [PubMed] [Google Scholar]
- 108. Klemmt PA, Carver JG, Kennedy SH, Koninckx PR, Mardon HJ. Stromal cells from endometriotic lesions and endometrium from women with endometriosis have reduced decidualization capacity. Fertil Steril. 2006;85:564–572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bulun SE, Monsavais D, Pavone ME, et al. Role of estrogen receptor-β in endometriosis. Semin Reprod Med. 2012;30:39–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Jeong JW, Lee HS, Lee KY, et al. Mig-6 modulates uterine steroid hormone responsiveness and exhibits altered expression in endometrial disease. Proc Natl Acad Sci USA. 2009;106:8677–8682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Jones A, Teschendorff AE, Li Q, et al. Role of DNA methylation and epigenetic silencing of HAND2 in endometrial cancer development. PLoS Med. 2013;10:e1001551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Lessey BA, Damjanovich L, Coutifaris C, Castelbaum A, Albelda SM, Buck CA. Integrin adhesion molecules in the human endometrium. Correlation with the normal and abnormal menstrual cycle. J Clin Invest. 1992;90:188–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Yang JZ, Ajonuma LC, Tsang LL, et al. Differential expression and localization of CFTR and ENaC in mouse endometrium during pre-implantation. Cell Biol Int. 2004;28:433–439 [DOI] [PubMed] [Google Scholar]
- 114. Salker MS, Christian M, Steel JH, et al. Deregulation of the serum- and glucocorticoid-inducible kinase SGK1 in the endometrium causes reproductive failure. Nat Med. 2011;17:1509–1513 [DOI] [PubMed] [Google Scholar]
- 115. Yoshinaga K, Adams CE. Delayed implantation in the spayed, progesterone treated adult mouse. J Reprod Fertil. 1966;12:593–595 [DOI] [PubMed] [Google Scholar]
- 116. Renfree MB, Shaw G. Diapause. Annu Rev Physiol. 2000;62:353–375 [DOI] [PubMed] [Google Scholar]
- 117. Lee JE, Oh HA, Song H, et al. Autophagy regulates embryonic survival during delayed implantation. Endocrinology. 2011;152:2067–2075 [DOI] [PubMed] [Google Scholar]
- 118. Roberts RM, Chen Y, Ezashi T, Walker AM. Interferons and the maternal-conceptus dialog in mammals. Semin Cell Dev Biol. 2008;19:170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bazer FW. Pregnancy recognition signaling mechanisms in ruminants and pigs. J Anim Sci Biotechnol. 2013;4:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Fazleabas AT, Donnelly KM, Srinivasan S, Fortman JD, Miller JB. Modulation of the baboon (Papio anubis) uterine endometrium by chorionic gonadotrophin during the period of uterine receptivity. Proc Natl Acad Sci USA. 1999;96:2543–2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Laws MJ, Taylor RN, Sidell N, et al. Gap junction communication between uterine stromal cells plays a critical role in pregnancy-associated neovascularization and embryo survival. Development. 2008;135:2659–2668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Manaster I, Mandelboim O. The unique properties of uterine NK cells. Am J Reprod Immunol. 2010;63:434–444 [DOI] [PubMed] [Google Scholar]
- 123. Nancy P, Tagliani E, Tay CS, Asp P, Levy DE, Erlebacher A. Chemokine gene silencing in decidual stromal cells limits T cell access to the maternal-fetal interface. Science. 2012;336:1317–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. González IM, Martin PM, Burdsal C, et al. Leucine and arginine regulate trophoblast motility through mTOR-dependent and independent pathways in the preimplantation mouse embryo. Dev Biol. 2012;361:286–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Bazer FW, Kim J, Ka H, Johnson GA, Wu G, Song G. Select nutrients in the uterine lumen of sheep and pigs affect conceptus development. J Reprod Dev. 2012;58:180–188 [DOI] [PubMed] [Google Scholar]
- 126. Frolova AI, O'Neill K, Moley KH. Dehydroepiandrosterone inhibits glucose flux through the pentose phosphate pathway in human and mouse endometrial stromal cells, preventing decidualization and implantation. Mol Endocrinol. 2011;25:1444–1455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Kommagani R, Szwarc MM, Kovanci E, et al. Acceleration of the glycolytic flux by steroid receptor coactivator-2 is essential for endometrial decidualization. PLoS Genet. 2013;9:e1003900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Tan Y, Tan D, He M, et al. A model for implantation: coculture of blastocysts and uterine endometrium in mice. Biol Reprod. 2005;72:556–561 [DOI] [PubMed] [Google Scholar]
- 129. Grewal S, Carver JG, Ridley AJ, Mardon HJ. Implantation of the human embryo requires Rac1-dependent endometrial stromal cell migration. Proc Natl Acad Sci USA. 2008;105:16189–16194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ramathal C, Wang W, Hunt E, Bagchi IC, Bagchi MK. Transcription factor CCAAT enhancer-binding protein β (C/EBPβ) regulates the formation of a unique extracellular matrix that controls uterine stromal differentiation and embryo implantation. J Biol Chem. 2011;286:19860–19871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Garrido-Gómez T, Ruiz-Alonso M, Blesa D, Diaz-Gimeno P, Vilella F, Simón C. Profiling the gene signature of endometrial receptivity: clinical results. Fertil Steril. 2013;99:1078–1085 [DOI] [PubMed] [Google Scholar]
- 132. Lledo B, Turienzo A, Ortiz JA, et al. Negative effect of P72 polymorphism on p53 gene in IVF outcome in patients with repeated implantation failure and pregnancy loss. J Assist Reprod Genet. 2014;31:169–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Tamura I, Sato S, Okada M, et al. Importance of C/EBPβ binding and histone acetylation status in the promoter regions for induction of IGFBP-1, PRL, and Mn-SOD by cAMP in human endometrial stromal cells. Endocrinology. 2014;155:275–286 [DOI] [PubMed] [Google Scholar]
- 134. Houshdaran S, Zelenko Z, Irwin JC, Giudice LC. Human endometrial DNA methylome is cycle-dependent and is associated with gene expression regulation. Mol Endocrinol. 2014;28:1118–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]