

Abstract
Introduction
Calpains represent a family of neutral, calcium-dependent proteases, which modify the function of their target proteins by partial truncation. These proteases have been implicated in numerous cell functions, including cell division, proliferation, migration, and death. In the CNS, where μ-calpain and m-calpain are the main calpain isoforms, their activation has been linked to synaptic plasticity as well as to neurodegeneration. This review will focus on the role of calpains in synaptic plasticity and discuss the possibility of developing methods to manipulate calpain activity for therapeutic purposes.
Areas covered
This review covers the literature showing how calpains are implicated in synaptic plasticity and in a number of conditions associated with learning impairment. The possibility of developing new drugs targeting these enzymes for treating these conditions is discussed.
Expert opinion
As evidence accumulates that calpain activation participates in neurodegeneration and cancer, there is interest in developing therapeutic approaches using direct or indirect calpain inhibition. In particular, a peptide derived from the calpain truncation site of mGluR1α was shown to decrease neurodegeneration following neonatal hypoxia/ischemia. More selective approaches need to be developed to target calpain or some of its substrates for therapeutic indications associated with deregulation of synaptic plasticity.
Keywords: actin filament, calpain, cytoskeleton, long-term potentiation, synaptic plasticity
1. Introduction
Although calpain and its endogenous inhibitor, calpastatin, were identified in the brain in 1980, it has been difficult to determine the precise physiological function (s) of this neutral calcium-dependent protease in synaptic transmission and neuronal function. Based on the findings that calpain activity could truncate the cytoskeletal protein spectrin and regulate properties of glutamate receptors, we initially proposed in 1984 that calpain played a critical role in synaptic plasticity [1]. While a large body of work since then has supported such a role for calpain in long-term potentiation (LTP) of synaptic transmission at glutamatergic synapses (see [2,3] for reviews), it has been difficult to provide definitive evidence for the original hypothesis, and a detailed description of the mechanisms linking calpain activation to changes in synaptic efficacy is still lacking. In support of the original hypothesis, calpain inhibitors were shown to block LTP induction both in vitro, in acute and cultured hippocampal slices [4–6], and in vivo [7]. Conversely, rats or mice deficient in calpastatin, the endogenous inhibitor of calpain, exhibit enhanced LTP ([8], and Saido T., personal communication). However, while we had assumed that μ-calpain was the critical calpain isoform in LTP, calpain-1 (the large subunit of μ-calpain) knock-out mice did not exhibit any phenotype regarding LTP or learning and memory [9], suggesting that m-calpain could be the important calpain isoform in LTP induction. In support of this idea, we recently reported that down-regulating m-calpain using a novel Rabies-Virus Glycoprotein (RVG)-chimeric peptide, which enabled the transvascular delivery of small interfering RNA (siRNA) against calpain-2 (the large subunit of m-calpain) to the brain, resulted in impairment in LTP induction and in learning and memory [10]. Information regarding the mechanisms underlying calpain activation and the nature and role of its critical substrate(s) in the signaling cascade leading from NMDA receptor activation, which is a necessary step for LTP induction [11], to stable LTP has not been forthcoming. We initially argued that calpain-mediated degradation of spectrin was critical for changes in spine morphology following LTP induction, based on the links between the spectrin network and actin filaments [1]. Since the original hypothesis was proposed, many new findings regarding calpain properties, synaptic structure and the complexity of postsynaptic densities have been published and have considerably modified our understanding of the mechanisms underlying activity-dependent modifications of synaptic structure and function. The review will summarize the various elements that have recently been proposed to link patterns of electrical activity of the type that trigger changes in synaptic efficacy to calpain activation and its functional consequences, at the level of the cytoskeletal organization/reorganization as well as of the cellular processes involved in consolidation of synaptic modifications. We will first briefly review the properties of this family of calcium-dependent proteases and the major substrates that are targeted by calpains. We will then discuss the roles of calpains in the regulation of cell cytoskeleton, as well as of protein synthesis before reviewing the elements supporting a critical role for calpain in synaptic plasticity. Finally, the conclusions will highlight the implications of the understanding of the role of calpain in synaptic plasticity for a deeper understanding of the evolutionary mechanisms underlying memory processes and for the future development of new therapeutic approaches for treating learning and memory disorders.
2. Calpain properties
Calpains are a family of intracellular calcium-dependent neutral cysteine proteases. There are currently 15 isoforms of calpains identified in the human genome, of which calpain-1 and -2 are ubiquitously expressed, predominantly in mammalian brain and are the best-characterized isoforms. It is commonly agreed that the brain distribution of calpain-1 and -2 is ubiquitous and that they are present in both neurons and glia. In terms of subcellular localization, these two isoforms are not limited to a specific compartment although calpain-1 has been shown to be enriched in the mitochondrial intermembrane space [12].
Structurally, calpain-1 or calpain-2 interacts with a small regulatory subunit (calpain-S1, formerly known as calpain-4) to form functional heterodimeric proteins called μ- and m-calpain, respectively (Figure 1). The large catalytic subunit (calpain-1 or -2) contains four major domains and calpain-S1 contains two domains. Two naming systems for calpain domains are presented in Figure 1; domain numbers (I – VI) have been used conventionally while the other descriptive acronym naming system was recently proposed to help identifying domain function and structure [13]. Domain I is the N-terminal anchor helix region of the large subunit. Autolysis has been observed in this region during calpain activation by Ca2+, which in some cases further contributes to the dissociation of the large and small subunits [14]. Domain II, a.k.a. the protease (CysPc) domain, comprises two protease core domains (PC1 and PC2), which fuse to form an active cysteine catalytic region upon Ca2+ binding onto each core domain [15] and interact with substrates or calpastatin. Domain III, a.k.a. C2 domain-like (C2L) domain, is possibly involved in binding Ca2+ and phospholipids [16]. Domain IV (a.k.a. penta-EF-hand domain, PEF(L), L referring to large subunit) and Domain VI (a.k.a. PEF(S), S for small subunit), each containing five EF-hand motifs, not only bind Ca2+ but also contribute to the heterodimer formation [17]. Lastly, Domain V is the N-terminus of the small subunit and is rich in glycine, thus it is also called glycine-rich hydrophobic (GR) domain.
Figure 1. Schematic structure of a mammalian μ- or m-calpain.
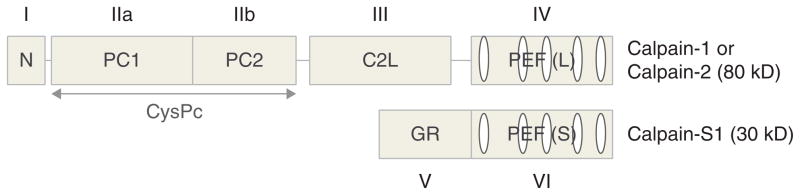
μ- and m-calpains are heterodimeric proteins, which share a common small regulatory subunit (calpain-S1, a.k.a. calpain-4, 30 kDa) and differ in the large catalytic subunit (calpain-1 and calpain-2, respectively; 80 kDa). Two domain nomenclature systems are presented here: domain numbers (I – VI) and descriptive acronyms. N, the N-terminal anchor helix region of the large subunit; CysPc, the protease domain, comprising two protease core domains (PC1 and PC2); C2L, C2 domain-like domain; PEF(L), penta-EF-hand domain, L referring to large subunit; PEF(S), penta-EF-hand domain, S for small subunit; GR, the glycine-rich hydrophobic domain at the N-terminus of the small subunit.
μ- and m-calpains were originally named after their in vitro calcium requirements. The concentration of calcium required for half-maximal activity in vitro is 3 – 50 μM for μ-calpain and 0.4 – 0.8 mM for m-calpain [18,19]. Besides calcium requirements, calpains are unique in that they can be activated at neutral pH and regulate the activity of target substrates by partial cleavage (nondigestive), while typical cellular proteases are usually independent of calcium and active in acidic compartments, and degrade their substrates completely.
The intracellular cytosolic calcium concentration is generally estimated to be 50 – 300 nM [20], much lower than its extracellular concentration, approximately 2 mM. In response to a stimulus, calcium is released from intracellular stores, such as the endoplasmic reticulum (ER) and mitochondria, or enters into cells through plasma membrane ionotropic receptors and voltage-gated Ca2+ channels. All these events rapidly increase intracellular calcium concentration 10 – 100-fold, and intracellular calcium concentration is estimated to rise to tens of μM at most. As a result, it is generally agreed that mM intracellular calcium concentrations cannot be reached, except under pathological conditions, such as following stroke or tissue damage. Although calcium levels required for μ-calpain activation are physiological under certain conditions, such as calcium spikes, this range of concentration does not clearly account for the activation of calpain observed in the regulation of cell cycle [21] or in apoptosis [22], nor do physiological calcium levels account for the activation of m-calpain. As a result, alternative in vivo activation mechanisms for calpains have been suggested. In particular, one group has found that m-calpain can be activated by extracellular signal-regulated kinase (ERK)-mediated direct phosphorylation at its serine 50 without increased intracellular Ca2+ concentration [23,24]. Furthermore, it appears that m-calpain can be equivalently activated by either ERK or high calcium concentrations in a murine fibroblast cell line [23]. We have also recently reported that both EGF and BDNF can activate m-calpain by ERK-mediated phosphorylation in dendritic spines of hippocampal neurons [25]. Therefore, calpain activation is likely to be regulated by several different mechanisms.
Calpain activation has been associated with a variety of cellular events, from cell adhesion and cell division to transcriptional regulation and LTP induction [26–30]. In addition, neuronal calpain activation has also been implicated in several chronic neurodegenerative conditions, including Alzheimer’s, Huntington’s and Parkinson’s diseases [31–33]. In our laboratory, we have also demonstrated that calpain-mediated cleavage of mGluR1α accounts, at least partially, for NMDA-induced excitotoxicity, as it can be blocked by using a fusion peptide consisting of the tat transduction domain and the amino acid sequence surrounding the calpain cleavage site of mGluR1α; this peptide prevents calpain-mediated cleavage of mGluR1α and is neuroprotective against kainic acid-mediated excitotoxicity and hypoxia/ischemia in hippocampal slices and in neonatal rats [34]. As many pathological conditions are associated with NMDA receptor-mediated excitotoxicity, and therefore with calpain activation, a specific calpain inhibitor may provide a potential treatment for these diseases. Currently, a number of calpain inhibitors are available [35] although the specificity of these synthetic calpain inhibitors is still in question. In particular, there are no inhibitors specific for μ-calpain or m-calpain. In addition to synthetic inhibitors, calpain has its own endogenous inhibitor, calpastatin, and as a result, calpastatin has been a focus of many studies. Although the activation mechanism of calpastatin itself and its specificity against different calpain isoforms are still indeterminate, the resolution of the crystal structure of calpastatin-bound calpain [36] might provide some new insights into the design of more specific calpain inhibitors.
3. Calpain targets
Calpain-mediated cleavage has been observed in cytoskeleton proteins, membrane-associated proteins, receptors/channels, scaffolding/anchoring proteins, protein kinases and phosphatases, as well as presynaptic proteins. However, it is important to stress the difference between in vitro substrates and in vivo substrates. While many proteins can certainly be cleaved by calpain in cell-free systems, the real calpain substrates are the proteins that are actually cleaved under various conditions following calpain activation in cells.
Spectrin isoform αII, also known as brain spectrin or α-fodrin, is the first-identified substrate of calpains and is the principle component of the neuronal sub-membrane cytoskeleton [37]. αII-spectrin is anchored to the plasma membrane and binds to actin, calmodulin, and microtubules. Cleavage of spectrin by calpain alters the dynamic organization of membrane domains and membrane trafficking events [38] and thus changes synaptic integrity and stability. Other preferred cytoskeletal substrates of calpains include: microtubule-associated proteins (MAPs), neurofilaments, actin [39–42], cortactin [43], and MARCKS [44].
NMDA receptor-mediated signals are critical for both neuronal plasticity and pathophysiological events, such as excitotoxicity [45,46]. It has been shown that the C-terminal domains of three NMDA receptor subunits (GluN2A, GluN2B, and GluN2C) can be truncated by calpain-mediated proteolysis, which may change NMDA receptor levels and functions at synapses [47]. Similarly, the subunits (GluA1, GluA2, and GluA3) of AMPA receptors, which mediate the majority of fast excitatory neurotransmission in mammalian brain, are targets of calpain as well [48,49]. Although the functional consequences of calpain cleavage of these receptors are not clearly defined, it has generally been assumed that calpains participate in the regulation of NMDA and AMPA receptor levels on the surface of dendritic spines. Other synaptic membrane-associated proteins that are preferentially targeted by calpains are IP3 receptors, L-type Ca2+ channels and Na+/Ca2+ exchangers [50–52].
Besides cytoskeleton proteins and membrane receptors, there are three major postsynaptic density (PSD) scaffolding proteins reported to be calpain substrates: PSD-95, SAP97, and GRIP1 [53–55]. PSD-95 is a major membrane-associated guanylate kinase (MAGUK) that binds to the C-terminal domains of GluN2A and GluA1 in mature neurons [56]. SAP97 and GRIP1 are involved in synaptic trafficking, anchoring, and/or stabilization of AMPA receptor subunits [57]. Therefore, cleavage of these PSD proteins by calpains may alter synaptic receptor stability and abundance. We also recently reported that calpain cleaves stargazin, a member of the Transmembrane AMPA Receptor Associated Proteins (TARPs), which participates in AMPA receptor trafficking and targeting [58]. All these findings indicate that calpain activation could participate in the regulation of ionotropic and metabotropic glutamate receptors in postsynaptic structures, and could modify their number, localization, and functional properties. Furthermore, as calpain-mediated truncation of target proteins is often regulated by their state of phosphorylation, this mechanism could provide an important degree of cross-talk between calpain activation and activation of protein kinases and phosphatases, which are also abundantly represented in postsynaptic structures.
Calpain also cleaves synaptic protein kinases and phosphatases. One of the best-studied calpain substrates is CaMKIIα (calcium/calmodulin-dependent protein kinase-II α chain). CaMKIIα is an important calmodulin-dependent enzymes in neurons and abundant in postsynaptic membranes. It phosphorylates various substrates, including AMPA receptors, NMDA receptors and calcium channels [59,60]. After cleavage by calpain, CaMKIIα loses its auto-inhibitory domain, and thus becomes irreversibly activated [61]. The same pattern of cleavage by calpain is also observed in other calcium-dependent enzymes, such as PKC and calcineurin A [62,63]. Since CaMKIIα and PKC have been proposed to be both necessary [64–66] and sufficient [67,68] for LTP induction, their activation induced by calpain-mediated cleavage could provide another line of evidence that calpain plays a major role in participating in LTP induction. As mentioned above, m-calpain can be phosphorylated by ERK and by PKA, with the former resulting in activation, while the latter inactivates m-calpain [23,25]. Whether ERK-induced m-calpain phosphorylation still requires calcium for activation is still debated, as it has been proposed that such phosphorylation results in m-calpain binding to PIP2 [24]. There is also significant evidence that m-calpain is dephosphorylated by protein phosphatase-2, PP2 [69]. Another phosphatase, calcineurin, a.k.a. PP2B, is also a calpain substrate and is activated by calpain-mediated truncation [70]. Calcineurin has also been shown to play an important role in synaptic plasticity as well as neuronal degeneration [71]. Thus, regulation of calpain activity by phosphorylation/dephosphorylation reactions produces a very complex network of reactions, which can participate in numerous functional adaptations at glutamatergic synapses (Figure 2).
Figure 2. Complex regulation of calpain activity by protein kinases and phosphatases.

As discussed in the text, calpain is activated by ERK-mediated phosphorylation and inactivated by PP2A-mediated dephosphorylation. In addition, calpain is also inactivated by PKA-mediated phosphorylation. Considering the numerous cross-talks between various protein kinases and phosphatases, calpain is subjected to extremely complex regulation by phosphorylation/dephosphorylation reactions.
Previous studies have suggested some important presynaptic roles for calpain. For example, calpain-mediated truncation of the presynaptic protein, amphiphysin I, which plays a key role in clathrin-mediated endocytosis of synaptic vesicles [72–74], has been shown to inhibit vesicle endocytosis in hippocampal slices during neural hyperexcitation [75]. In another study, amphiphysin I was shown to be cleaved by both μ- and m-calpain in vitro. Applying the calpain inhibitor, leupeptin, to cultured hippocampal neurons was reported to reduce mEPSC frequency [76]. In addition, several other presynaptic calpain substrates have been identified, such as the SNARE proteins SNAP-25 and SNAP-23, which are important for synaptic vesicle fusion and exocytosis [77]. In cultured hippocampal neurons, calpain activity was different in distinct neuronal populations, with a significantly higher level of activity in GABAergic interneurons; moreover, calpain contributed to reduced SNAP-25 expression in differentiated GABAergic interneurons and mediated activity-dependent SNAP-25 cleavage in vivo [78]. Since calpain-mediated SNAP-25 truncation also correlated with a reduction in SNARE function and thus inhibition of neurotransmitter release [77], it is possible that calpain functions to limit GABA release by constitutively cleaving SNAP-25 in GABAergic neurons. This mechanism could be particularly important during the postnatal period since we have previously reported that calpain appears to be tonically activated, as shown by the high levels of calpain-specific spectrin breakdown products present in both interneurons and pyramidal neurons in hippocampus [79]. Furthermore, during this period, GABA is depolarizing and thus calpain activation could represent a mechanism to prevent excessive depolarization during this period.
4. Calpain-mediated regulation of cytoskeleton
Our original hypothesis postulated that calpain, via digestion of subsynaptic structural proteins including spectrin, served to disassemble extant cytoskeleton, and thereby cleared the way for the construction of new actin networks and morphological changes to the synapse. Results obtained over the last 20 years have suggested that the protease plays a broader role than we originally envisioned and, in particular, that it helps orchestrate the sequence and timing of signaling cascades that disassemble, assemble and then stabilize newly formed actin filaments in the minutes following LTP induction.
One possibility was suggested by the discovery that BDNF stimulates calpain activation through ERK-mediated phosphorylation. This event could terminate the stabilization sequences that occur during the first 10 min post-theta burst stimulation (TBS) and, in addition, set in motion activities required for later stages of consolidation. It is now clear that phosphorylation-induced m-calpain activation plays a critical role in cytoskeletal remodeling in a variety of cell types under physiological conditions [80–82]. Furthermore, both theta-burst and NMDA receptor stimulations produced calpain activation as evidenced by the accumulation of a selective spectrin breakdown product generated by calpain-mediated proteolysis [83]. We also provided evidence that BDNF, through TrkB receptor activation, stimulates m-calpain through ERK-mediated phosphorylation [25]. Interestingly, integrin activation results in calpain activation and, depending on the state of phosphorylation of the integrin cytoplasmic domain, induces either inhibition of RhoA and cell spreading or activation of RhoA and cell retraction [84]. It is therefore tempting to envision a similar type of molecular switch in dendritic spines and to equate spreading to potentiation and retraction to depression. Interestingly, RhoA and its effectors, Rho kinase (ROCK), LIM-kinase, and cofilin, a constitutively active actin severing protein, are directly linked to actin polymerization. Two other members of the Rho family, Rac and Cdc42, and their effector p21-activated kinase (PAK), also play a pivotal role in reorganizing the cytoskeleton across cell types and experimental paradigms [85]. RhoA has been shown to be a calpain substrate, thus providing a clear path to link calpain activation to actin polymerization [86]. As calpain degrades integrins and adaptor proteins needed for their activation and signaling [84,87,88], this provides for the existence of a negative feedback regulatory loop in which integrin activation leads to calpain activation followed by integrin degradation and return to resting state for calpain. Furthermore, this would require the rapid synthesis of integrin proteins to reestablish the normal status of the dendritic spine. Another mechanism that could play a significant role in modifying the structure and function of synaptic contacts is related to the fact that by truncating focal adhesion kinase (FAK) [88–90], calpain could modify adhesion properties of dendritic spines, possibly indirectly influencing presynaptic terminals, although this mechanism has not yet been extensively studied in neurons.
We postulate that interactions between calpain, cortactin, slingshot (SSH, a phosphatase), LIM-kinases and cofilin are also implicated in activity-dependent synaptic actin network reorganization. The actin cytoskeleton represents a highly regulated complex in dynamic equilibrium between polymerized and depolymerized actin. Among the numerous regulatory elements participating in this equilibrium, the ADF/cofilin family of proteins plays a central role, as it determines the rate of elongation or retraction of actin filaments [91–96]. Activation of cofilin kinases, LIMK1 and LIMK2, leads to cofilin phosphorylation and inactivation, thereby disrupting cofilin-mediated depolymerization of actin filaments in vivo and in vitro [90,97]. In contrast, SSH dephosphorylates and reactivates cofilin [98]. SSH itself is regulated/inactivated by PAK-mediated phosphorylation and by binding to F-actin. Another actin interacting protein is cortactin, which together with Arp2/3 are potent activators of actin polymerization [99]. Cortactin is an m-calpain substrate [43], and previous studies have shown that distribution of cortactin in spines and dendrites in hippocampus is regulated by activation of NMDA receptors as well as BDNF [100]. Like cofilin, cortactin is also regulated by phosphorylation, which changes its susceptibility to calpain-mediated truncation [101]. Interestingly, during the developmental period, calpain-mediated cortactin truncation limits cortactin levels in neurites and repress protrusive activity and neurite extension [102]. Cortactin is also subject to modification by acetylation; whether this modification alters its function and stability remains unknown. Thus, much more needs to be understood regarding the role of calpain-mediated cortactin truncation in cytoskeletal reorganization in adult dendritic spines.
5. Calpain-mediated regulation of local protein synthesis
The question of the participation of proteases and therefore protein degradation in LTP is ineluctably linked to the question of protein synthesis. The role of protein synthesis in LTP has been the subject of intense and often controversial investigation, in parallel to its role in memory formation/consolidation [103]. A recent issue of “Neurobiology of Learning and Memory” is devoted to this question (NLM, 89, 3, 2008). It was initially thought that both transcription and translation played a significant role in LTP maintenance [104–106]. After the discovery that a significant number of mRNAs were present in dendrites, and that the protein synthesis machinery was present in dendrites and possibly in dendritic spines [107,108], a number of studies have provided evidence that LTP was associated with local protein synthesis [107,109–112]. Paul Gold summarized the current views by stating (statements in brackets are my addition) “there is agreement that protein synthesis is important for memory [and LTP] formation, but disagreement on the question of whether new protein synthesis triggered by an event [or LTP-inducing tetanus] is important for the formation of memory for that event [and LTP consolidation]” [113].
Numerous studies have shown that protein synthesis inhibitors prevent LTP formation when present at the time of tetanus, but not when applied after high frequency stimulation (see [104], for a review). The key questions that are still debated are: i) What are the relative contributions of local or global protein synthesis to LTP? ii) What are the proteins that are locally synthesized and what are their contributions to LTP? iii) What are the mechanisms linking LTP-inducing stimulation protocols to local protein synthesis? Significant progress has been made following the discovery that BDNF, which has been shown to play a critical role in LTP stabilization [114–117], could activate local protein synthesis by stimulating the mammalian/mechanistic target of rapamycin (mTOR), and activating spine eukaryotic initiation factor 4E (eIF4E) [118]. Among the proteins generally considered to be synthesized in dendritic spines following LTP induction, Arc and CaMKII exhibit increased dendritic synthesis following BDNF treatment in vitro and in vivo [119–121]. Two major signaling pathways have been linked to the regulation of local protein synthesis, the ERK-MAP kinase pathway and the PI3 kinase pathway [122]. Abundant evidence has indicated a crucial role for the ERK-MAP kinase pathway in synaptic plasticity as well as in learning and memory (see [123,124] for recent reviews).
Two recent manuscripts have further implicated calpain in synaptic plasticity through effects on well-characterized signaling cascades. First, Shimizu et al. reported that calpain, by degrading a Ras inhibitor called the suprachiasmatic nucleus circadian oscillatory protein (SCOP), regulated ERK activation [125], thereby providing a positive feedback loop, maintaining high level of calpain activation for extended periods of time after its initial activation. The second study reported that calpain, by truncating β-catenin, was a critical link between synaptic events and transcriptional regulation, as the truncated β-catenin functioned as a transcription factor [26]. Furthermore, calpain contributes, via known mechanisms, to mTOR-dependent local protein synthesis [125]. Calpain could accordingly be a molecular switch that drives the shift from early to late phases of consolidation (Figure 3).
Figure 3. Multiple functions of calpain activation in various stages of LTP.

Calpain can be activated by several signals: calcium influx from NMDA receptors, integrin activation, and BDNF→ TrkB→ ERK-mediated phosphorylation. Once activated, calpain can result in rapid modifications of glutamate receptors and synaptic structure; it can also participate in the regulation of dendritic spine cytoskeleton; finally, by regulating local protein synthesis and transcription factors, it can be involved in long-term consolidation.
Our evolving idea is that there is a pool of rapidly turning over proteins that play critical roles in the regulation of synaptic transmission and spine cytoskeleton, and that some of these are calpain substrates. If this notion is correct, it could reconcile several of the above-mentioned discrepancies and help resolving the debate regarding the role of protein synthesis in synaptic plasticity, learning, and memory. Thus, interfering with protein synthesis would directly impact some of the same mechanisms that are involved in LTP and learning and memory, and likewise, triggering LTP would result in protein degradation, which would require protein synthesis to replenish the degraded proteins. As mentioned earlier, the Ras inhibitor, SCOP is degraded by calpain following BDNF treatment of cultured neurons [125]. Those results indicated that BDNF-induced calpain activation positively regulated the RAS/MEK/CREB signaling pathway and participated in hippocampus-dependent learning tasks. The existence of the calpain–SCOP–CREB cascade provides a plausible link between calpain activation and transcriptional regulation of genes that could participate in synaptic plasticity and memory formation. In addition, recent results from our laboratory indicate that calpain inhibition prevents BDNF-mediated stimulation of local protein synthesis (Briz et al., submitted).
6. Calpain-mediated regulation of synaptic plasticity
Almost 30 years ago, we proposed that a brief episode of high frequency stimulation caused an increase in postsynaptic calcium concentration and resulted in calpain activation [1]. We proposed that calpain cleaved the cytoskeletal protein, spectrin, allowing insertion of glutamate receptors in postsynaptic membranes and resulting in morphological changes in dendritic spines, which could provide for a long-lasting enhancement of synaptic efficacy, or LTP [1]. In support of this hypothesis, LTP was blocked by administration of calpain inhibitors [4,5,7,126] and by administration of antisense oligo-nucleotides to decrease calpain-1 levels in cultured hippocampal slices [127]. As we pointed out at the time, the involvement of a protease as a critical regulatory step had several implications. Because proteases produce irreversible modifications of their substrate proteins, and in the case of calpain, functional modifications resulting from partial truncation [128], their activation results in alterations of cell functions that last for the duration of the lifetime of these proteins. It was thus conceivable that calpain activation could result in several functional changes that could exhibit different time-courses depending on the turnover of the various proteins truncated by calpain. This feature could therefore account for various stages in memory formation, as truncation of proteins with fast turnover would be responsible for short-term functional modifications, whereas truncation of proteins with long half-lives would produce longer-lasting changes. Beyond these changes directly related to protein half-lives, we postulated that calpain activation could result in very long-lasting functional modifications of synaptic function by indirectly contributing to structural modifications that could be resistant to protein turnover, such as changing the size or shape of dendritic spines. As we concluded our 1984 paper “if calpain activation is triggered by modest levels of calcium and produces irreversible changes in synaptic chemistry, then its activation would seem to be both a likely event and one that should produce lasting changes in the operation of neuronal circuitries”; as discussed below, evidence collected since then accords with this conclusion and has provided a wealth of details that were missing at the time. As mentioned previously, the discovery that BDNF stimulates calpain activation through ERK-mediated phosphorylation provided a mechanism linking LTP induction to calpain activation during the 10 min following TBS stimulation, since this time window corresponds to the period of increased BDNF release [129,130]. This event could then terminate the stabilization sequences that occur during the first 10 min post-TBS and, in addition, set in motion activities required for later stages of consolidation. All these findings suggest that various signaling cascades are involved in the various phases of LTP. Activation of NMDA receptors followed by calcium influx could trigger a number of calcium-dependent cascades, including calpain and CaMKII and the resulting partial proteolysis and phosphorylation of several targets for these two enzymes. These initial cascades would be responsible for LTP induction and presumably last a few minutes. BDNF release and integrin activation would then trigger a second series of intracellular cascades, including stimulation of local protein synthesis, and these events would be critical for LTP stabilization. These events presumably could last up to 1 h after TBS. As mentioned above, calpain could be a molecular switch that drives the shift from the early to late phases of consolidation (Figure 3).
7. Conclusions
Recent studies indicate that the postsynaptic machinery that is responsible for activity-dependent synaptic modifications evolve very early in evolution and is present, at least in some primitive form, in unicellular eukaryotes and expanded with further evolutionary steps [131]. Likewise, the calpain family evolved very early in evolution with a massive expansion of the calpain family in unicellular eukaryotes [132]. Not surprisingly, this family of neutral proteases participates in numerous cellular functions and thereby in a large array of physiological as well as pathological processes. In particular, calpains are probably playing a critical role in cell motility, cell division and cell proliferation. The involvement of calpains in these processes is likely to account for the known role of calpains in cancer, which has been recently reviewed in this series [133]. Interestingly, the same machinery that is triggered by various growth factors and extracellular signals and leading to modifications of cell adhesion properties and cell migratory properties is likely to play a similar role in mechanisms involved in synaptic plasticity (Figure 4). As discussed above, synaptic plasticity requires a complex set of cellular modifications, including loss of cell adhesion, cytoskeletal disassembly and reassembly, as well as local regulation of protein synthesis. We will discuss elsewhere the striking parallelism between the role of calpain in cancer and in synaptic plasticity, but here it is important to evaluate whether the tools that have been developed to understand and to treat cancer can be used to understand synaptic plasticity and to treat disorders that are associated with abnormal synaptic plasticity. In particular, calpains have been proposed to be potential therapeutic targets for retarding development and migration of cancer cells. It would therefore seems reasonable to propose that calpain inhibitors could be appropriate to treat neurological or neuropsychiatric disorders associated with deregulated calpain activity, or low levels of a number of calpain substrates. Calpain inhibition could be beneficial either directly, by rescuing the deficiency of a protein due to a defective gene, or indirectly, by preventing the degradation of a protein under pathological conditions. For the direct pathway, this could be particularly beneficial in cases of haplo-insufficiency in which one allele is defective, resulting in lower levels of the corresponding protein. In a rare neurological disorder, Lissencephaly, mutations in Lis1 result in brain malformation, mental retardation and seizure activity. As the protein encoded by Lis1 is a calpain substrate, it has been proposed that treatment with a calpain inhibitor could revert some of the symptoms of the disorder [134]. Similarly, calpain inhibition has been proposed as a potential treatment for Machado-Joseph disease, the most frequently found dominantly-inherited cerebellar ataxia, in which calpain-mediated cleavage of the overexpressed ataxin 3 protein generates toxic fragments [135]. Finally calpain has been repeatedly proposed as a potential target for treating Alzheimer’s disease [136–138]. However, as previously discussed regarding the use of calpain inhibitors for treating cancer [133], the multiple roles of calpain in normal physiological conditions impose serious limitations for the therapeutic use of “pan-calpain” inhibitors. As we previously reported, the use of peptides or peptide analogs preventing calpain-mediated truncation of specific target proteins would appear to be a much better strategy for developing therapeutic treatments of specific disorders inasmuch as these specific targets play a preponderant role in these disorders [139].
Figure 4. Comparison of the roles of calpain in tumorigenesis and synaptic plasticity.

By activating ubiquitously distributed cellular cascades participating in cell adhesion, migration, and cell morphogenesis, calpain activation can participate in a wide variety of basic cellular processes, including tumor formation and dissemination, as well as in neurite extension and synaptic plasticity.
8. Expert opinion
It is now clear that calpains are critically implicated in synaptic plasticity and play therefore critical roles not only during postnatal development, but also in adult learning and memory. It is also clear that overactivation of calpain is likely involved in a number of neurological and neuropsychiatric disorders, and in particular in neurodegenerative diseases. These multiple roles of calpains lead to the conundrum that while the direct use of calpain inhibitors to correct or treat these disorders appears attractive, such a use would be extremely challenging. Not only do we lack selective inhibitors for specific calpain isoforms, but the number of physiological functions regulated by this class of enzymes makes it clear that the use of calpain inhibitors will produce a very large number of detrimental side effects. While considerable progress has been made regarding the understanding of the roles of calpains in brain function and in different pathological conditions, much more remains to be done to be able to devise strategies that can overcome the challenges of using cal-pain inhibition as therapeutic strategies. In particular, we need to better understand the various signaling cascades affected by calpain activation under different physiological or pathological conditions. Moreover, we need to better understand the multiple regulatory processes involved in calpain activation and inhibition. As discussed in the review, the links between phosphorylation/dephosphorylation events and calpain activity might produce interesting strategies to provide for a fine regulation of calpain activity. In addition, we also propose that it should be possible to identify for each disorder where calpains are involved a small subset of calpain substrates whose degradation plays a critical role in the pathology. If this were to be the case, it should be possible to design specific tools to interfere with the degradation of these substrates by calpain. As mentioned above, this strategy was successful in the case of excitotoxicity, as the use of a small peptide comprising the sequence of mGluR1α around the calpain truncation site and the tat transduction domain was able to protect against neurodegeneration in several animal models of brain injury. Future efforts should therefore be directed at the identification of these specific calpain targets in various disorders associated with impairment of synaptic plasticity. Another potential approach for the treatment of chronic disorders might be to identify a dose of calpain inhibitor sufficient to slow down the basal rate of calpain activity while allowing brief bursts of calpain activation potentially involved in physiological processes. These are all attractive areas for further research and they should lead not only to a better understanding of calpain function and regulation in brain, but also to a variety of therapeutic approaches for disorders in which calpain deregulation participates in the pathology.
Article highlights.
Calpains play critical role in synaptic plasticity by partial truncation of several pre- and postsynaptic proteins.
Calpains regulate cytoskeleton assembly and disassembly thereby participating in activity-dependent modifications of synaptic structure and function.
Calpain deregulation is involved in several neurological and psychiatric disorders.
Direct targeting of calpains does not appear to be a viable approach to treat these disorders.
Identification of key calpain substrates in specific brain diseases is required to further develop calpain-based therapies.
This box summarizes key points contained in the article.
Acknowledgments
This work was supported by grants P01NS045260-01 from NINDS (PI: CM Gall), and grant R01NS057128 from NINDS to M Baudry. X Bi is also supported by funds from the Daljit and Elaine Sarkaria Chair.
Footnotes
Declaration of interest
The authors declare no other conflict of interest.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1••.Lynch G, Baudry M. The biochemistry of memory: a new and specific hypothesis. Science. 1984;224:1057–63. doi: 10.1126/science.6144182. This was the first publication proposing a critical role for calpain in learning and memory. [DOI] [PubMed] [Google Scholar]
- 2•.Liu J, Liu MC, Wang KK. Physiological and pathological actions of calpains in glutamatergic neurons. Sci Signal. 2008;1:tr3. doi: 10.1126/scisignal.123tr3. Interesting review of the roles of calpains in plasticity and neurodegeneration. [DOI] [PubMed] [Google Scholar]
- 3•.Wu HY, Lynch DR. Calpain and synaptic function. Mol Neurobiol. 2006;33:215–36. doi: 10.1385/MN:33:3:215. Interesting review of the roles of calpains in plasticity and neurodegeneration. [DOI] [PubMed] [Google Scholar]
- 4.Denny JB, Polan-Curtain J, Ghuman A, et al. Calpain inhibitors block long-term potentiation. Brain Res. 1990;534:317–20. doi: 10.1016/0006-8993(90)90148-5. [DOI] [PubMed] [Google Scholar]
- 5.del Cerro S, Larson J, Oliver MW, et al. Development of hippocampal long-term potentiation is reduced by recently introduced calpain inhibitors. Brain Res. 1990;530:91–5. doi: 10.1016/0006-8993(90)90660-4. [DOI] [PubMed] [Google Scholar]
- 6.Oliver MW, Baudry M, Lynch G. The protease inhibitor leupeptin interferes with the development of LTP in hippocampal slices. Brain Res. 1989;505:233–8. doi: 10.1016/0006-8993(89)91448-0. [DOI] [PubMed] [Google Scholar]
- 7.Staubli U, Larson J, Thibault O, et al. Chronic administration of a thiolproteinase inhibitor blocks long-term potentiation of synaptic responses. Brain Res. 1988;444:153–8. doi: 10.1016/0006-8993(88)90922-5. [DOI] [PubMed] [Google Scholar]
- 8.Muller D, Molinari I, Soldati L, et al. A genetic deficiency in calpastatin and isovalerylcarnitine treatment is associated with enhanced hippocampal long-term potentiation. Synapse. 1995;19:37–45. doi: 10.1002/syn.890190106. [DOI] [PubMed] [Google Scholar]
- 9•.Grammer M, Kuchay S, Chishti A, et al. Lack of phenotype for LTP and fear conditioning learning in calpain 1 knock-out mice. Neurobiol Learn Mem. 2005;84:222–7. doi: 10.1016/j.nlm.2005.07.007. This article raised the critical question of the respective roles of μ- and m-calpain in plasticity and learning and memory. [DOI] [PubMed] [Google Scholar]
- 10.Zadran S, Akopian G, Zadran H, et al. RVG-mediated Calpain2 gene silencing in the brain impairs learning and memory. Neuromolecular Med. 2012 doi: 10.1007/s12017-012-8196-8. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 11.Morris RG, Anderson E, Lynch GS, et al. Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature. 1986;319:774–6. doi: 10.1038/319774a0. [DOI] [PubMed] [Google Scholar]
- 12.Cao G, Xing J, Xiao X, et al. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J Neurosci. 2007;27:9278–93. doi: 10.1523/JNEUROSCI.2826-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorimachi H, Hata S, Ono Y. Impact of genetic insights into calpain biology. J Biochem. 2011;150:23–37. doi: 10.1093/jb/mvr070. [DOI] [PubMed] [Google Scholar]
- 14.Nakagawa K, Masumoto H, Sorimachi H, et al. Dissociation of m-calpain subunits occurs after autolysis of the N-terminus of the catalytic subunit, and is not required for activation. J Biochem. 2001;130:605–11. doi: 10.1093/oxfordjournals.jbchem.a003025. [DOI] [PubMed] [Google Scholar]
- 15.Moldoveanu T, Jia Z, Davies PL. Calpain activation by cooperative Ca2+ binding at two non-EF-hand sites. J Biol Chem. 2004;279:6106–14. doi: 10.1074/jbc.M310460200. [DOI] [PubMed] [Google Scholar]
- 16.Tompa P, Emori Y, Sorimachi H, et al. Domain III of calpain is a ca2+-regulated phospholipid-binding domain. Biochem Biophys Res Commun. 2001;280:1333–9. doi: 10.1006/bbrc.2001.4279. [DOI] [PubMed] [Google Scholar]
- 17.Hosfield CM, Elce JS, Davies PL, et al. Crystal structure of calpain reveals the structural basis for Ca(2+)-dependent protease activity and a novel mode of enzyme activation. Embo J. 1999;18:6880–9. doi: 10.1093/emboj/18.24.6880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cong J, Goll DE, Peterson AM, et al. The role of autolysis in activity of the Ca2+-dependent proteinases (mu-calpain and m-calpain) J Biol Chem. 1989;264:10096–103. [PubMed] [Google Scholar]
- 19.Kapprell HP, Goll DE. Effect of Ca2+ on binding of the calpains to calpastatin. J Biol Chem. 1989;264:17888–96. [PubMed] [Google Scholar]
- 20.Maravall M, Mainen ZF, Sabatini BL, et al. Estimating intracellular calcium concentrations and buffering without wavelength ratioing. Biophys J. 2000;78:2655–67. doi: 10.1016/S0006-3495(00)76809-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schollmeyer JE. Calpain II involvement in mitosis. Science. 1988;240:911–13. doi: 10.1126/science.2834825. [DOI] [PubMed] [Google Scholar]
- 22.Sarin A, Adams DH, Henkart PA. Protease inhibitors selectively block T cell receptor-triggered programmed cell death in a murine T cell hybridoma and activated peripheral T cells. J Exp Med. 1993;178:1693–700. doi: 10.1084/jem.178.5.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Glading A, Bodnar RJ, Reynolds IJ, et al. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol Cell Biol. 2004;24:2499–512. doi: 10.1128/MCB.24.6.2499-2512.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shao H, Chou J, Baty CJ, et al. Spatial localization of m-calpain to the plasma membrane by phosphoinositide biphosphate binding during epidermal growth factor receptor-mediated activation. Mol Cell Biol. 2006;26:5481–96. doi: 10.1128/MCB.02243-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25•.Zadran S, Jourdi H, Rostamiani K, et al. Brain-derived neurotrophic factor and epidermal growth factor activate neuronal m-calpain via mitogen-activated protein kinase-dependent phosphorylation. J Neurosci. 2010;30:1086–95. doi: 10.1523/JNEUROSCI.5120-09.2010. This article showed that BDNF activates m-calpain through ERK-mediated phosphorylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abe K, Takeichi M. NMDA-receptor activation induces calpain-mediated beta-catenin cleavages for triggering gene expression. Neuron. 2007;53:387–97. doi: 10.1016/j.neuron.2007.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Franco SJ, Huttenlocher A. Regulating cell migration: calpains make the cut. J Cell Sci. 2005;118:3829–38. doi: 10.1242/jcs.02562. [DOI] [PubMed] [Google Scholar]
- 28.Glading A, Lauffenburger DA, Wells A. Cutting to the chase: calpain proteases in cell motility. Trends Cell Biol. 2002;12:46–54. doi: 10.1016/s0962-8924(01)02179-1. [DOI] [PubMed] [Google Scholar]
- 29.Tomimatsu Y, Idemoto S, Moriguchi S, et al. Proteases involved in long-term potentiation. Life Sci. 2002;72:355–61. doi: 10.1016/s0024-3205(02)02285-3. [DOI] [PubMed] [Google Scholar]
- 30.Wang KK. Calpain and caspase: can you tell the difference? Trends Neurosci. 2000;23:20–6. doi: 10.1016/s0166-2236(99)01479-4. [DOI] [PubMed] [Google Scholar]
- 31.Crocker SJ, Smith PD, Jackson-Lewis V, et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J Neurosci. 2003;23:4081–91. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gafni J, Ellerby LM. Calpain activation in Huntington’s disease. J Neurosci. 2002;22:4842–9. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33•.Saito K, Elce JS, Hamos JE, et al. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA. 1993;90:2628–32. doi: 10.1073/pnas.90.7.2628. Interesting paper on the role of calpain in Alzheimer’s disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34•.Xu W, Wong TP, Chery N, et al. Calpain-mediated mGluR1alpha truncation: a key step in excitotoxicity. Neuron. 2007;53:399–412. doi: 10.1016/j.neuron.2006.12.020. Interesting paper showing the potential use of a peptide to specifically inhibit calpain-mediated truncation of a specific calpain target. [DOI] [PubMed] [Google Scholar]
- 35•.Donkor IO. Calpain inhibitors: a survey of compounds reported in the patent and scientific literature. Expert Opin Ther Patents. 2011;21:601–36. doi: 10.1517/13543776.2011.568480. Previous review related to the application of calpain inhibitors for various indications. [DOI] [PubMed] [Google Scholar]
- 36.Hanna RA, Campbell RL, Davies PL. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature. 2008;456:409–12. doi: 10.1038/nature07451. [DOI] [PubMed] [Google Scholar]
- 37.Carlin RK, Bartelt DC, Siekevitz P. Identification of fodrin as a major calmodulin-binding protein in postsynaptic density preparations. J Cell Biol. 1983;96:443–8. doi: 10.1083/jcb.96.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennett V. Spectrin-based membrane skeleton: a multipotential adaptor between plasma membrane and cytoplasm. Physiol Rev. 1990;70:1029–65. doi: 10.1152/physrev.1990.70.4.1029. [DOI] [PubMed] [Google Scholar]
- 39.Banik NL, Matzelle DC, Gantt-Wilford G, et al. Increased calpain content and progressive degradation of neurofilament protein in spinal cord injury. Brain Res. 1997;752:301–6. doi: 10.1016/s0006-8993(96)01488-6. [DOI] [PubMed] [Google Scholar]
- 40.Fischer I, Romano-Clarke G, Grynspan F. Calpain-mediated proteolysis of microtubule associated proteins MAP1B and MAP2 in developing brain. Neurochem Res. 1991;16:891–8. doi: 10.1007/BF00965538. [DOI] [PubMed] [Google Scholar]
- 41.Johnson GV, Litersky JM, Jope RS. Degradation of microtubule-associated protein 2 and brain spectrin by calpain: a comparative study. J Neurochem. 1991;56:1630–8. doi: 10.1111/j.1471-4159.1991.tb02061.x. [DOI] [PubMed] [Google Scholar]
- 42.Potter DA, Tirnauer JS, Janssen R, et al. Calpain regulates actin remodeling during cell spreading. J Cell Biol. 1998;141:647–62. doi: 10.1083/jcb.141.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perrin BJ, Amann KJ, Huttenlocher A. Proteolysis of cortactin by calpain regulates membrane protrusion during cell migration. Mol Biol Cell. 2006;17:239–50. doi: 10.1091/mbc.E05-06-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dulong S, Goudenege S, Vuillier-Devillers K, et al. Myristoylated alanine-rich C kinase substrate (MARCKS) is involved in myoblast fusion through its regulation by protein kinase Calpha and calpain proteolytic cleavage. Biochem J. 2004;382:1015–23. doi: 10.1042/BJ20040347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carroll RC, Zukin RS. NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci. 2002;25:571–7. doi: 10.1016/s0166-2236(02)02272-5. [DOI] [PubMed] [Google Scholar]
- 46.Lynch DR, Guttmann RP. Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. J Pharmacol Exp Ther. 2002;300:717–23. doi: 10.1124/jpet.300.3.717. [DOI] [PubMed] [Google Scholar]
- 47.Wu HY, Yuen EY, Lu YF, et al. Regulation of N-methyl-D-aspartate receptors by calpain in cortical neurons. J Biol Chem. 2005;280:21588–93. doi: 10.1074/jbc.M501603200. [DOI] [PubMed] [Google Scholar]
- 48•.Bi X, Chen J, Dang S, et al. Characterization of calpain-mediated proteolysis of GluR1 subunits of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate receptors in rat brain. J Neurochem. 1997;68:1484–94. doi: 10.1046/j.1471-4159.1997.68041484.x. First report indicating that calpain activation could truncate the C-terminal domain of AMPA receptors. [DOI] [PubMed] [Google Scholar]
- 49.Lu X, Rong Y, Bi R, et al. Calpain-mediated truncation of rat brain AMPA receptors increases their Triton X-100 solubility. Brain Res. 2000;863:143–50. doi: 10.1016/s0006-8993(00)02112-0. [DOI] [PubMed] [Google Scholar]
- 50.Bano D, Young KW, Guerin CJ, et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–85. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- 51.Hell JW, Westenbroek RE, Breeze LJ, et al. N-methyl-D-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci USA. 1996;93:3362–7. doi: 10.1073/pnas.93.8.3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Magnusson A, Haug LS, Walaas SI, et al. Calcium-induced degradation of the inositol (1,4,5)-trisphosphate receptor/Ca(2+)-channel. FEBS Lett. 1993;323:229–32. doi: 10.1016/0014-5793(93)81345-z. [DOI] [PubMed] [Google Scholar]
- 53.Dong YN, Waxman EA, Lynch DR. Interactions of postsynaptic density-95 and the NMDA receptor 2 subunit control calpain-mediated cleavage of the NMDA receptor. J Neurosci. 2004;24:11035–45. doi: 10.1523/JNEUROSCI.3722-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jourdi H, Lu X, Yanagihara T, et al. Prolonged positive modulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors induces calpain-mediated PSD-95/Dlg/ZO-1 protein degradation and AMPA receptor down-regulation in cultured hippocampal slices. J Pharmacol Exp Ther. 2005;314:16–26. doi: 10.1124/jpet.105.083873. [DOI] [PubMed] [Google Scholar]
- 55.Lu X, Rong Y, Baudry M. Calpain-mediated degradation of PSD-95 in developing and adult rat brain. Neurosci Lett. 2000;286:149–53. doi: 10.1016/s0304-3940(00)01101-0. [DOI] [PubMed] [Google Scholar]
- 56.Sans N, Petralia RS, Wang YX, et al. A developmental change in NMDA receptor-associated proteins at hippocampal synapses. J Neurosci. 2000;20:1260–71. doi: 10.1523/JNEUROSCI.20-03-01260.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jourdi H, Iwakura Y, Narisawa-Saito M, et al. Brain-derived neurotrophic factor signal enhances and maintains the expression of AMPA receptor-associated PDZ proteins in developing cortical neurons. Dev Biol. 2003;263:216–30. doi: 10.1016/j.ydbio.2003.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu L, Rostamiani K, Hsu YT, et al. Calpain-mediated regulation of stargazin in adult rat brain. Neuroscience. 2011;178:13–20. doi: 10.1016/j.neuroscience.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukunaga K, Soderling TR, Miyamoto E. Activation of Ca2+/calmodulin-dependent protein kinase II and protein kinase C by glutamate in cultured rat hippocampal neurons. J Biol Chem. 1992;267:22527–33. [PubMed] [Google Scholar]
- 60.Tan SE, Wenthold RJ, Soderling TR. Phosphorylation of AMPA-type glutamate receptors by calcium/calmodulin-dependent protein kinase II and protein kinase C in cultured hippocampal neurons. J Neurosci. 1994;14:1123–9. doi: 10.1523/JNEUROSCI.14-03-01123.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hajimohammadreza I, Raser KJ, Nath R, et al. Neuronal nitric oxide synthase and calmodulin-dependent protein kinase IIalpha undergo neurotoxin-induced proteolysis. J Neurochem. 1997;69:1006–13. doi: 10.1046/j.1471-4159.1997.69031006.x. [DOI] [PubMed] [Google Scholar]
- 62.Hrabetova S, Sacktor TC. Bidirectional regulation of protein kinase M zeta in the maintenance of long-term potentiation and long-term depression. J Neurosci. 1996;16:5324–33. doi: 10.1523/JNEUROSCI.16-17-05324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu HY, Tomizawa K, Oda Y, et al. Critical role of calpain-mediated cleavage of calcineurin in excitotoxic neurodegeneration. J Biol Chem. 2004;279:4929–40. doi: 10.1074/jbc.M309767200. [DOI] [PubMed] [Google Scholar]
- 64.Denny JB, Polan-Curtain J, Rodriguez S, et al. Evidence that protein kinase M does not maintain long-term potentiation. Brain Res. 1990;534:201–8. doi: 10.1016/0006-8993(90)90130-4. [DOI] [PubMed] [Google Scholar]
- 65.Malinow R, Schulman H, Tsien RW. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science. 1989;245:862–6. doi: 10.1126/science.2549638. [DOI] [PubMed] [Google Scholar]
- 66.Otmakhov N, Griffith LC, Lisman JE. Postsynaptic inhibitors of calcium/calmodulin-dependent protein kinase type II block induction but not maintenance of pairing-induced long-term potentiation. J Neurosci. 1997;17:5357–65. doi: 10.1523/JNEUROSCI.17-14-05357.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hu GY, Hvalby O, Walaas SI, et al. Protein kinase C injection into hippocampal pyramidal cells elicits features of long term potentiation. Nature. 1987;328:426–9. doi: 10.1038/328426a0. [DOI] [PubMed] [Google Scholar]
- 68.Lledo PM, Hjelmstad GO, Mukherji S, et al. Calcium/calmodulin-dependent kinase II and long-term potentiation enhance synaptic transmission by the same mechanism. Proc Natl Acad Sci USA. 1995;92:11175–9. doi: 10.1073/pnas.92.24.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu L, Deng X. Suppression of cancer cell migration and invasion by protein phosphatase 2A through dephosphorylation of mu-and m-calpains. J Biol Chem. 2006;281:35567–75. doi: 10.1074/jbc.M607702200. [DOI] [PubMed] [Google Scholar]
- 70.Tallant EA, Brumley LM, Wallace RW. Activation of a calmodulin-dependent phosphatase by a Ca2+-dependent protease. Biochemistry. 1988;27:2205–11. doi: 10.1021/bi00406a059. [DOI] [PubMed] [Google Scholar]
- 71.Baumgartel K, Mansuy IM. Neural functions of calcineurin in synaptic plasticity and memory. Learn Mem. 2012;19:375–84. doi: 10.1101/lm.027201.112. [DOI] [PubMed] [Google Scholar]
- 72.Evergren E, Marcucci M, Tomilin N, et al. Amphiphysin is a component of clathrin coats formed during synaptic vesicle recycling at the lamprey giant synapse. Traffic. 2004;5:514–28. doi: 10.1111/j.1398-9219.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 73.Wigge P, McMahon HT. The amphiphysin family of proteins and their role in endocytosis at the synapse. Trends Neurosci. 1998;21:339–44. doi: 10.1016/s0166-2236(98)01264-8. [DOI] [PubMed] [Google Scholar]
- 74.Zhang B, Zelhof AC. Amphiphysins: raising the BAR for synaptic vesicle recycling and membrane dynamics. Bin-Amphiphysin-Rvsp Traffic. 2002;3:452–60. doi: 10.1034/j.1600-0854.2002.30702.x. [DOI] [PubMed] [Google Scholar]
- 75.Wu Y, Liang S, Oda Y, et al. Truncations of amphiphysin I by calpain inhibit vesicle endocytosis during neural hyperexcitation. Embo J. 2007;26:2981–90. doi: 10.1038/sj.emboj.7601741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76••.Di Rosa G, Odrijin T, Nixon RA, et al. Calpain inhibitors: a treatment for Alzheimer’s disease. J Mol Neurosci. 2002;19:135–41. doi: 10.1007/s12031-002-0024-4. First article suggesting the use of calpain inhibitors for the treatment of Alzheimer’s disease. [DOI] [PubMed] [Google Scholar]
- 77.Ando K, Kudo Y, Takahashi M. Negative regulation of neurotransmitter release by calpain: a possible involvement of specific SNAP-25 cleavage. J Neurochem. 2005;94:651–8. doi: 10.1111/j.1471-4159.2005.03160.x. [DOI] [PubMed] [Google Scholar]
- 78.Grumelli C, Berghuis P, Pozzi D, et al. Calpain activity contributes to the control of SNAP-25 levels in neurons. Mol Cell Neurosci. 2008;39:314–23. doi: 10.1016/j.mcn.2008.07.011. [DOI] [PubMed] [Google Scholar]
- 79.Bi X, Chen J, Baudry M. Developmental changes in calpain activity, GluR1 receptors and in the effect of kainic acid treatment in rat brain. Neuroscience. 1997;81:1123–35. doi: 10.1016/s0306-4522(97)00218-2. [DOI] [PubMed] [Google Scholar]
- 80.Carragher NO, Frame MC. Calpain: a role in cell transformation and migration. Int J Biochem Cell Biol. 2002;34:1539–43. doi: 10.1016/s1357-2725(02)00069-9. [DOI] [PubMed] [Google Scholar]
- 81.Lebart MC, Benyamin Y. Calpain involvement in the remodeling of cytoskeletal anchorage complexes. FEBS J. 2006;273:3415–26. doi: 10.1111/j.1742-4658.2006.05350.x. [DOI] [PubMed] [Google Scholar]
- 82.Perrin BJ, Huttenlocher A. Calpain. Int J Biochem Cell Biol. 2002;34:722–5. doi: 10.1016/s1357-2725(02)00009-2. [DOI] [PubMed] [Google Scholar]
- 83.Vanderklish P, Saido TC, Gall C, et al. Proteolysis of spectrin by calpain accompanies theta-burst stimulation in cultured hippocampal slices. Mol Brain Res. 1995;32:25–35. doi: 10.1016/0169-328x(95)00057-y. [DOI] [PubMed] [Google Scholar]
- 84•.Flevaris P, Stojanovic A, Gong H, et al. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179:553–65. doi: 10.1083/jcb.200703185. Interesting review proposing a dual role for calpain in cell motility. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Szczepanowska J. Involvement of Rac/Cdc42/PAK pathway in cytoskeletal rearrangements. Acta Biochim Pol. 2009;56:225–34. [PubMed] [Google Scholar]
- 86.Kulkarni S, Goll DE, Fox JE. Calpain cleaves RhoA generating a dominant-negative form that inhibits integrin-induced actin filament assembly and cell spreading. J Biol Chem. 2002;277:24435–41. doi: 10.1074/jbc.M203457200. [DOI] [PubMed] [Google Scholar]
- 87.Du X, Saido TC, Tsubuki S, et al. Calpain cleavage of the cytoplasmic domain of the integrin beta 3 subunit. J Biol Chem. 1995;270:26146–51. doi: 10.1074/jbc.270.44.26146. [DOI] [PubMed] [Google Scholar]
- 88.Sawhney RS, Cookson MM, Omar Y, et al. Integrin alpha2-mediated ERK and calpain activation play a critical role in cell adhesion and motility via focal adhesion kinase signaling: identification of a novel signaling pathway. J Biol Chem. 2006;281:8497–510. doi: 10.1074/jbc.M600787200. [DOI] [PubMed] [Google Scholar]
- 89•.Chan KT, Bennin DA, Huttenlocher A. Regulation of adhesion dynamics by calpain-mediated proteolysis of focal adhesion kinase (FAK) J Biol Chem. 2010;285:11418–26. doi: 10.1074/jbc.M109.090746. Interesting paper describing the role of calpain in regulating cell adhesion through FAK truncation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shi Y, Pontrello CG, DeFea KA, et al. Focal adhesion kinase acts downstream of EphB receptors to maintain mature dendritic spines by regulating cofilin activity. J Neurosci. 2009;29:8129–42. doi: 10.1523/JNEUROSCI.4681-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Faivre-Sarrailh C, Lena JY, Had L, et al. Location of profilin at presynaptic sites in the cerebellar cortex; implication for the regulation of the actin-polymerization state during axonal elongation and synaptogenesis. J Neurocytol. 1993;22:1060–72. doi: 10.1007/BF01235749. [DOI] [PubMed] [Google Scholar]
- 92.Gungabissoon RA, Bamburg JR. Regulation of growth cone actin dynamics by ADF/cofilin. J Histochem Cytochem. 2003;51:411–20. doi: 10.1177/002215540305100402. [DOI] [PubMed] [Google Scholar]
- 93.Hatada Y, Wu F, Sun ZY, et al. Presynaptic morphological changes associated with long-term synaptic facilitation are triggered by actin polymerization at preexisting varicositis. J Neurosci. 2000;20:RC82. doi: 10.1523/JNEUROSCI.20-13-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kuhn TB, Meberg PJ, Brown MD, et al. Regulating actin dynamics in neuronal growth cones by ADF/cofilin and rho family GTPases. J Neurobiol. 2000;44:126–44. [PubMed] [Google Scholar]
- 95.Pham H, Yu H, Laski FA. Cofilin/ADF is required for retinal elongation and morphogenesis of the Drosophila rhabdomere. Dev Biol. 2008;318:82–91. doi: 10.1016/j.ydbio.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosenmund C, Westbrook GL. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron. 1993;10:805–14. doi: 10.1016/0896-6273(93)90197-y. [DOI] [PubMed] [Google Scholar]
- 97.Meng Y, Zhang Y, Tregoubov V, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–33. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 98.Nishita M, Tomizawa C, Yamamoto M, et al. Spatial and temporal regulation of cofilin activity by LIM kinase and Slingshot is critical for directional cell migration. J Cell Biol. 2005;171:349–59. doi: 10.1083/jcb.200504029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ammer AG, Weed SA. Cortactin branches out: roles in regulating protrusive actin dynamics. Cell Motil Cytoskeleton. 2008;65:687–707. doi: 10.1002/cm.20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hering H, Sheng M. Activity-dependent redistribution and essential role of cortactin in dendritic spine morphogenesis. J Neurosci. 2003;23:11759–69. doi: 10.1523/JNEUROSCI.23-37-11759.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Huang C, Tandon NN, Greco NJ, et al. Proteolysis of platelet cortactin by calpain. J Biol Chem. 1997;272:19248–52. doi: 10.1074/jbc.272.31.19248. [DOI] [PubMed] [Google Scholar]
- 102.Mingorance-Le Meur A, O’Connor TP. Neurite consolidation is an active process requiring constant repression of protrusive activity. EMBO J. 2009;28:248–60. doi: 10.1038/emboj.2008.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Routtenberg A, Rekart JL. Post-translational protein modification as the substrate for long-lasting memory. Trends Neurosci. 2005;28:12–19. doi: 10.1016/j.tins.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 104.Kelleher RJ, III, Govindarajan A, Tonegawa S. Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron. 2004;44:59–73. doi: 10.1016/j.neuron.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 105.Frey U, Morris RG. Synaptic tagging: implications for late maintenance of hippocampal long-term potentiation. Trends Neurosci. 1998;21:181–8. doi: 10.1016/s0166-2236(97)01189-2. [DOI] [PubMed] [Google Scholar]
- 106.Bailey CH, Bartsch D, Kandel ER. Toward a molecular definition of long-term memory storage. Proc Natl Acad Sci USA. 1996;93:13445–52. doi: 10.1073/pnas.93.24.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schuman EM, Dynes JL, Steward O. Synaptic regulation of translation of dendritic mRNAs. J Neurosci. 2006;26:7143–6. doi: 10.1523/JNEUROSCI.1796-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Steward O, Schuman EM. Compartmentalized synthesis and degradation of proteins in neurons. Neuron. 2003;40:347–59. doi: 10.1016/s0896-6273(03)00635-4. [DOI] [PubMed] [Google Scholar]
- 109.Pfeiffer BE, Huber KM. Current advances in local protein synthesis and synaptic plasticity. J Neurosci. 2006;26:7147–50. doi: 10.1523/JNEUROSCI.1797-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tang SJ, Reis G, Kang H, et al. A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA. 2002;99:467–72. doi: 10.1073/pnas.012605299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nguyen PV. Protein synthesis during LTP: linking synaptic activity to translation. Trends Neurosci. 2002;25:180. doi: 10.1016/s0166-2236(02)02166-5. [DOI] [PubMed] [Google Scholar]
- 112.Bradshaw KD, Emptage NJ, Bliss TV. A role for dendritic protein synthesis in hippocampal late LTP. Eur J Neurosci. 2003;18:3150–2. doi: 10.1111/j.1460-9568.2003.03054.x. [DOI] [PubMed] [Google Scholar]
- 113.Gold PE. Protein synthesis inhibition and memory: formation vs amnesia. Neurobiol Learn Mem. 2008;89:201–11. doi: 10.1016/j.nlm.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kramar EA, Lin B, Lin CY, et al. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24:5151–61. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115•.Lynch G, Rex CS, Gall CM. LTP consolidation: substrates, explanatory power, and functional significance. Neuropharmacology. 2007;52:12–23. doi: 10.1016/j.neuropharm.2006.07.027. Excellent review of the mechanisms involved in long-term potentiation. [DOI] [PubMed] [Google Scholar]
- 116.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–62. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 117.Schuman EM. Neurotrophin regulation of synaptic transmission. Curr Opin Neurobiol. 1999;9:105–9. doi: 10.1016/s0959-4388(99)80013-0. [DOI] [PubMed] [Google Scholar]
- 118.Kanhema T, Dagestad G, Panja D, et al. Dual regulation of translation initiation and peptide chain elongation during BDNF-induced LTP in vivo: evidence for compartment-specific translation control. J Neurochem. 2006;99:1328–37. doi: 10.1111/j.1471-4159.2006.04158.x. [DOI] [PubMed] [Google Scholar]
- 119.Soule J, Messaoudi E, Bramham CR. Brain-derived neurotrophic factor and control of synaptic consolidation in the adult brain. Biochem Soc Trans. 2006;34:600–4. doi: 10.1042/BST0340600. [DOI] [PubMed] [Google Scholar]
- 120.Rao VR, Pintchovski SA, Chin J, et al. AMPA receptors regulate transcription of the plasticity-related immediate-early gene Arc. Nat Neurosci. 2006;9:887–95. doi: 10.1038/nn1708. [DOI] [PubMed] [Google Scholar]
- 121.Bramham CR, Messaoudi E. BDNF function in adult synaptic plasticity: the synaptic consolidation hypothesis. Prog Neurobiol. 2005;76:99–125. doi: 10.1016/j.pneurobio.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 122.Sutton MA, Schuman EM. Local translational control in dendrites and its role in long-term synaptic plasticity. J Neurobiol. 2005;64:116–31. doi: 10.1002/neu.20152. [DOI] [PubMed] [Google Scholar]
- 123.Giovannini MG. The role of the extracellular signal-regulated kinase pathway in memory encoding. Rev Neurosci. 2006;17:619–34. doi: 10.1515/revneuro.2006.17.6.619. [DOI] [PubMed] [Google Scholar]
- 124.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–17. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 125.Shimizu K, Phan T, Mansuy IM, et al. Proteolytic degradation of SCOP in the hippocampus contributes to activation of MAP kinase and memory. Cell. 2007;128:1219–29. doi: 10.1016/j.cell.2006.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Oliver MW, Baudry M, Lynch G. The protease inhibitor leupeptin interferes with the development of LTP in hippocampal slices. Brain Res. 1989;505:233–8. doi: 10.1016/0006-8993(89)91448-0. [DOI] [PubMed] [Google Scholar]
- 127.Vanderklish P, Bednarski E, Lynch G. Translational suppression of calpain blocks long-term potentiation. Learn Mem. 1996;3:209–17. doi: 10.1101/lm.3.2-3.209. [DOI] [PubMed] [Google Scholar]
- 128•.Murachi T. Calcium-dependent proteinases and specific inhibitors: calpain and calpastatin. Biochem Soc Symp. 1984;49:149–67. Interesting review by the “grandfather” of calpain. [PubMed] [Google Scholar]
- 129.Gartner A, Staiger V. Neurotrophin secretion from hippocampal neurons evoked by long-term-potentiation-inducing electrical stimulation patterns. Proc Natl Acad Sci USA. 2002;99:6386–91. doi: 10.1073/pnas.092129699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Aicardi G, Argilli E, Cappello S, et al. Induction of long-term potentiation and depression is reflected by corresponding changes in secretion of endogenous brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 2004;101:15788–92. doi: 10.1073/pnas.0406960101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Emes RD, Grant SG. The human postsynaptic density shares conserved elements with proteomes of unicellular eukaryotes and prokaryotes. Front Neurosci. 2011;5:44. doi: 10.3389/fnins.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132••.Zhao S, Liang Z, Demko V, et al. Massive expansion of the calpain gene family in unicellular eukaryotes. BMC Evol Biol. 2012;12:193. doi: 10.1186/1471-2148-12-193. Important review of the evolutionary origin of the calpain family. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133•.Leloup L, Wells A. Calpains as potential anti-cancer targets. Expert Opin Ther Targets. 2011;15:309–23. doi: 10.1517/14728222.2011.553611. Previous review of the potential use of calpain inhibitors for the treatment of cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yamada M, Hirotsune S, Wynshaw-Boris A. A novel strategy for therapeutic intervention for the genetic disease: preventing proteolytic cleavage using small chemical compound. Int J Biochem Cell Biol. 2010;42:1401–7. doi: 10.1016/j.biocel.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Simoes AT, Goncalves N, Koeppen A, et al. Calpastatin-mediated inhibition of calpains in the mouse brain prevents mutant ataxin 3 proteolysis, nuclear localization and aggregation, relieving Machado-Joseph disease. Brain. 2012;135:2428–39. doi: 10.1093/brain/aws177. [DOI] [PubMed] [Google Scholar]
- 136.Higuchi M, Tomioka M, Takano J, et al. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J Biol Chem. 2005;280:15229–37. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- 137.Vosler PS, Brennan CS, Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138•.Zatz M, Starling A. Calpains and disease. N Engl J Med. 2005;352:2413–23. doi: 10.1056/NEJMra043361. Review of the roles of calpains in multiple diseases. [DOI] [PubMed] [Google Scholar]
- 139.Wei X, Miou Z, Baudry M. Neuroprotection by cell permeable TAT-mGluR1 peptide in ischemia: synergy between carrier and cargo sequences. Neuroscientist. 2008;14:409–14. doi: 10.1177/1073858407309762. [DOI] [PubMed] [Google Scholar]