

Abstract
Background & Aims
Colon cancers with high-frequency microsatellite instability (MSI-H) develop frameshift mutations in tumor suppressors as part of their pathogenesis. ACVR2 is mutated at its exon 10 polyadenine tract in >80% of MSI-H colon cancers, coinciding with loss of protein. ACVR2 transmits the growth effects of activin via phosphorylation of SMAD proteins to affect gene transcription. The functional effect of activin in colon cancers has not been studied. We developed and characterized a cell model in which we studied how activin signaling affects growth.
Methods
hMLH1 and ACVR2 mutant HCT116 cells were previously stably transferred with chromosome 2 (HCT116+chr2), restoring a single regulated copy of wild-type ACVR2 but not hMLH1. Both HCT116+chr2 and parental HCT116 cells (as well as HEC59 and ACVR2 and hMSH2 complemented HEC59+chr2 cells) were assessed for genetic complementation and biologic function.
Results
HCT116+chr2 cells and HEC59+chr2 cells, but not ACVR2-mutant HCT116 or HEC59 cells, acquired wild-type ACVR2 as well as expression of ACVR2 wild-type messenger RNA. Complemented ACVR2 protein complexed with ACVR1 with activin treatment, generating nuclear phosphoSMAD2 and activin-specific gene transcription. ACVR2-restored cells showed decreased growth and reduced S phase but increased cellular migration following activin treatment. ACVR2 small interfering RNA reversed these effects in complemented cells.
Conclusions
ACVR2-complemented MSI-H colon cancers restore activin-SMAD signaling, decrease growth, and slow their cell cycle following ligand stimulation but show increased cellular migration. Activin is growth suppressive and enhances migration similar to transforming growth factor β in colon cancer, indicating that abrogation of the effects of activin contribute to the pathogenesis of MSI-H colon cancers.
Approximately 15% of colon cancers display microsatellite instability and have a unique genetic makeup consisting of frameshifted target genes that drive their pathogenesis. Key growth and apoptosis regulators, such as TGFBR2 and BAX, contain coding microsatellites that become mutated with subsequent loss of transforming growth factor (TGF)-β signaling or apoptotic responses, respectively.1 More recently, ACVR2 was shown to be mutated in the majority of high-frequency microsatellite instability (MSI-H) colon cancers, with loss of protein expression.2 However, the functional consequences of loss of activin signaling in MSI-H colon cancer are unknown.
Activin is a member of the TGF-β superfamily that can regulate cell differentiation, proliferation, and apoptosis in a variety of cancer cell types.3,4 Similar to TGF-β, activin utilizes 2 types of cell surface receptors to transduce its signal. Both activin receptor 1 (ACVR1) and activin receptor 2 (ACVR2), as well as ACVR2B, are trans-membrane proteins with ligand-binding activity in the extracellular domain and serine/threonine kinases in their cytoplasmic domain. ACVR2 is located on chromosome 2 and harbors 2 coding polyadenine tracts. ACVR2B, located on chromosome 3, is 69% identical to ACVR2 and has similar binding characteristics to ACVR2 but does not contain any polyadenine tract. Control of expression of both receptors appears to occur in a tissue-and gene-specific manner during human development.5 Potential ligands for ACVR2 include activin A, activin B, and inhibin A, but not TGF-β1.6 Activin A is secreted by most cell types, is commercially available, and is the activin ligand most commonly used for studies published in the literature.
Activin signaling induces growth inhibition in T47D human breast cancer cells3,7 and LNCaP prostate cancer cells4 and induces apoptosis in hematopoietic and myeloid leukemia cells.8,9 Cell adhesion proteins are up-regulated through activin signaling in chicken limb buds, suggesting a role in cell anchoring.10 In normal keratinocytes, activin induces expression of Mad1, a c-MYC proto-oncogene antagonist.11 While Mad1 overexpression inhibits the activity of activin and TGF-β–induced transcriptional stimulation in a variety of mammalian cells, overexpression of Mad3 potentiates activin-induced transcription.12 Additionally, activin signaling may cause direct down-regulation of the proto-oncogene c-MYC in prostate cancer cells4 as well as an increase in hypophosphorylated retinoblastoma protein in hepatoma cell lines, indicative of decreased S-phase entry.13 In the same model, p21 induction was observed following activin treatment. Defective activin signaling drives carcinogenesis, and blocking activin signaling in breast cancer cells abrogates activin-mediated growth arrest.3 Activin has been implicated in tumor formation of the pituitary and ovaries,14,15 and loss-of-function mutations in ACVR1 have been described in human pituitary tumors.16 Somatic mutations in ACVR1 have been demonstrated in pancreatic cancer,17 and somatic mutation and loss of heterozygosity at the ACVR2 locus have been described in pancreatic carcinoma.18 Mutated ACVR2 abolishes activin-mediated erythroid differentiation,19 further underscoring its role of a potential tumor suppressor.
Mutant ACVR2 has been implicated in colon carcinogenesis. Through a genome-wide search, 58% of 46 colon cancer cell lines with MSI-H were found to harbor mutant ACVR2.20 A second study revealed that 92% of 24 colon cancer cell lines and xenografts with MSI-H were ACVR2 mutated,18 all primarily due to a frameshift in the A8 tract of exon 10. We demonstrated a similarly high mutational rate in primary colon cancer specimens, accompanied by loss of ACVR2 protein expression in the majority of colon cancers.2 Our primary human tumor data underscore the potential importance of activin signaling in colon cancer. To date, little is known about the mechanisms and cellular effects of activin signaling in colon cancer. Here, we explored the effects of ACVR2 restoration in cell models of MSI-H in which ACVR2 is mutated. We found that activin is growth suppressive in our single-copy, gene-complemented colon cancer cell models and enhances cellular migration. These findings are similar to the effects of TGF-β in colon cancer cells and indicate that loss of activin signaling contributes to the pathogenesis of MSI-H colon cancers.
Materials and Methods
Ligand
Activin A (R&D, Minneapolis, MN) was reconstituted in phosphate-buffered saline (PBS) and used at a final concentration of 25 ng/mL as described elsewhere.21
Antibodies
Rabbit anti-TyrGly mACVR2 (482–494) (a generous gift from W. Vale, Salk Institute), with its target epitope in the C-terminus region of ACVR2 (beyond the resulting truncation from frameshift in exon 10), was used for Western blotting at 1:800; rabbit anti-TyrGly ACVR1 (474–494) and rabbit anti-TyrGly ACVR2B (524–536) (also gifts from W. Vale, Salk Institute) were both used at 1:800; pSMAD2 (Cell Signaling, Beverly, MA) was used at 1:250; SMAD2 (Upstate, Lake Placid, NY) was used at 1:400; and α-tubulin and α-histone (Santa Cruz Biotechnology, Santa Cruz, CA) were used at 1:250 and 1:500, respectively
Cell Lines
Both ACVR2-mutated HCT116 cells and ACVR2-restored HCT116+chr2 cells were created as described previously.22 We also utilized another cell line, the MSI-H endometrial cell line HEC59, as well as its microsatellite stable and ACVR2-restored counterpart HEC59+chr2,23 the TGF-β–responsive human colon carcinoma cell line FET,24 and the ACVR2- and ACVR1-expressing pancreatic cancer CAPAN-1 cells.25 While the results shown are all from one clone of the respective cell lines, experiments were repeated using multiple clones.
Tissue Culture
HCT116 colon cancer and CAPAN-1 pancreatic cancer cells were maintained in Iscove’s modified Dulbec-co’s medium (Invitrogen Corp, Carlsbad, CA) with 10% fetal bovine serum and penicillin G (100 U/mL)/streptomycin (100 μg/mL) (Invitrogen Corp) as supplements. HEC59 and FET cells were maintained in F12/Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum. G418 sulfate (CellGro; Media Tech Inc, Herndon, VA) at 400 μg/mL was added to cultures of HCT116+chr2 and HEC59+chr2 cells to maintain the transferred chromosome 2, which contains an antibiotic selection marker.22,23
ACVR2 Genotyping
We amplified the 2 coding A8 microsatellites of ACVR2 in exon 3 (forward, 5′-TCTGCTTATTTATAG-GACTGATTGTG-3′; reverse, 5′-CGCTGTGTGACTTC-CATCTC-3′) and exon 10 (forward, 5′-GTTGCCATT-TGAGGAGGAAA; reverse, 5′-GCATGTTTCTGCC-AATAATCTC) as previously described.2 DNA was amplified in a thermocycler (MJ Research, Waltham, MA) in a reaction containing the primers, buffer, DNA template, deoxynucleotides, magnesium, and Taq polymerase. After polymerase chain reaction (PCR), the product bands were electrophoresed on a 6% polyacryl-amide gel, dried, and viewed with a phosphorimager (Molecular Dynamics, Sunnyvale, CA) for wild-type or frameshift mutated bands between the cell lines.
Transcriptional Expression of ACVR2 and Activin A
RNA was extracted from HCT116, HCT116+ chr2, HEC59, and HEC59+chr2 cells using a TRIzol-based protocol. Briefly, 1 mL of TRIzol was added per 1 × 107 cells and incubated for 5 minutes. Then, 200 μL of chloroform was added, followed by a centrifugation for 15 minutes with transfer of the aqueous phase to a fresh tube. RNA was precipitated using an equal amount of isopropanol, followed by ethanol washes and drying of the pellet. The concentration of RNA dissolved in DEPC-treated water was assessed using a Beckman Coulter DU640B spectrophotometer (Beckman Coulter, Fullerton, CA). We performed reverse-transcription (RT)-PCR using first an oligo dT for incubation at 37°C and then specific primers for exon 10 (forward, 5′-GTTGCCATT-TGAGGAGGAAA-3′; reverse, 5′-GCATGTTTCTGC-CAATAATCTC-3′) of ACVR2 or activin A (forward, 5′-CAGGATGCCCTTGCTTTGGCTGAGA-3′; reverse, 5′-CCCACATGAAGCTTTCTGATCGCGT-3′) for PCR amplification in a thermocycler (MJ Research) in a reaction containing the primers, buffer, DNA template, deoxynucleotides, magnesium, and Taq polymerase. We subcloned the resulting complementary DNA fragments for ACVR2 utilizing a TA cloning vector (Invitrogen Corp) as per the manufacturer’s protocol. Fifty clones per cell line were then individually sequenced to determine the prevalence of mutated and wild-type ACVR2 transcripts.
DNA Sequencing
PCR products were identified on a 2% agarose gel and DNA extracted from agarose using a gel extraction kit (Qiagen, Valencia, CA) followed by purification using Microcon YM30 columns (Millipore, Bedford, MA). The purified eluate was then used in the sequencing reaction with ABI Prism matrix (Applied Biosystems, Foster City, CA) with either the specific forward or reverse primer for exon 10 of ACVR2 as per the manufacturer’s protocol. Another purification using Centri SEP columns (Prince-ton Separations, Adelphia, NJ) was performed before direct sequencing in an ABI Automated Sequencer (Applied Biosystems).
Immunoprecipitation and Western Blotting
To detect presence of ACVR1, ACVR2, or ACVR2B in the colon cancer cell lines, we immunoprecipitated these receptors, followed by Western blotting. Briefly, HCT116, HCT116+chr2, and ACVR1/ACVR2 wild-type CAPAN-1 cells (as well as HEC59 and HEC59+chr2 cells) were grown to 60% confluency. Fresh media containing activin (25 ng/mL) was added for 4 hours. Protein lysates were obtained using 1 mL immunoprecipitation lysis buffer (2 mmol/L EDTA, 50 μmol/L Tris, pH 7.5, 250 mmol/L NaCl, 1% Triton X-100, 1 mmol/L phenylmethylsulfonyl fluoride [PMSF], 10 μg/mL aprotinin, 5 μg/mL leupeptin, 10 μg/mL soybean, 5 μg/mL antipen, 0.05% sodium dodecyl sulfate) or hypotonic lysis buffer (10 mmol/L Tris-HCl, pH 7.5, 0.1 mol/L EDTA, 0.2% Nonidet P40, 3 mmol/L MgCl, 40 mmol/L NaF, 5 mmol/L glycerolphosphate, 10 μg/mL aprotinin, 0.5 mmol/L benzimidine, 5 μg/mL leupeptin, 1 mmol/L sodium orthovanadate, 0.5 mmol/L PMSF). For immunoprecipitation, lysates were centrifuged for 15 minutes at 5°C and 13,000 rpm and the supernatant was quantified using spectroscopy. A total of 800 μg of protein lysate was used along with 50 μg of rabbit immunoglobulin G as a negative control and incubated overnight with 50 μg of primary antibody and 5 μL of 100 mmol/L PMSF. Fifty microliters of agarose beads (Upstate) and an additional 5 μL PMSF were added, and the suspension was rotated for 3 hours at 4°C. The samples were then washed with PBS, electrophoresed on an 8.5% polyacrylamide loading and 3.9% stacking gel, and Western blotting performed using standard procedures using several PBS-Tween 0.1% washes with appropriate secondary antibody incubation. Blotted proteins were detected with horseradish peroxidase–linked secondary antibodies (Sigma Chemical Co, St Louis, MO) followed by enhanced chemiluminescence detection (Amersham, Little Chalfont, England).
Coimmunoprecipitation
The formation of a heterodimeric ACVR1/ACVR2 complex was assayed by coimmunoprecipitation techniques. Briefly, cells were treated for immunoprecipitation and lysed in freshly made coimmunoprecipitation buffer (2 μg/mL leupeptin, 1 mmol/L PMSF, 50 mmol/L Tris-HCl, 150 mmol/L NaCl, 0.5% Nonidet P40, 5 ng/mL pepstatin). Following overnight incubation with anti-ACVR1, Western blotting was performed with anti-ACVR2 antibodies.
Nuclear and Cytoplasmic Fractionation
Hypotonic lysis buffer (10 mmol/L Tris-HCl, pH 7.4, 3 mmol/L MgCl2, 0.1% Nonidet P40) with added protease and phosphatase inhibitors were used to separate the nuclear-enriched fraction (pellet or P1) from the other compartments (supernatant or S1). Trafficking of pSMAD2 proteins to the nucleus after activin treatment was studied by Western blotting of nuclear and cytoplasmic fractions utilizing antibodies to SMAD2 (Santa Cruz Biotechnology) and phosphoSMAD2 (Santa Cruz Biotechnology) as well as histone H1 (Santa Cruz Biotechnology) and α-tubulin (Sigma Chemical Co) as loading controls and to ensure the purity of each fraction.
Transactivation Assay
Transient transfection of colon cancer cells with the activin specific p(ARE)3-Luc plasmid with a vector expressing FAST1 transcription factors (generous gift from Malcolm Whitman, Harvard Medical School, Boston, MA) was performed to assess the effects of activin on transcription. FAST1 has been shown to associate with SMAD2/SMAD4, coined the activin responsive factor, and bind to an enhancer element called activin responsive element to specify developmental gene expression.26 Reporter vectors (1 μg/well) and the pRL-TK vectors (20 ng/well) were transiently delivered by TransFectin (Pro-mega, Madison, WI) in 12-well plates with a 3:1 ratio of vector to DNA. Twelve to 16 hours posttransfection, cells were treated with 20 ng/mL of activin. Luciferase activity was measured by a dual-luciferase kit (Promega) 16–20 hours after the treatment, and normalization was performed using the Renilla luciferase activity expressed by the cotransfected pRL-TK vector.
Northern Blotting
ACVR2-mutant HCT116 cells and ACVR2-restored HCT116+chr2 cells were treated with activin A (R&D) at 25 ng/mL for 4 hours. RNA was then prepared using a TRIzol/isopropanol-based extraction. Twenty micrograms of RNA per sample was run on a 1.5% agarose gel containing formaldehyde and blotted using a standard protocol. A c-MYC probe was generated by enzymatic digestion from a c-MYC-ER construct (kindly provided by Bruno Amati, Palo Alto, CA) and labeled with 32P using a random primer DNA labeling system (Invitrogen Corp). Hybridization was then performed overnight in 7% sodium dodecyl sulfate hybridization solution. The membrane was exposed to a phosphorimager screen (Molecular Dynamics/Amersham, Piscataway, NJ) after standard washes. Standardization of c-MYC transcripts/glyceralde-3-phosphate dehydrogenase transcripts was performed following a second hybridization with a PCR-generated probe using specific primers for the housekeeping gene GAPDH (forward, 5′-ACCACAGTCCATGC-CATCAC-3′; reverse, 5′-TCCACCACCCTGTTGCTGTA-3′).
3-(4,5-Dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium Bromide Growth Assay
Cells were plated at a density of 10,000–20,000 cells/well in 48-well plates at 6 wells per treatment (control vs activin [25 ng/mL]). The compound 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; MP Biomedicals, Aurora, OH) was added to a concentration of 0.04 mg/mL medium, and the cells were then incubated for 3 hours. Color change indicative of metabolic activity was measured spectrophotometrically. For this, cells were stained for 3 hours with MTT, the reaction product was released by lysis with sodium do-decyl sulfate, and absorbency was detected at 570λ using a Beckman Coulter DU640B spectrophotometer (Beckman Coulter).
ACVR2 Messenger RNA Knockdown via Specific Small Interfering RNA
Transient transfection of HCT116+chr2 colon cancer cells with specific small interfering RNA (siRNA) to ACVR2 was performed to assess for dependence of signaling and functional assays on the wild-type ACVR2 complemented HCT116+chr2 cells. One day before transfection, exponentially growing HCT116+chr2 cells were plated onto 12-well plates at 9 × 104 cells/well. The following day, 100 μmol/L of validated siRNA to activin A receptor type II (Ambion, Austin, TX) was transiently delivered by Lipofectamine 2000 (Invitrogen Corp) at a ratio of 1:1 of siRNA to transfection reagent in OPTI-MEM reduced serum-free media (Gibco, Carlsbad, CA). Forty-eight hours posttransfection, HCT116+chr2 cells were lysed for RNA and protein extraction or assayed for growth with activin A. GAPDH siRNA (Ambion) was also used as a control for specific gene knockdown.
Cell Cycle Analysis
Cell cycle analysis was performed after 24-hour treatment with 25 ng/mL of activin using a FACScan (Becton Dickinson, San Jose, CA). Briefly, cells were grown on 10-cm dishes until 50% confluent. After 24 hours of serum starvation, 10% serum was added with activin (25 ng/mL) for 48 hours. The cells were harvested using 0.05% trypsin, washed with PBS, and resuspended in 0.6 mL of PBS and 1.0 mL of 100% ethanol. The cells were fixed overnight at 4°C. The next day, the cells were centrifuged, washed in PBS, and centrifuged again for 5 minutes. The pellet was resuspended in 0.5 mL of a PBS solution containing 40 U/mL ribonuclease A and 50 μg/mL propidium iodide. The cells were incubated at 37°C for 1 hour and then placed on ice until analysis with a Beckman Coulter Elite Flow Cytometer Multicycle (Phoenix Flow, San Diego, CA).
Migration Assay
HCT116, HEC59, HCT116+chr2, HEC59+chr2, and FET cells were each grown to 80% confluency in a 6-well dish. A wound was created crosswise using a sterile 1000-μL pipette tip, followed by a wash with 1× PBS to remove detached cells. Then, cells were treated with activin (25 ng/mL) in a series of 6 wells in serum-free media, and migration at the corresponding wound site (right above the cross) was documented using an Axiovert 2000 microscope with an AxioCAM HRC Camera (both Zeiss Microimaging, Thornwood, NY) at 0, 1, 2, 4, and 24 hours. Wound distance was then measured in the middle of the image, and the mean and standard deviation of each treatment were determined.
Transwell Migration Assay
After coating Corning Costar Transwell 24-well plates (8-μm pores; Corning, Corning, NY) with 0.1% fibronectin (Sigma Chemical Co) and blocking with 1% bovine serum albumin in 1× PBS for 1 hour at 37°C, HCT116 and HCT116+chr2 cells were seeded in triplicate at 0.5 × 105 cells per well in serum-free media containing 1% bovine serum albumin with or without activin (25 ng/mL). Cells were then allowed to migrate for 4 hours. After removal of media from both the chamber and the Transwell, and followed by 3 washes with 1×PBS, the chamber was gently wiped with a cotton swab. Migrated cells were fixed in 100% methanol for 1 hour and then allowed to air dry overnight. Cell staining was performed with a modified Giemsa stain (Sigma Chemical Co) at 1:20 for 1 hour. After carefully rinsing the Transwell and the chamber with water, images were captured using an Axiovert 2000 microscope with an Axio-CAM HRC Camera (both Zeiss Microimaging) at 10× magnification. Images were taken from 5 microscopic fields at the center of each well, and the stained cells on the bottom of the chamber were counted.
Statistical Analysis
Student t test was used for quantification of growth, cell cycle, and migration analysis, with a P value of .05 indicating statistical significance.
Results
ACVR2 Is Complemented by Stable Chromosome 2 Transfer in MSI-H, ACVR2-Mutant Cells
We tested several microsatellite stable (HCT116+ chr3, FET, SW480, HEC59+chr2) and unstable (HCT116, HCT116+chr2, HEC59) cell lines for mutations in the coding A8 microsatellite of exon 3 and exon 10 of ACVR2. None of the cell lines tested revealed a frameshift in the coding polyadenine tract of exon 3 (data not shown). As expected, the mismatch repair–proficient colon cancer cell lines SW480 and FET possessed wild-type A8 in exon 10. The mismatch repair–deficient colon cancer cell line HCT116, as well as the mismatch repair–deficient endometrial cancer line HEC59, revealed a frameshift mutation from A8 to A7 in exon 10 of ACVR2, with no wild-type A8 present (Figure 1A). Stable transfer of chromosome 2, which harbors a single copy of wild-type ACVR2, restored the wild-type A8 band in HCT116+chr2 and HEC59+chr2 cells (Figure 1A). Transfer of chromosome 3 to HCT116 cells (HCT116+chr3), which harbor the missing mismatch repair gene hMLH1 (Table 1), did not lead to the appearance of a wild-type ACVR2 A8 exon 10 band. Thus, despite missing hMLH1, HCT116+chr2 cells contain a single copy of a stably transferred wild-type ACVR2 in addition to 2 native mutant copies of the gene.
Figure 1.

Chromosome 2 complementation shows the presence of the wild-type ACVR2 allele and leads to expression of wild-type transcripts in HCT116+chr2 and HEC59+chr2 cells. (A) Frameshift mutation of exon 10 of ACVR2 was assessed by amplification of the DNA of the cell line using 32P-labeled primers. Note acquirement of the wild-type A8 allele in chromosome 2–complemented cells. FET and SW480 are microsatellite stable colon cancer cell lines. (B) RNA from each cell line was reverse transcribed, and the complementary DNAs were sub-cloned and sequenced to assess for ACVR2 expression. Note the acquisition of wild-type A8 transcripts (or T8 on the reverse strand) with chromosome 2 complementation.
Table 1.
Cell Lines Used in This Study
| Cell line | Key affected genes | Key restored genes |
|---|---|---|
| HCT116 | ACVR2, TGFBR2, hMLH1 | — |
| HCT116+chr2 | TGFBR2, hMLH1 | ACVR2 |
| HCT116+chr3 | ACVR2 | TGFBR2, hMLH1 |
| FET | None | — |
| SW480 | SMAD4 | — |
| HEC59 | hMSH2, ACVR2, TGFBR2 | — |
| HEC59+chr2 | TGFBR2 | ACVR2, hMSH2 |
Complementation With Chromosome 2 Is Sufficient to Lead to Wild-Type ACVR2 Transcript Expression
To demonstrate intact transcription from the complemented ACVR2 gene, 50 complementary DNA clones containing the polyadenine tract within exon 10 of ACVR2 were generated from HCT116 and HCT116+chr2 cells and sequenced. As expected, all clones from the mismatch repair–deficient and ACVR2-mutant HCT116 cells revealed frameshift mutations with no wild-type A8 sequences. Complementation with chromosome 2 in HCT116+chr2 cells led to expression of the wild-type transcript in 20% of the clones despite lacking the MMR protein hMLH1 (Figure 1B). In comparison, while the mismatch repair–deficient endometrial cell line HEC59 expressed 50 transcripts with a frameshift mutation, 33% of the chromosome 2–complemented and hMSH2- and ACVR2-restored HEC59+chr2 cells expressed the wild-type A8 tract (Figure 1B). This is at the expected ratio of 33% if transcription occurs from the single wild-type and 2 mutant ACVR2 genes.
ACVR2 Is Expressed in HCT116+chr2 Cells and Colocalizes With ACVR1 Upon Activin Treatment
To establish expression of ACVR2 protein following complementation with chromosome 2, we immuno-precipitated ACVR1 and ACVR2 in HCT116 and HCT116+chr2 cells. While both cell lines express ACVR1, ACVR2B is not expressed (data not shown) and ACVR2 protein is only expressed in the chromosome 2–complemented HCT116+chr2 cells (Figure 2A). In our coimmunoprecipitation experiments, there appears to be some ACVR2 associated with ACVR1 in a preformed complex, possibly even when ACVR2 is mutated. This complex is further induced upon activin treatment in control CAPAN-1 cells and in ACVR2-complemented HCT116+chr2 cells but not in ACVR2-mutated HCT116 cells (Figure 2B).
Figure 2.

Complemented ACVR2 causes expression of ACVR2 protein that complexes with ACVR1 with activin stimulation and phosphorylates SMAD2 for its nuclear translocation. (A) Immunoprecipitated ACVR1 protein is present in parental and chromosome 2–complemented cells, but immunoprecipitated ACVR2 protein is only present in chromosome 2–complemented cells. (B) ACVR2 protein coimmunoprecipitated with ACVR1 antibody in HCT116+chr2 and control CAPAN cells, but not in ACVR2-mutant HCT116 cells. Activin stimulation further increased ACVR1/ACVR2 complex formation. (C) SMAD2 is phosphorylated after activin stimulation minimally in HCT116 cells but is markedly activated in ACVR2-complemented HCT116+chr2 cells and translocates into the nucleus. Histone and tubulin blots were performed to indicate the relative purity of the nuclear and cytoplasmic fractions, respectively.
SMAD2 Is Phosphorylated and Translocates to the Nucleus Following Activin Treatment in ACVR2-Restored Cells
To establish whether signaling is restored with ACVR2 complementation, we prepared nuclear and cytoplasmic extracts from HCT116 cells and ACVR2-restored HCT116+chr2 cells following 4 hours of activin treatment. After electrophoretic separation, Western blotting was performed using SMAD2 as well as phosphoSMAD2 specific antibodies. While total SMAD2 was present in the cytosol and in the nucleus, phosphorylated SMAD2 was solely located in the nucleus. Even ACVR2-mutated HCT116 cells revealed a modest induction of pSMAD2 following ligand treatment (to the levels of unstimulated HCT116+chr2 cells, and independent of ACVR2 stimulation), which was significantly pronounced after activin treatment only in the ACVR2-restored HCT116+chr2 cells (Figure 2C). Similarly, HEC59+chr2 cells showed a pronounced pSMAD2 induction following activin treatment from baseline nuclear presence of pSMAD2 (data not shown).
SMAD-Induced Activin-Specific Transactivation Is Restored in ACVR2-Complemented Cells
To assess the effect of activin on SMAD-specific transcriptional activation, we performed a reporter assay using the pARE-Luc reporter, which is indicative of activin-specific transcriptional activity.26 Following 24 hours of activin treatment, there was an 11-fold increase in luciferase activity in the ACVR2-restored HCT116+ chr2 cells compared with the ACVR2-mutated HCT116 cells (Figure 3A).
Figure 3.
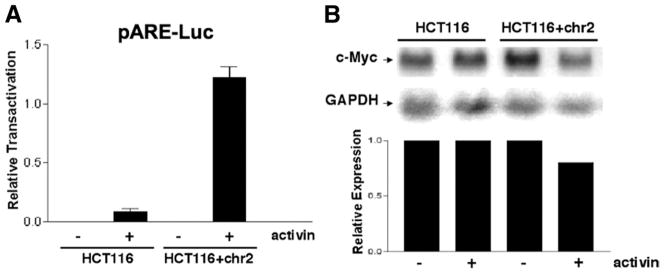
Chromosome 2 complementation restores activin-induced transactivation. (A) The pARE-Luc reporter was transfected into cells before stimulation with activin. (B) Northern blotting for c-Myc expression, known to be down-regulated by activin, was performed using a generated c-Myc probe. A GAPDH probe was used as a loading control, and the relative expression of c-Myc to GAPDH is shown in the bar graph.
Activin Induces c-MYC Down-regulation in ACVR2-Restored Cells
The effect of restored ACVR2 and SMAD signaling was assessed by Northern blotting of c-MYC, a known TGF-β and activin transcriptional target gene.4 While treatment with activin did not change c-MYC expression in the ACVR2 mutant HCT116 cells, c-MYC was down-regulated in the ACVR2-complemented HCT116+chr2 cells (Figure 3B). A more pronounced decrease in c-myc expression was seen in the activin-responsive FET colon cancer cell line following activin treatment (data not shown).
Cell Growth Is Suppressed Following Activin Treatment in ACVR2-Complemented Cells
To discern whether treatment with activin induces changes in cellular growth in cancer cells with intact activin signaling, we treated HCT116, HEC59, and ACVR2-restored HCT116+chr2 and HEC59+chr2 cells with activin. Activin caused a decrease in growth in the ACVR2-complemented cell lines, but cells harboring mutations in ACVR2 displayed no change in their growth pattern (Figure 4). The activin-sensitive FET colon cancer cells were used as positive control for comparison.
Figure 4.
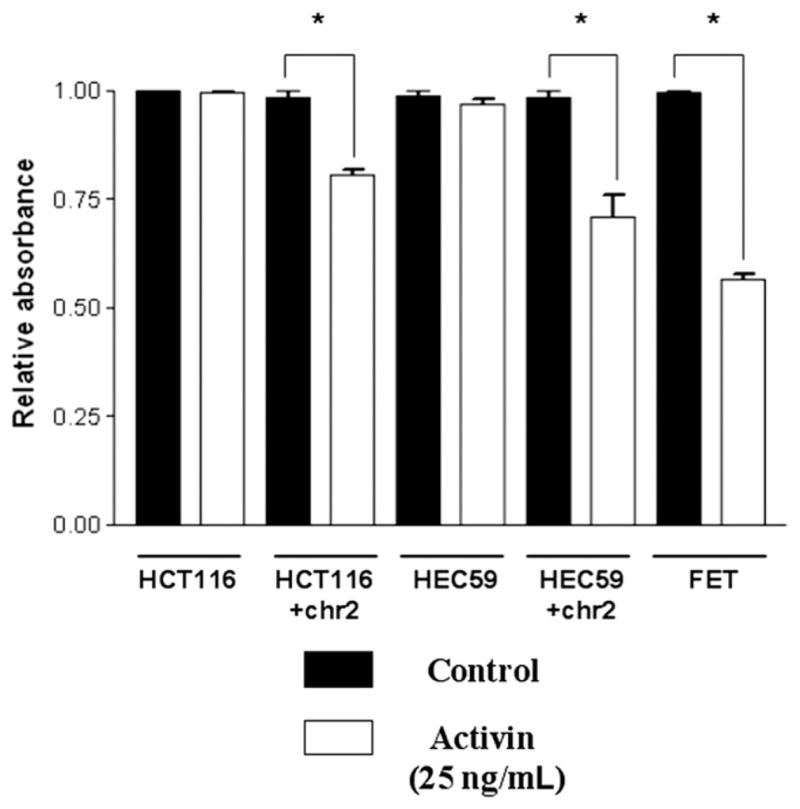
Activin is growth suppressive in chromosome 2–complemented cells. The MTT assay, which approximates cell growth, demonstrates a reduction of growth with activin in the chromosome 2–complemented cells only. Activin-responsive FET colon cancer cells were used as a positive control (*P < .05).
Activin A Is Expressed in ACVR2-Complemented HCT116+chr2 Cells, Demonstrating the Potential for Autocrine-Induced Growth Suppression In Vivo
Our laboratory has previously reported an in vivo determination of growth of HCT116, HCT116+chr2, and HCT116+chr3 cells in a nude mouse model.27 At that time, it was not known what could modulate growth of the chromosome-complemented cell lines, because the main goal of that experiment was to determine if hMLH1-complemented HCT116+chr3 cells would continue to be tumorigenic. The results of the experiment showed that HCT116, HCT116+chr2, and HCT116+chr3 cells were tumorigenic in mice, with measurable tumors detected by days 6–7 in HCT116-injected mice and tumors detected at days 8–9 for HCT116+chr2- and HCT116+chr3-injected mice.27 However, both HCT116+chr2 and HCT116+chr3 cells produced tumors that grew at a significantly lower rate than the parental cell line HCT116. We now believe that restored activin signaling in ACVR2-complemented HCT116+chr2 cells and restored TGF-β signaling in TGFBR2-complemented HCT116+chr3 cells are at least partially responsible for their reduced growth when compared with parental HCT116 cells (Table 1). To ascertain whether this hypothesis is a potential mechanism for reduced growth in HCT116+chr2 cells, we assessed if there could be autocrine production of activin A in HCT116+chr2 cells. We amplified activin A messenger RNA using semiquantitative RT-PCR. Both HCT116 and HCT116+chr2 cells expressed activin A ligand (Figure 5), but the effects of activin A would only be expected to be realized in the ACVR2-restored HCT116+chr2 cells.
Figure 5.
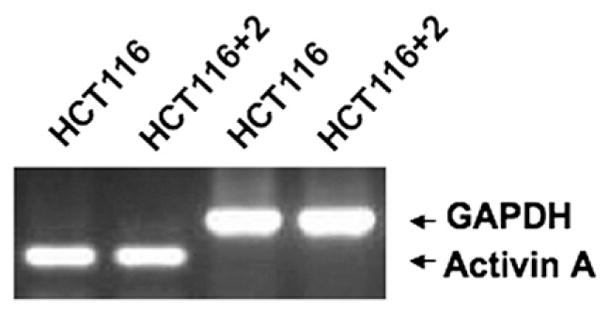
Both HCT116 and HCT116+chr2 cells transcribe native activin A ligand. RT-PCR was performed on extracted RNA from the cells utilizing specific activin A oligonucleotides for amplification. GAPDH was used as a loading control.
Use of ACVR2-Specific siRNA in HCT116+chr2 Cells Shows That Activin-Induced Signaling and Growth Suppression Are ACVR2 Dependent
To establish that the effects of activin observed in HCT116+chr2 cells are specific to ACVR2 complementation and not other genes located on chromosome 2, we performed knockdown experiments using ACVR2-specific siRNA (Figure 6). We were able to knock down ACVR2 transcripts to none-detectable levels by RT-PCR techniques with the ACVR2-specific siRNA, which was more effective than our knockdown of the housekeeping gene messenger RNA of GAPDH (Figure 6A). We then assessed the ACVR2 siRNA knockdown HCT116+chr2 cells for phosphoSMAD2 signaling as well as activin-induced growth suppression. Following knockdown of ACVR2, activin-induced phosphoSMAD2 activation and nuclear translocation were muted when compared with mock siRNA-transfected HCT116+chr2 cells treated with activin (Figure 6B). Furthermore, knockdown of ACVR2 in ACVR2-complemented HCT116+chr2 cells prohibited activin-induced growth suppression, completely reversing the effects seen in mock siRNA-transfected HCT116+chr2 cells (Figure 6C). Thus, in addition to the activin-specific signaling and growth suppression seen in ACVR2-complemented HCT116+chr2 cells, siRNA knockdown of the complemented ACVR2 gene abrogated the effects.
Figure 6.

ACVR2 silencing reverses activin signaling and growth effects observed in the ACVR2-complemented HCT116+chr2 cells. (A) ACVR2 messenger RNA expression is knocked down in ACVR2-complemented HCT116+chr2 colon cancer cells using ACVR2-specific siRNA. RT-PCR followed by agarose gel electrophoresis show the specificity of siRNA to knock down GAPDH and ACVR2 mRNA. dH2O, distilled water only; C, control; S, scrambled siRNA; KD, gene-specific knockdown via siRNA treatment. (B) ACVR2 knockdown abolished activin-induced pSMAD2 activation and its nuclear translocation in ACVR2-complemented HCT116+chr2 cells. KD, ACVR2-specific siRNA knockdown; Mock, treatment without any specific siRNA. Histone and tubulin Western blots were performed to indicate the relative purity of the nuclear and cytoplasmic fractions, respectively. (C) Activin-induced growth suppression is abrogated by ACVR2 knockdown in ACVR2-complemented cells by the MTT assay (*P < .05).
Activin Decreases the S-phase Cell Cycle Fraction in ACVR2-Restored Cells
To determine the effects of activin on the cell cycle, we studied cell cycle distribution of ACVR2-deficient HCT116 cells and ACVR2-restored HCT116+chr2 cells following activin treatment. While there was no change in the proportions of cells in G1, S, and G2 phase in ACVR2-mutant cells following activin treatment, ACVR2-restored cells displayed a decrease in S-phase following activin treatment. (Figure 7A). In ACVR2-restored HEC59+chr2 cells, a concomitant increase in G1 phase was observed (Figure 7B).
Figure 7.

Activin slows the S phase in chromosome 2–complemented cells. Cell cycle analysis revealed no changes after activin treatment without chromosome 2 complementation. (A) HCI116 and HCT116+chr2 cells. (B) HEC59 and HEC59+ chr2 cells. (*P < .05).
Intact Activin Signaling Enhances Migration in Colon Cancer Cells
To assess a potential effect on cellular migration with activin signaling akin to that observed for TGF-β, we treated ACVR2-mutant HCT116 and ACVR2-restored HCT116+chr2 cells with activin in a cellular wound assay and a Transwell migration assay. While HCT116 cells did not display enhanced migration across a wound in response to activin, cell motility of HCT116+chr2 cells was markedly enhanced over ACVR2-mutated control cells (Figure 8A and B) and became evident as early as 1 hour after activin treatment. Similarly, ACVR2-restored HCT16+chr2 cells had significantly more cells that migrated through pores in a Transwell in response to activin than ACVR2-mutant HCT116 cells (Figure 8C). There was an upward trend that did not reach significance when comparing untreated HCT116 with untreated HCT116+chr2 cells, which might be reflective of autocrine activin signaling with a restored pathway. Thus, both the cellular wound assay and the Transwell migration assay indicate that activin increases cell migration when activin signaling is intact.
Figure 8.

Activin enhances cell migration in chromosome 2–complemented cells. (A) Photomicrographs from the wound assay are shown. (B) Graphical depiction of the data from the photomicrographs in A. (C) Data from a Transwell migration assay (8-μm pore size) are shown. Note that activin increased migration in both the wound assay and the Transwell migration assay (*P < .05).
Discussion
The biological effects of activin in colon cancer have not been previously appreciated and recently came to light when the ACVR2 receptor was discovered to be mutated in the majority of MSI-H colon cancers.2,20 Here, we demonstrated a novel model system in which a single physiologically regulated copy of ACVR2 was complemented in MSI-H colon cancer cells. We found that the classic activin-SMAD signaling pathway is restored, is transcriptionally responsive, and, importantly, induces growth suppression. Although not the major focus of this study, activin appears to be growth suppressive in ACVR2-restored endometrial cancer cells as well. These findings underscore the role of activin in braking cell growth in these colon cancer and endometrial cancer cells.
We found that activin signaling is reconstituted in MSI colon cancer cell lines with one copy of wild-type ACVR2. We further showed that ACVR2/ACVR1 exists in a pre-formed complex, possibly even if ACVR2 is expressed in its truncated form. This in accordance with the previous observation that only mutations that involve the activin-binding site of ACVR2 located in the extracellular domain result in loss of ACVR1 binding.28 Further, truncated type 2 and type 1 receptors that lack their entire intracellular domain were found to cross-link to the ligand in a cooperative manner.29 Frameshift mutation of exon 10 is downstream of the extracellular domain and affects the cytosolic domain of the receptor, therefore maintaining its ability to bind ligand and form a heteromeric complex with ACVR1. It is also possible that, with ACVR2 restoration, some activin signaling through the ACVR2/ ACVR1 complex occurs from autocrine activin A production. Indeed, in ACVR2-complemented HCT116+chr2 cells, activin A is transcribed, the cells have some pre-formed ACVR2/ACVR1 complexes, and there is a minimal nonstimulated amount of phosphoSMAD2 present on Western blots. Additional ACVR2/ACVR1 complex induction occurs in the ACVR2-restored colon cancer cells following ligand binding, likely representing recruitment of additional ACVR1 receptors and resulting in enhanced pSMAD2 activation.
Activin has been shown to regulate growth in myeloid and hematopoietic cells,8,9 but there are limited data in epithelial cells, particularly colon cancer cells. In breast cancer cells, activin is growth suppressive,7 similar to what we found in our reconstituted colon cancer cell model. While our model has the advantage of being physiologic, with only one copy of ACVR2 restored as opposed to an overexpression model, this may have led to modest growth effects. The fact that we compared cells with and without ligand in multiple clones of 2 different models implicates ACVR2 as the target on chromosome 2. This is further confirmed by our ACVR2-selective knockdown experiments, which showed a reversal of the activininduced signaling and growth suppression. The mechanism of growth suppression, however, is not entirely clear. Antagonism of c-MYC is implicated because c-MYC is down-regulated by activin in prostate cancer cells4; in keratinocytes, Mad1, a negative regulator of c-MYC, is induced.11 We found a down-regulation of c-MYC in our ACVR2-restored model, indicating c-MYC may be regulated by activin in colon cancer cells as well. Cellular growth clearly appears to be regulated by activin-induced SMAD-dependent signaling because cells mutant for ACVR2 do not show response to activin. Both pituitary and pancreatic tumors harbor loss-of-function mutations in activin signaling components16,17 and, with our current data, underscore the role of activin signaling in tumor suppressor suppression. Intact activin signaling also appears to reduce the size and volume of colon cancers.30
In MSI-H colon cancers, both TGF-β and activin are abrogated due to frameshift mutations in their type II receptor.2 The loss of both of these signaling pathways must be beneficial for tumor growth. Both ACVR2-restored and TGFBR2-restored colon cancer cells are tumorigenic in nude mice, but both complemented cells grow slower than its parental counterpart.27 Both TGF-β and activin use the same intracellular SMAD proteins (SMAD 2 and 3) to transmit their signal to the nucleus. It is not clear to date how each respective ligand differentiates its signal through these identical SMADs, but we speculate that both ligands may have an additive effect in their growth response because both are inactivated in MSI-H colon cancers.
While ligands from the TGF-β family, such as TGF-β, activin, and the BMPs, are believed to signal in a similar manner, the activin/ACVR2 interface is distinct from the TGF-β/TGFBR2 interface,29 possibly alluding to functional differences. This is further underscored by the finding that ACVR2 regulates many genes related to the TGF-β pathway as well as some genes that have not been associated with TGF-β signaling.31
Despite the growth suppression observed in our ACVR2-restored cellular models, activin enhances cellular motility. Our finding is congruent with the fact that migration of normal colon epithelial cells is enhanced by TGF-β32 and with the recent observation of somatic acquisition of activating mutations in TGFBR1 in metastases and not from the source of primary colon cancers,33 as well as worse survival with wild-type TGFBR2 in patients with MSI-H colon cancers.34 Additionally, the non-invasive activin-sensitive colon cancer cell line FET did not show enhanced migration with activin treatment (data not shown), while the complemented HCT116+chr2 colon cancer cells, which are invasive,35 did.
In contrast to an ACVR2-overexpression model,31 complemented HCT116+chr2 cells produce physiologic levels of ACVR2 and remain tumorigenic in nude mice,27 indicating that the presence of wild-type ACVR2 does not abrogate tumor growth and invasion in these cells. However, when compared with parental HCT116 cells, mean tumor volume was significantly less in the HCT116+chr2 cells, which can now be explained by the complementation of ACVR2 in conjunction with activin A production.27 Furthermore, neuroblastoma cells with restored activin expression exhibit diminished proliferation but an unchanged lung metastasis rate,36 and invasion is enhanced by activin in ovarian cancer cell lines.37 Taken together, activin may promote cell migration and possibly contribute to the metastatic process.
In conclusion, our single-copy ACVR2-complemented MSI-H cell models revealed normal expression of ACVR2 and induced activin-specific transcriptional responses, with the biologic response of growth suppression, slower S-phase cycling, and enhanced migration. These responses are similar to that of intact TGF-β signaling in colon cancer, and loss of these activin responses contributes to the pathogenesis of MSI-H colon cancer.
Acknowledgments
Supported by US Public Health Service grants T32-DK07202 and K08-DK074019 (to B.H.J.), T32-HL07212 (to S.E.B.), and CA90231 and DK067287 (to J.M.C.); the Foundation for Digestive Diseases and Nutrition Fellow to Faculty Transition Award (to B.H.J.); and the VA Research Service (J.M.C.).
The authors thank C. Richard Boland, MD, and Minoru Koi, PhD, for developing the cells used in this report.
Abbreviations used in this paper
- MSI-H
high-frequency microsatellite instability
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PMSF
phenylmethylsulfonyl fluoride
- RT-PCR
reverse-transcription polymerase chain reaction
- siRNA
small interfering RNA
- TGF
transforming growth factor
References
- 1.Carethers JM, Boland CR. Neoplasia of the gastrointestinal tract. In: Yamada T, Alperts DH, Kaplowitz N, Laine L, Owyang C, Powell DW, editors. Textbook of gastroenterology. 4. Philadelphia, PA: Lippincott-Raven; 2003. pp. 557–583. [Google Scholar]
- 2.Jung B, Doctolero RT, Tajima A, Nguyen AK, Keku T, Sandler RS, Carethers JM. Loss of activin receptor type 2 protein expression in microsatellite unstable colon cancers. Gastroenterology. 2004;126:654–659. doi: 10.1053/j.gastro.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 3.Cocolakis E, Lemay S, Ali S, Lebrun JJ. The p38 MAPK pathway is required for cell growth inhibition of human breast cancer cells in response to activin. J Biol Chem. 2001;276:18430–18436. doi: 10.1074/jbc.M010768200. [DOI] [PubMed] [Google Scholar]
- 4.Zhang Z, Zhao Y, Batres Y, Lin MF, Ying SY. Regulation of growth and prostatic marker expression by activin A in an androgen-sensitive prostate cancer cell line LNCAP. Biochem Biophys Res Commun. 1997;234:362–365. doi: 10.1006/bbrc.1997.6649. [DOI] [PubMed] [Google Scholar]
- 5.Hilden K, Tuuri T, Eramaa M, Ritvos O. Expression of type II activin receptor genes during differentiation of human K562 cells and cDNA cloning of the human type IIB activin receptor. Blood. 1994;83:2163–2170. [PubMed] [Google Scholar]
- 6.Mathews LS, Vale WW. Expression cloning of an activin receptor, a predicted transmembrane serine kinase. Cell. 1991;65:973–982. doi: 10.1016/0092-8674(91)90549-e. [DOI] [PubMed] [Google Scholar]
- 7.Burdette JE, Jeruss JS, Kurley SJ, Lee EJ, Woodruff TK. Activin A mediates growth inhibition and cell cycle arrest through Smads in human breast cancer cells. Cancer Res. 2005;65:7968–7975. doi: 10.1158/0008-5472.CAN-04-3553. [DOI] [PubMed] [Google Scholar]
- 8.Valderrama-Carvajal H, Cocolakis E, Lacerte A, Lee EH, Krystal G, Ali S, Lebrun JJ. Activin/TGF-beta induce apoptosis through Smad-dependent expression of the lipid phosphatase SHIP. Nat Cell Biol. 2002;4:963–969. doi: 10.1038/ncb885. [DOI] [PubMed] [Google Scholar]
- 9.Fukuchi Y, Kizaki M, Yamato K, Kawamura C, Umezawa A, Hata J, Nishihara T, Ikeda Y. Mcl-1, an early-induction molecule, modulates activin A-induced apoptosis and differentiation of CML cells. Oncogene. 2001;20:704–713. doi: 10.1038/sj.onc.1204142. [DOI] [PubMed] [Google Scholar]
- 10.Jiang TX, Yi JR, Ying SY, Chuong CM. Activin enhances chondrogenesis of limb bud cells: stimulation of precartilaginous mesenchymal condensations and expression of NCAM. Dev Biol. 1993;155:545–557. doi: 10.1006/dbio.1993.1051. [DOI] [PubMed] [Google Scholar]
- 11.Werner S, Beer HD, Mauch C, Luscher B. The Mad1 transcription factor is a novel target of activin and TGF-beta action in keratinocytes: possible role of Mad1 in wound repair and psoriasis. Oncogene. 2001;20:7494–7504. doi: 10.1038/sj.onc.1204937. [DOI] [PubMed] [Google Scholar]
- 12.Chen Y, Lebrun JJ, Vale W. Regulation of transforming growth factor beta- and activin-induced transcription by mammalian Mad proteins. Proc Natl Acad Sci U S A. 1996;93:12992–12997. doi: 10.1073/pnas.93.23.12992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zauberman A, Oren M, Zipori D. Involvement of p21(WAF1/Cip1), CDK4 and Rb in activin A mediated signaling leading to hepatoma cell growth inhibition. Oncogene. 1997;15:1705–1711. doi: 10.1038/sj.onc.1201348. [DOI] [PubMed] [Google Scholar]
- 14.Danila DC, Inder WJ, Zhang X, Alexander JM, Swearingen B, Hedley-Whyte ET, Klibanski A. Activin effects on neoplastic proliferation of human pituitary tumors. J Clin Endocrinol Metab. 2000;85:1009–1015. doi: 10.1210/jcem.85.3.6473. [DOI] [PubMed] [Google Scholar]
- 15.Zheng W, Luo MP, Welt C, Lambert-Messerlian G, Sung CJ, Zhang Z, Ying SY, Schneyer AL, Lauchlan SC, Felix JC. Imbalanced expression of inhibin and activin subunits in primary epithelial ovarian cancer. Gynecol Oncol. 1998;69:23–31. doi: 10.1006/gyno.1998.4958. [DOI] [PubMed] [Google Scholar]
- 16.Alexander JM, Bikkal HA, Zervas NT, Laws ER, Jr, Klibanski A. Tumor-specific expression and alternate splicing of messenger ribonucleic acid encoding activin/transforming growth factor-beta receptors in human pituitary adenomas. J Clin Endocrinol Metab. 1996;81:783–790. doi: 10.1210/jcem.81.2.8636304. [DOI] [PubMed] [Google Scholar]
- 17.Su GH, Bansal R, Murphy KM, Montgomery E, Yeo CJ, Hruban RH, Kern SE. ACVR1B (ALK4, activin receptor type 1B) gene mutations in pancreatic carcinoma. Proc Natl Acad Sci U S A. 2001;98:3254–3257. doi: 10.1073/pnas.051484398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hempen PM, Zhang L, Bansal RK, Iacobuzio-Donahue CA, Murphy KM, Maitra A, Vogelstein B, Whitehead RH, Markowitz SD, Willson JK, Yeo CJ, Hruban RH, Kern SE. Evidence of selection for clones having genetic inactivation of the activin A type II receptor (ACVR2) gene in gastrointestinal cancers. Cancer Res. 2003;63:994–999. [PubMed] [Google Scholar]
- 19.Liu F, Shao LE, Yu J. Truncated activin type II receptor inhibits erythroid differentiation in K562 cells. J Cell Biochem. 2000;78:24–33. [PubMed] [Google Scholar]
- 20.Mori Y, Yin J, Rashid A, Leggett BA, Young J, Simms L, Kuehl PM, Langenberg P, Meltzer SJ, Stine OC. Instabilotyping: comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res. 2001;61:6046–6049. [PubMed] [Google Scholar]
- 21.Piek E, Van Dinther M, Parks WT, Sallee JM, Bottinger EP, Roberts AB, Ten Dijke P. RLP, a novel Ras-like protein, is an immediate-early transforming growth factor-beta (TGF-beta) target gene that negatively regulates transcriptional activity induced by TGF-beta. Biochem J. 2004;383:187–199. doi: 10.1042/BJ20040774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR. Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res. 1994;54:4308–4312. [PubMed] [Google Scholar]
- 23.Umar A, Koi M, Risinger JI, Glaab WE, Tindall KR, Kolodner RD, Boland CR, Barrett JC, Kunkel TA. Correction of hypermutability, N-methyl-N′-nitro-N-nitrosoguanidine resistance, and defective DNA mismatch repair by introducing chromosome 2 into human tumor cells with mutations in MSH2 and MSH6. Cancer Res. 1997;57:3949–3955. [PubMed] [Google Scholar]
- 24.Gong J, Ammanamanchi S, Ko TC, Brattain MG. Transforming growth factor beta 1 increases the stability of p21/WAF1/CIP1 protein and inhibits CDK2 kinase activity in human colon carcinoma FET cells. Cancer Res. 2003;63:3340–3346. [PubMed] [Google Scholar]
- 25.Kleeff J, Ishiwata T, Friess H, Buchler MW, Korc M. Concomitant over-expression of activin/inhibin beta subunits and their receptors in human pancreatic cancer. Int J Cancer. 1998;77:860–868. doi: 10.1002/(sici)1097-0215(19980911)77:6<860::aid-ijc11>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 26.Weisberg E, Winnier GE, Chen X, Farnsworth CL, Hogan BL, Whitman M. A mouse homologue of FAST-1 transduces TGF beta superfamily signals and is expressed during early embryogenesis. Mech Dev. 1998;79:17–27. doi: 10.1016/s0925-4773(98)00160-9. [DOI] [PubMed] [Google Scholar]
- 27.Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray PC, Greenwald J, Blount AL, Kunitake KS, Donaldson CJ, Choe S, Vale W. Identification of a binding site on the type II activin receptor for activin and inhibin. J Biol Chem. 2000;275:3206–3212. doi: 10.1074/jbc.275.5.3206. [DOI] [PubMed] [Google Scholar]
- 29.Greenwald J, Groppe J, Gray P, Wiater E, Kwiatkowski W, Vale W, Choe S. The BMP7/ActRII extracellular domain complex provides new insights into the cooperative nature of receptor assembly. Mol Cell. 2003;11:605–617. doi: 10.1016/s1097-2765(03)00094-7. [DOI] [PubMed] [Google Scholar]
- 30.Jung B, Smith EJ, Doctolero RT, Gervaz P, Alonso JC, Miyai K, Keku T, Sandler RS, Carethers JM. Influence of target gene mutations on survival, stage and histology in sporadic microsatellite unstable colon cancers. Int J Cancer. 2006;118:2509–2513. doi: 10.1002/ijc.21710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deacu E, Mori Y, Sato F, Yin J, Olaru A, Sterian A, Xu Y, Wang S, Schulmann K, Berki A, Kan T, Abraham JM, Meltzer SJ. Activin type II receptor restoration in ACVR2-deficient colon cancer cells induces transforming growth factor-beta response pathway genes. Cancer Res. 2004;64:7690–7696. doi: 10.1158/0008-5472.CAN-04-2082. [DOI] [PubMed] [Google Scholar]
- 32.McKaig BC, Makh SS, Hawkey CJ, Podolsky DK, Mahida YR. Normal human colonic subepithelial myofibroblasts enhance epithelial migration (restitution) via TGF-beta3. Am J Physiol. 1999;276:G1087–G1093. doi: 10.1152/ajpgi.1999.276.5.G1087. [DOI] [PubMed] [Google Scholar]
- 33.Pasche B, Knobloch TJ, Bian Y, Liu J, Phukan S, Rosman D, Kaklamani V, Baddi L, Siddiqui FS, Frankel W, Prior TW, Schuller DE, Agrawal A, Lang J, Dolan ME, Vokes EE, Lane WS, Huang CC, Caldes T, Di Cristofano A, Hampel H, Nilsson I, von Heijne G, Fodde R, Murty VV, de la Chapelle A, Weghorst CM. Somatic acquisition and signaling of TGFBR1*6A in cancer. JAMA. 2005;294:1634–1646. doi: 10.1001/jama.294.13.1634. [DOI] [PubMed] [Google Scholar]
- 34.Watanabe T, Wu TT, Catalano PJ, Ueki T, Satriano R, Haller DG, Benson AB, 3rd, Hamilton SR. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344:1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schlechte W, Brattain M, Boyd D. Invasion of extracellular matrix by cultured colon cancer cells: dependence on urokinase receptor display. Cancer Commun. 1990;2:173–179. [PubMed] [Google Scholar]
- 36.Panopoulou E, Murphy C, Rasmussen H, Bagli E, Rofstad EK, Fotsis T. Activin A suppresses neuroblastoma xenograft tumor growth via antimitotic and antiangiogenic mechanisms. Cancer Res. 2005;65:1877–1886. doi: 10.1158/0008-5472.CAN-04-2828. [DOI] [PubMed] [Google Scholar]
- 37.Steller MD, Shaw TJ, Vanderhyden BC, Ethier JF. Inhibin resistance is associated with aggressive tumorigenicity of ovarian cancer cells. Mol Cancer Res. 2005;3:50–61. [PubMed] [Google Scholar]