

Abstract
The diagnosis and treatment of soft tissue sarcomas (STS) have been difficult. Of the diverse histological subtypes, undifferentiated pleomorphic sarcoma (UPS) is particularly difficult to diagnose accurately, and its classification per se is still controversial. Recent advances in genomic technologies provide an excellent way to address such problems. However, it is often difficult, if not impossible, to identify definitive disease-associated genes using genome-wide analysis alone, primarily because of multiple testing problems. In the present study, we analyzed microarray data from 88 STS patients using a combination method that used knowledge-based filtering and a simulation based on the integration of multiple statistics to reduce multiple testing problems. We identified 25 genes, including hypoxia-related genes (e.g., MIF, SCD1, P4HA1, ENO1, and STAT1) and cell cycle- and DNA repair-related genes (e.g., TACC3, PRDX1, PRKDC, and H2AFY). These genes showed significant differential expression among histological subtypes, including UPS, and showed associations with overall survival. STAT1 showed a strong association with overall survival in UPS patients (logrank p = 1.84×10−6 and adjusted p value 2.99×10−3 after the permutation test). According to the literature, the 25 genes selected are useful not only as markers of differential diagnosis but also as prognostic/predictive markers and/or therapeutic targets for STS. Our combination method can identify genes that are potential prognostic/predictive factors and/or therapeutic targets in STS and possibly in other cancers. These disease-associated genes deserve further preclinical and clinical validation.
Introduction
Recent advances in genomic technologies offer an excellent opportunity to determine the complete biological characteristics of neoplastic tissues, resulting in improved diagnosis, treatment selection, rational classification based on molecular carcinogenesis, and identification of therapeutic targets. The diagnosis and treatment of soft tissue sarcomas (STS) have been difficult because STSs comprise a group of highly heterogeneous tumors in terms of histopathology, molecular signature, histological grade, and primary site. These tumors have generally been classified into subtypes according to their histological resemblance to normal tissue. The Fédération Francaise des Centres de Lutte Contre le Cancer (FNCLCC) grading system was defined more than 20 years ago and is still the most commonly used grading system for STS [1], [2]. Treatment of STS is based on both histological subtype and histological grade. The understanding gained regarding the molecular pathology of cancer in recent decades suggests that some tumor types exhibit stand-alone recurrent genetic aberrations, such as chromosomal translocations, that result in gene fusions, e.g., SYT-SSX in synovial sarcoma (SS) [3], TLS-CHOP in myxoid/round cell liposarcoma (MLS) [4], and KIF5B-RET in lung adenocarcinoma [5], or somatic mutations, e.g., KIT in gastrointestinal stromal tumors (GIST) [6] and 26 mutated genes (TP53, KRAS, EGFR, and 23 other genes) in lung adenocarcinoma [7]. The molecular markers specific to each tumor type are useful for tumor classification [8]. In contrast, several malignant tumors, such as malignant fibrous histiocytoma (MFH), are characterized by numerous nonrecurrent, complex chromosomal aberrations, and they frequently show overlapping histological features and immunophenotypes that are difficult for pathologists to interpret [9]. In particular, the diagnosis of MFH has been a controversial issue [10]–[13]. MFH is the most common soft tissue sarcoma in adults. It has a wide range of histological subtypes [13]. For this reason, discrimination between MFH and other STSs is difficult, but this discrimination is necessary because there are significant differences in the 5-year survival rates of the STS subtypes [14]: 100% for well-differentiated liposarcoma (WLS), 71% for synovial sarcoma (SS), 46% for pleomorphic MFH, and 92% for myxofibrosarcoma (MFS). MFH was renamed undifferentiated pleomorphic sarcoma (UPS) in 2002 by the World Health Organization (WHO) [15]. MFS was considered a subtype of MFH before this classification; WHO reclassified MFS as another subtype of STS [15]. Discrimination between UPS and MFS is particularly difficult [14] because of their histological similarities and because of the considerable heterogeneity of UPS [13]. UPS was previously characterized by global gene expression analysis using analysis of variance (ANOVA) and clustering analysis [13]. Although some possible prognostic factors were identified, the list of factors was not complete because the study was conducted without information on patient outcomes. In the present study, we hypothesized that some genes can serve both as diagnostic markers for histological subtyping and as prognostic markers of overall survival in STS. We used a combination of statistical and bioinformatic methods to identify those genes.
Many statistical and bioinformatic methods have been proposed for global biological information analysis in the past 3 decades. For example, basic local alignment search tool (BLAST) [16], ClustalW [17], BLAST-based algorithm for the identification of upstream ORFs with conserved amino acid sequences (BAIUCAS) [18], and G4 DNA motif region finder by R (G4MR-FindeR) [19] have been used for sequence analysis; hierarchical clustering [20], fuzzy k-means [21], and fuzzy adaptive resonance theory (FuzzyART) [22], [23] have been used for gene cluster analysis; gene set enrichment analysis (GSEA) [24], modified signal-to-noise (S2N′) [25], and projective adaptive resonance theory (PART) [26], [27] have been used for gene selection; fuzzy neural network (FNN) [28], [29] and boosted fuzzy classifier with a SWEEP operator (BFCS) [30]–[32] have been used for the construction of prediction models; and IntPath [33] and Stringent DDI-based Prediction [34] were used for analysis of pathways and protein–protein interactions. The use of statistical or bioinformatic analysis is practical and useful for clinical diagnosis [35]–[37] and the identification of marker genes [38]–[43]. In the present study, we focused on microarray data analysis; however, the analysis of data obtained using next-generation sequencing technologies [44] is a subject of an upcoming project.
Global analysis of gene expression is a powerful method for the identification of prognostic/predictive factors and/or therapeutic targets. However, it is often difficult, if not impossible, to identify definitive disease-associated genes using genome-wide analysis alone, primarily because of multiple testing problems. In this situation, knowledge-based approaches, such as knowledge-based fuzzy adaptive resonance theory (KB-FuzzyART) [45] and knowledge-based single nucleotide polymorphism (KB-SNP) [46], [47], are effective and interpretable [48]–[50]. Online Mendelian Inheritance in Man (OMIM) is a continuously updated catalog of human genes and genetic disorders and traits. In the present study, we used OMIM as a knowledge source for narrowing the list of candidate genes and applied the OMIM-based method to gene expression data from STS patients. Thus, we identified 25 genes that showed significant differential expression among histological subtypes, including UPS, and showed associations with overall survival. According to the literature, these genes are useful not only as diagnostic markers for the discrimination of molecular pathway-based subtypes but also as prognostic/predictive markers and/or therapeutic targets for STS. Moreover, these genes are useful for understanding the mechanisms underlying tumor progression or metastasis and for the rational design of anticancer therapeutics. Therefore, our combination method of knowledge-based filtering and simulation based on the integration of multiple statistics can identify potential prognostic/predictive factors and/or therapeutic targets in STS and possibly in other cancers.
Materials and Methods
Ethics statement
The study was conducted according to the principles expressed in the Declaration of Helsinki. The ethics committee of the National Cancer Center approved the study protocol. All patients provided written informed consent.
Patients and tumor samples
The characteristics of the 88 STS patients (20 with UPS, 15 with MFS, 17 with SS, 20 with myxoid liposarcoma [MLS], 6 with leiomyosarcoma [LMS], 5 with fibrosarcoma [FS], and 5 with a malignant peripheral nerve sheath tumor [MPNST]) enrolled in this study are shown in Table 1. All patients had received a histological diagnosis of primary soft tissue tumor at the National Cancer Center Hospital, Tokyo, between 1996 and 2002 [51], as shown in Table S1. Tumor samples were obtained at the time of excision and were cryopreserved in liquid nitrogen.
Table 1. Characteristics of the 88 patients with soft tissue sarcoma.
| Characteristics | STS patients (n = 88) | |
| Gender | Male | 46 |
| Female | 42 | |
| Age | Median | 54 |
| MAD | 19 | |
| Histological type | UPS | 20 |
| MLS | 20 | |
| SS | 17 | |
| MFS | 15 | |
| LMS | 6 | |
| FS | 5 | |
| MPNST | 5 | |
| Histological grade | 1 | 14 |
| 2 | 23 | |
| 3 | 51 | |
| Relapse events | Metastasis | 43 |
STS: soft tissue sarcoma, MAD: Median absolute deviation, UPS: undifferentiated pleomorphic sarcoma, MLS: myxoid liposarcoma, SS: synovial sarcoma, MFS: myxofibrosarcoma, LMS: leiomyosarcoma, FS: fibrosarcoma, MPNST: malignant peripheral nerve sheath tumor.
Microarray analysis
For RNA extraction, trained pathologists carefully excised the tissue samples from the main tumor, leaving a margin free from the surrounding nontumorous tissue. The elimination of nontumorous stromal cells is necessary for gene expression analysis of carcinomas because tumor tissues contain a significant number of nontumorous stromal cells, including fibroblasts, endothelial cells, and inflammation-associated cells. STS contains non-tumorous stromal cells that are difficult to exclude because STS originates from mesenchymal cells. However, in STS, the tumor tissue contains very few non-tumorous stromal cells and therefore unlikely to confound the analysis. Hence, laser microdissection was not performed in this study. Total RNA samples extracted from the bulk tissue specimens were labeled with biotin and hybridized to high-density oligonucleotide microarrays (Human Genome U133A 2.0 Array; Affymetrix, Santa Clara, CA, USA) comprising 22,283 probe sets representing 18,400 transcripts, according to the manufacturer’s instructions. The scanned array data were processed using the Affymetrix Microarray Suite v.5.1 software (MAS5), which scaled the average intensity of all the genes on each array to the target signal of 1000. The microarray data from the present study are available in the Genome Medicine Database of Japan (GeMDBJ) [52] (https://gemdbj.nibio.go.jp/dgdb/) under the accession number EXPR058P.
Data preprocessing
We excluded 68 control probe sets and 2343 genes that were subject to cross-hybridization according to NetAffx Annotation (www.affymetrix.com). Furthermore, we excluded those genes for which more than 50% (44/88) of the samples showed an absent call (i.e., the detection call determined by MAS5 based on the p value of the one-sided Wilcoxon signed-rank test; an absent call corresponds to p≥0.065, which is the default threshold in MAS5). An absent call indicates that the expression signal was undetectable. Genes showing low variance, i.e., a signal range value (95th percentile to 5th percentile) of less than 2000, were excluded [40]. Furthermore, we conducted an OMIM-based reduction of the number of candidate genes. In total, 1412 genes were selected, to which we applied log-transformation or binarization using the median value as a threshold for each gene, as shown in Fig. 1. The 2 types of datasets, log-transformed and binarized, were used for ANOVA and the logrank test, respectively.
Figure 1. A schematic of gene selection and the simulation based on the permutation test.

(A) The knowledge (OMIM)-based method. The list of OMIM numbers related to cancer (e.g., cancer, carcinoma, sarcoma, tumor, and neoplasm) was selected and converted into Affymetrix probe IDs in Ensembl. (B) Prefiltering of probe sets. This procedure was based on the number of absent calls and the range of signals. A signal range (95th percentile to 5th percentile) of >2000 was used as a percentile filter. Furthermore, we excluded probe sets for which the number of absent calls was >50% (44/88). Probe sets related to cancer were selected using the OMIM-based method. (C) Integration of survival analysis and discriminant analysis. (D) Clinical data from all patients were permutated. Permutated data for 72 STS patients (20 UPS, 15 MFS, 20 MLS, and 17 SS patients) were extracted from the permutated data of all patients. For these data, p values (p 1) were calculated by applying ANOVA to the log-transformed gene expression data to discriminate among UPS, MFS, MLS, and SS. In addition, permutated data from 88 patients were used for survival analysis. For these data, p values (p 2) were calculated by applying the logrank test to the binarized gene expression data to analyze the outcomes in the STS group. The integrated statistic p′ was defined as p 1×p 2. The lowest p′ value was selected for each repetition. This procedure was repeated 100,000 times, and an empirical null distribution was constructed. Using the distribution, the actual p′ value obtained from the real data was converted to the adjusted p value (based on the correction for multiple testing problems).
Simulation based on the combination of a permutation test and the integration of multiple statistics
We previously proposed a statistical simulation based on a permutation test and the integration of multiple statistics [51]. This method was used in the present study. We first calculated p values using ANOVA to discriminate among histological subtypes, including UPS, MFS, SS, and MLS. We also calculated p values by means of the logrank test in the survival analysis of all STS patients in relation to the 1412 filtered genes. We defined the integrated statistic p′ as p 1×p 2, where p 1 is the p value from ANOVA and p 2 is the p value from the logrank test. The same STS patients (n = 72; 20 UPS, 15 MFS, 17 SS, and 20 MLS patients) were used in both of these tests. The integrated statistic p′ could be underestimated by the use of 72 common samples. Therefore, to cancel this influence, we conducted a simulation based on the permutation test, as shown in Fig. 1, to estimate the adjusted p′ values as well as the multiple testing problems.
Statistical analysis
The median value of the gene expression signals for each gene was calculated, and the patients were distributed into 2 groups using the median value as a threshold for each gene. Logrank tests [53] were performed for overall survival of STS patients for each gene. We also calculated Spearman’s rank correlation coefficients to assess the relationships between gene expression signals and histological grades [54] or incidence of tumor metastases. We considered data obtained after 50 months of follow-up as censored data in the analysis of the logrank test, similar to the procedure followed in our previous study [51]. Kaplan-Meier curves [55] based on histological subtype were constructed for all STS patients.
OMIM
OMIM is a continuously updated catalog of human genes and genetic disorders and traits, with a focus on the molecular relationship between genetic variation and phenotypic expression. The list of MIM gene accession numbers associated with keywords related to cancer was obtained from the OMIM website (http://www.omim.org/). We used several keywords related to cancer, including “cancer,” “carcinoma,” “sarcoma,” “tumor,” and “neoplasm,” to create the MIM gene accession number list. There were 4394 MIM gene accession numbers, as shown in Table S2. The final MIM gene accession number list was obtained on January 10, 2014.
Ensembl
Ensembl is a joint project between EMBL-EBI and the Sanger Centre to develop software that produces and maintains automatic annotation of eukaryotic genomes [56]. We converted MIM numbers to the Affymetrix probe set IDs of the Human Genome U133A 2.0 Array using information retrieved from Ensembl on January 10, 2014. There were 5155 Affymetrix probe set IDs, as shown in Table S3.
Principal component analysis (PCA)
We used PCA to reduce the gene expression profile data to a two-dimensional dataset. PCA was first proposed in 1901 by Pearson [57]. This method is a statistical procedure that uses orthogonal transformation to convert a set of observations of possibly correlated variables into a set of values of linearly uncorrelated variables called principal components (PCs). The number of PCs is less than or equal to the number of original variables. This transformation is defined in such a way that the first PC has the greatest possible variance.
Multiple testing correction
The Bonferroni correction is a method used to address the problem of multiple comparisons (also known as the multiple testing problem). It is considered the simplest and most conservative method for control of the family-wise error rate (FWER). False discovery rate (FDR) controlling procedures, such as the Benjamini-Hochberg (BH) method [58], are more powerful (i.e., less conservative) than the FWER procedures, but their use increases the likelihood of false positives within the rejected hypothesis. In the present study, the BH method was used to calculate the q value. The q value is defined as an FDR analog of the p value.
Heatmap and hierarchical clustering analyses
A heatmap was created using the R program (function heatmap.2 in Package gplots) for the log-transformed and scaled gene expression data of selected genes. Hierarchical clustering was also conducted using the Euclidean distance and complete linkage (default parameters of function heatmap.2).
Results
Kaplan-Meier curves for 4 histological subtypes
Kaplan-Meier curves based on a histological subtype were constructed for all STS patients, as shown in Fig. 2. This figure shows that MFS had a good prognosis, MLS and SS had intermediate prognoses, and UPS had a poor prognosis. Although the logrank test yielded statistically significant results (p<0.05) in histological types, we conducted gene expression analysis to select molecular markers for more accurate diagnosis in accordance with the analysis.
Figure 2. Kaplan-Meier curves for 4 histological types of STS.
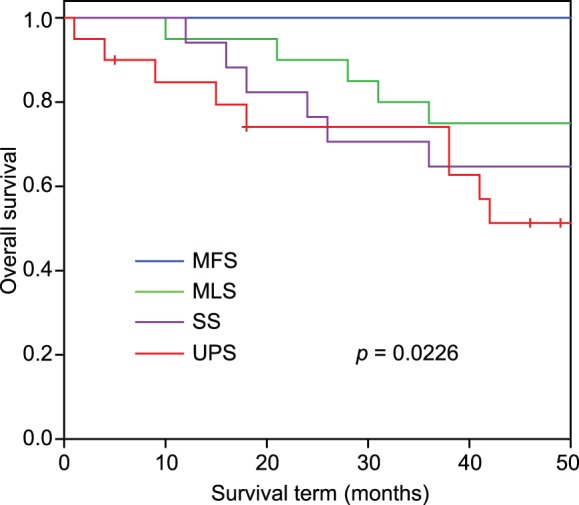
P value was calculated by logrank test. UPS: undifferentiated pleomorphic sarcoma, MLS: myxoid liposarcoma, SS: synovial sarcoma, MFS: myxofibrosarcoma.
Extraction of genes that are both diagnostic and prognostic markers, by means of a simulation using the permutation test
To extract genes that are both diagnostic markers (for discrimination of histological subtypes) and prognostic markers (of overall survival in STS), we applied a simulation based on the combination of a permutation test and the integration of multiple statistics into 1412 prefiltered probe sets of microarray data obtained from STS patients. As shown in Table 2, 29 probe sets, representing 25 genes, were extracted (adjusted p value <0.05).
Table 2. Genes extracted using the simulation based on the permutation test.
| Affymetrix probe ID | Accession no. | Gene symbol | p value | Integrated statistics p′ | Adjusted p value | |
| ANOVA | Log-rank test | |||||
| 200832_s_at | AB032261 | SCD1 | 2.47E-06 | 6.06E-03 | 1.50E-08 | 6.70E-04 |
| 200887_s_at | NM_007315 | STAT1 | 1.17E-04 | 1.91E-02 | 2.24E-06 | 3.59E-02 |
| 201231_s_at | NM_001428 | ENO1/MBP1 | 2.27E-08 | 1.06E-03 | 2.40E-11 | <1.00E-05 |
| 201508_at | NM_001552 | IGFBP4 | 3.21E-06 | 4.01E-02 | 1.29E-07 | 3.76E-03 |
| 202236_s_at | NM_003051 | SLC16A1/MCT1 | 1.12E-04 | 6.93E-04 | 7.77E-08 | 2.34E-03 |
| 202870_s_at | NM_001255 | CDC20 | 9.26E-07 | 6.28E-03 | 5.81E-09 | 2.90E-04 |
| 203065_s_at | NM_001753 | CAV1 | 1.33E-10 | 3.28E-02 | 4.35E-12 | <1.00E-05 |
| 203323_at | BF197655 | CAV2 | 5.67E-10 | 2.35E-02 | 1.33E-11 | <1.00E-05 |
| 203554_x_at | NM_004219 | PTTG1 | 7.33E-09 | 5.64E-03 | 4.13E-11 | <1.00E-05 |
| 207011_s_at | NM_002821 | PTK7 | 2.57E-07 | 1.89E-02 | 4.86E-09 | 2.70E-04 |
| 207168_s_at | NM_004893 | H2AFY/H2AX | 2.83E-05 | 1.80E-02 | 5.11E-07 | 1.19E-02 |
| 207543_s_at | NM_000917 | P4HA1 | 1.06E-08 | 5.73E-04 | 6.06E-12 | <1.00E-05 |
| 208680_at | L19184 | PRDX1 | 5.73E-08 | 1.64E-02 | 9.37E-10 | 6.00E-05 |
| 208694_at | U47077 | PRKDC/DNA-PKcs | 1.71E-04 | 1.31E-02 | 2.25E-06 | 3.60E-02 |
| 208767_s_at | AW149681 | LAPTM4B | 5.47E-05 | 1.65E-02 | 9.04E-07 | 1.81E-02 |
| 209030_s_at | NM_014333 | CADM1/TSLC1 | 1.80E-10 | 4.20E-02 | 7.59E-12 | <1.00E-05 |
| 209031_at | AL519710 | CADM1/TSLC1 | 2.10E-11 | 5.68E-03 | 1.19E-13 | <1.00E-05 |
| 209543_s_at | M81104 | CD34 | 2.66E-06 | 1.54E-02 | 4.10E-08 | 1.33E-03 |
| 210495_x_at | AF130095 | FN1 | 3.90E-08 | 1.78E-02 | 6.96E-10 | 2.00E-05 |
| 210559_s_at | D88357 | CDK1/CDC2 | 7.69E-07 | 4.30E-02 | 3.31E-08 | 1.14E-03 |
| 212097_at | AU147399 | CAV1 | 1.54E-09 | 2.95E-03 | 4.53E-12 | <1.00E-05 |
| 212464_s_at | X02761 | FN1 | 1.93E-08 | 1.78E-02 | 3.44E-10 | 1.00E-05 |
| 217294_s_at | U88968 | ENO1/MBP1 | 8.81E-08 | 2.33E-02 | 2.05E-09 | 1.50E-04 |
| 217871_s_at | NM_002415 | MIF | 5.67E-08 | 1.46E-02 | 8.29E-10 | 5.00E-05 |
| 218308_at | NM_006342 | TACC3 | 2.82E-05 | 2.26E-02 | 6.38E-07 | 1.40E-02 |
| 218502_s_at | NM_014112 | TRPS1 | 1.48E-18 | 3.99E-02 | 5.90E-20 | <1.00E-05 |
| 218755_at | NM_005733 | KIF20A/MKlp2 | 3.01E-06 | 2.02E-02 | 6.08E-08 | 1.94E-03 |
| 219918_s_at | NM_018123 | ASPM | 1.22E-05 | 1.64E-02 | 2.00E-07 | 5.51E-03 |
| 220942_x_at | NM_014367 | FAM162A/HGTD-P | 4.44E-05 | 3.21E-02 | 1.42E-06 | 2.56E-02 |
Adjusted p values were calculated using the permutation test (100,000 repeats).
Association analysis of the histological grade (or metastasis status) and gene expression data for the 25 selected genes
We next used Spearman’s rank correlation analysis to analyze the association between the gene expression level in STS patients and the histological grade (or metastasis status), as shown in Table 3. Table 3 shows that genes with positive ρ were upregulated in highly malignant tumors, whereas genes with negative ρ were downregulated in highly malignant tumors. The expression levels of almost all of the 25 genes were associated with either the histological grade or metastasis. However, stearoyl-CoA desaturase 1 (SCD1) and signal transducer and activator of transcription 1 (STAT1) were not associated with either the histological grade (SCD1: ρ = −0.0191, p = 0.860; STAT1: ρ = −0.146, p = 0.173) or metastasis (SCD1: ρ = 0.0237, p = 0.826; STAT1: ρ = −0.177, p = 0.0995). This result indicates that SCD1 and STAT1 expression levels can be related to the overall survival of STS patients but not to metastasis. Therefore, these data suggest that SCD1 and STAT1 expression levels can be used in combination with the histological grade to predict the survival of STS patients.
Table 3. Correlation analysis based on Spearman’s rank correlation coefficient between gene expression data and the histological grade (or metastasis status).
| Affymetrixprobe ID | Accession no. | Gene symbol | With histological grade | With metastasis | ||
| ρ | p value | ρ | p value | |||
| 200832_s_at | AB032261 | SCD1 | −0.0191 | 8.60E-01 | 0.0237 | 8.26E-01 |
| 200887_s_at | NM_007315 | STAT1 | −0.146 | 1.73E-01 | −0.177 | 9.95E-02 |
| 201231_s_at | NM_001428 | ENO1/MBP1 | 0.356 | 6.66E-04 | 0.247 | 2.01E-02 |
| 201508_at | NM_001552 | IGFBP4 | −0.247 | 2.04E-02 | −0.211 | 4.87E-02 |
| 202236_s_at | NM_003051 | SLC16A1/MCT1 | 0.400 | 1.12E-04 | 0.341 | 1.17E-03 |
| 202870_s_at | NM_001255 | CDC20 | 0.413 | 6.27E-05 | 0.204 | 5.65E-02 |
| 203065_s_at | NM_001753 | CAV1 | −0.250 | 1.87E-02 | −0.159 | 1.39E-01 |
| 203323_at | BF197655 | CAV2 | −0.363 | 5.11E-04 | −0.094 | 3.82E-01 |
| 203554_x_at | NM_004219 | PTTG1 | 0.402 | 1.05E-04 | 0.132 | 2.20E-01 |
| 207011_s_at | NM_002821 | PTK7 | 0.265 | 1.26E-02 | 0.232 | 2.95E-02 |
| 207168_s_at | NM_004893 | H2AFY/H2AX | 0.411 | 7.03E-05 | 0.161 | 1.35E-01 |
| 207543_s_at | NM_000917 | P4HA1 | 0.449 | 1.12E-05 | 0.424 | 3.89E-05 |
| 208680_at | L19184 | PRDX1 | 0.258 | 1.51E-02 | 0.111 | 3.05E-01 |
| 208694_at | U47077 | PRKDC/DNA-PKcs | 0.409 | 7.64E-05 | 0.229 | 3.21E-02 |
| 208767_s_at | AW149681 | LAPTM4B | 0.329 | 1.75E-03 | 0.130 | 2.27E-01 |
| 209030_s_at | NM_014333 | CADM1/TSLC1 | 0.196 | 6.70E-02 | 0.136 | 2.05E-01 |
| 209031_at | AL519710 | CADM1/TSLC1 | 0.231 | 3.03E-02 | 0.143 | 1.85E-01 |
| 209543_s_at | M81104 | CD34 | −0.363 | 5.11E-04 | −0.239 | 2.52E-02 |
| 210495_x_at | AF130095 | FN1 | 0.286 | 6.99E-03 | 0.096 | 3.73E-01 |
| 210559_s_at | D88357 | CDK1/CDC2 | 0.435 | 2.34E-05 | 0.259 | 1.50E-02 |
| 212097_at | AU147399 | CAV1 | −0.237 | 2.64E-02 | −0.163 | 1.28E-01 |
| 212464_s_at | X02761 | FN1 | 0.286 | 6.99E-03 | 0.0944 | 3.82E-01 |
| 217294_s_at | U88968 | ENO1/MBP1 | 0.387 | 1.97E-04 | 0.187 | 8.03E-02 |
| 217871_s_at | NM_002415 | MIF | 0.421 | 4.41E-05 | 0.308 | 3.47E-03 |
| 218308_at | NM_006342 | TACC3 | 0.333 | 1.52E-03 | 0.136 | 2.05E-01 |
| 218502_s_at | NM_014112 | TRPS1 | 0.276 | 9.23E-03 | 0.242 | 2.31E-02 |
| 218755_at | NM_005733 | KIF20A/MKlp2 | 0.407 | 8.35E-05 | 0.162 | 1.31E-01 |
| 219918_s_at | NM_018123 | ASPM | 0.399 | 1.16E-04 | 0.204 | 5.71E-02 |
| 220942_x_at | NM_014367 | FAM162A/HGTD-P | 0.151 | 1.60E-01 | 0.239 | 2.47E-02 |
Hierarchical clustering based on the gene expression pattern of the 25 selected genes
We performed hierarchical clustering for the 29 selected probe sets, representing 25 genes and 4 histological subtypes (UPS, MFS, MLS, and SS), as shown in Fig. 3. The genes were roughly classified into 4 clusters (clusters A, B, C, and D). Almost all genes were upregulated in both UPS and MFS. In addition, genes in cluster A were upregulated in SS, and genes in cluster D were upregulated in MLS.
Figure 3. Heatmap and hierarchical clustering analyses.

Twenty-nine probe sets were extracted using a simulation based on the permutation test (with adjusted p<0.05). The 29 probe sets were roughly divided into 4 clusters (clusters A–D). Columns represent probe sets, and rows represent samples. Red and green indicate high and low expression, respectively. UPS: undifferentiated pleomorphic sarcoma, MLS: myxoid liposarcoma, SS: synovial sarcoma, MFS: myxofibrosarcoma.
Analysis of the distribution of histological subtypes based on gene expression levels
We performed PCA to calculate the first and the second PCs using the 29 probe sets. Detailed information on PCA, including eigenvector, standard deviation, proportion of variance, and cumulative proportion, is provided in Tables S4 and S5. The distribution of the 4 histological subtypes of STS on the 2 axes is shown in Fig. 4. The 4 histological subtypes were clearly classified into 3 clusters (SS, MLS, and UPS+MFS). This result indicated that UPS and MFS had histological similarities and similar gene expression patterns. Therefore, to discriminate between UPS and MFS, we applied Welch’s t test and the BH method to the gene expression data of the 29 probe sets, as shown in Table 4. We extracted 9 probe sets, representing 8 genes (q value <0.05): enolase 1 (ENO1)/c-myc-promoter binding protein-1 (MBP1); prolyl 4-hydroxylase subunit alpha-1 (P4HA1); peroxiredoxin 1 (PRDX1); CD34; family with sequence similarity 162, member A (FAM162A)/human growth and transformation-dependent protein (HGTD-P); protein tyrosine kinase 7 (PTK7); and macrophage migration inhibitory factor (MIF). We performed PCA to calculate the first and the second PCs from these 9 probe sets. Detailed information of PCA, including eigenvector, standard deviation, proportion of variance, and cumulative proportion, are shown in Table S5. The distribution of the 2 histological subtypes, UPS and MFS, on the 2 axes is shown in Fig. 5. UPS and MFS were classified into approximately 2 clusters. For the contribution of this classification, MIF, ENO1/MBP1, and CD34 contributed to the top 3 largest coefficients for PC1, PTK7, PRDX1, and ENO1/MBP1 contributed to the top 3 largest coefficients for PC2, and only SCD1 contributed to the largest coefficients for PC3, as shown in Table S5. MIF, ENO1/MBP1, and SCD1 were extracted in our previous study [51]. We also applied Welch’s t test and the BH method to the gene expression data from the 29 probe sets to discriminate UPS from SS and UPS from MLS, as shown in Table 4.
Figure 4. Principal component analysis using 29 probe sets for 4 histological types.
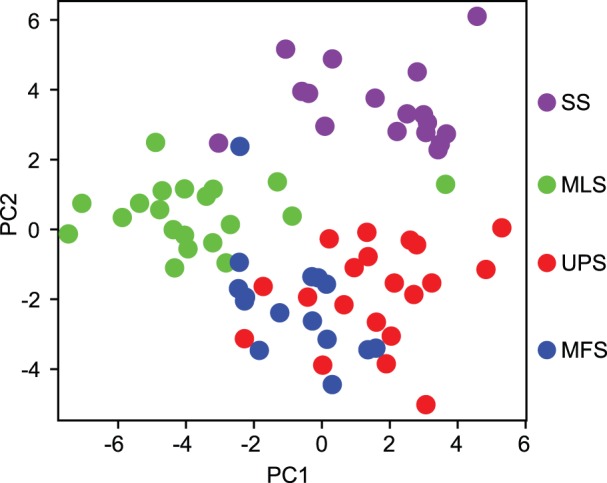
The x-axis and y-axis represent the first and second principal components (PC1 and PC2), respectively. Each dot represents a sample colored according to its histological type. UPS: undifferentiated pleomorphic sarcoma, MLS: myxoid liposarcoma, SS: synovial sarcoma, MFS: myxofibrosarcoma.
Table 4. Pairwise comparison between histological types using Welch’s t test for 29 probe sets.
| Affymetrixprobe ID | Accessionno. | Gene symbol | UPS vs. MFS | UPS vs. SS | UPS vs. MLS | ||||||
| p value | q value | p value | q value | p value | q value | ||||||
| 200832_s_at | AB032261 | SCD1 | 7.36E-05 | * | 8.87E-04 | 1.06E-03 | * | 2.56E-03 | 3.52E-01 | 4.26E-01 | |
| 200887_s_at | NM_007315 | STAT1 | 2.81E-01 | 4.07E-01 | 1.54E-03 | * | 3.19E-03 | 2.04E-01 | 2.69E-01 | ||
| 201231_s_at | NM_001428 | ENO1/MBP1 | 1.06E-04 | * | 8.87E-04 | 4.73E-08 | * | 6.85E-07 | 4.27E-06 | * | 1.42E-05 |
| 201508_at | NM_001552 | IGFBP4 | 4.21E-02 | 1.15E-01 | 7.39E-03 | * | 1.13E-02 | 7.25E-02 | 1.00E-01 | ||
| 202236_s_at | NM_003051 | SLC16A1/MCT1 | 1.54E-01 | 2.80E-01 | 3.92E-01 | 4.06E-01 | 6.49E-04 | * | 1.25E-03 | ||
| 202870_s_at | NM_001255 | CDC20 | 2.10E-01 | 3.58E-01 | 1.23E-03 | * | 2.74E-03 | 6.26E-06 | * | 1.78E-05 | |
| 203065_s_at | NM_001753 | CAV1 | 8.76E-01 | 8.76E-01 | 5.56E-07 | * | 2.69E-06 | 5.31E-01 | 5.93E-01 | ||
| 203323_at | BF197655 | CAV2 | 8.45E-01 | 8.75E-01 | 6.14E-05 | * | 1.98E-04 | 1.26E-03 | * | 2.15E-03 | |
| 203554_x_at | NM_004219 | PTTG1 | 3.76E-01 | 4.96E-01 | 8.95E-05 | * | 2.60E-04 | 1.59E-08 | * | 2.31E-07 | |
| 207011_s_at | NM_002821 | PTK7 | 6.14E-03 | * | 2.23E-02 | 4.21E-03 | * | 6.78E-03 | 9.19E-01 | 9.19E-01 | |
| 207168_s_at | NM_004893 | H2AFY/H2AX | 4.37E-02 | 1.15E-01 | 1.18E-01 | 1.37E-01 | 6.75E-06 | * | 1.78E-05 | ||
| 207543_s_at | NM_000917 | P4HA1 | 1.22E-04 | * | 8.87E-04 | 2.64E-02 | * | 3.48E-02 | 2.51E-03 | * | 4.05E-03 |
| 208680_at | L19184 | PRDX1 | 1.84E-03 | * | 7.61E-03 | 5.31E-05 | * | 1.93E-04 | 1.36E-08 | * | 2.31E-07 |
| 208694_at | U47077 | PRKDC/DNA-PKcs | 5.49E-02 | 1.33E-01 | 9.76E-01 | 9.76E-01 | 1.13E-03 | * | 2.06E-03 | ||
| 208767_s_at | AW149681 | LAPTM4B | 4.20E-01 | 5.30E-01 | 3.73E-02 | * | 4.60E-02 | 8.30E-03 | * | 1.27E-02 | |
| 209030_s_at | NM_014333 | CADM1/TSLC1 | 2.49E-01 | 3.80E-01 | 2.81E-07 | * | 1.82E-06 | 6.43E-01 | 6.66E-01 | ||
| 209031_at | AL519710 | CADM1/TSLC1 | 6.04E-02 | 1.35E-01 | 2.67E-07 | * | 1.82E-06 | 2.71E-01 | 3.42E-01 | ||
| 209543_s_at | M81104 | CD34 | 8.73E-03 | * | 2.81E-02 | 1.78E-01 | 1.91E-01 | 3.97E-05 | * | 8.22E-05 | |
| 210495_x_at | AF130095 | FN1 | 4.83E-01 | 5.61E-01 | 2.50E-03 | * | 4.27E-03 | 3.53E-06 | * | 1.42E-05 | |
| 210559_s_at | D88357 | CDK1/CDC2 | 7.05E-02 | 1.46E-01 | 2.35E-02 | * | 3.24E-02 | 3.57E-06 | * | 1.42E-05 | |
| 212097_at | AU147399 | CAV1 | 6.43E-01 | 6.91E-01 | 3.14E-07 | * | 1.82E-06 | 4.16E-01 | 4.83E-01 | ||
| 212464_s_at | X02761 | FN1 | 5.22E-01 | 5.83E-01 | 2.33E-03 | * | 4.22E-03 | 2.07E-06 | * | 1.20E-05 | |
| 217294_s_at | U88968 | ENO1/MBP1 | 4.24E-04 | * | 2.46E-03 | 4.07E-05 | * | 1.69E-04 | 1.55E-07 | * | 1.50E-06 |
| 217871_s_at | NM_002415 | MIF | 5.31E-06 | * | 1.54E-04 | 1.38E-01 | 1.54E-01 | 1.35E-05 | * | 3.27E-05 | |
| 218308_at | NM_006342 | TACC3 | 2.36E-01 | 3.80E-01 | 7.67E-04 | * | 2.02E-03 | 2.91E-05 | * | 6.49E-05 | |
| 218502_s_at | NM_014112 | TRPS1 | 3.64E-01 | 4.96E-01 | 5.21E-11 | * | 1.51E-09 | 1.85E-02 | * | 2.68E-02 | |
| 218755_at | NM_005733 | KIF20A/MKlp2 | 4.44E-01 | 5.37E-01 | 9.97E-03 | * | 1.45E-02 | 4.41E-06 | * | 1.42E-05 | |
| 219918_s_at | NM_018123 | ASPM | 1.11E-01 | 2.15E-01 | 2.25E-03 | * | 4.22E-03 | 7.89E-07 | * | 5.72E-06 | |
| 220942_x_at | NM_014367 | FAM162A/HGTD-P | 1.39E-03 | * | 6.70E-03 | 3.81E-02 | * | 4.60E-02 | 6.23E-01 | 6.66E-01 | |
*q <0.05. The p value was calculated using Welch’s t test, and the q value was calculated from the p value by means of the Benjamini-Hochberg method for the correction of multiple testing problems.
Figure 5. Principal component analysis using 9 probe sets for UPS and MFS.
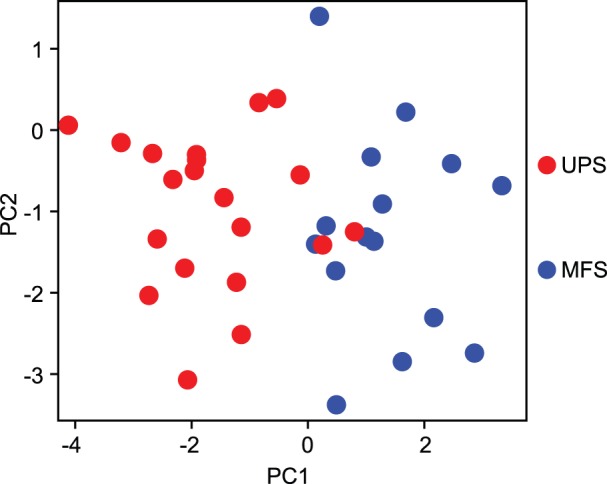
The x-axis and y-axis represent the first and second principal components (PC1 and PC2), respectively. Each dot represents a sample colored according to its histological type. UPS: undifferentiated pleomorphic sarcoma, MLS: myxoid liposarcoma, SS: synovial sarcoma, MFS: myxofibrosarcoma.
Classification of the 25 genes based on pairwise comparison of histological subtypes
We classified the 25 genes into 7 groups on the basis of 3 comparisons (UPS vs. MFS, UPS vs. SS, and UPS vs. MLS), as shown in Fig. 6. Only 3 genes, ENO1/MBP1, P4HA1, and PRDX1, were commonly selected (genes that were selected in the UPS vs. MFS comparison were also selected in the UPS vs. SS or UPS vs. MLS comparison). Furthermore, we compared the 25 genes selected in our study with the genes involved in the complexity index in sarcomas (CINSARC) [59] because the use of CINSARC (composed of 67 genes) instead of the FNCLCC grading system [1], [2] was recently proposed for predicting metastasis in STS [59]. In this comparison, only 4 common genes, that is, pituitary tumor-transforming 1 (PTTG1), abnormal spindle-like microcephaly-associated protein (ASPM), cell-division cycle protein 20 (CDC20), and kinesin family member 20A (KIF20A)/mitotic kinesin-like protein 2 (MKlp2), were extracted. The differential expression of these 4 genes was statistically significant (q <0.05) for UPS vs. SS and for UPS vs. MLS, but not for UPS vs. MFS. These 4 genes belonged to cluster B, as shown in Fig. 3. Consequently, the 25 genes were classified into 7 groups on the basis of pairwise comparisons of histological subtypes, as shown in Fig. 4.
Figure 6. A Venn diagram of gene classification based on pairwise comparisons of histological types using Welch’s t test.
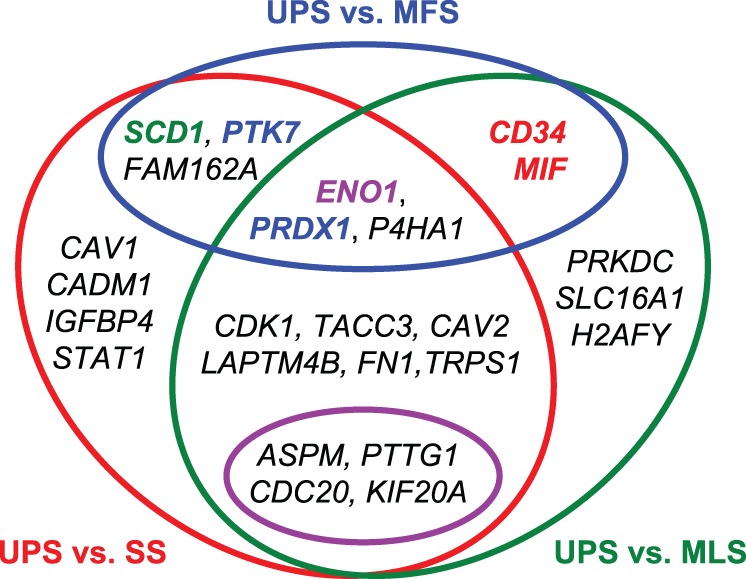
Genes inside the red circle were statistically significant (q <0.05 calculated using Welch’s t test and the BH method) in the comparison of UPS with SS. Genes inside the green oval were statistically significant (q <0.05) in the comparison of UPS with MLS. Genes inside the blue oval were statistically significant (q <0.05) in the comparison of UPS and MFS. Genes inside the pink oval are common to CINSARC and our 25-gene set. For PCA of the 9-probe set, MIF and CD34 highlighted in red were the first and third largest contributing coefficients to PC1, respectively. PTK7 and PRDX1 highlighted in blue were the first and second largest contributing coefficients to PC2, respectively. ENO1/MBP1 highlighted in purple was the second largest contributing coefficient to PC1 and the third largest contributing coefficient to PC2. SCD1 highlighted in green was the largest contributing coefficient to PC3.
Survival analysis in UPS patients
We used the logrank test to analyze the survival of UPS patients. We selected the best p value for various thresholds (30th, 40th, 50th, 60th, 70th, and 80th percentiles) of gene expression signals in UPS patients for each probe set when the gene expression signals were binarized. Adjusted p values were obtained by adjusting the data for the multiple testing problem (6 thresholds×29 probe sets) based on the permutation test, as shown in Table S6. Only STAT1 showed a statistically significant association with survival in UPS (logrank p value 1.84×10−6 and adjusted p value 2.99×10−3 after the permutation test). Fig. 7 shows that STAT1-positive and STAT1-negative groups had clearly different survival curves based on the Kaplan-Meier method.
Figure 7. The Kaplan-Meier curve and the logrank test for STAT1 in UPS patients.
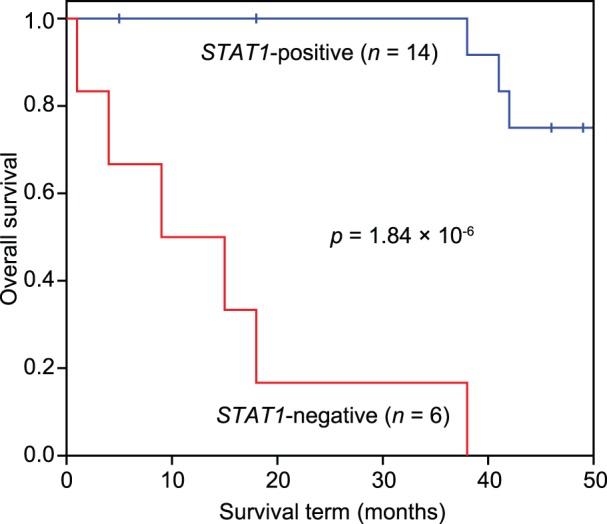
The STAT1-positive group (STAT1 expression level >4871.5) consisted of 14 patients (blue line), and the STAT1-negative group consisted of 6 patients (red line). A hazard ratio (exp(B) = 30.2) was calculated using the Cox proportional hazards model.
Discussion
In the present study, we conducted a simulation based on a permutation test to extract genes that are both diagnostic markers (for discrimination of histological subtypes) and prognostic markers (for overall survival in STS). As shown in Table 2, 25 genes were extracted, and their adjusted p values were statistically significant (adjusted p<0.05). We analyzed studies related to these 25 genes and found many reports suggesting that these 25 genes are effective prognostic/predictive factors or therapeutic targets, as shown in Table S7, according to the literature (See Supplementary Discussion).
Although we did not try to identify the molecular mechanisms behind the 25 selected genes, several published studies have examined pathways related to these 25 genes, as shown in Table S7 and Fig. 8. These 25 genes are roughly classified into 4 types, namely, hypoxia-related genes (MIF, SCD1, P4HA1, ENO1/MBP1, FAM162A/HGTD-P, SLC16A1/MCT1, FN1, and STAT1), cell cycle- and DNA repair-related genes (ASPM, CDK1/CDC2, CDC20, KIF20A/MKlp2, PTTG1, TACC3, PRDX1, PRKDC/DNA-PKcs, and H2AFY/H2AX), growth factor signal transduction-related genes, and other genes. Cell cycle- and DNA repair-related genes, hypoxia-induced genes, and growth factor signal transduction-related genes are key players in tumor growth, angiogenesis, metabolism, invasion, and metastasis in various types of cancer. In fact, these processes are attenuated by the inhibition or silencing of many of these 25 genes, as shown in Table S7. These genes are therefore possible prognostic/predictive markers and/or therapeutic targets.
Figure 8. A hypothetical regulation model of metabolic and signaling control in highly malignant STS.

(A) Signaling pathways, excluding cell cycle and DNA repair. (B) Cell cycle and DNA repair pathways. The pink oval indicates the genes selected in the present study. MUFA, monounsaturated fatty acid; SFA, saturated fatty acid; SCD1, stearoyl-CoA desaturase 1; MIF, macrophage migration inhibitory factor; CXCR, CXC chemokine receptor; PI3K, phosphoinositide 3-kinase; MAPK, extracellular signal-regulated kinase; ERK, mitogen-activated protein kinase; PTTG1, pituitary tumor-transforming 1; ASPM, abnormal spindle-like microcephaly-associated protein; CDC20, cell division cycle protein 20; KIF20A, kinesin family member 20A; ENO1, enolase 1; P4HA, prolyl 4-hydroxylase subunit α; PRDX1, peroxiredoxin 1; FAM162A, family with sequence similarity 162, member A; STAT1, signal transducer and activator of transcription 1; CDK1, cyclin-dependent kinase 1; TACC3, transforming, acidic coiled-coil containing protein 3; PRKDC, protein kinase, DNA-activated, catalytic polypeptide; H2AFY, H2A histone family, member Y; SLC16A1, solute carrier family 16, member 1; VEGF, vascular endothelial growth factor; HIF, hypoxia inducible factor; PLOD2, procollagen-lysine,2-oxoglutarate 5-dioxygenase 2; NF-κB, nuclear factor-kappa B.
STAT1 expression was found to be strongly associated with survival in UPS patients. STAT1 interacts directly with p53 and induces cell growth arrest and apoptosis, as shown in Fig. 8. Although STAT1 is repressed by HIF-1, the STAT1-positive group among the UPS patients had a better prognosis, even when hypoxia-related genes were upregulated. Therefore, STAT1 is a possible novel, independent prognostic/predictive factor of STS, particularly UPS.
In the diagnosis of STS, classification of UPS is the most controversial topic. Among the 25 selected genes, hypoxia-related genes (MIF, SCD1, P4HA1, ENO1/MBP1, FAM162A/HGTD-P, SLC16A1/MCT1, FN1, and STAT1) are present in this study. In particular, the genes MIF, SCD1, P4HA1, ENO1/MBP1, and FAM162A/HGTD-P are differentially expressed between UPS and MFS, as shown in Fig. 6 and Table 4. Furthermore, STAT1 is a prognostic marker in UPS patients, as shown in Fig. 7. Therefore, these hypoxia-related genes are promising prognostic and therapeutic targets and, if validated, may improve the treatment/diagnosis of this type of cancer. Further research is needed regarding the hypoxia-related pathways in highly malignant STS.
We manually constructed a hypothetical regulation model (Figure 8) of metabolic and signaling control in highly malignant STS. Nevertheless, according to the literature, a part of these networks could be automatically predicted by pathway and interaction analyses. For example, pathways of the cell cycle and the DNA damage response were identified by IntPath [33], [60], [61] with statistical significance (q value <0.05), as shown in Table S8. Interaction networks of the cell cycle (ASPM, CDK1, CDC20, KIF20A, PTTG1, PRKDC, and TACC3) and HIF-1 (MIF, ENO1, and PRDX1) were identified by means of STRING [62], as shown in Fig. S1. Nonetheless, these tools should be used with appropriate parameters [34], [60], [61]. Such tools are more effective methods when large numbers of candidate genes are extracted.
In summary, we analyzed microarray gene expression data from 88 STS patients using a combination method involving knowledge-based filtering and a simulation based on the integration of multiple statistics to reduce multiple testing problems. Our combination method automatically identified 25 genes in the gene expression data from STS. These genes showed significant differential expression between different histological subtypes, including UPS, and showed associations with survival in STS. Furthermore, we conducted a bibliographic survey in terms of cancer progression for the 25 identified genes, and substantial evidence was uncovered in the literature. These genes were roughly classified into 4 types, namely, hypoxia-related genes, cell cycle- and DNA repair-related genes, growth factor signal transduction-related genes, and other genes. STAT1 showed a statistically significant association with the survival of UPS patients (logrank adjusted p = 0.00299). Although only a few studies have investigated the association of these genes with survival in STS, many recent studies have reported that these genes are prognostic factors and/or therapeutic targets in other types of cancers. Therefore, these results suggest that our combination method is capable of identifying genes that are potential prognostic/predictive factors and/or therapeutic targets in STS and possibly in other cancers. These disease-associated genes deserve further preclinical and clinical validation.
Supporting Information
The pathways predicted by STRING from the 25 selected genes.
(PDF)
Clinical data of the 88 patients with soft tissue sarcoma. UPS: undifferentiated pleomorphic sarcoma, MLS: myxoid liposarcoma, SS: synovial sarcoma, MFS: myxofibrosarcoma, LMS: leiomyosarcoma, FS: fibrosarcoma, MPNST: malignant peripheral nerve sheath tumor, Tumor metastasis indicates the incidence of tumor metastasis in STS patients.
(XLS)
The MIM number list.
(XLS)
Selected Affymetrix probe IDs.
(XLS)
Information on PCA, including the eigenvector, standard deviation, proportion of variance, and cumulative proportion for 29 probe sets. PCA: principal component analysis, PC: principal components.
(XLS)
Information on PCA, including the eigenvector, standard deviation, proportion of variance, and cumulative proportion for 9 probe sets. PCA: principal component analysis, PC: principal components.
(XLS)
Survival analysis in UPS using the logrank test. Adjusted p values were calculated using the permutation test (100,000 repeats) from logrank p values.
(XLS)
Gene or pathway annotations and likelihood as prognostic/predictive factors and/or therapeutic targets. Adjusted p values were calculated using the permutation test (100,000 repeats) from logrank p values.
(XLS)
Pathway analysis in IntPath. k: genes from the overlap between genes in the list and genes in the pathway, n: the number of genes in the input gene list, m: the number of genes in the identified pathways, N: the total number of genes. The p values were calculated using the hypergeometric test; the q values were calculated from the p values using the Benjamini-Hochberg (BH) method.
(XLS)
(PDF)
Acknowledgments
The authors thank Professor Yasunori Machida (Nagoya University, Japan) and the Laboratory Head Hitoshi Ichikawa (National Cancer Center Research Institute, Japan) for the helpful discussions.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported in part by the Ministry of Education, Culture, Sports, Science, and Technology of Japan (MEXT): Grants-in-Aid for Scientific Research for Young Scientists (B) (nos. 21710211 and 24710222 to H.T.) and Grant-in-Aid for Scientific Research on Innovative Areas (no. 26114703 to H.T.). This work was also supported by the Advanced Research for Medical Products Mining Program of the National Institute of Biomedical Innovation (NIBIO ID10-41), the Futaba Electronics Memorial Foundation, the Research Foundation for the Electrotechnology of Chubu, and the Nakajima Foundation. The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of this manuscript.
References
- 1. Trojani M, Contesso G, Coindre JM, Rouesse J, Bui NB, et al. (1984) Soft-tissue sarcomas of adults; study of pathological prognostic variables and definition of a histopathological grading system. Int J Cancer 33: 37–42. [DOI] [PubMed] [Google Scholar]
- 2. Guillou L, Coindre JM, Bonichon F, Nguyen BB, Terrier P, et al. (1997) Comparative study of the National Cancer Institute and French Federation of Cancer Centers Sarcoma Group grading systems in a population of 410 adult patients with soft tissue sarcoma. J Clin Oncol 15: 350–362. [DOI] [PubMed] [Google Scholar]
- 3. Clark J, Rocques PJ, Crew AJ, Gill S, Shipley J, et al. (1994) Identification of novel genes, SYT and SSX, involved in the t(X;18)(p11.2;q11.2) translocation found in human synovial sarcoma. Nat Genet 7: 502–508. [DOI] [PubMed] [Google Scholar]
- 4. Antonescu CR, Elahi A, Humphrey M, Lui MY, Healey JH, et al. (2000) Specificity of TLS-CHOP rearrangement for classic myxoid/round cell liposarcoma: absence in predominantly myxoid well-differentiated liposarcomas. J Mol Diagn 2: 132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kohno T, Ichikawa H, Totoki Y, Yasuda K, Hiramoto M, et al. (2012) KIF5B-RET fusions in lung adenocarcinoma. Nat Med 18: 375–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lux ML, Rubin BP, Biase TL, Chen CJ, Maclure T, et al. (2000) KIT extracellular and kinase domain mutations in gastrointestinal stromal tumors. Am J Pathol 156: 791–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, et al. (2008) Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455: 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Helman LJ, Meltzer P (2003) Mechanisms of sarcoma development. Nat Rev Cancer 3: 685–694. [DOI] [PubMed] [Google Scholar]
- 9. Hasegawa T, Yamamoto S, Nojima T, Hirose T, Nikaido T, et al. (2002) Validity and reproducibility of histologic diagnosis and grading for adult soft-tissue sarcomas. Hum Pathol 33: 111–115. [DOI] [PubMed] [Google Scholar]
- 10. Fletcher CD (1992) Pleomorphic malignant fibrous histiocytoma: fact or fiction? A critical reappraisal based on 159 tumors diagnosed as pleomorphic sarcoma. Am J Surg Pathol 16: 213–228. [PubMed] [Google Scholar]
- 11. Hollowood K, Fletcher CD (1995) Malignant fibrous histiocytoma: morphologic pattern or pathologic entity? Semin Diagn Pathol 12: 210–220. [PubMed] [Google Scholar]
- 12. Fletcher CD, Gustafson P, Rydholm A, Willen H, Akerman M (2001) Clinicopathologic re-evaluation of 100 malignant fibrous histiocytomas: prognostic relevance of subclassification. J Clin Oncol 19: 3045–3050. [DOI] [PubMed] [Google Scholar]
- 13. Nakayama R, Nemoto T, Takahashi H, Ohta T, Kawai A, et al. (2007) Gene expression analysis of soft tissue sarcomas: characterization and reclassification of malignant fibrous histiocytoma. Mod Pathol 20: 749–759. [DOI] [PubMed] [Google Scholar]
- 14. Takahashi H, Nemoto T, Yoshida T, Honda H, Hasegawa T (2006) Cancer diagnosis marker extraction for soft tissue sarcomas based on gene expression profiling data by using projective adaptive resonance theory (PART) filtering method. BMC Bioinformatics 7: 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fletcher CDM, Unni KK, Mertens F, editors (2002) Pathology and Genetics of Tumors of Soft Tissue and Bone. Lyon: IARC Press.
- 16. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 17. Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Takahashi H, Takahashi A, Naito S, Onouchi H (2012) BAIUCAS: a novel BLAST-based algorithm for the identification of upstream open reading frames with conserved amino acid sequences and its application to the Arabidopsis thaliana genome. Bioinformatics 28: 2231–2241. [DOI] [PubMed] [Google Scholar]
- 19. Takahashi H, Nakagawa A, Kojima S, Takahashi A, Cha BY, et al. (2012) Discovery of novel rules for G-quadruplex-forming sequences in plants by using bioinformatics methods. J Biosci Bioeng 114: 570–575. [DOI] [PubMed] [Google Scholar]
- 20. Eisen MB, Spellman PT, Brown PO, Botstein D (1998) Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA 95: 14863–14868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Arima C, Hakamada K, Okamoto M, Hanai T (2008) Modified fuzzy gap statistic for estimating preferable number of clusters in fuzzy k-means clustering. J Biosci Bioeng 105: 273–281. [DOI] [PubMed] [Google Scholar]
- 22. Tomida S, Hanai T, Honda H, Kobayashi T (2002) Analysis of expression profile using fuzzy adaptive resonance theory. Bioinformatics 18: 1073–1083. [DOI] [PubMed] [Google Scholar]
- 23. Takahashi H, Tomida S, Kobayashi T, Honda H (2003) Inference of common genetic network using fuzzy adaptive resonance theory associated matrix method. J Biosci Bioeng 96: 154–160. [PubMed] [Google Scholar]
- 24. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Takahashi H, Honda H (2006) Modified signal-to-noise: a new simple and practical gene filtering approach based on the concept of projective adaptive resonance theory (PART) filtering method. Bioinformatics 22: 1662–1664. [DOI] [PubMed] [Google Scholar]
- 26. Takahashi H, Kobayashi T, Honda H (2005) Construction of robust prognostic predictors by using projective adaptive resonance theory as a gene filtering method. Bioinformatics 21: 179–186. [DOI] [PubMed] [Google Scholar]
- 27. Kawamura T, Takahashi H, Honda H (2008) Proposal of new gene filtering method, BagPART, for gene expression analysis with small sample. J Biosci Bioeng 105: 81–84. [DOI] [PubMed] [Google Scholar]
- 28. Ando T, Suguro M, Hanai T, Kobayashi T, Honda H, et al. (2002) Fuzzy neural network applied to gene expression profiling for predicting the prognosis of diffuse large B-cell lymphoma. Jpn J Cancer Res 93: 1207–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Takahashi H, Masuda K, Ando T, Kobayashi T, Honda H (2004) Prognostic predictor with multiple fuzzy neural models using expression profiles from DNA microarray for metastases of breast cancer. J Biosci Bioeng 98: 193–199. [DOI] [PubMed] [Google Scholar]
- 30. Takahashi H, Honda H (2005) A new reliable cancer diagnosis method using boosted fuzzy classifier with a SWEEP operator method. J Chem Eng Jpn 38: 763–773. [Google Scholar]
- 31. Takahashi H, Murase Y, Kobayashi T, Honda H (2007) New cancer diagnosis modeling using boosting and projective adaptive resonance theory with improved reliable index. Biochem Eng J 33: 100–109. [Google Scholar]
- 32. Takahashi H, Honda H (2006) Prediction of peptide binding to major histocompatibility complex class II molecules through use of boosted fuzzy classifier with SWEEP operator method. J Biosci Bioeng 101: 137–141. [DOI] [PubMed] [Google Scholar]
- 33. Zhou H, Jin J, Zhang H, Yi B, Wozniak M, et al. (2012) IntPath–an integrated pathway gene relationship database for model organisms and important pathogens. BMC Syst Biol 6 Suppl 2 S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou H, Wong L (2011) Comparative analysis and assessment of M. tuberculosis H37Rv protein-protein interaction datasets. BMC Genomics 12 Suppl 3 S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kotooka N, Komatsu A, Takahashi H, Nonaka M, Kawaguchi C, et al. (2013) Predictive value of high-molecular weight adiponectin in subjects with a higher risk of the development of metabolic syndrome: From a population based 5-year follow-up data. Int J Cardiol 167: 1068–1070. [DOI] [PubMed] [Google Scholar]
- 36. Takahashi H, Honda H (2006) Lymphoma prognostication from expression profiling using a combination method of boosting and projective adaptive resonance theory. J Chem Eng Jpn 39: 767–771. [Google Scholar]
- 37. Takahashi H, Aoyagi K, Nakanishi Y, Sasaki H, Yoshida T, et al. (2006) Classification of intramural metastases and lymph node metastases of esophageal cancer from gene expression based on boosting and projective adaptive resonance theory. J Biosci Bioeng 102: 46–52. [DOI] [PubMed] [Google Scholar]
- 38. Matsuo N, Mase H, Makino M, Takahashi H, Banno H (2009) Identification of ENHANCER OF SHOOT REGENERATION 1-upregulated genes during in vitro shoot regeneration. Plant Biotechnol 26: 385–393. [Google Scholar]
- 39. Yajima I, Kumasaka MY, Naito Y, Yoshikawa T, Takahashi H, et al. (2012) Reduced GNG2 expression levels in mouse malignant melanomas and human melanoma cell lines. Am J Cancer Res 2: 322–329. [PMC free article] [PubMed] [Google Scholar]
- 40. Sano M, Aoyagi K, Takahashi H, Kawamura T, Mabuchi T, et al. (2010) Forkhead box A1 transcriptional pathway in KRT7-expressing esophageal squamous cell carcinomas with extensive lymph node metastasis. Int J Oncol 36: 321–330. [PubMed] [Google Scholar]
- 41. Chiba Y, Mineta K, Hirai MY, Suzuki Y, Kanaya S, et al. (2013) Changes in mRNA stability associated with cold stress in Arabidopsis cells. Plant Cell Physiol 54: 180–194. [DOI] [PubMed] [Google Scholar]
- 42. Nakagawa A, Takahashi H, Kojima S, Sato N, Ohga K, et al. (2012) Berberine enhances defects in the establishment of leaf polarity in asymmetric leaves1 and asymmetric leaves2 of Arabidopsis thaliana. Plant Mol Biol 79: 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoshimura K, Mori T, Yokoyama K, Koike Y, Tanabe N, et al. (2011) Identification of alternative splicing events regulated by an Arabidopsis serine/arginine-like protein, atSR45a, in response to high-light stress using a tiling array. Plant Cell Physiol 52: 1786–1805. [DOI] [PubMed] [Google Scholar]
- 44. Portal D, Zhou H, Zhao B, Kharchenko PV, Lowry E, et al. (2013) Epstein-Barr virus nuclear antigen leader protein localizes to promoters and enhancers with cell transcription factors and EBNA2. Proc Natl Acad Sci USA 110: 18537–18542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takahashi H, Iwakawa H, Nakao S, Ojio T, Morishita R, et al. (2008) Knowledge-based fuzzy adaptive resonance theory and its application to the analysis of gene expression in plants. J Biosci Bioeng 106: 587–593. [DOI] [PubMed] [Google Scholar]
- 46. Takahashi H, Kaniwa N, Saito Y, Sai K, Hamaguchi T, et al. (2013) Identification of a candidate single-nucleotide polymorphism related to chemotherapeutic response through a combination of knowledge-based algorithm and hypothesis-free genomic data. J Biosci Bioeng 116: 768–773. [DOI] [PubMed] [Google Scholar]
- 47.Takahashi H, Sai K, Saito Y, Kaniwa N, Matsumura Y, et al.. (2014) Application of a combination of a knowledge-based algorithm and 2-stage screening to hypothesis-free genomic data on irinotecan-treated patients for identification of a candidate single nucleotide polymorphism related to an adverse effect. PLoS One (DOI: 10.1371/journal.pone.0105160) [DOI] [PMC free article] [PubMed]
- 48. Takahashi H, Iwakawa H, Ishibashi N, Kojima S, Matsumura Y, et al. (2013) Meta-analyses of microarrays of arabidopsis asymmetric leaves1 (as1), as2 and their modifying mutants reveal a critical role for the ETT pathway in stabilization of adaxial-abaxial patterning and cell division during leaf development. Plant Cell Physiol 54: 418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kojima S, Iwasaki M, Takahashi H, Imai T, Matsumura Y, et al. (2011) Asymmetric leaves2 and Elongator, a histone acetyltransferase complex, mediate the establishment of polarity in leaves of Arabidopsis thaliana . Plant Cell Physiol 52: 1259–1273. [DOI] [PubMed] [Google Scholar]
- 50. Iwasaki M, Takahashi H, Iwakawa H, Nakagawa A, Ishikawa T, et al. (2013) Dual regulation of ETTIN (ARF3) gene expression by AS1–AS2, which maintains the DNA methylation level, is involved in stabilization of leaf adaxial-abaxial partitioning in Arabidopsis. Development 140: 1958–1969. [DOI] [PubMed] [Google Scholar]
- 51. Takahashi H, Nakayama R, Hayashi S, Nemoto T, Murase Y, et al. (2013) Macrophage migration inhibitory factor and stearoyl-CoA desaturase 1: potential prognostic markers for soft tissue sarcomas based on bioinformatics analyses. PLoS One 8: e78250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yoshida T, Ono H, Kuchiba A, Saeki N, Sakamoto H (2010) Genome-wide germline analyses on cancer susceptibility and GeMDBJ database: Gastric cancer as an example. Cancer Sci 101: 1582–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mantel N (1966) Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemoth Rep 50: 163–170. [PubMed] [Google Scholar]
- 54. Hasegawa T, Yokoyama R, Lee YH, Shimoda T, Beppu Y, et al. (2000) Prognostic relevance of a histological grading system using MIB-1 for adult soft-tissue sarcoma. Oncology 58: 66–74. [DOI] [PubMed] [Google Scholar]
- 55. Kaplan EL, Meier P (1958) Nonparametric estimation from incomplete observations. J Amer Statist Assn 53: 457–481. [Google Scholar]
- 56. Flicek P, Amode MR, Barrell D, Beal K, Billis K, et al. (2014) Ensembl 2014. Nucleic Acids Res 42: D749–D755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pearson K (1901) On Lines and Planes of Closest Fit to Systems of Points in Space. Philos Mag 2 559–572.
- 58. Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc serB 57: 298–300. [Google Scholar]
- 59. Chibon F, Lagarde P, Salas S, Perot G, Brouste V, et al. (2010) Validated prediction of clinical outcome in sarcomas and multiple types of cancer on the basis of a gene expression signature related to genome complexity. Nat Med 16: 781–787. [DOI] [PubMed] [Google Scholar]
- 60. Zhou H, Gao S, Nguyen NN, Fan M, Jin J, et al. (2014) Stringent homology-based prediction of H. sapiens-M. tuberculosis H37Rv protein-protein interactions. Biol Direct 9: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhou H, Rezaei J, Hugo W, Gao S, Jin J, et al. (2013) Stringent DDI-based prediction of H. sapiens-M. tuberculosis H37Rv protein-protein interactions. BMC Syst Biol 7 Suppl 6 S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, et al. (2013) STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res 41: D808–D815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The pathways predicted by STRING from the 25 selected genes.
(PDF)
Clinical data of the 88 patients with soft tissue sarcoma. UPS: undifferentiated pleomorphic sarcoma, MLS: myxoid liposarcoma, SS: synovial sarcoma, MFS: myxofibrosarcoma, LMS: leiomyosarcoma, FS: fibrosarcoma, MPNST: malignant peripheral nerve sheath tumor, Tumor metastasis indicates the incidence of tumor metastasis in STS patients.
(XLS)
The MIM number list.
(XLS)
Selected Affymetrix probe IDs.
(XLS)
Information on PCA, including the eigenvector, standard deviation, proportion of variance, and cumulative proportion for 29 probe sets. PCA: principal component analysis, PC: principal components.
(XLS)
Information on PCA, including the eigenvector, standard deviation, proportion of variance, and cumulative proportion for 9 probe sets. PCA: principal component analysis, PC: principal components.
(XLS)
Survival analysis in UPS using the logrank test. Adjusted p values were calculated using the permutation test (100,000 repeats) from logrank p values.
(XLS)
Gene or pathway annotations and likelihood as prognostic/predictive factors and/or therapeutic targets. Adjusted p values were calculated using the permutation test (100,000 repeats) from logrank p values.
(XLS)
Pathway analysis in IntPath. k: genes from the overlap between genes in the list and genes in the pathway, n: the number of genes in the input gene list, m: the number of genes in the identified pathways, N: the total number of genes. The p values were calculated using the hypergeometric test; the q values were calculated from the p values using the Benjamini-Hochberg (BH) method.
(XLS)
(PDF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.