

Abstract
Background
Bacterial sortases are transpeptidases that covalently anchor surface proteins to the peptidoglycan of the Gram-positive cell wall. Sortase protein anchoring is mediated by a conserved cell wall sorting signal on the anchored protein, comprising of a C-terminal recognition sequence containing an “LPXTG-like” motif, followed by a hydrophobic domain and a positively charged tail.
Results
We report that Clostridium difficile strain 630 encodes a single sortase (SrtB). A FRET-based assay was used to confirm that recombinant SrtB catalyzes the cleavage of fluorescently labelled peptides containing (S/P)PXTG motifs. Strain 630 encodes seven predicted cell wall proteins with the (S/P)PXTG sorting motif, four of which are conserved across all five C. difficile lineages and include potential adhesins and cell wall hydrolases. Replacement of the predicted catalytic cysteine residue at position 209 with alanine abolishes SrtB activity, as does addition of the cysteine protease inhibitor MTSET to the reaction. Mass spectrometry reveals the cleavage site to be between the threonine and glycine residues of the (S/P)PXTG peptide. Small-molecule inhibitors identified through an in silico screen inhibit SrtB enzymatic activity to a greater degree than MTSET.
Conclusions
These results demonstrate for the first time that C. difficile encodes a single sortase enzyme, which cleaves motifs containing (S/P)PXTG in-vitro. The activity of the sortase can be inhibited by mutation of a cysteine residue in the predicted active site and by small-molecule inhibitors.
Electronic supplementary material
The online version of this article (doi:10.1186/s12866-014-0219-1) contains supplementary material, which is available to authorized users.
Keywords: Clostridium difficile, Sortase, Cysteine protease, Fluorescence resonance energy transfer (FRET), Enzyme kinetics, Enzyme inhibitors
Background
Sortases are membrane-bound cysteine transpeptidases that anchor surface proteins to the peptidoglycan cell wall in Gram-positive bacteria. Surface proteins anchored via sortases are often essential virulence factors important in colonization and invasion, evasion of the host immune system, and nutrient acquisition. The sorting process is mediated by a conserved C-terminal cell wall sorting signal on the anchored protein, comprised of a C-terminal recognition sequence (often LPXTG, where X is any amino acid), followed closely by a hydrophobic transmembrane domain and a positively charged tail [1]. A conserved catalytic cysteine residue of the sortase cleaves the LPXTG motif of the polypeptide between the threonine and glycine residues and covalently attaches the protein to the peptidoglycan [2–6].
There are six described sortase families, A-F, that share amino acid similarity [7]. All catalyze similar transpeptidation reactions, but recognize different substrate motifs and serve different functions within the cell. Class A sortases (SrtA), such as the prototypical Staphylococcus aureus Sortase A (SaSrtA), are considered housekeeping sortases as they are capable of anchoring many functionally distinct proteins to the cell wall. SaSrtA, which recognizes an LPXTG motif, is responsible for anchoring a variety of surface proteins involved in adherence and immune response evasion, and is essential for virulence in animal models [8,9]. SrtA orthologues have been found in the genomes of almost all Gram-positive bacteria [8,10–16]. Class B sortases are functionally different from class A in their substrate specificity. In S. aureus and B. anthracis, the sortase B gene (srtB) is part of an iron-regulated locus isd (iron-responsive surface determinant) responsible for heme-iron transport, and anchors the iron transporter protein, IsdC, by recognizing an NPQTN motif [17,18]. Though mutating srtB has no effect on establishing infection, SaSrtB is required for persistence of the bacterium in mice [17].
Clostridium difficile, an anaerobic Gram-positive, spore-forming bacillus, is the leading cause of hospital-acquired infectious diarrhea in North America and Europe. Infection with C. difficile can result in a range of clinical presentations, from mild self-limiting diarrhea to the life-threatening pseudomembranous colitis (PMC), known collectively as C. difficile infection (CDI) [19]. MLST studies have identified that the C. difficile population structure forms at least five distinct lineages that are all associated with CDI [20–22]. Complications of severe CDI can lead to toxic megacolon, bowel perforation, sepsis and death in up to 25% of cases [23]. Broad-spectrum antibiotic usage is the greatest risk factor for development of CDI due to the consequent disruption of the intestinal microflora. Treatment of CDI with metronidazole and vancomycin can exacerbate the problem by continuing to disrupt the intestinal microflora. This leaves the patient susceptible to relapse or re-infection. Approximately one third of patients experience CDI relapse following treatment, and those who relapse have a greater risk of succumbing to the infection [23]. A current imperative is the development of therapies that selectively target C. difficile, whilst leaving the intestinal microflora intact.
The C. difficile reference strain 630 encodes a single predicted sortase, CD630_27180, which has high amino-acid similarity with SrtB of S. aureus and B. anthracis [24]. A second sortase encoded within the genome is interrupted by a stop codon prior to the catalytic cysteine and is considered a pseudogene. Thus, in contrast to other Gram-positive bacteria, C. difficile appears to have only a single functional sortase. As such, a compound that inhibits the activity of C. difficile sortase could target the pathogen without disrupting the numerous Gram-negative bacteria that make up the intestinal flora.
In this study, we demonstrate that the predicted sortase encoded by CD630_27180 recognizes and cleaves an (S/P)PXTG motif between the threonine and glycine residues. The cleavage of this motif is dependent on the conserved cysteine residue at position 209 in the predicted active site of the sortase. We have also identified seven putative sortase substrates, all of which contain the (S/P)PXTG motif. These substrates are conserved among the five C. difficile lineages and include potential adhesins, a 5’ nucleotidase, and cell wall hydrolases. Furthermore, we identified a number of small-molecule inhibitors by means of an in silico screen that inhibit the activity of the C. difficile SrtB.
Results
Conservation of the catalytically active residues of sortase
The genome sequence of C. difficile strain 630 previously indicated the presence of a single copy of a sortase-like protein, encoded by CD630_27180, based on the presence of the sortase catalytic motif TLXTC [24]. We performed BLAST searches (BlastP) to reveal the protein encoded by CD630_27180 shares 32% and 34% amino acid identity with SrtB from S. aureus (SaSrtB) and B. anthracis (BaSrtB), respectively. In addition to the TLXTC active site, the catalytically essential histidine (His120 in SaSrtA) and arginine (R197 in SaSrtA) residues [3,25,26] are conserved in the C. difficile SrtB. A structural prediction analysis of SrtB was performed using Phyre2 Protein Fold Recognition Server (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) [27], and the resulting alignment suggests a high level of conservation between the predicted secondary structure of SrtB and the known crystal structure of the BaSrtB [28] (Figure 1). Expression of C. difficile SrtB was analysed in vitro using RT-PCR analysis on strain 630, which confirmed that CD2718 is actively transcribed during early exponential, late exponential and stationary phases (Additional file 1: Figure S1).
Figure 1.

Predicted C. difficile SrtB secondary structure . A structural alignment between the known crystal structure of BaSrtB [28] and the predicted structure of C. difficile SrtB using the Phyre2 Protein Fold Recognition Server suggests a high degree of structural conservation. Top: C. difficile SrtB predicted secondary structure and sequence. Bottom: BaSrtB sequence and known structure. Arrows indicate beta sheets, and striped rectangles indicate alpha helixes. Amino acid positions relative to start position are indicated. The sortase active site signature sequence TLXTC is boxed, as are the conserved essential histidine and arginine residues.
The C. difficile population structure forms at least five distinct clonal lineages that are all associated with human infection [20–22]. To determine whether SrtB is conserved between C. difficile strains, representatives for each of the five distinct clades were chosen for analysis based on the availability of a fully annotated sequence: C. difficile strains 630 for Clade 1, R20291 and CD196 (RT027) for Clade 2 [29], M68 and CF5 (RT017) for Clade 3 [20], CD305 (RT023) for Clade 4 (unpublished, WTSI), and M120 (RT078) for Clade 5 [20]. BLAST searches of these representative strains show that srtB is conserved across all five C. difficile lineages. A second sortase-like gene in the 630 genome, classified as a pseudogene because of an in frame stop codon prior to the catalytic cysteine, is absent from the other four C. difficile lineages.
Bioinformatic prediction of sortase substrates
A bioinformatics approach was used for the preliminary identification of sortase substrate proteins in C. difficile strain 630. The predicted recognition sequence for CD630_27180 has been proposed to be (S/P)PXTG by Pallen et al. [11], and recently to also include the sequence NVQTG, found in the surface- associated collagen binding protein CbpA, by Tulli et al. [30]. Putative proteins were screened for the patterns (S/P)PXTG and NVQTG at the C-terminus [11,30]. Putative candidates were then assessed for the known features of a sortase substrate: a predicted N-terminal signal peptide sequence, and a cell wall sorting signal comprising of a potential transmembrane domain following the sortase recognition sequence, and at least two consecutive basic residues (arginine or lysine) at the C-terminus [31–33].
Eight proteins satisfied our definition of a sortase substrate in strain 630 (Table 1). The newly described C. difficile collagen binding protein A, CbpA, is the only protein containing the proposed NVQTG motif [30]. The remaining proteins contained one of four observed variations of the (S/P)PXTG motif: SPKTG, PPKTG, and SPSTG and SPQTG. These predicted C. difficile sortase substrates are a diverse range of surface proteins that include putative cell wall hydrolases, putative adhesins, a collagen-binding protein, and a 5’ nucleotidase/phosphoesterase (Table 1). Transcriptional analysis performed by RT-PCR confirmed that all eight predicted substrate proteins are transcribed during growth in vitro (Additional file 1: Figure S1B-I). The eight predicted substrates are transcribed during all three growth phases examined, with the exception of CD630_25370 and CD630_32460, which do not appear to be transcribed during stationary phase. Four of these putative substrates are conserved across all five C. difficile lineages: CD630_01830, CD630_25370, CD630_27680, and CD630_28310.
Table 1.
Identification of putative C. difficile SrtB substrates in strain 630
| Protein | Function | C-terminal sorting signal |
|---|---|---|
| CD630_01830 | Putative cell wall hydrolase | MIHSPSTGKTVSVTSINSSYYTARFVTA KRIL |
| CD630_03860 | Putative cell surface protein, collagen binding protein | PSDSPKTGDNTNLYGLLALLLTSGAGLAGIFFY KRRKMKKS |
| CD630_25370 | Putative membrane-associated 5'-nucleotidase/phosphoesterase | KEKSPKTGDLGFSNSIIIFIVSSTLICLLNFNQKELKDKKSK |
| CD630_27680 | Putative cell-wall hydrolase | FIHSPQTGDVVKVTSMAPGTNYA RRLITATRVLQ |
| CD630_28310 | Putative adhesion, collagen binding protein | PPVPPKTGDSTTIIGEILLVIGAIVGLIVL RRNKNTN |
| CD630_31450 | Collagen binding protein, CbpA | VGQNVQTGDQSNIMLDLALMFISLFFLI KNLTNKYLRRK |
| CD630_32460 | Putative surface protein | IVKSPKTGDETQLMSYVFISVIAICGLAYQCKIKRN |
| CD630_33920 | Putative cell surface protein, collagen binding protein | PSDSPKTGDSTNLMAFIVMLLVSGGGLAGTYLY KRRKMKKS |
Bold = predicted sortase recognition sequence.
Bold and Italic = hydrophobic residues.
Italics only = positively charged residues.
Purified C. difficile SrtB cleaves (S/P)PXTG peptides
To determine whether C. difficile SrtB cleaves putative substrates at the predicted motifs, FRET peptides were designed based on the variations observed in the predicted (S/P)PXTG motif (Table 2). Two residues upstream of the motif were included, and two glycine residues were incorporated downstream, as this has been previously shown to improve sortase cleavage efficiency in vitro [34]. Fluorescence of the Edans fluorophore within the peptides is blocked when in close proximity to the fluorescent quencher Dabcyl [35]. When the peptide is cleaved, the Edans fluorophore is separated from Dabcyl, and a fluorescent signal is observed.
Table 2.
FRET peptide details
| Peptide sequence* | Description |
|---|---|
| d-IHSPSTGGG-e | Based on CD0183 sequence |
| d-IHGSSTPGG-e | Control for above peptide |
| d-SDSPKTGGG-e | Based on CD0386, CD3392 sequence |
| d-SDGSKTPGG-e | Control for above peptide |
| d-IHSPQTGGG-e | Based on CD2768 sequence |
| d-IHGSQTPGG-e | Control for above peptide |
| d-PVPPKTGGG-e | Based on CD2831 sequence |
| d-PVGPKTPGG-e | Control for above peptide |
| d-GQNVQTGGG-e | Based on CbpA sequence |
| d-QALPETGGG-e | SaSrtA peptide |
| d-NPQTN-e | SaSrtB peptide |
| d-IHSPSTGKT-e | Based on CD0183 sequence |
| d-SDSPKTGDN-e | Based on CD0386 sequence |
| d-IHSPQTGDV-e | Based on CD2768 sequence |
| d-PVPPKTGDS-e | Based on CD2831 sequence |
*Where d is Dabcyl (4-([4-(dimethylamino)phenyl]azo)-benzoyl) and e is Edans (5-((2-Aminoethyl)amino)naphthalene-1-sulfonic acid).
The N-terminal transmembrane domain of C. difficile SrtB (residues 2–25) was replaced with a six-histidine tag (SrtBΔN26) to improve soluble protein yield. SrtBΔN26 was expressed in E. coli NiCo21(DE3) and purified by nickel affinity chromatography from cleared lysates (Figure 2). Purified SrtBΔN26 was then incubated with a FRET peptide containing the SPKTG sequence. An increase in fluorescence was observed over time, indicating that cleavage of the SPKTG peptide occurred in the presence of SrtBΔN26 over 48 hours (Figure 3). In addition to the SPKTG motif, SrtBΔN26 also cleaved peptides containing the predicted substrate sequences PPKTG, SPSTG, and SPQTG (Figure 4). SrtBΔN26 failed to cleave the scrambled peptide sequences GSKTP, GPKTP, GSSTP and GSQTP (Figure 4). Interestingly, SrtBΔN26 failed to cleave peptides containing the LPETG and NPQTN motifs of SaSrtA and SaSrtB, respectively, and also failed to cleave the proposed sortase recognition motif NVQTG found in the C. difficile collagen binding protein, CbpA [30] (Figure 4).
Figure 2.
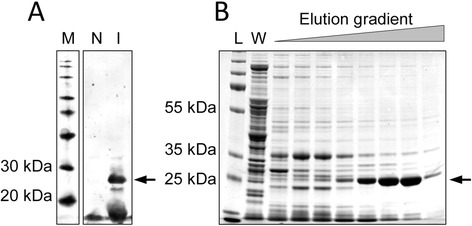
Expression and purification of SrtB ΔN26 . E. coli NiCo21(DE3) expressing SrtBΔN26, in which the N-terminal membrane anchor has been replaced with a six-histidine tag, were lysed by sonication and cleared lysates purified by nickel affinity chromatography. A. Anti-his western testing for expression of SrtBΔN26. Lane M: molecular mass marker, N: whole cell lysate of non-induced culture, I: whole cell lysate of culture induced with 1 mM IPTG. B. Coomassie-stained SDS-PAGE analysis of SrtBΔN26 purification over an imidazole gradient. Lane L: molecular mass marker, W: column wash, imidazole gradient indicated by grey triangle, arrows indicate the SrtBΔN26 protein.
Figure 3.
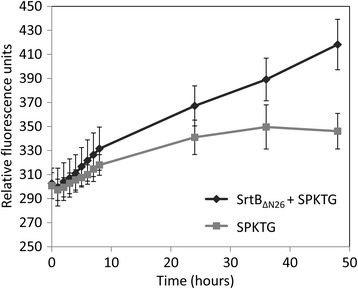
Cleavage of SPKTG peptide by recombinant SrtB ΔN26 . Purified recombinant SrtBΔN26 was incubated with a FRET peptide containing the SPKTG motif and fluorescence measured every hour for the first eight hours, and also at 24 h, 36 h, and 48 h. An increase in relative fluorescence units (arbitrary units) was observed, compared with the peptide incubated alone, indicating that SrtBΔN26 mediated cleavage of the SPKTG peptide occurred over a period of 48 hours. Error bars represent the standard error of the mean.
Figure 4.
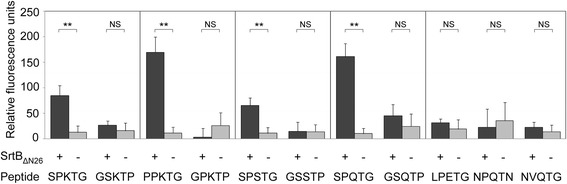
SrtB ΔN26 substrate specificity. Purified recombinant SrtBΔN26 protein was incubated with a range of peptide sequences to investigate its substrate specificity. The motifs SPKTG, PPKTG, SPSTG and SPQTG were all recognized and cleaved following incubation with SrtBΔN26. The scrambled peptide sequences GSKTP, GPKTG, GSSTP, and GSQTP serve as controls for the cleavage specificity of SrtBΔN26. The sequences LPETG and NPQTN, corresponding to the motifs recognized by S. aureus sortase A and B, respectively, do not appear to be substrates for SrtBΔN26. SrtBΔN26 also failed to cleave the proposed sorting signal NVQTG from recently characterized collagen binding protein, CbpA. Bars indicate the mean, and error bars represent the standard error (**corresponds to p < 0.01).
Analysis of FRET reaction
To investigate the importance of the cysteine residue in the proposed active site of C. difficile SrtB, site-directed mutagenesis was used to replace the cysteine residue at position 209 with an alanine. When the resulting mutant protein SrtBΔN26,C209A was incubated with the FRET peptides, the fluorescent signal fell below the limits of detection (Figure 5), indicating that the cysteine residue at position 209 was essential for the activity of the C. difficile SrtB. Cleavage in the FRET-based assay was also inhibited by the addition of MTSET (Figure 5), a known cysteine protease inhibitor and inhibitor of sortase function in S. aureus [36,37] and B. anthracis [15].
Figure 5.
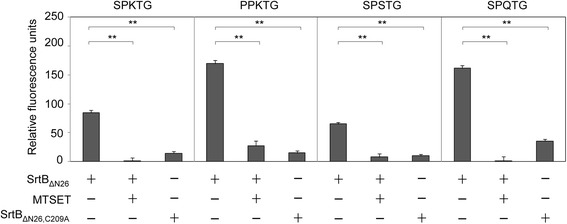
SrtB ΔN26 activity requires a cysteine residue at position 209. To determine if SrtBΔN26 activity depended on the cysteine residue at position 209, a C209A substitution was made to create SrtBΔN26,C209A. This enzyme was inactive against the FRET peptides when compared with SrtBΔN26. Addition at 5 mM of the cysteine protease inhibitor, MTSET, to the reaction also eliminates activity (**corresponds to p < 0.01).
The cleavage of the SPKTG, PPKTG, and SPQTG motifs was enhanced at least two-fold by the addition of the two native amino acids immediately downstream of this sequon (data not shown). Analysis of the FRET reaction with these modified peptides revealed that SrtBΔN26, cleaves these peptides between the T and G residues. MALDI analysis of d-PVPPKTGDS-e peptide incubated with SrtBΔN26 results in a peptide with a mass of 889 Da, corresponding to the fragment d-PVPPKT-OH (Figure 6, top). The peptide control, incubated without SrtBΔN26, lacked this fragment (Figure 6, bottom). Cleavage between the T and G residues for the d-SDSPKTGDN-e and d-IHSPQTGDV-e peptides was also confirmed (data not shown), indicating that C. difficile SrtB cleaves the (S/P)PXTG motif between the same residues as other functional sortases [4,15,38,39].
Figure 6.
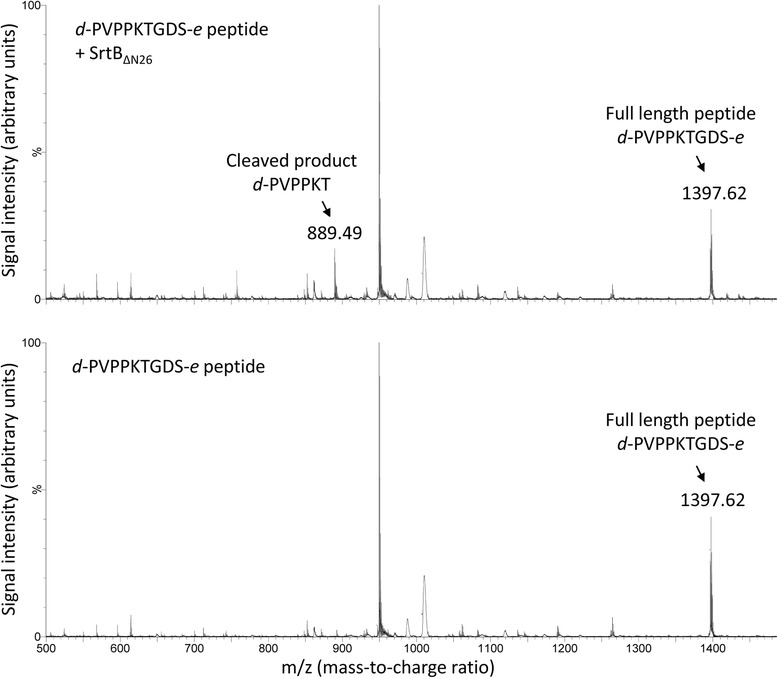
SrtB ΔN26 cleaves FRET peptides between T and G residues. MALDI analysis of FRET reaction products revealed a fragment of mass 889.46, corresponding to the predicted mass of d-PVPPKT-OH (top) when d-PVPPKTGDS-e was incubated with SrtBΔN26. This fragment was absent in the mock treated peptide sample (bottom), indicating that SrtBΔN26 cleaves the d-PVPPKTGDS-e between the T and G residues.
Kinetic measurements of SrtB activity
In order to calculate the in vitro kinetic parameters of SrtBΔN26 for the d-SDSPKTGDN-e and d-PVPPKTGDS-e peptides, we performed a kinetic analysis of the sortase-catalyzed hydrolysis reaction. Figure 7A shows the progress curves of the SrtBΔN26 catalyzed hydrolysis reactions at various d-SDSPKTGDN-e concentrations. For each progress curve, the amount of fluorescent product (after conversion from RFU to concentration) was approximately 5% of the initial substrate concentration. Within the time period analyzed, the progress curves are linear, so the steady state rate (V) was determined by fitting the data to a linear function. Figure 7B shows V plotted against the concentration of the peptide. Non-linear regression of these data fitted to a modified Michaelis-Menten equation incorporating substrate inhibition (Equation 1):
| 1 |
Figure 7.
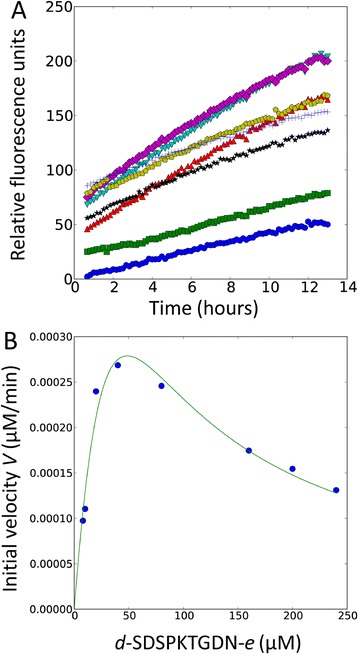
Kinetic parameters of SrtB
ΔN26
. In order to determine the in vitro kinetic parameters of SrtBΔN26 for the SPKTG and PPKTG motifs, we performed a kinetic analysis of the sortase-catalyzed hydrolysis reaction. A. Progress curves of the SrtBΔN26-catalyzed hydrolysis reactions at various concentrations of d-SDSPKTGDN-e [8 (blue ●), 10 (green ▪), 20 (red ▲), 40 (teal ▼), 80 (purple ♦), 160 (yellow  ), 200 (black ★), and 240 μM (blue +). The steady state rate (V) was determined by fitting the data to a linear function. B. Plot of V against the concentration of the peptide [S]. Nonlinear regression of these data fitted to Equation 1 resulted in a K
m of 74.7 ± 48.2 μM for d-SDSPKTGDN-e. SrtBΔN26 is subject to substrate inhibition at peptide concentrations > 30 μM, which is not expected to be physiologically relevant.
), 200 (black ★), and 240 μM (blue +). The steady state rate (V) was determined by fitting the data to a linear function. B. Plot of V against the concentration of the peptide [S]. Nonlinear regression of these data fitted to Equation 1 resulted in a K
m of 74.7 ± 48.2 μM for d-SDSPKTGDN-e. SrtBΔN26 is subject to substrate inhibition at peptide concentrations > 30 μM, which is not expected to be physiologically relevant.
Using SciPy 0.11.0 in Python 2.7.3, where Vmax is the apparent maximal enzymatic velocity, Km is the apparent Michaelis constant, and Ki is the apparent inhibitor dissociation constant for unproductive substrate binding. This resulted in a Km of 74.7 ± 48.2 μM and a Kcat of 1.1×10−3 ± 6×10−4 min−1 for d-SDSPKTGDN-e (Figure 7B). This analysis was performed for d-PVPPKTGDS-e, resulting in a Km of 53.3 ± 25.6 μM and a Kcat of 8.3×10−4 ± 3×10−4 min−1. SrtBΔN26 is subject to substrate inhibition; at peptide concentrations greater than 30 μM, the rate of SrtBΔN26 activity decreases. Substrate inhibition has previously been observed for other sortase enzymes in vitro, and is not expected to be physiologically relevant [40].
Inhibiting SrtB activity
We sought to determine whether C. difficile SrtB could be specifically targeted using small-molecule inhibitors. The published crystal structure of the B. anthracis SrtB (BaSrtB) [28] was used as a template for the selection of potential C. difficile SrtB inhibitors. These orthologous proteins show 70% identity and 90% similarity at the active site, and their differences are confined to the periphery of the active site. The proprietary LeadBuilder virtual-screening method (Domainex Ltd) was used to interrogate the PROTOCATS database of potential protease inhibitors with pharmacophoric and docking filters derived from analysis of the BaSrtB crystal structure. PROTOCATS comprises 80,000 commercially-available compounds that may form reversible transition-state-like complexes with protease enzymes. Compounds in PROTOCATS contain a carbonyl group which is activated to make a fully reversible complex with the active-site serine/cysteine group by virtue of adjacent moderately electron-withdrawing substituents, which are not leaving groups. Some examples of these functional groups are α-ketoamides and aryl ketones. Figure 8A shows one of the identified compounds docking within the active site structure of BaSrtB.
Figure 8.
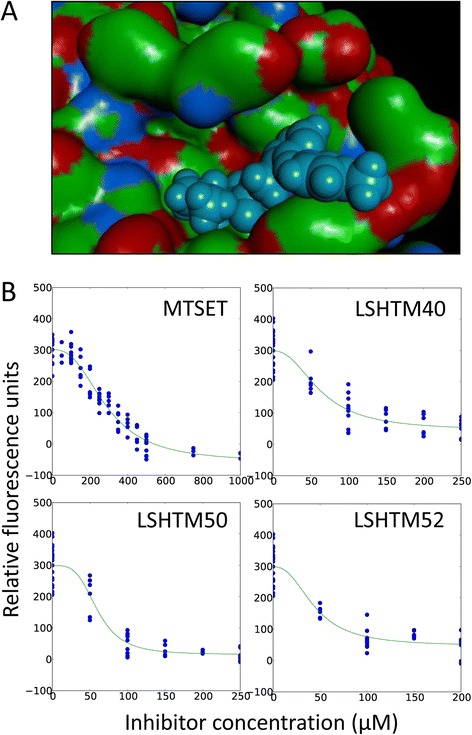
SrtB ΔN26 activity can be inhibited by rationally designed inhibitors. The proprietary LeadBuilder virtual-screening method (Domainex Ltd) was used to screen a database of 80,000 potential protease inhibitors, PROTOCATS, with pharmacophoric and docking filters derived from analysis of the BaSrtB crystal structure [28]. A. Space filling model showing one of the hit compounds fitting into the active site of BaSrtB and interacting with the catalytic cysteine residue. B. MTSET and the hits from the virtual screen were tested in the FRET-based assay at varying concentrations to screen for inhibition of SrtBΔN26 mediated cleavage of d-PVPPKTGDS-e. The most effective compounds were LSHTM40, LSHTM50, and LSHTM52, which had IC50 values of 63.1 ± 8.8, 60.1 ± 4.7 and 44.1 ± 6.9 μM, respectively. The IC50 for MTSET was 286.7 ± 16.6 μM, indicating its inhibitory effect on SrtBΔN26 is less potent than the three identified compounds.
Compounds identified in this screen as potential SrtB inhibitors were tested alongside the cysteine protease inhibitor MTSET at a range of concentrations in the FRET-based assay using the d-PVPPKTGDS-e peptide to compare IC50 values. Addition of MTSET reduced SrtBΔN26 activity to below the limits of detection at concentrations of 500 μM and greater. MTSET exhibited an IC50 of 286.7 ± 16.6 μM (Figure 8B). A panel of potential C. difficile SrtB inhibitors were screened for inhibition of SrtBΔN26 activity. The most effective of the 62 compounds were LSHTM40, LSHTM50, and LSHTM52. They had IC50 values below 100 μM (Figure 8B, Table 3), at 63.1 ± 8.8 μM, 60.1 ± 4.7 μM, and 44.1 ± 6.9 μM, respectively, showing a good efficacy against C. difficile SrtB activity.
Table 3.
Structure of most effective inhibitors of SrtB ΔN26
| Compound | Structure | IC50 |
|---|---|---|
| LSHTM-0040 |
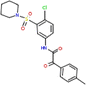
|
63.1 ± 8.8 μM |
| LSHTM-0050 |
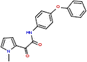
|
60.1 ± 4.7 μM |
| LSHTM-0052 |
|
44.1 ± 6.9 μM |
Discussion
C. difficile infection is invariably associated with the disruption of the normal intestinal microflora by the administration of broad spectrum antibiotics. Thus there is a pressing need to develop therapies that selectively target C. difficile while leaving the intestinal microflora intact. The C. difficile reference strain 630 encodes a single predicted sortase, CD630_27180, which has strong amino acid similarity with SrtB of S. aureus and B. anthracis [24]. Sortase substrates frequently contribute toward pathogenesis via their involvement in attachment to specific tissues during infection [17,41–44], as well as the bacteria’s ability to evade the immune response of the host [32,36]. Sortases, although not essential for growth or viability of the organism, are often essential for virulence in Gram-positive organisms; inactivation of sortases reduces colonization in mice [8,13,44,45], and decreases adhesion and invasion in vitro [8,10,14,46,47]. Sortases and their substrates are considered promising targets for the development of new anti-infective compounds [10,14,48]. Unusually for Gram-positive bacteria, C. difficile appears to possess a single sortase enzyme that is likely to be important for the viability of the pathogen as we have been unable to construct a C. difficile strain 630 SrtB defined mutant (unpublished data). Inhibiting the C. difficile sortase could prove to be a strategy to specifically target C. difficile.
In this study, we cloned, expressed and characterized the sortase encoded by CD630_27180 of C. difficile 630, a predicted class B sortase (SrtB). Sortase nomenclature is based on sequence similarity to the known classes of sortase, A-F [7]. Sortases of class B typically are involved in heme-iron uptake and tend to be expressed in operons with their substrates [17,18]. Genes encoding class A sortases are not found in proximity to their substrates, which consist of a variety of surface proteins with diverse biological functions. Several exceptions to these rules have already been described, notably a class B sortase that polymerizes pilin subunits in S. pyogenes [49], and a class E sortase from C. diphtheriae that serves a housekeeping function [50]. The potential C. difficile sortase substrates identified in this paper comprise a diverse range of surface proteins, suggesting that SrtB may serve as a housekeeping sortase in C. difficile, a function usually reserved for class A sortases.
These potential sortase substrates in C. difficile strain 630 comprise of seven proteins, all containing an (S/P)PXTG motif, that are predicted to be surface localized and are conserved across C. difficile strains. Recently it was proposed that a C. difficile collagen binding protein, CbpA, may be sorted to the cell surface by sortase recognizing an NVQTG motif [30]. In this study, we developed a FRET-based assay to demonstrate that SrtB of C. difficile recognizes and cleaves the (S/P)PXTG motif between the threonine and glycine residues, and that cleavage is dependent on a single cysteine residue at position 209. SrtBΔN26 does not appear to cleave the S. aureus SrtA and SrtB motifs, LPXTG and NPQTN, respectively, nor the NVQTG motif in vitro, suggesting that CbpA from C. difficile may be attached to the cell surface by another mechanism.
The FRET-based assay enabled us to determine kinetic parameters for the recombinant C. difficile SrtB. Although the catalytic activity appears low, low catalytic efficiency is observed for most sortases in vitro [40,51]. The kinetic and cleavage data we report for SrtBΔN26 is consistent with this trend. In vivo, the sorting motifs are part of a larger protein, and the transpeptidation substrates are part of a cell wall precursor or mature peptidoglycan [5,6,39]. The transpeptidation reaction has been observed in vitro for sortases from bacteria with a Lys-type peptidoglycan, where cross-linking occurs through a peptide bridge [52,53] such as S. aureus and Streptococcus species [4,40,54], but not for bacteria with Dap-type peptidoglycan such as Bacillus with direct cross-linkages through m-diaminopimelic acid [55]. The likely cell wall anchor of the C. difficile SrtB substrates is the diaminopimelic acid cross-link [56], similar to Bacillus. When transpeptidation is observed in vitro, the cleavage efficiency of sortase increases.
This study revealed that recombinant SrtBΔN26 cleaves the (S/P)PXTG motifs with varying levels of efficiency, cleaving the sequences PPKTG and SPQTG with the greatest efficiency. Apparent preferential cleavage efficiency of certain substrate sequences in vitro has been observed in other sortases. For example, in B. anthracis, BaSrtA cleaves LPXTG peptides more readily than a peptide containing the sequence LPNTA [15]. The biological significance of this peptide sequence preference is unknown.
Small-molecule inhibitors with activity against SrtA and SrtB have been reported that prevent cleavage of fluorescently-labelled peptide compounds by sortase in vitro [57]. These compounds inhibit cell adhesion to fibronectin, yet, they have no effect on in vitro growth. Inhibitors tested against SrtA, SrtB and SrtC in B. anthracis irreversibly modified the active cysteine residue [58]. Several compounds identified in this study had an inhibitory effect on C. difficile SrtB activity. However, these lead compounds had no direct effect on in vitro C. difficile growth (data not shown), which is consistent with observations in S. aureus [57]. Inhibition of bacterial growth is not considered vital in the development of sortase-based drug therapies. In both Staphylococcus and Bacillus, sortase inhibitors show good suitability for further development as therapeutics despite their lack of bactericidal activity. When mice challenged with S. aureus were treated with sortase inhibitor compounds, infection rates and mortality were reduced [59], despite these compounds having no effect on staphylococcal growth [57]. The use of in silico approaches such as the LeadBuilder method employed by this study to screen databases of putative small-molecule inhibitors for further analysis has been validated. Further analysis of the structural similarities between the hit compounds could lead to a refinement of SrtB inhibitor design and increased potency in vitro.
Conclusions
In conclusion, we demonstrate that C. difficile encodes a single sortase, SrtB, with in vitro activity. We have confirmed the C. difficile SrtB recognition sequence as (S/P)PXTG, and show that C. difficile SrtB cleaves the (S/P)PXTG motif within peptides between the threonine and glycine residues. The cysteine residue within the predicted active site is essential for activity of the enzyme, and the cleavage of fluorescently-labelled peptides can be inhibited by MTSET, a known cysteine protease inhibitor. SrtB inhibitors identified through our in silico screen show a greater level of efficacy then MTSET at inhibiting the protease activity of C. difficile SrtB. Such inhibitors provide a significant step in successfully identifying C. difficile SrtB inhibitor compounds, which can be further refined to enhance their efficacy, and may contribute towards the development of novel selective therapeutics against CDI.
Methods
Bacterial culture
C. difficile strain 630 [24] was cultured on Brazier’s agar (BioConnections) supplemented with 4% egg yolk (BioConnections) and 1% defibrinated horse blood (TCS Biosciences Ltd.). Liquid cultures were grown in brain heart infusion broth (Oxoid Ltd.) supplemented with 0.05% L-cysteine (BHIS broth). All media was supplemented with C. difficile antibiotic supplement (250 μg/ml D-cycloserine and 8 μg/ml cefoxitin, BioConnections). C. difficile cultures were incubated at 37°C for 24–48 hours in a Whitley MG500 anaerobic workstation (Don Whitley Scientific Ltd.).
One Shot Top10® (Invitrogen) and XL-1 Blue (Agilent) Escherichia coli were used for all cloning steps, and NiCo21(DE3) E. coli (NEB) was used for the expression of recombinant proteins [60]. E. coli strains were grown at 37°C on Luria-Bertani (LB) agar (Novagen) or in LB broth (Difco). Media was supplemented with 100 μg/ml ampicillin or 50 μg/ml kanamycin as required.
Genomic DNA isolation
Genomic DNA was isolated from C. difficile strain 630 [24,61] by phenol chloroform extraction as previously described [29] and used as a template for cloning. The annotated genome sequences from C. difficile strains R20291 and CD196 (RT027) [29], M68 and CF5 (RT017) [20], M120 (RT078) [20], and CD305 (RT023) (unpublished, Wellcome Trust Sanger Institute) were used for analysis.
Identification of sortase substrates
All proteins encoded by C. difficile strain 630 [24,61] were searched for the patterns (S/P)PXTG [11] and NVQTG [30] positioned 17–45 amino acid residues from the C-terminus [31]. The resulting candidate protein list was assessed for the known features of a sortase substrate: (i) a suitable N-terminal signal peptide sequence, as predicted by SignalP 4.1 Server (http://www.cbs.dtu.dk/services/SignalP/) [62], (ii) a potential transmembrane segment following the C-terminal “LPXTG-like” sequence, as predicted by TMHMM Server v. 2.0 (http://www.cbs.dtu.dk/services/TMHMM/) [63], and (iii) at least two consecutive basic residues (arginine or lysine) at the C-terminus [31–33].
Genetic manipulation
A list of primers and plasmids used in this study can be found in Tables 4 and 5, respectively. The coding sequence for srtB (CD630_27180) was codon-optimized for expression in E. coli by Celtek Bioscience, LLC (Nashville, TN, USA). The resulting fragment was cloned into the NcoI/XhoI sites of pET28a using primers pET_3 and pET_4 to create pET28a-srtB. To improve soluble SrtB yield, the N-terminal transmembrane anchor domain (residues 2–25) was replaced with a six-histidine tag. The truncated gene srtBΔN26 was amplified from pET28a-srtB using primers pET_17 and pET_16, and cloned into the NcoI/XhoI sites of pET28a to create pET28a-srtBΔN26. The mutant protein SrtBΔN26,C209A was generated using the QuikChange Site-Directed Mutagenesis kit (Agilent) in accordance with the manufacturer’s instructions using pET28a-srtBΔN26 as a template and primers C209A and C209A_antisense. Successful construction of recombinant plasmids was confirmed by DNA sequencing using primers T7F and T7R (Source BioScience).
Table 4.
Primers used in this study
| Primer | Sequence |
|---|---|
| pET_3 | gatattccatggatgaagaaactgtaccgtatcgttatc |
| pET_4 | gatgagctcgaggatcagacgaccgtggataacc |
| pET_17 | gatataccatggatgcaccaccaccaccaccactctaaactgaccaaatacaaccacgacac |
| pET_16 | gatgagctcgagttagatcagacgaccgtggataac |
| C209A | tcgttaccctgtctaccgccacctacgaattcgacg |
| C209A_antisense | cgtcgaattcgtaggtggcggtagacagggtaacga |
| T7F | taatacgactcactataggg |
| T7R | gctagttattgctcagcgg |
Table 5.
Plasmids used in this study
| Plasmid | Description | Reference |
|---|---|---|
| pQE30Xa- srtB | Codon optimized srtB, synthesized and cloned in pQE30xa | Obtained from Celtek Bioscience, LLC |
| pET28a | Commercial protein expression vector | Provided by Neil Fairweather |
| pET28a- srtB | Codon optimized srtB cloned in pET28a | This work |
| pET28a- srtB ΔN26 | srtB with residues 2–25 replaced with a six-histidine tag | This work |
| pET28a- srtB ΔN26,C209A | Same as above, with C209A substitution | This work |
RT-PCR analysis
Total RNA was isolated from C. difficile 630 grown in BHIS at early exponential phase (t = 3 hours, OD600 = 0.4-0.5), late exponential phase (t = 5 hours, OD600 = 0.8-0.9), and stationary phase (t = 24 hours, OD600 = 0.6-0.8) using RNAprotect Bacteria Reagent (Qiagen) and the FastRNA Pro Blue Kit (MP Biomedicals LLC., Illkirch, France) in accordance with the manufacturer’s instructions. Genomic DNA was removed from total RNA samples using TURBO DNase (Life Technologies). Equal amounts of RNA were reverse transcribed into complementary DNA (cDNA) for expression analysis. Briefly, one μg random primers (Invitrogen) and 40 units RNasin Plus RNase inhibitor (Promega) was added to one μg RNA in a final volume of 10 μl, and incubated at 65°C for 10 min followed by room temperature for 30 min. The following first-strand mixture was added for cDNA synthesis: four μl of 5x first-strand buffer (Invitrogen), two μl 0.1 M DTT (Invitrogen), two μl 10 mM dNTP mix (New England BioLabs), and 1.5 μl Superscript II (Invitrogen). The reaction mixture was incubated at 25°C for 10 minutes, 42°C for 1 h, and finally 70°C for 15 minutes. RT-PCRs were performed with gene specific primers (Additional file 2: Table S1) using cDNA as a template.
Purification of recombinant protein
Expression constructs were transformed into E. coli NiCo21(DE3) (NEB). Cultures grown at 37°C were induced for expression with 1 mM IPTG when the OD600 reached 0.6, and harvested after 5 hours. Cell pellets were resuspended in lysis buffer [1× Bugbuster (Novagen), 50 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole, 1 mM DTT, 1 mg/ml lysozyme, and 25 U/ml Benzonase nuclease (Novagen) (pH 7.5)]. Lysates were sonicated on ice for 2 min (15 sec on/off) at 50% Vibra Cell™ high intensity ultrasonic processor (Jencon, Leighton Buzzard, Bedfordshire, UK) before centrifugation at 10,000 rpm for 45 min. The supernatant was passed through a 0.22 μM filter before applying to a 1 ml HisTrap HP column (GE Healthcare), pre-equilibrated with buffer (50 mM NaH2PO4, 300 mM NaCl, 40 mM imidazole, 1 mM DTT, pH 7.5). SrtBΔN26 was eluted with an imidazole gradient (40 – 500 mM) over 25 column volumes. Fractions containing SrtBΔN26 (as identified by SDS-PAGE) were pooled and injected onto a HiLoad 16/60 Superdex 200 column (GE Healthcare) pre-equilibrated with buffer F (5 mM CaCl2, 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM DTT). Eluted fractions containing SrtBΔN26 were pooled and concentrated using an Amicon Ultra-15 (10 kDa) centrifuge filter unit (Millipore). Protein samples were quantified using Bradford reagent (Thermo Scientific) and analyzed by SDS-PAGE. The mutant protein SrtBΔN26,C209A was expressed and purified following the above method. Expression of SrtBΔN26 and SrtBΔN26,C209A was confirmed by MALDI fingerprinting.
Immunoblotting
Samples were resolved on Novex NuPage 10% Bis-Tris SDS-PAGE gels (Invitrogen) before transferring to Hybond-C Extra nitrocellulose (GE Healthcare). Membranes were probed with rabbit antiserum directed against 6xHis-tag (1:5000, Abcam), followed by goat anti-rabbit IRDye conjugated secondary antibody (1:7500, LI-COR Biotechnology). Blots were visualized using an Odyssey near-infrared imager (LI-COR Biotechnology).
In vitro analysis of sortase activity
SrtBΔN26 activity was monitored using a fluorescence resonance energy transfer (FRET) assay [58] in buffer F (5 mM CaCl2, 50 mM Tris–HCl (pH 7.5), 150 mM NaCl, and 1 mM DTT). Fluorescently self-quenched peptides tagged with 5-((2-Aminoethyl)amino)naphthalene-1-sulfonic acid (Edans) as a fluorophore and 4-([4-(Dimethylamino)phenyl]azo)benzoic acid (Dabcyl) as a quencher [35], and containing the predicted sorting signals of SrtB were purchased from Protein Peptide Research Ltd and solubilized in DMSO (Table 2). The FRET-based assay was performed in a final volume of 100 μl buffer F containing 10 μM SrtBΔN26 and 20 μM fluorogenic peptide in clear-bottomed, black polystyrene 384-well plates (Nunc). Plates were incubated for 48 hours at 37°C, during which fluorescence (excitation = 340 nm, emission = 490 nm) was measured using a SpectraMax M3 plate reader (Molecular Devices). Five mM 2-(trimethylamonium)ethylmethanethiosulfonate (MTSET, Affymetrix) was added to the reaction as indicated. Each experiment was performed in triplicate with a minimum of three biological replicates, and the results are presented as the means and the standard error of the data obtained. The two-tailed Student’s T-test was used to analyze the data. MALDI analysis of FRET reaction samples was performed by the Protein and Nucleic Acid Chemistry Facility (University of Cambridge) to determine exact cleavage site within each peptide.
Kinetic measurements
Kinetic data for SrtBΔN26 were obtained by incubating varying concentrations of peptide (8, 10, 20, 40, 80, 160, 200 and 240 μM) with 10 μM SrtBΔN26. All reactions were performed as described above, with fluorescence monitored every ten minutes over a 13 hour period. To correlate fluorescence signal, expressed as arbitrary relative fluorescence units (RFU), with the concentration of product formed, standard curves of the fluorophore Edans were collected. The linear segment of the fluorophore standard curve generated a conversion ratio of 703.9 RFU/ μM Edans. Initial velocities (V) were determined from the progress curves and plotted against substrate concentration [S]. The data were fitted to a modified version of the Michaelis-Menten equation incorporating substrate inhibition using SciPy 0.11.0 in Python 2.7.3, where Vmax is the maximal enzymatic velocity, Km is the Michaelis constant, and Ki is the inhibitor dissociation constant for unproductive substrate binding. All data points were collected in triplicate, and the overall assay was run in duplicate.
Identification of SrtB inhibitors
The proprietary LeadBuilder virtual screening method (Domainex, Ltd) was used to interrogate a database (PROTOCATS) of 80,000 potential compounds which had been pre-selected as protease inhibitors. The virtual screening protocol used pharmacophoric and docking filters derived from analysis of the BaSrtB crystal structure (with which the C. difficile SrtB shows 70% identity and 90% similarity at the active site). Sixty-two compounds identified in this screen as potential SrtB inhibitors were obtained from Enamine, ChemBridge, and Key Organics, and solubilized in DMSO. Selected compounds and MTSET were incubated with 10 μM SrtBΔN26 at a range of concentrations in the FRET-based assay conditions described above, so that final DMSO concentrations were ≤ 3.75%, a concentration shown to have no significant effect on control fluorescence (data not shown). IC50 values were determined by non-linear least squares fit to a four parameter sigmoidal function using SciPy 0.11.0 in Python 2.7.3.
Acknowledgments
We thank Jun Wheeler for MALDI mass spectrometry fingerprinting analysis of recombinant proteins; Mark Donahue for assistance with data analysis; Hayley Angove and Wendy Savory for assistance with development of the FRET-based assay and sortase protein expression, respectively. We thank Neil Fairweather, Johann Peltier, Helen A. Shaw and Madeleine Moule for critical reading of the manuscript.
Funding
This research was supported by funding from Wellcome Trust grant number 086418/Z/ and MRC grant number 499 94717.
Abbreviations
- SrtA
Sortase A
- SrtB
Sortase B
- FRET
Fluorescence resonance energy transfer
- MTSET
2-(trimethylamonium)ethylmethanethiosulfonate
- Dabcyl or d
4-([4-(dimethylamino)phenyl]azo)-benzoyl
- Edans or e
5-((2-Aminoethyl)amino)naphthalene-1-sulfonic acid
- CDI
C. difficile infection
- IPTG
Isopropyl β-D-1-thiogalactopyranoside
- RFU
Relative fluorescence units
Additional files
RT-PCR analysis in C. difficile strain 630 of CD2718 and its predicted substrates. PCR reactions were performed with 630 cDNA that was prepared from cultures grown to early exponential (E), late exponential (L) and stationary phase (S). M = Hyperladder I (Bioline), G = 630 genomic DNA, W = dH2O. A “+“indicates cDNA reaction with added reverse transcriptase, “-“ indicates cDNA reaction without added reverse transcriptase (control for DNA depletion of RNA sample).
Primers used for RT-PCR analysis.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Laboratory work: EHD; experimental design: EHD, LFD, EV, SFC, MRM, EL, TRP, BWW; writing of manuscript: EHD, LFD, BWW. All authors read and approved the final manuscript.
Contributor Information
Elizabeth H Donahue, Email: elizabeth.donahue@lshtm.ac.uk.
Lisa F Dawson, Email: lisa.dawson@lshtm.ac.uk.
Esmeralda Valiente, Email: esmeralda.valiente@lshtm.ac.uk.
Stuart Firth-Clark, Email: stuart.firth-clark@domainex.co.uk.
Meriel R Major, Email: meriel.major@domainex.co.uk.
Eddy Littler, Email: eddy.littler@domainex.co.uk.
Trevor R Perrior, Email: trevor.perrior@domainex.co.uk.
Brendan W Wren, Email: brendan.wren@lshtm.ac.uk.
References
- 1.Mazmanian SK, Ton-That H, Schneewind O: Sortase-catalysed anchoring of surface proteins to the cell wall ofStaphylococcus aureus.Mol Microbiol 2001, 40(5):1049–1057. [DOI] [PubMed]
- 2.Ton-That H, Faull KF, Schneewind O. Anchor structure of staphylococcal surface proteins. A branched peptide that links the carboxyl terminus of proteins to the cell wall. J Biol Chem. 1997;272(35):22285–22292. doi: 10.1074/jbc.272.35.22285. [DOI] [PubMed] [Google Scholar]
- 3.Ton-That H, Mazmanian SK, Alksne L, Schneewind O: Anchoring of surface proteins to the cell wall ofStaphylococcus aureus. Cysteine 184 and histidine 120 of sortase form a thiolate-imidazolium ion pair for catalysis.J Biol Chem 2002, 277(9):7447–7452. [DOI] [PubMed]
- 4.Ton-That H, Mazmanian SK, Faull KF, Schneewind O: Anchoring of surface proteins to the cell wall ofStaphylococcus aureus. Sortase catalyzedin vitrotranspeptidation reaction using LPXTG peptide and NH(2)-Gly(3) substrates.J Biol Chem 2000, 275(13):9876–9881. [DOI] [PubMed]
- 5.Perry AM, Ton-That H, Mazmanian SK, Schneewind O: Anchoring of surface proteins to the cell wall ofStaphylococcus aureus.III. Lipid II is an in vivo peptidoglycan substrate for sortase-catalyzed surface protein anchoring.J Biol Chem 2002, 277(18):16241–16248. [DOI] [PubMed]
- 6.Ruzin A, Severin A, Ritacco F, Tabei K, Singh G, Bradford PA, Siegel MM, Projan SJ, Shlaes DM. Further evidence that a cell wall precursor [C(55)-MurNAc-(peptide)-GlcNAc] serves as an acceptor in a sorting reaction. J Bacteriol. 2002;184(8):2141–2147. doi: 10.1128/JB.184.8.2141-2147.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spirig T, Weiner EM, Clubb RT. Sortase enzymes in Gram-positive bacteria. Mol Microbiol. 2011;82:1044–1059. doi: 10.1111/j.1365-2958.2011.07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mazmanian SK, Liu G, Jensen ER, Lenoy E, Schneewind O: Staphylococcus aureussortase mutants defective in the display of surface proteins and in the pathogenesis of animal infections.Proc Natl Acad Sci U S A 2000, 97(10):5510–5515. [DOI] [PMC free article] [PubMed]
- 9.Mazmanian SK, Liu G, Ton-That H, Schneewind O: Staphylococcus aureussortase, an enzyme that anchors surface proteins to the cell wall.Science 1999, 285(5428):760–763. [DOI] [PubMed]
- 10.Kharat AS, Tomasz A: Inactivation of thesrtAgene affects localization of surface proteins and decreases adhesion ofStreptococcus pneumoniaeto human pharyngeal cellsin vitro.Infect Immun 2003, 71(5):2758–2765. [DOI] [PMC free article] [PubMed]
- 11.Pallen MJ, Lam AC, Antonio M, Dunbar K. An embarrassment of sortases - a richness of substrates? Trends Microbiol. 2001;9(3):97–102. doi: 10.1016/S0966-842X(01)01956-4. [DOI] [PubMed] [Google Scholar]
- 12.Barnett TC, Scott JR: Differential recognition of surface proteins inStreptococcus pyogenesby two sortase gene homologs.J Bacteriol 2002, 184(8):2181–2191. [DOI] [PMC free article] [PubMed]
- 13.Bierne H, Mazmanian SK, Trost M, Pucciarelli MG, Liu G, Dehoux P, Jansch L, Garcia-del Portillo F, Schneewind O, Cossart P: Inactivation of thesrtAgene inListeria monocytogenesinhibits anchoring of surface proteins and affects virulence.Mol Microbiol 2002, 43(4):869–881. [DOI] [PubMed]
- 14.Garandeau C, Reglier-Poupet H, Dubail I, Beretti JL, Berche P, Charbit A: The sortase SrtA ofListeria monocytogenesis involved in processing of internalin and in virulence.Infect Immun 2002, 70(3):1382–1390. [DOI] [PMC free article] [PubMed]
- 15.Gaspar AH, Marraffini LA, Glass EM, Debord KL, Ton-That H, Schneewind O: Bacillus anthracissortase A (SrtA) anchors LPXTG motif-containing surface proteins to the cell wall envelope.J Bacteriol 2005, 187(13):4646–4655. [DOI] [PMC free article] [PubMed]
- 16.Swaminathan A, Mandlik A, Swierczynski A, Gaspar A, Das A, Ton-That H: Housekeeping sortase facilitates the cell wall anchoring of pilus polymers inCorynebacterium diphtheriae.Mol Microbiol 2007, 66(4):961–974. [DOI] [PMC free article] [PubMed]
- 17.Mazmanian SK, Ton-That H, Su K, Schneewind O: An iron-regulated sortase anchors a class of surface protein duringStaphylococcus aureuspathogenesis.Proc Natl Acad Sci U S A 2002, 99(4):2293–2298. [DOI] [PMC free article] [PubMed]
- 18.Maresso AW, Chapa TJ, Schneewind O: Surface protein IsdC and Sortase B are required for heme-iron scavenging ofBacillus anthracis.J Bacteriol 2006, 188(23):8145–8152. [DOI] [PMC free article] [PubMed]
- 19.Rupnik M, Wilcox MH, Gerding DN: Clostridium difficileinfection: new developments in epidemiology and pathogenesis.Nat Rev Microbiol 2009, 7(7):526–536. [DOI] [PubMed]
- 20.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HM, Quail MA, Rance R, Brooks K, Churcher C, Harris D, Bentley SD, Burrows C, Clark L, Corton C, Murray V, Rose G, Thurston S, van Tonder A, Walker D, Wren BW, Dougan G, Parkhill J: Evolutionary dynamics ofClostridium difficileover short and long time scales.Proc Natl Acad Sci U S A 2010, 107(16):7527–7532. [DOI] [PMC free article] [PubMed]
- 21.Dingle KE, Griffiths D, Didelot X, Evans J, Vaughan A, Kachrimanidou M, Stoesser N, Jolley KA, Golubchik T, Harding RM, Peto TE, Fawley, Walker AS, Wilcox M, Crook DW: ClinicalClostridium difficile: clonality and pathogenicity locus diversity.PLoS One 2011, 6(5):e19993. [DOI] [PMC free article] [PubMed]
- 22.Stabler RA, Dawson LF, Valiente E, Cairns MD, Martin MJ, Donahue EH, Riley TV, Songer JG, Kuijper EJ, Dingle KE, Wren BW: Macro and micro diversity ofClostridium difficileisolates from diverse sources and geographical locations.PLoS One 2012, 7(3):e31559. [DOI] [PMC free article] [PubMed]
- 23.Cleary RK: Clostridium difficile-associated diarrhea and colitis - Clinical manifestations, diagnosis and treatment.Dis Colon Rectum 1998, 41(11):1435–1449. [DOI] [PubMed]
- 24.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, et al: The multidrug-resistant human pathogenClostridium difficilehas a highly mobile, mosaic genome.Nat Genet 2006, 38(7):779–786. [DOI] [PubMed]
- 25.Liew CK, Smith BT, Pilpa R, Suree N, Ilangovan U, Connolly KM, Jung ME, Clubb RT: Localization and mutagenesis of the sorting signal binding site on sortase A fromStaphylococcus aureus.FEBS Lett 2004, 571(1–3):221–226. [DOI] [PubMed]
- 26.Marraffini LA, Ton-That H, Zong Y, Narayana SV, Schneewind O: Anchoring of surface proteins to the cell wall ofStaphylococcus aureus.A conserved arginine residue is required for efficient catalysis of sortase A.J Biol Chem 2004, 279(36):37763–37770. [DOI] [PubMed]
- 27.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4(3):363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 28.Zhang R, Wu R, Joachimiak G, Mazmanian SK, Missiakas DM, Gornicki P, Schneewind O, Joachimiak A: Structures of sortase B fromStaphylococcus aureusandBacillus anthracisreveal catalytic amino acid triad in the active site.Structure 2004, 12(7):1147–1156. [DOI] [PMC free article] [PubMed]
- 29.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW: Comparative genome and phenotypic analysis ofClostridium difficile027 strains provides insight into the evolution of a hypervirulent bacterium.Genome Biol 2009, 10(9):R102. [DOI] [PMC free article] [PubMed]
- 30.Tulli L, Marchi S, Petracca R, Shaw HA, Fairweather NF, Scarselli M, Soriani M, Leuzzi R: CbpA: a novel surface exposed adhesin ofClostridium difficiletargeting human collagen.Cell Microbiol 2013, 15(10):1674–1687. [DOI] [PubMed]
- 31.Comfort D, Clubb RT. A comparative genome analysis identifies distinct sorting pathways in gram-positive bacteria. Infect Immun. 2004;72(5):2710–2722. doi: 10.1128/IAI.72.5.2710-2722.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneewind O, Mihaylova-Petkov D, Model P. Cell wall sorting signals in surface proteins of gram-positive bacteria. EMBO J. 1993;12(12):4803–4811. doi: 10.1002/j.1460-2075.1993.tb06169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janulczyk R, Rasmussen M. Improved pattern for genome-based screening identifies novel cell wall-attached proteins in gram-positive bacteria. Infect Immun. 2001;69(6):4019–4026. doi: 10.1128/IAI.69.6.4019-4026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pritz S, Wolf Y, Kraetke O, Klose J, Bienert M, Beyermann M. Synthesis of biologically active peptide nucleic acid-peptide conjugates by sortase-mediated ligation. J Org Chem. 2007;72(10):3909–3912. doi: 10.1021/jo062331l. [DOI] [PubMed] [Google Scholar]
- 35.Matayoshi ED, Wang GT, Krafft GA, Erickson J. Novel fluorogenic substrates for assaying retroviral proteases by resonance energy transfer. Science. 1990;247(4945):954–958. doi: 10.1126/science.2106161. [DOI] [PubMed] [Google Scholar]
- 36.Ton-That H, Liu G, Mazmanian SK, Faull KF, Schneewind O: Purification and characterization of sortase, the transpeptidase that cleaves surface proteins ofStaphylococcus aureusat the LPXTG motif.Proc Natl Acad Sci U S A 1999, 96(22):12424–12429. [DOI] [PMC free article] [PubMed]
- 37.Ton-That H, Schneewind O. Anchor structure of staphylococcal surface proteins. IV. Inhibitors of the cell wall sorting reaction. J Biol Chem. 1999;274(34):24316–24320. doi: 10.1074/jbc.274.34.24316. [DOI] [PubMed] [Google Scholar]
- 38.Dhar G, Faull KF, Schneewind O: Anchor structure of cell wall surface proteins inListeria monocytogenes.Biochemistry (Mosc) 2000, 39(13):3725–3733. [DOI] [PubMed]
- 39.Marraffini LA, Schneewind O. Anchor structure of staphylococcal surface proteins. V. Anchor structure of the sortase B substrate IsdC. J Biol Chem. 2005;280(16):16263–16271. doi: 10.1074/jbc.M500071200. [DOI] [PubMed] [Google Scholar]
- 40.Race PR, Bentley ML, Melvin JA, Crow A, Hughes RK, Smith WD, Sessions RB, Kehoe MA, McCafferty DG, Banfield MJ: Crystal structure ofStreptococcus pyogenessortase A: implications for sortase mechanism.J Biol Chem 2009, 284(11):6924–6933. [DOI] [PMC free article] [PubMed]
- 41.McDevitt D, Francois P, Vaudaux P, Foster TJ: Molecular characterization of the clumping factor (fibrinogen receptor) ofStaphylococcus aureus.Mol Microbiol 1994, 11(2):237–248. [DOI] [PubMed]
- 42.Ni Eidhin D, Perkins S, Francois P, Vaudaux P, Hook M, Foster TJ: Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin ofStaphylococcus aureus.Mol Microbiol 1998, 30(2):245–257. [DOI] [PubMed]
- 43.Patti JM, Jonsson H, Guss B, Switalski LM, Wiberg K, Lindberg M, Hook M: Molecular characterization and expression of a gene encoding aStaphylococcus aureuscollagen adhesin.J Biol Chem 1992, 267(7):4766–4772. [PubMed]
- 44.Cheng AG, Kim HK, Burts ML, Krausz T, Schneewind O, Missiakas DM: Genetic requirements forStaphylococcus aureusabscess formation and persistence in host tissues.FASEB J 2009, 23(10):3393–3404. [DOI] [PMC free article] [PubMed]
- 45.Weiss WJ, Lenoy E, Murphy T, Tardio L, Burgio P, Projan SJ, Schneewind O, Alksne L: Effect ofsrtAandsrtBgene expression on the virulence ofStaphylococcus aureusin animal models of infection.J Antimicrob Chemother 2004, 53(3):480–486. [DOI] [PubMed]
- 46.Bolken TC, Franke CA, Jones KF, Zeller GO, Jones CH, Dutton EK, Hruby DE: Inactivation of thesrtAgene inStreptococcus gordoniiinhibits cell wall anchoring of surface proteins and decreasesin vitroandin vivoadhesion.Infect Immun 2001, 69(1):75–80. [DOI] [PMC free article] [PubMed]
- 47.Mandlik A, Swierczynski A, Das A, Ton-That H: Corynebacterium diphtheriaeemploys specific minor pilins to target human pharyngeal epithelial cells.Mol Microbiol 2007, 64(1):111–124. [DOI] [PMC free article] [PubMed]
- 48.Jonsson IM, Mazmanian SK, Schneewind O, Bremell T, Tarkowski A: The role ofStaphylococcus aureussortase A and sortase B in murine arthritis.Microbes Infect 2003, 5(9):775–780. [DOI] [PubMed]
- 49.Kang HJ, Coulibaly F, Proft T, Baker EN: Crystal structure of Spy0129, aStreptococcus pyogenesclass B sortase involved in pilus assembly.PLoS One 2011, 6(1):e15969. [DOI] [PMC free article] [PubMed]
- 50.Chang C, Mandlik A, Das A, Ton-That H: Cell surface display of minor pilin adhesins in the form of a simple heterodimeric assembly inCorynebacterium diphtheriae.Mol Microbiol 2011, 79(5):1236–1247. [DOI] [PMC free article] [PubMed]
- 51.Frankel BA, Kruger RG, Robinson DE, Kelleher NL, McCafferty DG: Staphylococcus aureussortase transpeptidase SrtA: insight into the kinetic mechanism and evidence for a reverse protonation catalytic mechanism.Biochemistry (Mosc) 2005, 44(33):11188–11200. [DOI] [PubMed]
- 52.Dziarski R. Peptidoglycan recognition proteins (PGRPs) Mol Immunol. 2004;40(12):877–886. doi: 10.1016/j.molimm.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 53.Schleifer KH, Kandler O. Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol Rev. 1972;36(4):407–477. doi: 10.1128/br.36.4.407-477.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Necchi F, Nardi-Dei V, Biagini M, Assfalg M, Nuccitelli A, Cozzi R, Norais N, Telford JL, Rinaudo CD, Grandi G, Maione D. Sortase A substrate specificity in GBS pilus 2a cell wall anchoring. PLoS One. 2011;6(10):e25300. doi: 10.1371/journal.pone.0025300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Weiner EM, Robson S, Marohn M, Clubb RT: The Sortase A enzyme that attaches proteins to the cell wall ofBacillus anthraciscontains an unusual active site architecture.J Biol Chem 2010, 285(30):23433–23443. [DOI] [PMC free article] [PubMed]
- 56.Peltier J, Courtin P, El Meouche I, Lemee L, Chapot-Chartier MP, Pons JL: Clostridium difficilehas an original peptidoglycan structure with a high level of N-acetylglucosamine deacetylation and mainly 3–3 cross-links.J Biol Chem 2011, 286(33):29053–29062. [DOI] [PMC free article] [PubMed]
- 57.Oh KB, Oh MN, Kim JG, Shin DS, Shin J: Inhibition of sortase-mediatedStaphylococcus aureusadhesion to fibronectin via fibronectin-binding protein by sortase inhibitors.Appl Environ Microbiol 2006, 70(1):102–106. [DOI] [PubMed]
- 58.Maresso AW, Wu R, Kern JW, Zhang R, Janik D, Missiakas DM, Duban ME, Joachimiak A, Schneewind O. Activation of inhibitors by sortase triggers irreversible modification of the active site. J Biol Chem. 2007;282(32):23129–23139. doi: 10.1074/jbc.M701857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oh K-B, Nam K-W, Ahn H, Shin J, Kim S, Mar W: Therapeutic effect of (Z)-3-(2,5-dimethoxyphenyl)-2-(4-methoxyphenyl) acrylonitrile (DMMA) againstStaphylococcus aureusinfection in a murine model.Biochem Biophys Res Commun 2010, 396(2):440–444. [DOI] [PubMed]
- 60.Robichon C, Luo J, Causey TB, Benner JS, Samuelson JC: EngineeringEscherichia coliBL21(DE3) derivative strains to minimizeE. coliprotein contamination after purification by immobilized metal affinity chromatography.Appl Environ Microbiol 2011, 77(13):4634–4646. [DOI] [PMC free article] [PubMed]
- 61.Monot M, Boursaux-Eude C, Thibonnier M, Vallenet D, Moszer I, Medigue C, Martin-Verstraete I, Dupuy B: Reannotation of the genome sequence ofClostridium difficilestrain 630.J Med Microbiol 2011, 60(Pt 8):1193–1199. [DOI] [PubMed]
- 62.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 63.Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17(7):646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]