

Abstract
Summary Molecular inversion probes (MIPs) enable cost-effective multiplex targeted gene resequencing in large cohorts. However, the design of individual MIPs is a critical parameter governing the performance of this technology with respect to capture uniformity and specificity. MIPgen is a user-friendly package that simplifies the process of designing custom MIP assays to arbitrary targets. New logistic and SVM-derived models enable in silico predictions of assay success, and assay redesign exhibits improved coverage uniformity relative to previous methods, which in turn improves the utility of MIPs for cost-effective targeted sequencing for candidate gene validation and for diagnostic sequencing in a clinical setting.
Availability and implementation: MIPgen is implemented in C++. Source code and accompanying Python scripts are available at http://shendurelab.github.io/MIPGEN/.
Contact: shendure@uw.edu or boylee@uw.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
While rare variants and de novo mutations contribute to the genetic basis of complex diseases including intellectual disability (Vissers et al., 2010), autism spectrum disorders (O'Roak et al., 2012; Vissers et al., 2010), epilepsy (Epi4K Consortium et al., 2013) and congenital heart disease (Zaidi et al., 2013), the implication of individual genes in these phenotypes typically requires sequencing of large numbers of cases and controls. Molecular inversion probes [MIPs, also known as padlock probes (Nilsson et al., 1994)] have proven successful in a broad range of applications, including targeted genotyping (Hardenbol et al., 2003), DNA sequencing (O'Roak et al., 2012; Peidong et al., 2011; Porreca et al., 2007; Umbarger et al., 2013), assessing copy number and content (Nuttle et al., 2013; O'Roak et al., 2012; Schiffman et al., 2009), methylation patterns (Diep et al., 2012; Li et al., 2009), RNA allelotyping (Zhang et al., 2009) and detection of bacteria in clinical samples (Hyman et al., 2012). MIPs boast low amortized cost per sample and high scalability (O'Roak et al., 2012)—characteristics that may allow it to replace Sanger sequencing for clinical genetic testing (Umbarger et al., 2013). We recently built upon the MIP assay with the introduction of single-molecule MIPs or smMIPs: MIPs with molecular tags to track independent capture events (Hiatt et al., 2013). However, while genotyping accuracy and sensitivity for detecting low-frequency alleles have been enhanced, smMIPs do not address a key limitation: non-uniformity of capture efficiencies within probe sets. Early large-scale experiments (Porreca et al., 2007) that attempted to optimize targeting arm melting temperatures demonstrated substantial non-uniformity across target sites, with longer exons and GC extremes frequently failing capture. Dosing MIPs to compensate for dropout, known as repooling, enables significantly enhanced coverage (Diep et al., 2012), but collecting the empirical data for repooling lowers assay turnaround time and expends sequencing resources.
Previous studies with MIP (Porreca et al., 2007) and long padlock probe (LPP) (Peidong et al., 2011) assays were limited in their exploration of possible design remedies, including choosing only high-performing nucleotides at the MIP ligation junction, preferring low copy targeting arm sequences and prioritization based on oligonucleotide melting temperatures. Work by Deng et al. (2009) incorporated DNA folding metrics into the neural network-driven framework ppDesigner, and suggests that further modifications to MIP design and capture protocols could yield additional gains in coverage uniformity.
Here we describe an empirically trained design algorithm for MIP design that attains our goal of optimizing performance and reducing reliance on empirical testing for developing successful smMIP assays.
2 METHODS
MIPgen was implemented to facilitate MIP sequence design informed by statistical models of MIP performance, with both simplified user input and high extensibility. The models are derived from quantifying the performance of 12 000 randomly designed MIPs to arbitrary targets in the human exome. Using these models, MIPgen can objectively compare two candidate probe sequences in silico and curtail the number of suboptimal MIPs in the finished design. Each run of MIPgen consists minimally of an indexed reference genome, the desired range of target sizes (from 120 to 250 bp) and a BED file of the targeted regions.
To prepare for tiling targeted sequences with MIPs, queried targets are joined as needed into features that are sufficiently far apart to avoid redundancy of capture. The following steps are then applied to each of the features:
Sequences corresponding to the targeted regions are pulled from the reference genome directly from a fasta or using SAMtools.
SNPs in the targeted region are either retrieved by Tabix (Li, 2011) or read in from a local file in VCF format so that probe arms can be preferentially placed in non-polymorphic sites.
All possible targeting arms and insert sequences are tested for copy number to the reference genome using Burrows-Wheeler Aligner (BWA), and characteristics from all possible combinations of targeting arms are calculated for scoring by either the logistic regression or support vector regression model via A Library for Support Vector Machines (LIBSVM).
MIP selection is guided by scoring and continues until all targeted bases have been tiled. In the event that the targets cannot be tiled owing to low complexity or low specificity, a BED file of the untiled positions is printed in addition to the probes selected.
Optional parameters modulating behavior such as redundant tiling of targeted sites, degenerate molecular tags and the stringency of prioritizing low-scoring regions, detailed in the documentation, also alter handling of MIP tiling.
By iterating over the targeted sites and simultaneously traversing sequence while selecting probe designs, an optimal MIP tiling that covers all targeted bases can be produced. More details on the training set, including probe sequences (Supplementary Figure S1 and Supplementary Table S1), model characteristics (Supplementary Figures S2–6 and Supplementary Table S2) and MIPgen design algorithm are available in the Supplementary Material.
3 RESULTS
A set of eight genes that had previously been extensively characterized via MIP sequencing data (CHD8, TBL1XR1, TBR1, DYRK1A, ADNP, GRIN2B, PTEN and CTNNB1) plus an additional high GC target (SHANK3) were selected to test the models’ predictions of MIP performance, both on previously designed MIPs for these genes (O'Roak et al., 2012) as well as for a newly designed MIPgen set. The new design consisted of 402 smMIPs with complete tiling of the targeted sites, which were tested alongside the original MIP assay on control DNA. Predictions for the performance of the previous MIP designs were correlated with total read counts for both logistic scoring (ρ = 0.536) and SVR scoring (ρ = 0.540). For smMIPs, tagged read depth was slightly, but not significantly, more correlated with MIPgen scores than total read depth (ρ = 0.569, P > 0.05); unsurprisingly given this information, tagged and untagged read depths were highly correlated with each other (ρ = 0.900). Special attention was given to MIPs with <10% of the average coverage per MIP, as these are largely responsible for gaps in coverage. The scores were successful at detecting these low-performing MIPs, for both logistic (Area under the receiver operating characteristic curve of 0.827) and SVR (Support Vector Regression); (AUC = 0.864) models (Supplementary Figure S7). Logistic and SVR scores were only slightly more correlated with each other than with total read depth (Supplementary Figure S8).
We next analyzed the performance of the new MIP assay relative to that of the original set to ascertain the success of the new design algorithm. Average coverage per MIP in the new set increased 18% over the original set; however, the proportion of the 19 349 targeted bases <10% of the median per-base coverage (2668×) of the replicates remained unchanged: 23.7% for the original set and 23.8% for the redesigned set. Still, uniformity of coverage improved (Fig. 1), with the relative standard deviation of read depth per MIP dropping from 0.962 to 0.830. Scores continued to correlate with MIP performance in the redesigned set for both the logistic (ρ = 0.581) and SVR (ρ = 0.638) models (Fig. 1). The power to detect low-performing MIPs in the redesigned set was similarly accurate for the logistic model (AUC = 0.895) and for the SVR model (AUC = 0.926).
Fig. 1.
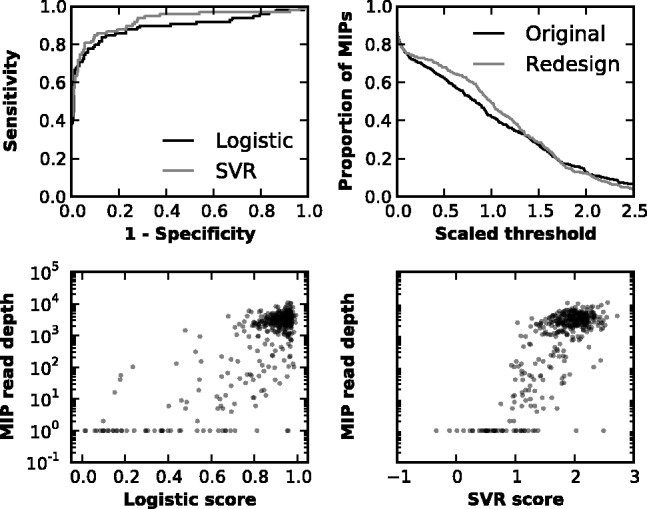
Model scores predict MIP performance. Both logistic and SVR modeling capture most of the variation in MIP performance. SVR scoring displays slightly greater power to discriminate adequately performing MIPs from poorly performing MIPs, as demonstrated by the higher AUC for the ROC curve conditioned on whether an MIP attained at least 10% of the median number of reads per MIP (upper left panel). Additionally, redesigning MIPs to the locus with MIPgen slightly increases the fraction of MIPs attaining levels of coverage at or below the level of average MIP coverage across sets (set to 1.0 in the upper right panel). Also shown are scatterplots of MIP scores versus realized read depth in the redesigned MIP set (lower panels)
Shearing protocols developed by Umbarger et al. (2013) substantially mitigated, but did not eliminate, coverage loss associated with poorly performing MIPs (Supplementary Figures S9 and S10). Furthermore, increasing the capture temperature from 60° to 65° did not resolve inadequate coverage of high-GC regions (Supplementary Figure S11). Of note, visual comparison of MIP coverage at these sites to coverage levels reported on the Exome Variant Server showed comparable coverage across targeted regions (Supplementary Figure S12). GC content is known to be a strong correlate of non-uniformity in both MIP capture and in-solution hybridization, and might underlie similarities in coverage patterns (Asan et al., 2011; Porreca et al., 2007; Sulonen et al., 2011).
4 FUTURE APPLICATIONS
MIPgen accurately predicts MIP and smMIP performance in silico, including identifying low-performing MIP and smMIP sequences. This core capability in turn enables a more effective process—also provided within the MIPgen package—for designing MIPs and smMIPs to arbitrary targets of interest. Even though we have not resolved the difficulties for MIPs in genes or regions with high-GC content, and the realized gains over prior design methodologies were relatively modest, MIPgen scores enable stratification of MIP targets a priori and in silico with reasonable discriminatory power. In the future, MIP scoring may be calibrated with new training sets that can account for advances in MIP protocols or match more specialized conditions that entail DNA of variable concentration or quality. Curated MIP/smMIP designs per gene may be indicated to facilitate comparison across studies and improve access to validated MIP/smMIP assays, and surveying MIPgen scores prior to committing to a MIP target may serve to maximize the economy of a MIP target enrichment approach. More generally, we anticipate that the user-friendly package for MIP and smMIP design provided in the MIPgen package will facilitate its broader use in both research and clinical applications.
Supplementary Material
Acknowledgements
The authors thank Joseph B. Hiatt, Jerrod J. Schwartz and Alexandra P. Lewis for support in developing the experimental protocols, and members of the Shendure and Eichler labs for helpful discussions.
Funding: This work was supported by a grant from the National Cancer Institute (CA160080 to J.S.).
Conflict of Interest: none declared.
REFERENCES
- Asan, et al. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011;12:R95. doi: 10.1186/gb-2011-12-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009;27:353–360. doi: 10.1038/nbt.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diep D, et al. Library-free methylation sequencing with bisulfite padlock probes. Nat. Methods. 2012;9:270–272. doi: 10.1038/nmeth.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epi4K Consortium et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardenbol P, et al. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol. 2003;21:673–678. doi: 10.1038/nbt821. [DOI] [PubMed] [Google Scholar]
- Hiatt JB, et al. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23:843–854. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman RW, et al. Molecular probe technology detects bacteria without culture. BMC Microbiol. 2012;12:29. doi: 10.1186/1471-2180-12-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011;27:718–719. doi: 10.1093/bioinformatics/btq671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JB, et al. Multiplex padlock targeted sequencing reveals human hypermutable CpG variations. Genes Dev. 2009;19:1606–1615. doi: 10.1101/gr.092213.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, et al. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science. 1994;265:2085–2088. doi: 10.1126/science.7522346. [DOI] [PubMed] [Google Scholar]
- Nuttle X, et al. Rapid and accurate large-scale genotyping of duplicated genes and discovery of interlocus gene conversions. Nat. Methods. 2013;10:903–909. doi: 10.1038/nmeth.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJB, et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peidong, et al. High-quality DNA sequence capture of 524 disease candidate genes. PNAS. 2011;108:6549–6554. doi: 10.1073/pnas.1018981108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porreca GJG, et al. Multiplex amplification of large sets of human exons. Nat. Methods. 2007;4:931–936. doi: 10.1038/nmeth1110. [DOI] [PubMed] [Google Scholar]
- Schiffman JD, et al. Molecular inversion probes reveal patterns of 9p21 deletion and copy number aberrations in childhood leukemia. Cancer Genet. Cytogenet. 2009;193:9–18. doi: 10.1016/j.cancergencyto.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulonen A-M, et al. Comparison of solution-based exome capture methods for next generation sequencing. Genome Biol. 2011;12:R94. doi: 10.1186/gb-2011-12-9-r94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbarger MA, et al. Next-generation carrier screening. Genet. Med. 2013;16:132–140. doi: 10.1038/gim.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vissers LELM, et al. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Zaidi S, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, et al. Digital RNA allelotyping reveals tissue-specific and allele-specific gene expression in human. Nat. Methods. 2009;6:613–618. doi: 10.1038/nmeth.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.