

Abstract
A need for more accurate and reliable radiation dosimetry has become increasingly important due to the possibility of a large-scale radiation emergency resulting from terrorism or nuclear accidents. Although traditional approaches provide accurate measurements, such methods usually require tedious effort and at least two days to complete. Therefore, we provide a new method for rapid prediction of radiation exposure. Eleven microarray datasets were classified into two groups based on their radiation doses and utilized as the training samples. For the two groups, Student's t-tests and resampling tests were used to identify biomarkers, and their gene expression ratios were used to develop a prediction model. The performance of the model was evaluated in four independent datasets, and Ingenuity pathway analysis was performed to characterize the associated biological functions. Our meta-analysis identified 29 biomarkers, showing approximately 90% and 80% accuracy in the training and validation samples. Furthermore, the 29 genes significantly participated in the regulation of cell cycle, and 19 of them are regulated by three well-known radiation-modulated transcription factors: TP53, FOXM1 and ERBB2. In conclusion, this study demonstrates a reliable method for identifying biomarkers across independent studies and high and reproducible prediction accuracy was demonstrated in both internal and external datasets.
Radiation exposure has become an important concern for human beings in everyone's daily life, because a person may receive irradiation from many different sources. For example, radiological weapons are able to cause radiation exposure to many people in a short time even if they are only small dirty bombs. A nuclear power plant may change into a dangerous radiation source when natural disasters happen, such as the devastating tsunami in Fukushima in Japan. In such cases, a large number of people will receive different amounts of radiation exposure and suffer a corresponding variety of ill effects.
Several literature reports have shown that distinct biological functions and damage patterns are triggered in cells in response to different doses of ionizing radiation (IR)1,2,3. Therefore, how to estimate the radiation exposure of IR-treated samples, which is called radiation dosimetry, has become a critical and urgent issue.
Several methods have been developed for assessing the radiation doses received by exposed samples4,5,6, but their applications may be limited in a severe radiation emergency. For instance, the dicentric assay requires much effort and at least three days to be completed6,7. Another popular approach is to observe the declining lymphocyte counts in the first 48 hours after irradiation4; however, such measurement still takes at least two days to be done, which may make it difficult to be used in a large-scale radiation emergency. Therefore, developing a new methodology to assess radiation exposure in tissue samples more quickly is necessary and beneficial for future applications.
With the advancement in high-throughput technologies, data from DNA microarrays and next-generation sequencing provide a good basis to address this issue. In recent years, several studies have demonstrated the success and effectiveness of predicting radiation doses based on gene expression levels7,8,9. For example, Paul et al. identified a 74-gene signature that can be utilized to predict four radiation doses from human peripheral blood7, and Dressman et al. reported a 25-gene signature to classify irradiated human samples based on the radiation doses8. Such studies had high prediction accuracies within their own datasets; however, challenges arise when trying to validate the predictions in independent datasets.
It is well known that biomarkers identified in one microarray dataset are usually irreproducible across studies, even if the investigated samples have similar clinical parameters10,11. The reasons for such high inconsistency in identified biomarkers across datasets may be attributed to different microarray platforms, various experimental protocols, and dissimilar statistical approaches. Consequently, the practical application of the identified biomarkers is limited. One possible approach to address this issue is to perform a large-scale meta-analysis of microarray data in the public domain12.
In this study, 11 microarray datasets from tissue samples irradiated by γ–rays were retrieved from the Gene Expression Omnibus (GEO) and utilized as an integrated training set. A set of 29 biomarkers associated with different radiation doses was identified based on several criteria, such as Student's t-test and resampling tests. Furthermore, a machine learning algorithm, called support vector machine (SVM), was used to develop a prediction model based on the genes' expression ratios. More than 80% accuracy was shown in classifying samples treated with higher and/or lower doses of radiation using 10,000 repetitions of 10-fold cross-validation in the training set. The prediction model showed similar accuracy in four independent datasets, suggesting its potential application to predict radiation doses across different studies.
Methods
Data collection and pre-processing
A protocol to identify differentially expressed (DE) genes associated with radiation dosage is illustrated in Figure 1. A total of 11 microarray datasets retrieved from the GEO13 were utilized as the training set (Table 1). To reduce the effects of confounding factors, only samples irradiated with γ–rays and collected 4–8 hours after radiation exposure were included. Furthermore, distinct cell lines were considered as different signatures and analyzed separately. Two pre-processing steps, including log2 transformation and quantile normalization, were performed to eliminate systematic biases across different platforms.
Figure 1. Flowchart for identification of differentially expressed genes associated with radiation doses and development of a prediction model.

The number of genes shown in the right dotted box denotes the union of genes across multiple signatures.
Table 1. Characteristics of training samples.
| Accession No.a | Sample No. | Cell type | Dose (Gy) | Time after radiation (h) |
|---|---|---|---|---|
| GSE26835 | 362 | Lymphoblast | 10.0 | 6 |
| GSE36720b | 9 | LNCap, PC3, DU145 | 10.0 | 6 |
| GSE8917 | 5 | Peripheral blood | 8.0 | 6 |
| GSE35372 | 6 | HL60, RV+ | 8.0 | 4 |
| GSE30043 | 3 | U87 | 8.5 | 4 |
| GSE23515c | 12 | Peripheral blood | 2.0 | 6 |
| GSE25772 | 4 | Fibroblast | 2.0 | 8 |
| GSE30044d | 3 | HEK | 2.0 | 4 |
| GSE8917 | 5 | Peripheral blood | 2.0 | 6 |
| GSE6971 | 4 | Fibroblast | 1.5 | 6 |
| GSE7075 | 6 | Fibroblast | 1.5 | 6 |
aGEO accession number.
bSamples treated with multiple-fraction irradiation were excluded.
cSamples from smokers were excluded.
dSamples treated with DNA minor groove binding ligand were excluded.
Identification of biomarkers associated with different radiation doses
Based on the radiation dosage, each microarray was classified as higher (≥8 Gy) or lower (≤2 Gy) dose. To reduce redundancy and difficulty in processing multiple probes targeting the same gene, only probes with the largest coefficient of variation (CV) were retained for further analyses. Student's t-test was performed to compare the gene expression levels in radiation-treated cells to those in untreated cells. The estimated false discovery rate (Q-value) was computed to address the issue of multiple test correction and only those genes with Q-values <0.1 were selected12,14. The formula for calculating Q-value is Q = (P*N)/I, where P is the P-value from the t-test, N is the total number of genes, and I is the ranking of the P-value among N (Figure 1: Step 2.1).
To exclude genes identified by chance, a resampling test was performed12. The number of DE genes for each signature was set from previous analyses, but genes in each DE signature were randomly selected. We repeated this procedure 10,000 times and tallied the number of signatures that each gene was selected as a DE gene to establish a null baseline. In other words, a null baseline of identifying one gene as a DE gene in the number of signatures was simulated after 10,000 repetitions. In this way, an empirical P-value for each gene was determined according to its number of significance in multiple signatures versus the null baseline. Only genes with empirical P-values less than 0.05 were included for the following analyses (Figure 1: Step 2.2).
To consider multiple signatures simultaneously, we adopted an approach to estimate the minimum meta-false discovery rate (mFDRMIN)12 as shown in the following equation:
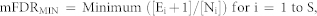 |
where S denotes the number of signatures, Ei denotes the number of DE genes selected at random, and Ni denotes the number of DE genes present in the ith signature, respectively. This measurement estimates the possibility of randomly identifying DE genes in multiple signatures (Figure 1: Step 3.1). Lastly, Student's t-test was used to select biomarkers showing significant differences between microarrays exposed to higher or lower dose radiation (Figure 1: Step 3.2, P < 0.05).
Development of a prediction model using support vector machine
After identification of the DE genes associated with radiation dosage, a machine learning algorithm, SVM15, was utilized to develop a classifier for predicting the radiation exposure of tissue samples (Figure 1: Step 4). For each biomarker identified in Step 3.2, the ratio of gene expression levels between radiation treated and untreated cells was used as an input variable for the SVM model. As shown in Table 1, for the samples treated with higher radiation dose, GSE26835 has 362 samples compared to only 23 samples in total from the other four datasets (GSE36720, GSE8917, GSE35372, GSE30043). In order to compensate the imbalanced sample size, we randomly selected 23 samples from dataset GSE26835, and combined them with the 23 samples from the other four datasets to develop the prediction model. A 10-fold cross-validation was performed for 10,000 iterations to calculate the accuracy and stability of prediction performance in the training samples. In addition, the reproducibility of the proposed classifier in independent datasets was evaluated by using several microarrays collected at different time points after irradiation (Table S1).
Results
Identification of biomarkers associated with radiation dosage
As shown in Figure 1, several filtering and selection steps were utilized to identify DE genes associated with radiation dosage. A signature was defined as one set of genes summarized from one cell line or one dataset. The results of Student's t-test (Step 2.1) showed that 5,949 genes in the higher-dose group and 11,413 genes in the lower-dose group had differential expression in at least one signature. To address multiple hypothesis testing and concurrently integrate different signatures, a resampling test following Rhodes's study12 was adopted and repeated 10,000 times (Step 2.2). As shown in Table 2, only 502 and 697 genes were found to be significantly DE (P < 0.05) in response to higher or lower doses of radiation. These results showed that more than 90% of the DE genes identified in Step 2.1 were excluded due to their high possibility of being selected randomly, suggesting that such a resampling test (Step 2.2) is useful to exclude false positive genes.
Table 2. Numbers of identified biomarkers in different steps in Figure 1.
| Higher Dose (≥8 Gy) | Lower Dose (≤2 Gy) | |||||
|---|---|---|---|---|---|---|
| # of signatures | # of DE genes (Q < 0.1: Step 2.1) | # of Sig genes (P < 0.05: Step 2.2) | mFDRMIN (Step 3.1) | # of DE genes (Q < 0.1: Step 2.1) | # of Sig genes (P < 0.05: Step 2.2) | mFDRMIN (Step 3.1) |
| 8 | 0 | 0 | -- | -- | -- | -- |
| 7 | 1 | 1 | 1.00 | -- | -- | -- |
| 6 | 5 | 5 | 0.20 | 0 | 0 | -- |
| 5 | 29 | 29 | 0.03 | 1 | 1 | 1.00 |
| 4 | 83 | 83 | 0.01 | 6 | 6 | 0.16 |
| 3 | 551 | 258 | 0.53 | 44 | 44 | 0.02 |
| 2 | 2,534 | 126 | 0.95 | 603 | 595 | 0.01 |
| 1 | 8,210 | 0 | 1.00 | 5,295 | 51 | 0.99 |
| Total | 11,413 | 502 | 5,949 | 697 | ||
#: Number; DE: differentially expressed; Sig: Significant; mFDRMIN: minimum meta-false discovery rate.
Next, we utilized an approach called mFDRMIN to determine the cutoff value of a biomarker in multiple signatures (Figure 1, Step 3.1). This study aimed to develop a prediction model, and large numbers of biomarkers would make it difficult to use the model for practical applications. Therefore, to considering the issues of practicability and false positive rates simultaneously, we determined that a biomarker must be identified in at least five signatures in response to higher-dose radiation and three signatures in response to lower-dose (Table 2). This cutoff of significance in multiple signatures was determined based on the low mFDR (Q ≤ 0.3) and an acceptable number of biomarkers (N < 100). The results indicated that 35 genes in higher dose and 51 genes in lower dose were significantly different from untreated cells (Table 3). Among them, Student's t-test was performed to exclude genes having no differences in expression in response to higher and lower doses of radiation (Figure 1, Step 3.2; P < 0.05). A set of 29 biomarkers showed significant differences at both higher and lower radiation doses, and thus all 29 biomarkers were included for developing the prediction model (Table 3).
Table 3. Identified biomarkers for samples treated by higher and/or lower radiation doses.
| Group (Gene No.) | Gene symbol |
|---|---|
| Higher Dose (35) | TPX2, ASPM, AURKB, CDCA3, CENPA, CENPF, AURKA, BIRC5, C12orf5, CCNB1, CCNF, CDC6, CDC20, CDCA2, CENPE, DLGAP5, E2F5, FAM83D, GTSE1, KIAA1333, KIF2C, KIF20A, MTIF2, MXD3, NDC80, NEK2, NIF3L1, PLK1, PRR11, PSRC1, RAD54L, RFC5, SPTBN1, TMEM19, TNFRSF10B |
| Lower Dose (51) | CDKN1A, FDXR, FHL2, GADD45A, MYC, TP53I3, TRIAP1, ACAP1, AEN, AFF1, APOBEC3H, BLOC1S2, BST1, C12orf5, C13orf30, C14orf128, COL27A1, CPT2, DDB2, ECHDC1, FANCE, FBXL4, FXYD2, GRIA1, IGFBP7, JAM2, LY9, MCM3, MDM2, MSH6, NMT2, NSRP1, NSUN7, PHLDA3, PITHD1, PLK2, PLK3, REV3L, RPS27L, SAP30, SESN1, SESN2, SLBP, SLFN11, SS18L2, TNFRSF10B, TNFSF4, TRIM22, VWCE, WHSC1L1, ZDHHC14 |
| High and Low Doses (29) | AEN, ASPM, AURKB, BIRC5, CCNB1, CCNF, CDC20, CDCA3, CENPA, CENPE, CENPF, DLGAP5, FBXL4, FDXR, FHL2, GTSE1, KIF20A, KIF2C, LY9, MXD3, NDC80, NEK2, PLK1, PLK2, PSRC1, RPS27L, SESN1, SLBP, TPX2 |
Internal 10-fold cross-validation of the 29 biomarkers
After identifying the final set of 29 biomarkers, a prediction model was developed based on the ratios of gene expression in radiation treated versus untreated cells using the SVM algorithm. A 10-fold cross-validation was repeated 10,000 times to evaluate the prediction performance and consistency in the training samples. As shown in Figure 2A, the average values of accuracy in the training samples were approximately 0.9 for the identified 29 biomarkers. The standard deviations were only around 0.03 for the 10,000 repetitions, suggesting that the prediction performances of the 29 biomarkers were highly consistent, even if the 23 samples from GSE26835 were used. In addition, a 10-fold cross-validation was performed by using all 419 samples shown in Table 1, and the prediction accuracy (0.86) was highly similar to that obtained from using 80 training samples. Therefore, these results demonstrated that the prediction model based on the 29 biomarkers was accurate in classifying samples treated with higher versus lower radiation doses.
Figure 2. Prediction performance of the three sets of biomarkers.

A 10-fold cross-validation was repeated 10,000 times and the accuracies in the samples treated with higher and/or lower radiation doses were plotted. (A) Training samples (46 higher-dose and 34 lower-dose). (B) External independent datasets (64 higher-dose and 30 lower-dose).
Comparisons with published signatures
To further evaluate the 29 biomarkers, their predictive performance was also compared with two published signatures for predicting radiation dosage7,8. Only the genes that existed in the training samples and showed no missing values were included in the analysis (Table S2). The same 10-fold cross-validation approach described previously was performed to investigate their performance. The 29 biomarkers identified in our approach showed significantly (P <0.05) better accuracy than the other two signatures (Figure 2A). Notably, the standard deviations were also not high for the two published studies and thus the random selection of samples in GSE26835 was not a critical factor in affecting the prediction performance.
External evaluations of the 29 biomarkers in independent datasets
To validate the prediction performance of the 29 biomarkers and the two published signatures, four microarray datasets collected within 3–6 hours after irradiation treatments were examined (Table S1). As described previously, a total of 10,000 analyses were performed to calculate the accuracy. The results are summarized in Figure 2B, which shows that the 29 biomarkers identified in this study were significantly better predictors than the genes obtained from Paul's study. The accuracy of the 29 biomarkers in the samples treated with higher-dose radiation was similar to that of the meta-genes obtained from Dressman's study. However, compared with the 29 identified biomarkers, Dressman's genes showed poor performance in the prediction of the samples treated with lower-dose radiation. The 29 identified biomarkers showed around 90% accuracy in classifying samples treated with higher-dose radiation, 65% accuracy in lower-dose radiation samples, and 83% accuracy overall.
Functional characterization of the 29 biomarkers
Ingenuity pathway analysis was used to explore possible upstream regulators and characterize associated biological pathways of the 29 biomarkers (Table S3). As shown in Figure 3, 19 out of the 29 biomarkers can be regulated by three genes including TP53, FOXM1, and ERBB2. All three regulators have been reported to have pivotal roles in modulating radiation responses16,17,18. In addition to transcription regulators, two major biological pathways—cell cycle regulation and the pyrimidine salvage pathway—were significantly enriched (P < 0.01). Several studies also have shown that irradiation is able to cause transcriptional changes in genes involved in these two functions16,19,20, suggesting that the 29 biomarkers not only can be used for predicting radiation doses but also participate in important functions in response to radiation exposure.
Figure 3. Gene-gene interaction networks of the 29 biomarkers.

Three possible upstream regulators were enriched. Direct evidence between two genes from previous literature reports is shown as a solid line and indirect evidence is depicted as a dashed line.
Discussion
A large-scale radiation emergency may affect many people at one time, and thus how to efficiently and accurately measure radiation exposure becomes an important issue. Several studies have identified predictive biomarkers for radiation dose using gene expression microarrays7,8,9. However, the generalizability (i.e., reproducibility) of those reported biomarkers was not evaluated and validated in external independent datasets, which makes them difficult to apply. To address this issue, this study developed a meta-analysis flowchart to select biomarkers across independent microarray datasets. The 29 identified biomarkers had approximately 90% accuracy in the training samples, which were composed of 11 datasets, and had more than 80% accuracy in four independent datasets, suggesting the effectiveness of our proposed method.
Two different approaches can be used to identify biomarkers for predicting radiation exposure across independent datasets. One possibility is to select one dataset as the training set and then validate the identified biomarkers in external samples21,22. Notably, the most important limitation of such an approach is that a dataset with a large sample size is required because potential biomarkers are selected based solely on it. However, the sizes of irradiated microarray datasets that are available in the published domain are usually not very large, which makes this approach difficult to execute. In addition, most of these datasets were investigated by distinct platforms, including both one-color and two-color systems. Challenges arise when trying to merge those datasets into one integrated meta-dataset due to their huge discrepancy and systematic biases. Therefore, we chose an alternative approach (Figure 1) in which multiple datasets were considered individually.
The 29 biomarkers identified by our methods were compared with two published sets of biomarkers shown in Table S2. Six and zero of the 29 biomarkers were in common with those obtained from Paul's study and Dressman's study, respectively, illustrating the fact that biomarkers identified in one microarray dataset rarely overlap with those reported in other studies11. As shown in Figure 2, the prediction performance of the 29 biomarkers was generally higher than that of Paul's and Dressman's studies, except the accuracy of external samples treated with higher radiation dose (91% in our study versus 95% in Dressman's). Notably, Dressman's biomarkers showed much better performance in classifying samples treated with higher radiation doses than classifying those with lower doses. One possible reason for this imbalance is that most of their original samples were irradiated with 200-1,350 cGy, which fell primarily into the higher radiation dose group in the current study. Since radiation doses in future samples are unknown, a balanced prediction performance is more practical for real-world applications.
There are some limitations of the prediction model developed in this study. First, the collection of microarray datasets analyzed in this study included only samples that were treated with γ–rays and harvested 3–8 hours later. It is well known that the radiation source plays an important role in driving downstream gene expression, and different signaling pathways are triggered in response to the different types of irradiation23,24. In addition, dissimilar genes and biological functions are induced in cells at different time points after radiation exposure25,26. To reduce potential variations in gene expression levels, a unified radiation source (γ–ray) and post-irradiation time period (3–6 hours) were utilized in the analyses. Therefore, the prediction model only showed about 55% accuracy in identifying radiation dose in samples harvested 24 hours after irradiation (Figure S1), suggesting this model is useful in classifying radiation exposures during the early-response period but not the late-response period. Furthermore, the prediction model classified samples into higher or lower radiation dose groups, instead of numerically estimating their exposed radiation dose. Although specific definition of exposed radiation dose in samples may provide better classification performance, the sizes of published irradiated microarrays are not sufficient to develop a prediction model. Meanwhile, some variations in gene expression levels were still observed, even if the samples were reclassified in the opposite group, especially the lower-dose ones. This may be attributed to the fact that transcriptional changes in response to radiation are not linear, and genes may be switched on/off after a certain threshold of radiation exposure1,27. However, more samples irradiated by different doses are required to develop a regression model across independent datasets.
Analyses of upstream regulators (Figure 3) and biological functions were performed to elucidate how the 29 biomarkers participate in the radiation response. Unsurprisingly, TP53 was a consistently significant regulator (P < 10−10) in the three-biomarker sets for samples treated with higher and/or lower radiation doses (Table S4). A previous report demonstrated that inhibition of FOXM1 expression can elevate radiation sensitivity in cells after 8 or 10 Gy γ–ray irradiation17, which is consistent with our finding that FOXM1 was the most significant regulator (P = 2.04 * 10−20) after higher-dose radiation exposure. As the second most significant regulator after higher-dose irradiation (P = 1.07 * 10−14), ERBB2 is able to reduce radiation induced apoptosis by activating NFκB-related signaling in cells exposed to 5 Gy18. In addition, the 29 biomarkers were significantly involved in cell cycle regulation and in the pyrimidine salvage pathway. Undoubtedly, the identified biomarkers were associated with these pathways because deoxyribonucleotide synthesis was observed in two irradiated cell lines with different radiosensitivities20. We conclude that these 29 biomarkers not only are regulated by well-known radiation modulators but also have important roles in response to radiation exposure.
Author Contributions
T.P.L. and E.Y.C. conceived and designed the experiments; T.P.L. and Y.Y.H. performed the experiments and analyzed the data. L.C.L., M.H.T. and E.Y.C. contributed reagents, materials, and/or analysis tools. T.P.L., L.C.L., M.H.T. and E.Y.C. wrote the paper.
Supplementary Material
Supplementary data
Acknowledgments
This study was supported partly by the grant from National Taiwan University, Taiwan (grant no. 103R8400).
References
- Lu T. P. et al. Distinct signaling pathways after higher or lower doses of radiation in three closely related human lymphoblast cell lines. Int J Radiat Oncol Biol Phys 76, 212–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L. H. et al. Gene expression profiles of normal human fibroblasts after exposure to ionizing radiation: a comparative study of low and high doses. Radiat Res 164, 17–26 (2005). [DOI] [PubMed] [Google Scholar]
- Short S. C. et al. Dose- and time-dependent changes in gene expression in human glioma cells after low radiation doses. Radiat Res 168, 199–208 (2007). [DOI] [PubMed] [Google Scholar]
- Donnelly E. H. et al. Acute radiation syndrome: assessment and management. South Med J 103, 541–6 (2010). [DOI] [PubMed] [Google Scholar]
- Straume T., Lucas J. N., Tucker J. D., Bigbee W. L. & Langlois R. G. Biodosimetry for a radiation worker using multiple assays. Health Phys 62, 122–30 (1992). [DOI] [PubMed] [Google Scholar]
- Blakely W. F., Salter C. A. & Prasanna P. G. Early-response biological dosimetry--recommended countermeasure enhancements for mass-casualty radiological incidents and terrorism. Health Phys 89, 494–504 (2005). [DOI] [PubMed] [Google Scholar]
- Paul S. & Amundson S. A. Development of gene expression signatures for practical radiation biodosimetry. Int J Radiat Oncol Biol Phys 71, 1236–1244 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dressman H. K. et al. Gene expression signatures that predict radiation exposure in mice and humans. PLoS Med 4, e106 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L. H. et al. Distinct transcriptome profiles identified in normal human bronchial epithelial cells after exposure to gamma-rays and different elemental particles of high Z and energy. BMC Genomics 14, 372 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T. P., Chuang E. Y. & Chen J. J. Identification of reproducible gene expression signatures in lung adenocarcinoma. BMC Bioinformatics 14, 371 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau S. K. et al. Three-gene prognostic classifier for early-stage non small-cell lung cancer. J Clin Oncol 25, 5562–9 (2007). [DOI] [PubMed] [Google Scholar]
- Rhodes D. R. et al. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci U S A 101, 9309–14 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R., Domrachev M. & Lash A. E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–10 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey J. D. & Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100, 9440–5 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey T. S. et al. Support vector machine classification and validation of cancer tissue samples using microarray expression data. Bioinformatics 16, 906–14 (2000). [DOI] [PubMed] [Google Scholar]
- Fei P. & El-Deiry W. S. P53 and radiation responses. Oncogene 22, 5774–83 (2003). [DOI] [PubMed] [Google Scholar]
- Halasi M. & Gartel A. L. Suppression of FOXM1 sensitizes human cancer cells to cell death induced by DNA-damage. PLoS One 7, e31761 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G. et al. Expression of ErbB2 enhances radiation-induced NF-kappaB activation. Oncogene 23, 535–45 (2004). [DOI] [PubMed] [Google Scholar]
- Teyssier F., Bay J. O., Dionet C. & Verrelle P. [Cell cycle regulation after exposure to ionizing radiation]. Bull Cancer 86, 345–57 (1999). [PubMed] [Google Scholar]
- Wei S. et al. Radiation-induced changes in nucleotide metabolism of two colon cancer cell lines with different radiosensitivities. Int J Radiat Biol 75, 1005–13 (1999). [DOI] [PubMed] [Google Scholar]
- Lu T. P. et al. Integrated analyses of copy number variations and gene expression in lung adenocarcinoma. PLoS One 6, e24829 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D. T. et al. Prognostic and predictive value of a malignancy-risk gene signature in early-stage non-small cell lung cancer. J Natl Cancer Inst 103, 1859–70 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikjoo H. & Lindborg L. RBE of low energy electrons and photons. Phys Med Biol 55, R65–109 (2010). [DOI] [PubMed] [Google Scholar]
- Asaithamby A. & Chen D. J. Mechanism of cluster DNA damage repair in response to high-atomic number and energy particles radiation. Mutat Res 711, 87–99 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K. H. et al. Time-course analysis of DNA damage response-related genes after in vitro radiation in H460 and H1229 lung cancer cell lines. Exp Mol Med 43, 419–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandhi S. A., Ming L., Ivanov V. N., Hei T. K. & Amundson S. A. Regulation of early signaling and gene expression in the alpha-particle and bystander response of IMR-90 human fibroblasts. BMC Med Genomics 3, 31 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoel D. G. & Li P. Threshold models in radiation carcinogenesis. Health Phys 75, 241–50 (1998). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data