

High-energy radical species are widespread in biology as transient reaction intermediates and free radicals. The control of these extremely reactive molecules is crucial to limit oxidative stress and promote healthy physiological function.[1] In some cases this reactivity can be harnessed by a system to do useful work.[2] Over two decades ago Rétey discussed how energy can be “borrowed” from an enzyme to effect an unlikely chemical change.[3] In the case of coenzyme B12-dependent enzymes,[4] this energy is “lent” in the form of radical intermediates, which are formed upon substrate binding by Co–C bond homolysis in the cofactor. The energy is then returned by radical pair recombination after turnover (Scheme 1).
Scheme 1.
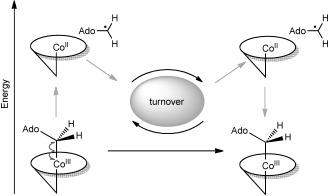
Energy “borrowed” from a coenzyme B12-dependent enzyme in the form of radicals. The high-energy 5′-deoxyadenosyl/cob(II)alamin radical pair is created upon substrate binding, and energy is returned during radical pair recombination after turnover.
The key is the way in which the protein generates the radicals and then prevents these high-energy intermediates from stabilising (i.e., dropping to a lower energy) through side reactions. Thus, the borrowed energy is channelled to the desired chemical change. Despite ample data demonstrating the role of radicals in coenzyme B12-dependent enzymes, there is less direct evidence for how the protein controls them.[5] Herein, we present data that suggest that a single point mutation can have a significant impact on Co–C bond homolysis and the control of the radical trajectory and reactivity in coenzyme B12-dependent ethanolamine ammonia lyase (EAL). The data show that the size and negative charge of an active-site glutamate (E287, Figure 1) contribute variously to 1) apparent substrate affinity, 2) promotion of rapid Co–C bond homolysis, 3) control of the reactivity of both the 5′-deoxyadenosyl and cob(II)alamin radicals, 4) the kinetics of the rate-limiting steps. The data also indicate that local flexibility in EAL might facilitate the approach of the charged E287 towards the polar ribose of the 5′-deoxyadenosyl, potentially offering a unified dynamic/electrostatic model of radical generation and control.
Figure 1.
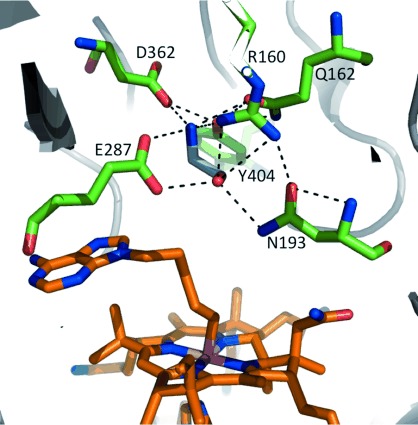
The active site of substrate-bound, adeninylpentylcobalamin-complexed ethanolamine ammonia lyase from E. coli (PDB ID: 3ABS). Residues (green) that form H-bonds with 2-aminoethanol (cyan) are highlighted, including those known to be of catalytic importance (E287 and R160). Adeninylpentylcobalamin (orange) is a coenzyme B12 derivative that allows the substrate to bind to EAL without Co–C bond homolysis.
After substrate binding to EAL, and Co–C bond homolysis, the 5′-deoxyadenosyl radical abstracts hydrogen from the α carbon of 2-aminoethanol to give the substrate radical. This undergoes intramolecular rearrangement—that is, a 1,2-shift of the amine—to the product-like radical, which abstracts hydrogen back from the 5′-deoxyadenosine and then dissociates into products (see Figure S1 in the Supporting Information). Kinetic isotope effect (KIE) and magnetic field effect experiments showed that Co–C bond homolysis and initial H-abstraction are kinetically coupled, and went some way to explaining the fleeting existence of the 5′-deoxyadenosyl radical.[6] Until crystal structures of EAL were published recently,[7] however, we had only limited insight into how the protein might both create and control the radical intermediates. These structures identified a mobile, active-site glutamate residue (E287) that only becomes localised upon substrate binding (Figure 1). We then proposed a dynamic “substrate trigger” mechanism for coupled Co–C bond homolysis and H abstraction, whereby substrate binding draws the negatively charged E287 past the polar ribose of the 5′-deoxyadenosyl, initiating homolysis and guiding the radical towards the substrate.[8] In support of this, we acquired ultrafast IR signals that suggest vibrational coupling between the protein and cofactor, and made direct observations of protein motions coupled to the reaction chemistry by stopped-flow FTIR. These motions occur both at the beginning and end of turnover, and accompany the homolysis and recombination processes illustrated in Scheme 1.[9]
To investigate the “substrate trigger” hypothesis set out above for Co–C bond homolysis in EAL, we made three separate single-point mutations (see the Experimental Section and Figure S2). The electrostatic contribution was assessed with E287Q (i.e., glutamine), which maintains the approximate volume and H-bonding of E287, whilst removing the formal negative charge. The contribution of residue size and proximity (H-bonding distance, van der Waals contact) were investigated by using the E287D variant, which incorporates the smaller, negatively charged aspartate. E287A was also investigated because it removes most of these interactions.
The peak of the αβ absorption band for free coenzyme B12 in aqueous buffer is at 525 nm and that for methylcobalamin is at 521 nm. These peaks were found to blue-shift by varying amounts when bound to wild-type (WT) EAL and the E287 variants (Figures 2 and S3 as well as Table S1). The magnitude of the shift is similar within error (∼4 nm) for coenzyme B12 bound to WT, E287D and E287Q. Such a shift suggests a more hydrophobic environment for the B12 chromophore when bound to the protein than in aqueous solution. This hydrophobic effect is enhanced greatly in E287A (a (15±2) nm blue shift). On the other hand, the average blue shift is ∼7 nm for protein-bound methylcobalamin and is comparable within error across the EAL variants. These data suggest a strong influence of E287 on coenzyme B12, through direct contact with the upper axial 5′-deoxyadenosyl that is not facilitated by the smaller upper axial methyl. Moreover, there is greater general variation between the spectra of B12 bound to the EAL variants than between those of protein-bound methylcobalamin (Figure S3 A and C). Contact between E287 and 5′-deoxyadenosyl might be the origin of the ultrafast vibrational coupling observed between photoexcited B12 and EAL.[9a]
Figure 2.
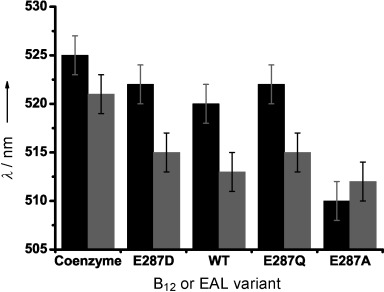
Peak wavelengths of the αβ absorption band for coenzyme B12 (5′-deoxyadenosylcobalamin, black bars) and methylcobalalmin (grey bars) both free in aqueous buffer and bound to WT EAL and the E287D/Q/A variants. The EAL variants are arranged from left to right in order of increasing hydrophobicity of residue 287 (Asp<Glu<Gln<Ala).[10] The ±2 nm error was based on the breadth of the peaks. See text for discussion.
Steady-state kinetic parameters (Table 1) were calculated for WT EAL and each E287 variant with 2-aminoethanol (see the Experimental Section and Figure S4). EAL protects against the efficient reaction between the cob(II)alamin radical and O2, both after Co–C bond homolysis through substrate binding[6b] and Co–C bond photolysis.[6c] Initially, therefore, these data were acquired under aerobic conditions. There is a substantial decrease in the apparent kcat in both E287D (∼100-fold) and E287Q (∼820-fold) compared to WT. This can be explained by Co–C bond homolysis and/or H-transfer from the substrate becoming at least partially rate limiting during turnover in E297D and E297Q. Alternatively, E287 could play an important role in radical rearrangement and/or the second H transfer, which are thought to be rate limiting in the WT (Figure S1).[11] No turnover was observed for E287A. Similar data from coenzyme B12-dependent ornithine 4,5-aminomutase (OAM) and methylmalonyl-CoA mutase (MCM) show a more modest decrease in kcat with equivalent variants, and retention of activity after the alanine mutation.[12] Moreover, the glutamine variant has a higher turnover number than the aspartate for both OAM and MCM, whereas the opposite is true for EAL (see later discussion).
Table 1.
Steady-state turnover kinetic parameters for the EAL wild-type and E287 variants with 2-aminoethanol at 298 K under aerobic and anaerobic conditions
| E287 | kcat [s−1] | Km [mm] | kcat/Km [m−1 s−1] | |
|---|---|---|---|---|
| +O2 | WT[a] | 33.5±0.62 | (1.97±0.12)×10−3 | (1.71±0.07)×107 |
| D | 0.34±0.01 | 2.02±0.18 | 170±11 | |
| Q | 0.041±0.007 | 1.47±0.19 | 28.7±3.3 | |
| A | – | – | – | |
| −O2 | D | 0.34±0.002 | 2.01±0.11 | 171±8 |
| Q | 0.059±0.004 | 1.42±0.09 | 41.8±2.3 | |
| A | – | – | – |
The WT EAL steady-state parameters are invariant in the presence and absence of oxygen.
The apparent substrate affinity is significantly lower in E287D and E287Q (Km is approximately three orders of magnitude higher). This is perhaps unsurprising considering that the crystal structure of EAL from E. coli shows two H-bonds between E287 and the substrate.[7] The Km values are very similar between the two active variants of EAL, despite the significantly lower kcat for E297Q. Electrostatics, therefore, has more of an influence on turnover than apparent affinity for substrate. The catalytic efficiency, kcat/Km, is reduced by approximately five orders of magnitude in the active EAL variants; this is substantial for a single point mutation.
When the steady-state data were acquired under anaerobic conditions, the kinetic parameters for E287D remained the same within error (Table 1). However, kcat for E287Q increased by ∼45 % with Km remaining the same within error. Oxygen, therefore, appears only to influence enzyme turnover. We also investigated the effect of oxygen on the pre-steady-state data by stopped-flow (see the Experimental Section). Figure 3 A shows the change in absorbance at 525 nm after rapidly mixing E287D with 2-aminoethanol under anaerobic (main panel) and aerobic (inset) conditions. The absorbance decrease at 525 nm in both cases is consistent with the conversion of 5′-deoxyadenosylcob(III)alamin to the cob(II)alamin radical during Co–C bond homolysis. Both data sets also follow the same single-phase kinetics: kobs=(7.29±0.18) s−1 (anaerobic) and (7.24±0.31) s−1 (aerobic). Figure 3 B shows the equivalent data for E287Q. Under anaerobic conditions, the accumulation of cob(II)alamin is again observed (main panel, kobs=(3.13±0.83) s−1). However, there is an increase in absorbance under aerobic conditions (inset), which is consistent with a reaction between cob(II)alamin and O2 that generates cob(III)alamin species with higher extinction coefficients at 525 nm than coenzyme B12.[13] The more hydrophobic environment produced by the glutamine in E287Q compared to either glutamate (WT) or aspartate (E287D) appears to allow oxygen to access the active site, thus quenching the cob(II)alamin radical.
Figure 3.
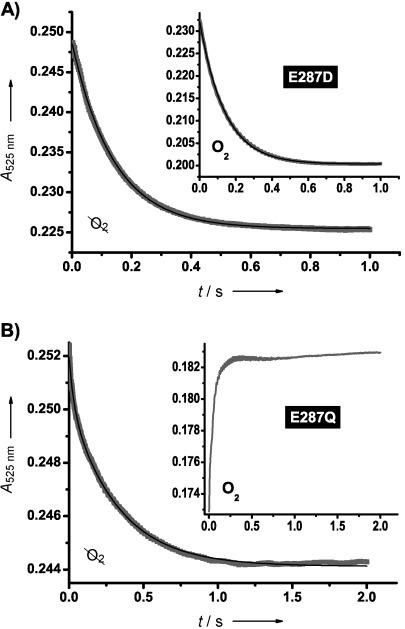
Pre-steady-state traces (grey points) and single exponential fits (black line) representing the change in absorbance at 525 nm after rapid mixing of 2-aminoethanol (60 mm final concentration) and EAL variants (30–40 μm final concentration) for A) E287D and B) E287Q. The main panels represent anaerobic reaction conditions, and insets represent aerobic. Unlike for E287D, the aerobic data for E287Q were not fit to an exponential function. Any difference in absolute absorbance reflects variations in active-site concentrations.
Photolysis of coenzyme B12 also generates the cob(II)alamin/5′-deoxyadenosyl radical pair. Under aerobic conditions, O2 is thought to react with both radicals.[13] The inset of Figure 3 B provides evidence for a similar oxidative reaction with cob(II)alamin following the substrate-driven, thermal Co–C bond homolysis by E287Q. Quenching of the cob(II)alamin radical in this way will prevent it from recombining with the 5′-deoxyadenosyl radical. If radical pair recombination is required after each turnover, one might expect either deactivation after a single turnover or a substantial reduction in turnover of E287Q under aerobic conditions. Instead, we only see a slight reduction in kcat upon the introduction of oxygen to the reaction sample, this suggests a significant proportion of radical pairs remain dissociated until the substrate is exhausted. Furthermore, it is unlikely that the reaction between O2 and 5′-deoxyadenosyl radical is a dominant aerobic pathway in this EAL variant. If it were, one would expect E287Q to deactivate immediately following homolysis under aerobic conditions because the organic radical is required for H abstraction from the substrate. Therefore, the size and H-bonding of the glutamine residue in E287Q provides at least partial control over the path of the 5′-deoxyadenoyl radical towards the substrate.
The kobs values for the anaerobic data from E287D ((7.29±0.18) s−1) and E287Q ((3.13±0.83) s−1) with 2-aminoethanol are around two orders of magnitude slower than kobs estimated for the WT at 298 K.[6b] Moreover, both kobs and the amplitude of the absorbance change at 525 nm for E287D are ∼2.5 times those of E287Q. This suggests that the equilibrium for Co–C bond homolysis is further towards the intact cofactor in E287Q. Unlike the WT data (at 278 K), which exhibit multiphasic kinetics,[6b] both anaerobic data sets in Figure 3 fit well to a single exponential function. Although the substantial rise in Km indicates that the apparent substrate affinity might be significantly compromised, this is unlikely to limit the pre-steady-state kinetics because the experiments were conducted under saturating substrate concentrations (Figure S5). The single-phase behaviour therefore suggests that one of the chemical steps that contribute to this signal (i.e., homolysis or H-transfer) has become sufficiently slow compared to the other to dominate the pre-steady-state kinetics.
The kobs values for E287D and E287Q are lower than kcat for the WT enzyme, but are ∼20 and ∼50 times higher, respectively, than their own kcat values. The homolysis/H-transfer steps that contribute to the pre-steady-state signal are therefore unlikely to be rate limiting for steady-state turnover in these variants. This indicates a substantial effect on subsequent steps in the catalytic cycle (Figure S1), which is supported by the fact that kobs for E287D and E287Q are closer in value than are their respective kcat values. EPR data indicate the substrate-like radical accumulates during turnover; this suggests that radical rearrangement could be rate limiting.[11a] However, a deuterium KIE of ∼7 has been reported on turnover of 2-aminoethanol by EAL, thus indicating that H transfer from the coenzyme to the product-like radical must also contribute, at least partially, to the overall rate.[11b, c] The effect on these steps warrants further investigation.
E287A does undergo substrate-induced Co–C bond homolysis, albeit very slowly, despite being ostensibly inactive (Figure 4). These data suggest that E287 is not an absolute requirement for Co–C bond homolysis in EAL, but that it is required for homolysis to happen with any efficiency. It also appears crucial for activity. Although E287A can trigger Co–C bond homolysis upon substrate binding, we did not observe turnover for this variant. The absence of charge and the small size of the alanine side chain appear to result in a loss of control over the high-energy 5′-deoxyadenosyl radical. The UV–visible spectra in Figure 4 show the accumulation of cob(II)alamin despite the fact that the data were acquired under aerobic conditions. The spectra are similar if acquired in the absence of oxygen (not shown). This is perhaps surprising considering the result in Figure 3 B, which shows a very efficient side reaction between the cob(II)alamin radical and O2 immediately following Co–C bond homolysis in E287Q. This side reaction is sufficiently rapid that it seems highly unlikely that product release is conditional. This apparent disparity may be explained by the fact that E287Q fills a similar space to E287 in the WT, whereas E287A leaves a void that can be occupied by neighbouring polar/charged side chains or water molecules. The result would be the retention of the hydrophilic environment around cob(II)alamin that limits the approach of O2.
Figure 4.
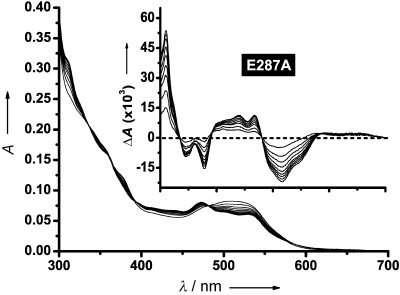
Main panel: UV–visible absorption spectra acquired every minute for 18 min after adding 2-aminoethanol (110 mm final concentration) to the E287A EAL variant (see the Experimental Section). Inset: difference spectra calculated by subtracting each subsequent spectrum from the initial spectrum. The x-axis in the inset is on the same scale as in the main panel.
The formal negative charges of the glutamate in WT EAL and aspartate in the E287D variant appear to play a more prominent role in EAL than in the mutases studied in ref. [12]. This is an interesting result because published computational data suggest that the electrostatic contribution to Co–C bond homolysis is dominant in coenzyme B12-dependent mutases.[14] The authors of ref. [12] argue that, assuming Co–C bond homolysis is rate limiting during turnover, the low kcat for the aspartate variant of both OAM and MCM might be explained by a decreased charge–dipole interaction between the aspartate and polar 5′-deoxyadenosyl. The magnitude of this interaction follows an inverse r2 relationship, in which r=distance between the residues and 5′-deoxyadenosyl. kcat for the glutamine variant, on the other hand, is higher because the closer dipole–dipole interaction is comparatively stronger. Wolthers et al. also suggest that the active sites of OAM and MCM are too rigid to compensate for this increased distance between the aspartate and the 5′-deoxyadenosyl of B12. Our pre-steady-state data indicate by contrast, the EAL active site is able to compensate and therefore might be more flexible. Such generalised local flexibility in EAL therefore provides a dynamic contribution to the electrostatic model of Co–C bond homolysis in B12-dependent enzymes.[14] The active site is able to compress, thus bringing the charged residue(s) into closer proximity to the polar 5′-deoxyadenosyl. kobs and Km are both affected significantly by E287 mutations. However, they are of a similar order between E287D and E287Q, thus suggesting a substantial impact of electrostatics on the chemistry later in the catalytic cycle, as demonstrated by the difference in kcat.
It is also possible that the apparent flexibility of EAL ensures that protection against the side reaction between cob(II)alamin and O2 is maintained in E287D. The aspartate is brought adequately close to the cofactor to keep the environment of the central Co hydrophilic. The fact that aerobic addition of substrate to E287A results in the accumulation of cob(II)alamin, with no evidence of reaction with O2, is again consistent with an active site that is flexible enough to compress and fill the void left by the glutamate-to-alanine substitution. These neighbouring residues might also facilitate substrate-triggered Co–C bond homolysis in E287A. A nearby arginine, R160 (Figure 1), has previously been shown as playing a critical role in EAL catalysis.[15] Again, the formal (positive) charge of R160 is significant, with R160K retaining activity with a modest drop in kcat/Km (∼180-fold) and R160A resulting in the accumulation of cob(II)alamin but not facilitating turnover. There was also evidence for the involvement of more than one residue from our stopped-flow FTIR data that estimated the movement of approximately seven peptide bonds alongside the pre-steady-state chemistry.[9a] Such a movement might not directly involve seven side chains, but does suggest a flexible, dynamic active site in EAL. This picture supports the model for Co–C bond homolysis in EAL proposed by Robertson et al.: coordinated protein motions that guide the cleavage of the Co–C bond along the reaction coordinate.[16]
Structural and computational studies indicate that large-scale motions help prime B12-dependent enzymes, such as OAM,[17] lysine 5,6-aminomutase,[18] diol dehydratase,[19] and MCM,[20] for radical-mediated catalysis. There is an increasing body of evidence that suggests that localised motions are then intimately involved in controlling the trajectory and reactivity of the activated radical intermediates (e.g., ref. [5b, 21]. However, EAL shows no evidence of large-scale motions upon substrate binding.[7] It therefore achieves the substrate trigger of Co–C bond homolysis and subsequent radical control by using active-site interactions and dynamics alone. This has been borne out here, with E287 apparently providing both electrostatic and dynamical contributions to Co–C bond homolysis and subsequent radical control in EAL.
Experimental Section
Refer to the Supporting Information for Experimental details.
Acknowledgments
We thank the University of Manchester, UK Biotechnology and Biological Sciences Research Council (BBSRC), Natural Science Foundation of China (No. 31071495) and Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions for funding. S.H. is a BBSRC David Phillips Fellow. N.S.S. holds a Royal Society Wolfson Merit Award and an Engineering and Physical Sciences Research Council (EPSRC) Established Career Fellowship.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1.Halliwell B, Gutteridge JMC. Free Radicals in Biology and Medicine. 4th ed. Oxford: University Press; 2007. [Google Scholar]
- 2.Golding BT, Buckel W. In: Encyclopedia of Radicals in Chemistry, Biology and Materials, Vol. 3. Chatgilialoglu C, Studer A, editors. Chichester: Wiley; 2012. pp. 1501–1546. [Google Scholar]
- 3.Rétey J. Angew. Chem. 1990;102:373–379. [Google Scholar]
- Angew. Chem. Int. Ed. Engl. 1990;29:355–361. [Google Scholar]
- 4a.Brown KL. Chem. Rev. 2005;105:2075–2149. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- 4b.Marsh ENG, Patterson DP, Li L. ChemBioChem. 2010;11:604–621. doi: 10.1002/cbic.200900777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c.Gruber K, Puffer B, Kräutler B. Chem. Soc. Rev. 2011;40:4346–4363. doi: 10.1039/c1cs15118e. [DOI] [PubMed] [Google Scholar]
- 5a.Dowling DP, Croft AK, Drennan CL. Annu. Rev. Biophys. 2012;41:403–427. doi: 10.1146/annurev-biophys-050511-102225. [DOI] [PubMed] [Google Scholar]
- 5b.Jones AR, Levy C, Hay S, Scrutton NS. FEBS J. 2013;280:2997–3008. doi: 10.1111/febs.12223. [DOI] [PubMed] [Google Scholar]
- 6a.Bandarian V, Reed GH. Biochemistry. 2000;39:12069–12075. doi: 10.1021/bi001014k. [DOI] [PubMed] [Google Scholar]
- 6b.Jones AR, Hay S, Woodward JR, Scrutton NS. J. Am. Chem. Soc. 2007;129:15718–15727. doi: 10.1021/ja077124x. [DOI] [PubMed] [Google Scholar]
- 6c.Jones AR, Woodward JR, Scrutton NS. J. Am. Chem. Soc. 2009;131:17246–17253. doi: 10.1021/ja9059238. [DOI] [PubMed] [Google Scholar]
- 7.Shibata N, Tamagaki H, Hieda N, Akita K, Komori H, Shomura Y, Terawaki S-i, Mori K, Yasuoka N, Higuchi Y, Toraya T. J. Biol. Chem. 2010;285:26484–26493. doi: 10.1074/jbc.M110.125112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones AR, Hardman SJO, Hay S, Scrutton NS. Angew. Chem. 2011;123:11035–11038. doi: 10.1002/anie.201105132. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2011;50:10843–10846. doi: 10.1002/anie.201105132. [DOI] [PubMed] [Google Scholar]
- 9a.Russell HJ, Jones AR, Hay S, Greetham GM, Towrie M, Scrutton NS. Angew. Chem. 2012;124:9440–9444. doi: 10.1002/anie.201202502. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:9306–9310. doi: 10.1002/anie.201202502. [DOI] [PubMed] [Google Scholar]
- 9b.Jones AR, Russell HJ, Greetham GM, Towrie M, Hay S, Scrutton NS. J. Phys. Chem. A. 2012;116:5586–5594. doi: 10.1021/jp304594d. [DOI] [PubMed] [Google Scholar]
- 10.Monera OD, Sereda TJ, Zhou NE, Kay CM, Hodges RS. J. Pept. Sci. 1995;1:319–329. doi: 10.1002/psc.310010507. [DOI] [PubMed] [Google Scholar]
- 11a.Bender G, Poyner RR, Reed GH. Biochemistry. 2008;47:11360–11366. doi: 10.1021/bi801316v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11b.Babior BM. J. Biol. Chem. 1969;244:449–456. [PubMed] [Google Scholar]
- 11c.Weisblat DA, Babior BM. J. Biol. Chem. 1971;246:6064–6071. [PubMed] [Google Scholar]
- 12.Makins C, Pickering AV, Mariani C, Wolthers KR. Biochemistry. 2013;52:878–888. doi: 10.1021/bi3012719. [DOI] [PubMed] [Google Scholar]
- 13.Schwartz PA, Frey PA. Biochemistry. 2007;46:7284. doi: 10.1021/bi700077v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma PK, Chu ZT, Olsson MHM, Warshel A. Proc. Natl. Acad. Sci. USA. 2007;104:9661–9666. doi: 10.1073/pnas.0702238104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun L, Groover OA, Canfield JM, Warncke K. Biochemistry. 2008;47:5523–5535. doi: 10.1021/bi702366e. [DOI] [PubMed] [Google Scholar]
- 16.Robertson WD, Wang M, Warncke K. J. Am. Chem. Soc. 2011;133:6968–6977. doi: 10.1021/ja107052p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17a.Wolthers KR, Levy C, Scrutton NS, Leys D. J. Biol. Chem. 2010;285:13942–13950. doi: 10.1074/jbc.M109.068908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b.Pang J, Li X, Morokuma K, Scrutton NS, Sutcliffe MJ. J. Am. Chem. Soc. 2012;134:2367–2377. doi: 10.1021/ja210417k. [DOI] [PubMed] [Google Scholar]
- 18.Berkovitch F, Behshad E, Tang K-H, Enns EA, Frey PA, Drennan CL. Proc. Natl. Acad. Sci. USA. 2004;101:15870–15875. doi: 10.1073/pnas.0407074101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibata N, Masuda J, Morimoto Y, Yasuoka N, Toraya T. Biochemistry. 2002;41:12607–12617. doi: 10.1021/bi026104z. [DOI] [PubMed] [Google Scholar]
- 20a.Mancia F, Evans PR. Structure. 1998;6:711–720. doi: 10.1016/s0969-2126(98)00073-2. [DOI] [PubMed] [Google Scholar]
- 20b.Mancia F, Smith GA, Evans PR. Biochemistry. 1999;38:7999–8005. doi: 10.1021/bi9903852. [DOI] [PubMed] [Google Scholar]
- 20c.Froese DS, Kochan G, Muniz JRC, Wu X, Gileadi C, Ugochukwu E, Krysztofinska E, Gravel RA, Oppermann U, Yue WW. J. Biol. Chem. 2010;285:38204–38213. doi: 10.1074/jbc.M110.177717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21a.Bucher D, Sandala GM, Durbeej B, Radom L, Smith DM. J. Am. Chem. Soc. 2012;134:1591–1599. doi: 10.1021/ja207809b. [DOI] [PubMed] [Google Scholar]
- 21b.Friedrich P, Baisch U, Harrington RW, Lyatuu F, Zhou K, Zelder F, McFarlane W, Buckel W, Golding BT. Chem. Eur. J. 2012;18:16114–16122. doi: 10.1002/chem.201201840. [DOI] [PubMed] [Google Scholar]
- 21c.Buckel W, Friedrich P, Golding BT. Angew. Chem. 2012;124:10114–10116. doi: 10.1002/anie.201205299. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:9974–9976. doi: 10.1002/anie.201205299. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information