

Background: Pyrin, ASC, and procaspase-1 associate to form an inflammasome that mediates inflammatory responses.
Results: Multiple binding sites on the pyrin domains of ASC and pyrin mediate their interaction.
Conclusion: Interaction between pyrin and ASC via multiple sites drives ASC clustering to form an inflammasome.
Significance: These findings provide insight into the interaction modes of pyrin domains and inflammasome assembly.
Keywords: Death Domain, Inflammation, Innate Immunity, Mutagenesis, Pattern Recognition Receptor (PRR), Protein Complex, Protein-Protein Interaction, Familial Mediterranean Fever, Inflammasome, Pyrin
Abstract
Inflammasomes are macromolecular complexes that mediate inflammatory and cell death responses to pathogens and cellular stress signals. Dysregulated inflammasome activation is associated with autoinflammatory syndromes and several common diseases. During inflammasome assembly, oligomerized cytosolic pattern recognition receptors recruit procaspase-1 and procaspase-8 via the adaptor protein ASC. Inflammasome assembly is mediated by pyrin domains (PYDs) and caspase recruitment domains, which are protein interaction domains of the death fold superfamily. However, the molecular details of their interactions are poorly understood. We have studied the interaction between ASC and pyrin PYDs that mediates ASC recruitment to the pyrin inflammasome, which is implicated in the pathogenesis of familial Mediterranean fever. We demonstrate that both the ASC and pyrin PYDs have multifaceted binding modes, involving three sites on pyrin PYD and two sites on ASC PYD. Molecular docking of pyrin-ASC PYD complexes showed that pyrin PYD can simultaneously interact with up to three ASC PYDs. Furthermore, ASC PYD can self-associate and interact with pyrin, consistent with previous reports that pyrin promotes ASC clustering to form a proinflammatory complex. Finally, the effects of familial Mediterranean fever-associated mutations, R42W and A89T, on structural and functional properties of pyrin PYD were investigated. The R42W mutation had a significant effect on structure and increased stability. Although the R42W mutant exhibited reduced interaction with ASC, it also bound less to the pyrin B-box domain responsible for autoinhibition and hence may be constitutively active. Our data give new insights into the binding modes of PYDs and inflammasome architecture.
Introduction
Autoinflammatory diseases characterized by unprovoked bouts of fever and systemic inflammation are primarily caused by genetic mutations in proteins that mediate an inflammatory response (1–3). The proinflammatory cytokine IL-1β plays a key role in the pathogenesis of these diseases and is generated by the assembly of caspase-1-activating inflammasome complexes that process proIL-1β into its active form (4, 5). Familial Mediterranean fever (FMF)3 is an autoinflammatory disease caused by mutations in the MEFV gene, which encodes pyrin (6, 7). Pyrin interacts with ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain (CARD)) to recruit procaspase-1 and form an inflammasome complex (8, 9). The complex formed is analogous to inflammasomes assembled by cytoplasmic pattern recognition receptors, including the NOD-like receptor (NLR) and PYHIN (pyrin and HIN domain-containing) protein families, in response to pathogens and cellular stress signals (10). Furthermore, the pyrin inflammasome is activated in response to challenge by Francisella novicida and Burkholderia cenocepacia (11, 12) and in response to p38 MAP kinase activation upon ribotoxic stress (13). However, there is evidence of an anti-inflammatory role for pyrin (14–16), suggesting that pyrin can have either a proinflammatory or anti-inflammatory role under different conditions.
Human pyrin is a multidomain protein comprised of 781 amino acids that is expressed in neutrophils, eosinophils, and monocytes (17, 18). Pyrin contains an N-terminal pyrin domain (PYD), a member of the death fold superfamily of protein interaction domains (19, 20), through which it interacts with ASC. In addition to an N-terminal PYD, pyrin contains a bZIP domain, a B-box zinc finger domain, a coiled-coil domain, and a B30.2/SPRY domain (6, 7, 21) (see Fig. 1A). The coiled-coil domain mediates oligomerization of pyrin into a trimer, thus forming a platform for oligomerization and activation of procaspase-1 upon recruitment to the complex (22). The B-box domain has been shown to interact intramolecularly with the PYD to maintain pyrin in a repressed state (22). One protein that can derepress pyrin is PSTPIP1 (proline-serine-threonine phosphatase-interacting protein 1), a cytoskeleton-associated protein that causes the autoinflammatory PAPA syndrome when mutated (23). Binding of PSTPIP1 to the pyrin B-box releases pyrin PYD and allows interaction with ASC (22). The role of the pyrin B30.2 domain is unclear; however, the majority of FMF-associated mutations map to this domain (24). Other FMF-associated mutations are spread throughout the protein with two mutations, R42W and A89T, located in pyrin PYD (24, 25). However, it is unclear whether these mutations are disease-causative.
FIGURE 1.

Identification of residues on pyrin PYD that are important for interaction with ASC PYD. A, schematic representation of a pyrin trimer indicating positions of the PYD (residues 1–92), bZIP domain (residues 266–270), B-box domain (residues 375–407), coiled-coil domain (CC, residues 408–594), and B30.2/SPRY domain (residues 598–774). The central pyrin molecule is shown as a linear schematic, whereas the molecules on either side illustrate the intramolecular interaction between the PYD and B-box. B, from left to right, ensemble of 10 NMR-derived structures of pyrin PYD superimposed over the backbone atoms (N, Cα, and C′) of residues 5–92; ribbon diagram of the lowest energy pyrin PYD structure illustrating the six α-helices and the α2-α3 loop, which is colored magenta; and superposition of the pyrin PYD (gray) and ASC PYD (Protein Data Bank code 1UCP, cyan) (32) structures for backbone Cα residues 5–92 of pyrin PYD and residues 4–91 of ASC PYD. C, ribbon representation of the pyrin PYD structure with residues mutated in this study shown in black. The residues mutated include Lys3, Leu10, and Glu14 (helix α1); Glu22, Lys25, and Gln29 (helix α2); Lys35 and His37 (α2-α3 loop); Arg42 and Gln46 (helix α3); Arg49 (α3-α4 loop); Val58 (helix α4); Glu63, Leu71, and Arg75 (helix α5); and Arg80, Glu84, and Arg88 (helix α6). D, purified His6-tagged WT and mutant pyrin PYDs were used for in vitro binding assays with purified bead-bound GST-ASC PYD or GST alone. Bound protein was eluted with SDS-PAGE sample buffer, subjected to SDS-PAGE, and then transferred to a PVDF membrane. The bound His6-tagged WT and mutant pyrin PYDs detected by immunoblotting with an anti-His antibody are shown above a Ponceau S stain from the same blot to detect GST-ASC PYD or GST alone. An amount representing 5% of the input of WT or mutant pyrin PYDs used for binding studies is also shown.
The adaptor protein ASC plays a central role in assembly of the pyrin inflammasome, as well as the inflammasomes formed by several NLR and PYHIN family proteins (10). ASC consists of an N-terminal PYD and a C-terminal CARD, which is also a death fold domain (21, 26). A homotypic interaction between ASC PYD and the N-terminal PYDs of pyrin, NLRP1, NLRP3, NLRP7, and AIM2 recruits ASC to inflammasomes, whereas ASC CARD recruits procaspase-1 via a homotypic CARD interaction (10). ASC PYD can also recruit procaspase-8 to inflammasomes to induce apoptotic cell death (27, 28). In addition to bridging the interaction between cytosolic receptors and caspases, ASC self-associates via its PYD and CARD domains to mediate further clustering of the inflammasome to form a compact speck (29–31).
Despite the critical role of PYD-mediated interactions in inflammasome assembly, the molecular details of these interactions are poorly characterized. Structures of several PYDs have been determined (32–40), indicating a six-helix bundle structure similar to other members of the death fold superfamily. However, PYDs typically have a short third helix (α3) and a long preceding loop (32, 33, 35). There is no structure available for a PYD complex, although recent biochemical analysis of ASC PYD self-association has given some insights into the interaction mode in the ASC PYD homodimer (30). Distinct positively and negatively charged surfaces of ASC PYD each contain a binding site, which interacts in the homodimer. However, it is unclear whether other PYD complexes will share a similar mode of interaction. Interestingly, the interface of the ASC PYD homodimer involves helix α3, which has been proposed to transition between a folded and unfolded state to regulate PYD function (34, 41).
In this study, we examined the interaction between the PYDs of pyrin and ASC, which is required for ASC recruitment to the pyrin inflammasome. We have identified multiple binding sites on both PYDs that are important for their interaction and show that the sites for ASC self-association overlap with the sites for pyrin PYD interaction. However, we also demonstrate that ASC can self-associate and interact with pyrin, which is consistent with a proinflammatory role for pyrin. In addition, the effect of the two FMF-associated mutations, R42W and A89T, on the structure and function of pyrin PYD was investigated. The R42W mutation in helix α3 stabilized the pyrin PYD, whereas the A89T mutation at the end of helix α6 had minimal effects on protein structure and stability. Neither mutation enhanced the interaction between pyrin and ASC, as might be expected to promote autoinflammation, but R42W was defective in the autoinhibitory interaction with the B-box domain and thus may be more easily activated. Finally, using our data, we rationalize models for the PYD interaction network at the pyrin inflammasome.
EXPERIMENTAL PROCEDURES
Plasmids
Plasmids expressing GST-ASC PYD (wild-type or mutant), His6-ASC PYD, and pyrin PYD-His6 have been described previously (30). Pyrin without the N-terminal PYD (pyrinΔPYD, residues 93–781) was cloned into pET-21a (Novagen) for in vitro translation. Full-length ASC (residues 1–195) was cloned into pGEX-4T-1 (GE Healthcare) for expression in Escherichia coli with an N-terminal GST tag. For expression in mammalian cells, full-length pyrin with a C-terminal Myc tag was cloned into pcDNA 3.1 (Invitrogen). Full-length PSTPIP1 with an N-terminal FLAG tag was cloned into pcDNA 3.1. Full-length ASC was expressed in mammalian cells from pcDNA3-ASC. Specific point mutations were introduced into pyrin PYD using the QuikChange site-directed mutagenesis approach (Stratagene). The sequences of all constructs were verified by automated DNA sequencing (Australian Genome Research Facility).
Protein Expression and Purification
His6- and GST-tagged proteins were expressed in E. coli and purified as previously described (30). For NMR studies, uniformly 15N- and 13C/15N-labeled proteins were expressed in minimal medium containing 15NH4Cl (1 g/liter) and 13C-glucose (2 g/liter) as the sole nitrogen and carbon sources (42). For in vitro binding studies, soluble His6-tagged pyrin PYD mutants (K3A, L10A, Q29A, K35A, H37A, R42A, Q46A, R49A, L71A, R80A, E84A, and R88A) were eluted with 50 mm Tris, pH 8, 1 m NaCl, and 250 mm imidazole, whereas the less soluble mutants (E14A, E22A, K25A, V58A, E63A, and R75A) were eluted with buffer also containing 0.5% Nonidet P-40 to improve solubility. Aliquots of wild-type pyrin PYD were eluted from Ni2+ affinity resin with or without 0.5% Nonidet P-40 to compare binding properties under similar buffer conditions. All proteins were then diluted 100-fold into binding buffer (50 mm HEPES, pH 7.4, 50 mm NaCl, 5 mm EDTA, 0.1% Nonidet P-40, 10% glycerol).
In Vitro Protein Interaction Assays
To identify residues important for pyrin PYD interaction, 200 μl of purified wild-type or mutant pyrin PYD was added to glutathione agarose-bound GST-ASC PYD, GST-ASC full-length, or GST alone in 800 μl of binding buffer. Samples were incubated overnight at 4 °C, and the resin was washed three times with 1 ml of wash buffer (50 mm Tris, pH 7.5, 200 mm NaCl, 0.5% Nonidet P-40). Bound proteins were analyzed by Western blotting. His6-tagged proteins were detected with a penta-His antibody (Qiagen), whereas GST and GST-ASC PYD were detected with Ponceau S stain. To identify residues important for ASC PYD interaction, purified wild-type pyrin PYD (200 μl) was incubated with glutathione agarose-bound GST-ASC PYD (wild-type or mutant) or GST alone as described above. Bound proteins were detected as described above. To test the effect of FMF-associated mutations R42W and A89T on binding of pyrin PYD to pyrinΔPYD, pyrinΔPYD was expressed using the TnT T7 coupled reticulocyte lysate system (Promega) in the presence of [35S]methionine and incubated with glutathione agarose-bound GST-pyrin PYD (wild-type or mutant) or GST alone in 150 μl of binding buffer containing 150 mm NaCl. Samples were incubated for 2 h at 4 °C and then washed three times with 500 μl of binding buffer. Bound proteins were resolved by SDS-PAGE. 35S-Labeled protein was detected by phosphorimaging, whereas GST and GST-pyrin PYD were detected with Coomassie stain. To test whether pyrin PYD and ASC PYD compete for binding to GST-ASC PYD, purified soluble ASC PYD was incubated with glutathione-agarose-bound GST-ASC PYD in the presence or absence of purified pyrin PYD. Bound proteins were detected by Western blotting as described above. To test the effect of ASC PYD self-association on ASC PYD interaction with pyrin, [35S]methionine-labeled pyrin was incubated with glutathione agarose-bound GST-ASC PYD or GST alone in the presence or absence of purified recombinant His6-tagged ASC PYD. Bound 35S-labeled pyrin was detected by phosphorimaging, whereas GST and GST-ASC PYD were detected with Coomassie stain.
Immunoprecipitation Assays
HEK 293T cells were maintained in DMEM/F-12 medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin. The cells were seeded in 10-cm dishes and transfected using Lipofectamine 2000 (Invitrogen). After incubation for 24 h, cell lysates were prepared as previously described (30), and pyrin-ASC or pyrin-ASC-PSTPIP1 complexes were immunoprecipitated using an anti-Myc antibody (Cell Signaling Technology) and immobilized on protein G-Sepharose (Sigma). Bead-bound proteins were eluted with SDS-PAGE sample buffer and analyzed by Western blotting using antibodies to pyrin (Enzo Life Sciences), ASC (Enzo Life Sciences), and the FLAG tag (Cell Signaling Technology).
NMR Spectroscopy
Samples for NMR contained 0.3–0.8 mm protein in 50 mm sodium phosphate, pH 4.0, and 150 mm NaCl. NMR experiments were acquired at 25 °C on either a Bruker Avance 750 MHz spectrometer equipped with a z-shielded gradient triple resonance probe or a Bruker Avance 900 MHz spectrometer equipped with a z-gradient triple resonance cryoprobe. NMR spectra were processed with NMRPipe (43) and analyzed using PIPP (44) or CcpNmr (45). Two-dimensional 1H-15N heteronuclear single-quantum coherence (HSQC) spectra were recorded for wild-type pyrin PYD and point mutants that affected interactions with ASC. Backbone resonance assignments of wild-type pyrin PYD and the R42W mutant were made using three-dimensional HNCO, HN(CA)CO, CBCA(CO)NH, HNCACB, and C(CO)NH experiments (46). Mutation-induced chemical shift changes were calculated using weighted average values, Δδav, with Δδav = {[Δδ(1HN)2 + (0.2Δδ (15N))2]/2}1/2 (47). Assignments for the backbone resonances of wild-type and R42W pyrin PYD have been deposited in the BioMagResBank database with the accession numbers 19875 and 19926, respectively.
Pyrin PYD Structure
The secondary structure elements in pyrin PYD were delineated from 13Cα, 13Cβ, 13CO, 15N, 1Hα, and 1HN chemical shifts with the program TALOS+ (48). Residues 1–3 at the N terminus and residues 94–100 at the C terminus (including the His6 tag) were estimated from TALOS+ to be disordered and excluded from subsequent CS-ROSETTA calculations (49). A total of 5000 all-atom ROSETTA models were generated, and the all-atom ROSETTA energies of these models were rescored based on agreement with the experimental chemical shifts. Convergence of the calculations was evaluated from the Cα root mean square deviation values of the 10 lowest energy models compared with the lowest energy model. The ensemble of 10 structures was validated using the PSVS software package (50), and the lowest energy model was selected as a representation of the three-dimensional structure of pyrin PYD. Images and electrostatic properties of the structure were generated using the PyMOL molecular graphics system (Schrödinger, LLC). Coordinates of the 10 pyrin PYD structures have been deposited in the Protein Data Bank with the accession number 2MPC.
HADDOCK Modeling
Modeling of pyrin-ASC PYD complexes was performed using the HADDOCK (High Ambiguity Driven protein-protein DOCKing) web server (51). The ASC PYD structure (32) (Protein Data Bank code 1UCP) and the pyrin PYD structure were entered as starting structures, and active residues were defined based on mutagenesis data from this study. For the first model, Glu14 on pyrin PYD and residues Lys21 and Arg41 on ASC PYD were defined as active. For the second model, Lys25 and Arg42 on pyrin PYD and Asp10, Gly13, Asp48, Asp51, and Asp54 on ASC PYD were defined as active. Finally, in the third model, Arg75 and Arg80 on pyrin PYD and Asp10, Glu13, Asp48, Asp51, and Asp54 on ASC PYD were defined as active. Passive residues were defined automatically. A total of 1000 complex structures were calculated using rigid body docking. The best structures, which were between 139 and 167 structures for the docking runs submitted, were subjected to further refinement and cluster analysis. The best structure from the lowest energy cluster from each docking run was chosen to represent each of the pyrin-ASC PYD complexes.
CD Spectroscopy
Purified pyrin PYD proteins were dialyzed against CD buffer (10 mm sodium phosphate, pH 4.0, 150 mm NaF) and diluted to a final concentration of 20 μm. Protein concentration was determined based on A280 measurements under denaturing conditions (52). CD spectra were acquired using a JASCO J-710 spectropolarimeter using a quartz cell with a 1-mm path length. Far-UV spectra were recorded at room temperature between 185 and 260 nm using a scan speed of 10 nm/min, 100 mdeg sensitivity, and a 4-s response time. Three scans were averaged, and the spectra were corrected using a buffer blank. The ellipticity is reported as mean residue ellipticity ([Θ]MRW, deg cm2 dmol−1) (53). The secondary structure content was calculated using the CDSSTR spectra deconvolution program (54) from the DichroWeb server (55).
For chemical denaturation experiments, proteins were diluted to a final concentration of 10 μm with different concentrations of urea (0–8 m in 0.25 m increments) prepared in CD buffer. After overnight incubation at room temperature, the CD signal at 222 nm was recorded at each concentration of urea for 30 s at 100 mdeg sensitivity and 4 s of response time. Thermodynamic parameters were obtained by fitting the observed ellipticity at varying concentrations of urea to the equation for a two-state mechanism (56). The fraction unfolded was calculated as previously described (57).
For thermal denaturation, purified proteins dialyzed against CD buffer were diluted 7.5-fold with 10 mm sodium phosphate, pH 7.5, and 150 mm NaF to a final protein concentration of 10 μm. The protein sample was heated within the CD spectropolarimeter from 25 to 95 °C using the attached water bath, whereas the CD signal at 222 nm was recorded at 1 °C intervals at 100 mdeg sensitivity and 2 s of response time. The fraction unfolded at each temperature was calculated as previously described (58).
RESULTS
Three Binding Sites on Pyrin PYD Mediate Interaction with ASC PYD
To identify the mode of interaction between pyrin and ASC, we mapped the interaction sites on both PYDs using a combination of mutagenesis and in vitro binding studies. The three-dimensional structure of pyrin PYD was calculated from the experimental NMR data using CS-ROSETTA (49). The six-helix bundle structure of pyrin PYD with a long α2-α3 loop (Fig. 1B) closely resembles previously determined PYD structures. In particular, the pyrin and ASC PYD (32) structures are very similar, with a root mean square deviation of 1.6 Å over 88 aligned Cα atoms (Fig. 1B). Surface mutations were introduced throughout pyrin PYD by mutating two to three residues per helix. However, only a single residue in helix α4 was mutated, because many residues in this short helix are buried. A total of 18 alanine point mutations (K3A, L10A, E14A, E22A, K25A, Q29A, K35A, H37A, R42A, Q46A, R49A, V58A, E63A, L71A, R75A, R80A, E84A, and R88A) were introduced into pyrin PYD (Fig. 1C).
Wild-type and mutant pyrin PYDs were expressed with a His6 tag and purified for binding studies with purified bead-bound GST-ASC PYD (Fig. 1D). Several mutants (E14A, E22A, K25A, V58A, E63A, and R75A) exhibited decreased soluble expression in E. coli compared with wild-type pyrin PYD; however, sufficient amounts were obtained for binding studies. When binding of the pyrin PYD mutants to GST-ASC PYD was tested, K25A completely abolished interaction with ASC, whereas several other mutations (L10A, E14A, R42A, L71A, R75A, and R80A) exhibited diminished interaction. Interestingly, there was also a subset of mutations, E22A, V58A, and E63A, which increased the interaction between pyrin and ASC PYDs (Fig. 1D). The residues that affected interaction with ASC PYD are distributed throughout the six helices of pyrin PYD (Fig. 2A). However, closer examination revealed that the mutations that diminished interactions are localized to three distinct clusters, consistent with the presence of a few discrete binding sites. Residues Leu10 and Glu14, which are located in helix α1, form one site, whereas residues Lys25 (helix α2) and Arg42 (helix α3) form a second site. Residues Leu71, Arg75 (helix α5), and Arg80 (helix α6) form a third site. Of the pyrin PYD residues that increased interaction with ASC when mutated, Glu22 is proximal to the Lys25 and Arg42 cluster and suggests that removal of this negatively charged residue promotes ASC interaction with the positively charged binding site 2. In contrast, residues Val58 (helix α4) and Glu63 (helix α5) are distant from the three binding sites, and thus the mechanism by which mutation of these residues promotes interaction with ASC is unclear. Interestingly, the residues on pyrin PYD that we have identified to be important for interaction localize to positively and negatively charged surface patches (Fig. 2B), consistent with charged residues playing an important role in pyrin PYD interactions.
FIGURE 2.
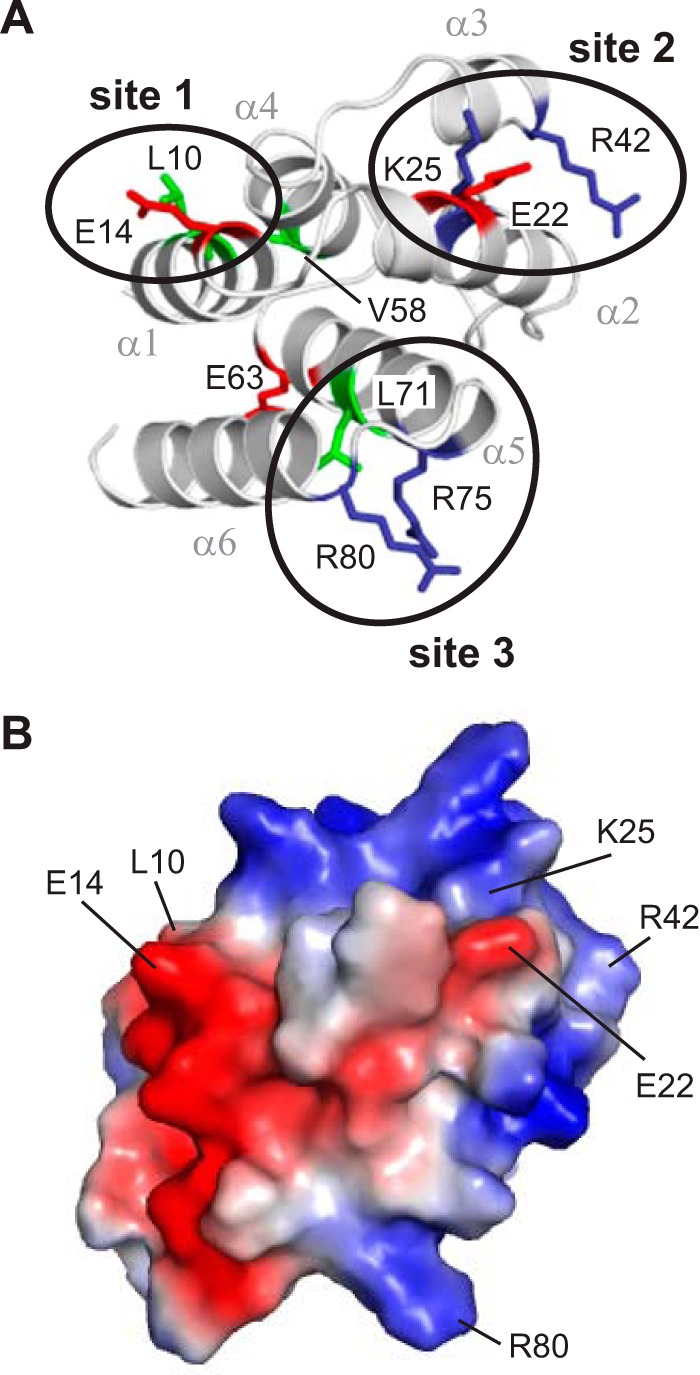
Residues important for interaction of pyrin PYD with ASC PYD localize to three clusters. A, ribbon representation of the pyrin PYD structure highlighting residues that affect interaction when mutated. Basic, acidic, and hydrophobic residues are colored blue, red, and green, respectively. Clusters of residues that form an interaction site are circled. B, surface electrostatic potential of pyrin PYD shown in the same orientation as in A and indicating residues important for interaction.
To confirm that the observed effects on binding were not due to perturbations in the pyrin PYD structure, the structural integrity of the mutants was verified using NMR spectroscopy. Assignment of the backbone resonances of wild-type pyrin PYD was accomplished using standard triple resonance experiments (46), and the two-dimensional 1H-15N HSQC spectra of 15N-labeled wild-type and mutant PYDs were compared. The majority of mutants (E14A, E22A, R42A, E63A, R75A, and R80A) displayed only minor shifts in residues that were close in sequence or space to the mutation site, indicating that the mutations do not perturb the structure (Fig. 3). In contrast, the L10A and L71A mutations induced numerous chemical shift changes throughout the protein (Fig. 4, A and B). Analysis of the chemical shift changes has been performed with caution, because without backbone resonance assignments for the L10A and L71A mutants, we cannot be certain of the identity of all peaks in their HSQC spectra. However, this analysis clearly revealed that many hydrophobic residues experienced significant chemical shift changes, suggesting that these mutants have altered helix packing (Fig. 4, C–F). The point mutations were initially designed using a homology model of pyrin PYD, but the NMR structure subsequently revealed that residues Leu10 and Leu71 contribute to the hydrophobic core of the protein. We were unable to test the effect of the K25A mutation because the protein was insoluble at the concentration required for NMR experiments and thus cannot rule out structural effects of this mutation on interactions of the pyrin PYD. Taken together, our data indicate that residues Glu14, Arg42, Arg75, and Arg80 that diminish interaction when mutated and do not perturb the structure of pyrin PYD are directly involved in interaction with ASC PYD. These findings are consistent with the presence of three distinct interaction sites on pyrin PYD.
FIGURE 3.
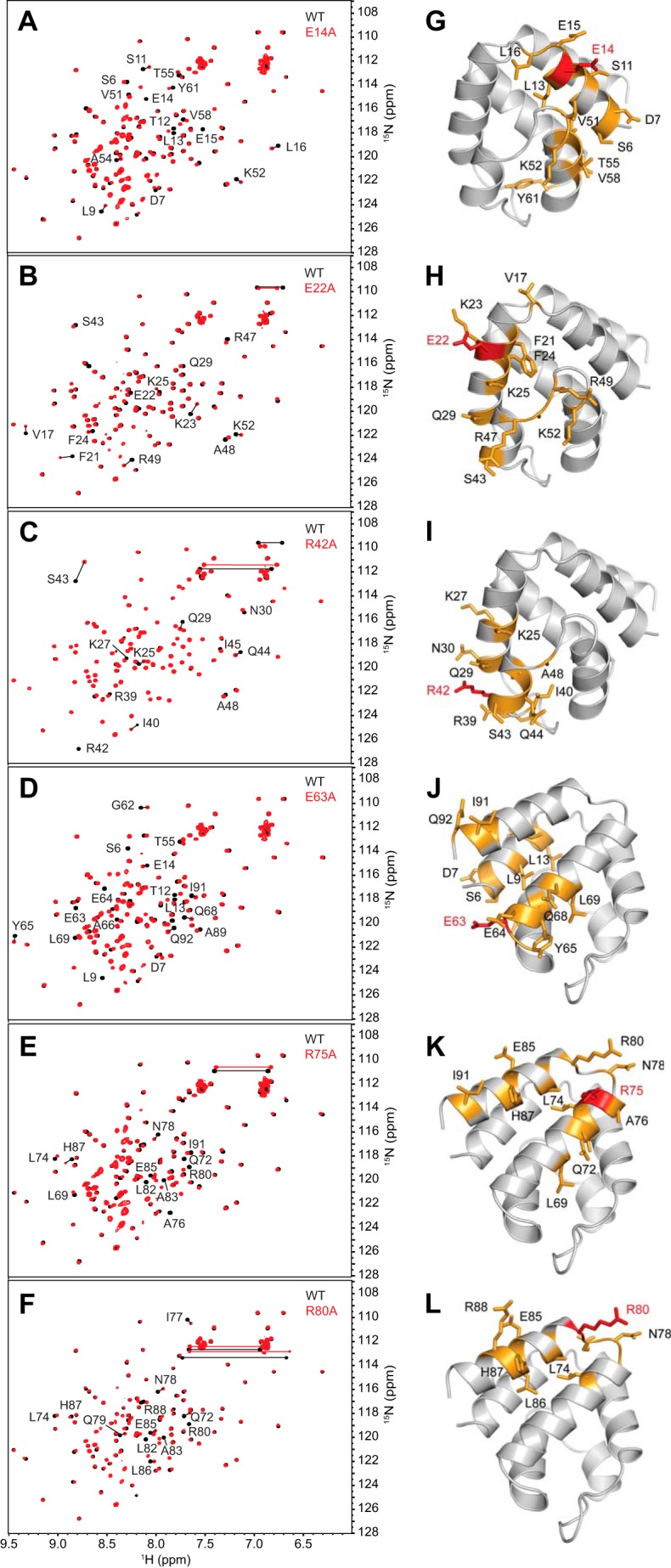
The majority of pyrin PYD mutant proteins retain structural integrity. A–F, overlay of the two-dimensional 1H-15N HSQC spectra of wild-type (black) and mutant (red) pyrin PYD proteins E14A, E22A, R42A, E63A, R75A, and R80A, respectively. All spectra were recorded in 50 mm sodium phosphate, pH 4, and 150 mm NaCl at 25 °C. Residues that show chemical shift changes are indicated with the one-letter amino acid code and sequence number. Horizontal lines connect peaks corresponding to side chain NH2 groups of Asn and Gln residues that exhibit changes in chemical shifts. G–L, ribbon diagrams of the pyrin PYD structure showing residues with their chemical shifts perturbed by the E14A, E22A, R42A, E63A, R75A, and R80A mutations, respectively. Mutated residues are colored red, and residues with chemical shift changes are colored orange, and their side chains are shown.
FIGURE 4.

Pyrin PYD mutations L10A and L71A perturb the protein structure. A and B, overlay of the two-dimensional 1H-15N HSQC spectra of wild-type (black) and mutant (red) pyrin PYD proteins L10A and L71A, respectively. All spectra were recorded in 50 mm sodium phosphate, pH 4, and 150 mm NaCl at 25 °C. Residues that show chemical shift changes are indicated with the one-letter amino acid code and sequence number. Horizontal lines connect peaks corresponding to side chain NH2 groups of Asn and Gln residues that exhibit changes in chemical shifts. C and E, histograms of the weighted backbone amide chemical shift changes (Δδav) versus residue number for the L10A and L71A mutants, respectively. The dashed line represents the mean Δδav value. D and F, ribbon diagrams of the pyrin PYD structure showing residues with chemical shifts perturbed by the L10A and L71A mutations, respectively. Mutated residues are colored red, and residues with chemical shift changes greater than the mean Δδav are colored orange, and their side chains are shown. For the L71A mutant, numerous residues in the C terminus experienced significant chemical shift changes such that the locations of these peaks in the HSQC spectrum (B) are uncertain. These residues are colored yellow.
Two Binding Sites on ASC PYD Mediate Interaction with Pyrin PYD
To identify the pyrin binding site on ASC PYD, we compared the binding of pyrin PYD to wild-type ASC PYD or a panel of 16 ASC PYD mutants, with individual point mutations that span the surface of ASC PYD (30). We found that five mutations, E13A, K21A, R41A, D48A, and D51A, abolished interaction with pyrin PYD (Fig. 5, A–C). In addition, mutations D10A, R38A, and D54A reduced binding to pyrin PYD. All of these mutations, except for R38A, do not perturb the ASC PYD structure as judged by NMR spectroscopy (30). However, the R38A mutation in the α2-α3 loop exhibited structural perturbation in the vicinity of the mutation (30). Residues Lys1 (helix α2) and Arg41 (helix α3) cluster together to form an interaction site in a positively charged patch, whereas residues Asp10, Glu13 (helix α1), Asp48 (α3-α4 loop), Asp51, and Asp54 (helix α4) cluster to form a second interaction site in a negatively charged patch (Fig. 5D).
FIGURE 5.
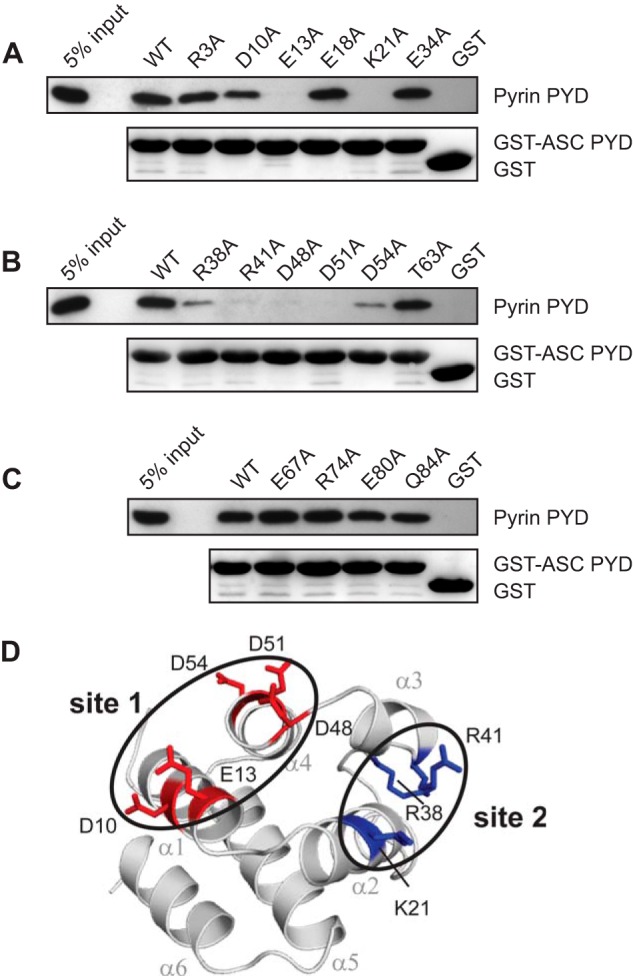
Identification of residues on ASC PYD that are important for interaction with pyrin PYD. A–C, purified bead-bound WT or mutant GST-ASC PYD or GST alone were used in binding assays with purified WT pyrin PYD. Bound protein was eluted with SDS-PAGE sample buffer, subjected to SDS-PAGE, and then transferred to a PVDF membrane. GST-ASC PYD and GST alone were detected with Ponceau S stain, whereas His6-tagged pyrin PYD was detected by immunoblotting with an anti-His antibody. An amount representing 5% of the input of WT pyrin PYD used for binding studies is shown. D, ribbon representation of ASC PYD (Protein Data Bank code 1UCP) (32) showing residues that affect binding when mutated. Residues important for interaction are colored blue (basic residues) and red (acidic residues). Residues important for interaction that cluster together are circled.
Two Interaction Sites on Pyrin PYD Are Similar to ASC PYD, but the Third Site Is Distinct
Amino acid sequence alignment of the pyrin and ASC PYDs showed that both PYDs have additional residues important for interaction that are not conserved (Fig. 6). First, although Glu13 of ASC PYD is conserved in pyrin PYD (Glu14) and is important for interaction, the proximal acidic residues Asp10, Asp48, Asp51, and Asp54 of ASC PYD (Fig. 5D) are not conserved in pyrin PYD. Nonetheless, our data are consistent with the existence of an interaction site in the vicinity of Glu14 on pyrin PYD. Second, Arg41 of ASC PYD is conserved in pyrin PYD (Arg42) and is important for interactions of both PYDs, consistent with a binding site in the vicinity of this residue. However, Lys21 of ASC PYD (helix α2), which clusters with Arg41 on the ASC PYD structure (Fig. 5D), is not conserved in pyrin PYD. There are three additional lysine residues in the vicinity of Lys21 on ASC PYD (Lys22, Lys24, and Lys26) that are conserved in pyrin PYD (Lys23, Lys25, and Lys27) (Fig. 6), and mutation of one of these residues, Lys25, disrupted interactions of pyrin PYD. Thus, the binding site on pyrin PYD that includes arg42 may include other lysine residues in helix α2. A third unique interaction site on pyrin PYD comprises basic residues Arg75 and Arg80 (Fig. 2A). Although Arg75 is conserved in ASC PYD (Arg74), mutation of this residue did not affect interaction of ASC PYD; thus this interaction site may not be critical for ASC PYD interactions. We conclude that two of the interaction sites identified on pyrin PYD are similar to the two interaction sites of ASC PYD; however, the third interaction site containing Arg75 and Arg80 is distinct. Notably, residues from all three sites are conserved to varying extents in other PYDs (Fig. 6).
FIGURE 6.
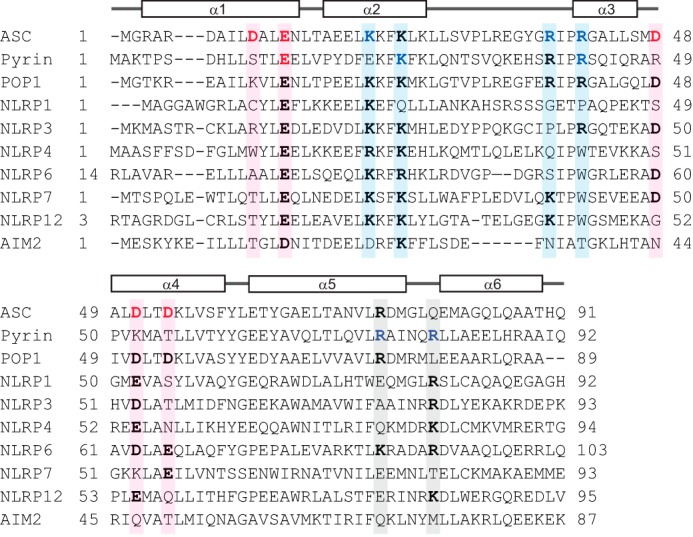
Amino acid sequence alignment of PYDs to determine conservation of residues that mediate interaction of pyrin and ASC PYDs. Sequence alignment of ASC PYD (NP_037390), pyrin PYD (AF018080), and the PYDs of other human proteins that interact with ASC including POP1 (NP_690865), NLRP1 (NP_127497.1), NLRP3 (AF468522.1), NLRP4 (NP_604393), NLRP6 (NP_612202), NLRP7 (AAI09126), NLRP12 (NP_653288), and AIM2 (AAH10940). Acidic and basic residues important for interaction of ASC PYD and pyrin PYD are indicated in red and blue, respectively. Sequence positions that contribute to binding sites 1, 2, and 3 are shaded in pink, blue, and gray, respectively. Residues corresponding to these binding sites that are conserved in other PYDs are shown in bold.
ASC PYD Can Self-associate and Interact with Pyrin
Because the residues important for interaction between ASC PYD and pyrin PYD overlap with those reported for ASC self-association (30), it was unclear whether ASC PYD self-association is a prerequisite for pyrin binding. To further investigate the role of ASC self-association in pyrin binding, we first tested the effect of pyrin PYD on ASC PYD self-association. When equimolar amounts of soluble pyrin PYD and soluble ASC PYD were incubated with GST-ASC PYD, we found that pyrin PYD inhibited ASC PYD self-association (Fig. 7A). This result suggests that the binding sites for ASC PYD self-association and pyrin PYD interaction coincide. To determine whether ASC can self-associate at the pyrin inflammasome, we tested the binding of full-length pyrin to GST-ASC PYD in the presence or absence of soluble ASC PYD (Fig. 7B). In this experiment, the interaction of full-length pyrin with GST-ASC PYD was promoted in the presence of soluble ASC PYD, indicating that ASC PYD can self-associate and interact with full-length pyrin. These data are consistent with the ability of ASC to cluster at the pyrin inflammasome (9).
FIGURE 7.
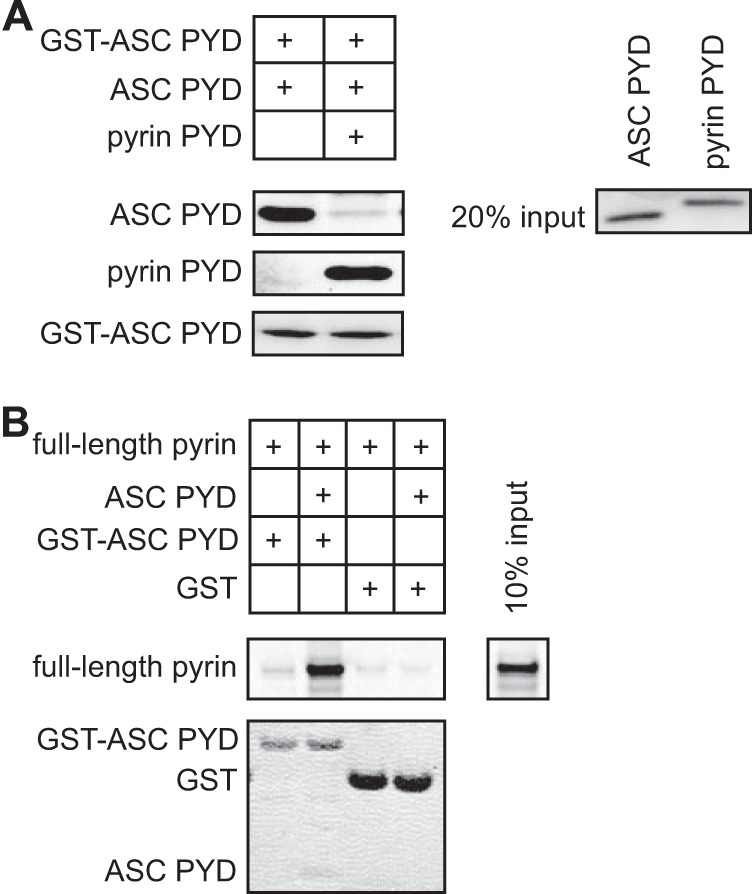
The interaction site on ASC PYD for self-association overlaps with the interaction site for pyrin PYD. A, in vitro binding of purified soluble ASC PYD to bead-bound GST-ASC PYD in the absence or presence of purified pyrin PYD. Bound ASC PYD was detected using an anti-ASC antibody, and bound pyrin PYD was detected using an anti-His antibody. GST-ASC PYD was detected using Ponceau S stain. An amount representing 20% of the input of ASC PYD and pyrin PYD detected by Coomassie stain is shown. B, binding of in vitro translated [35S]methionine-labeled pyrin to bead-bound GST-ASC PYD or GST in the absence or presence of purified soluble ASC PYD. Bound [35S]methionine-labeled pyrin was detected by phosphorimaging, whereas GST, GST-ASC PYD, and soluble ASC PYD were detected using Coomassie stain. An amount representing 10% of the input of 35S-labeled pyrin is shown.
Binding Modes of Pyrin and ASC PYDs
Models for the interaction between the pyrin and ASC PYDs were generated using each of the three binding sites identified on pyrin PYD (Fig. 2A) in the molecular docking program HADDOCK (51). Furthermore, we compared the resulting models with the three conserved interaction modes that have been identified in complexes of the structurally related death domain (DD) and CARD subfamilies (59–62). For docking studies, we surmised that clusters of oppositely charged residues on the two different PYDs would interact, and thus we defined the active sites for each of the interactions accordingly. When Glu14 (helix α1) in site 1 of pyrin PYD was defined as an active residue, and residues Lys21 (helix α2) and Arg41 (helix α3) were defined as active on ASC PYD, analysis of the resulting model (Fig. 8A) suggests an interaction that is similar to a death fold domain type I interaction. The type I interaction typically involves helices α1 and α4 of one domain interacting with helices α2 and α3 of a second domain (20). However, the model does not support a role for helix α4 of pyrin PYD. When Lys25 (helix α2) and Arg42 (helix α3) in site 2 of pyrin PYD were defined as active residues, and Asp10 and Glu13 (helix α1), Asp48 (α3-α4 loop), and Asp51 and Asp54 (helix α4) were defined as active residues on ASC PYD, the resulting model also resembles a type I interaction (Fig. 8B). These first two models of a pyrin-ASC PYD complex are similar to the ASC PYD homodimer model, which is a type I interaction (30). This is not surprising given the similarity between the interaction sites. When Arg75 (helix α5) and Arg80 (helix α6) in site 3 of pyrin PYD were defined as active residues, and Asp10, Glu13, Asp48, Asp51, and Asp54 were again defined as active residues on ASC PYD, the resulting model (Fig. 8C) suggests that it is similar to a type II interaction that involves helices α5 and α6 of one domain interacting with helix α4 and the α4-α5 loop of another domain. Notably, the spatial arrangement of the three interaction sites on pyrin PYD indicates that it can engage in all three modes of interaction with ASC simultaneously without any steric hindrance (Fig. 8D). These data indicate that each pyrin PYD can recruit multiple ASC molecules to the inflammasome and subsequently drive ASC clustering and activation of procaspase-1 and/or procaspase-8.
FIGURE 8.
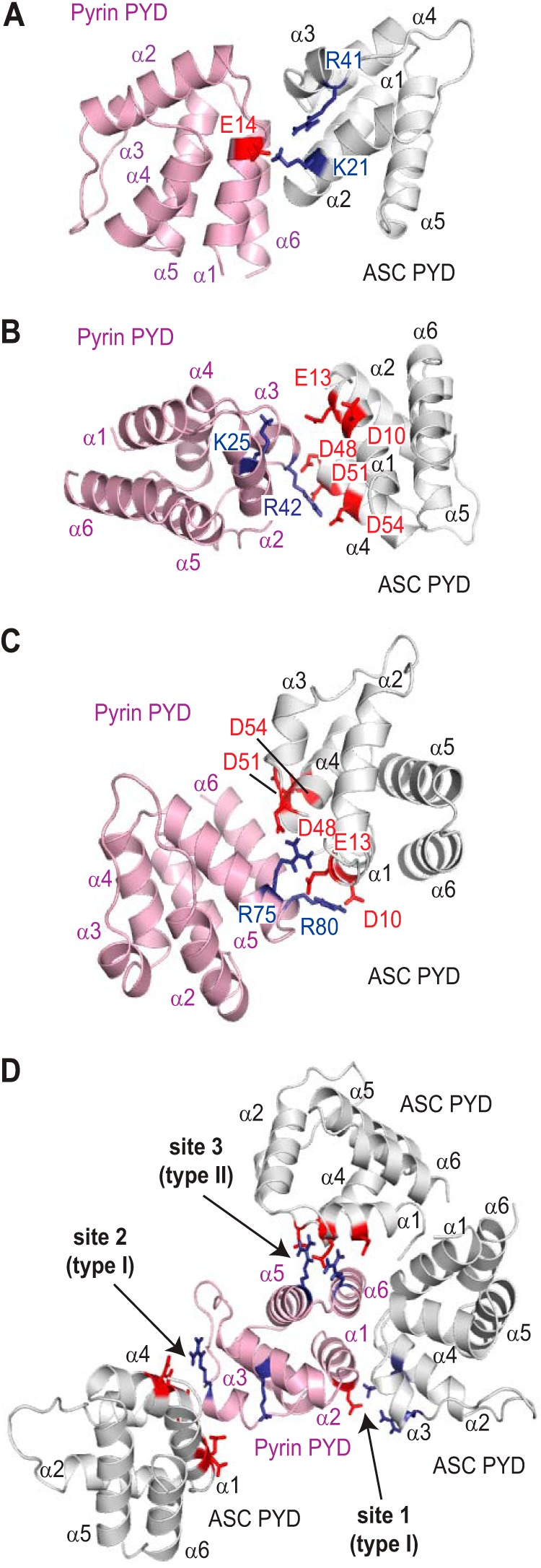
Binding modes of pyrin PYD with ASC PYD. A–C, models of ASC-pyrin PYD complexes generated using HADDOCK. In A, the active residue defined on pyrin PYD was site 1 (Glu14), and the active residues defined on ASC PYD were Lys21 and Arg41. In B, the active residues defined on pyrin PYD were site 2 (Lys25 and Arg42), and the active residues defined on ASC PYD were Asp10, Glu13, Asp48, Asp51, and Asp54. In C, the active residues defined on pyrin PYD were site 3 (Arg75 and Arg80), and the active residues defined on ASC PYD were Asp10, Glu13, Asp48, Asp51, and Asp54. D, the pyrin PYDs in the different models shown in A–C were superimposed to visualize how the three interaction sites on pyrin PYD may interact with multiple ASC PYDs. In all models, acidic residues are indicated in red, and basic residues are indicated in blue.
FMF-associated Pyrin PYD Mutants Retain Interaction with ASC
To gain further insights into assembly and function of the pyrin inflammasome, we tested the effect of the FMF-associated mutations R42W and A89T on pyrin PYD interactions. We first examined the effect of the mutations on interaction of the pyrin PYD in isolation. The R42W mutant had a decreased interaction with ASC PYD, whereas interaction of the A89T mutant was comparable with that of wild-type pyrin PYD (Fig. 9A). Similar results were obtained when interaction with full-length ASC was tested (Fig. 9B). Interestingly, the R42W mutation had a milder effect on ASC binding than R42A (Figs. 1D and 9C).
FIGURE 9.

FMF-associated mutations in pyrin PYD do not abrogate interaction with ASC. A and B, effect of FMF-associated mutations, R42W and A89T, on interaction of pyrin PYD with ASC PYD (A) or full-length ASC (B). C, comparative effect of the R42A and R42W pyrin PYD mutations on interaction with ASC PYD. In A–C, purified His6-tagged WT and mutant pyrin PYDs were used for in vitro binding assays with bead-bound GST-ASC PYD, GST-ASC (full-length) or GST alone. Bound protein was eluted with SDS-PAGE sample buffer, subjected to SDS-PAGE, and then transferred to a PVDF membrane. GST-ASC PYD, GST-ASC (full-length), and GST alone were detected with Ponceau S stain, whereas His6-tagged WT and mutant pyrin PYDs were detected by immunoblotting with an anti-His antibody. An amount representing 5% (A and B) or 10% (C) of the input of WT or mutant pyrin PYD used for binding studies is shown. D, effect of FMF-associated mutations on interaction between pyrin PYD and B-box domain. A pyrin construct lacking the N-terminal PYD (pyrinΔPYD) was in vitro translated in the presence of [35S]methionine and incubated with bead-bound WT or mutant pyrin PYD fused to GST or with GST alone. Bound proteins were resolved on an SDS-PAGE gel. GST, GST-ASC PYD, and GST-ASC were detected with Coomassie stain, whereas [35S]methionine-labeled pyrinΔPYD was detected by phosphorimaging. An amount representing 10% of the input of 35S-labeled pyrinΔPYD is also shown. E and F, effect of FMF-associated mutations on interaction of full-length pyrin with ASC. Plasmids expressing Myc-tagged WT or mutant pyrin or empty vector (V) were co-transfected with a plasmid expressing ASC into HEK 293T cells. Pyrin was immunoprecipitated (IP) with an anti-Myc antibody 24 h after transfection, and the immunoprecipitated complexes were analyzed by Western blotting. In E, a plasmid expressing FLAG-tagged PSTPIP1 was also co-transfected. Blots were probed with antibodies to ASC and pyrin and with an anti-FLAG antibody to detect PSTPIP1.
To examine the effect of the FMF mutations on the intramolecular interaction between pyrin PYD and the B-box domain, we generated an N-terminal truncation of pyrin lacking the PYD (pyrinΔPYD). This construct has previously been used to demonstrate interaction of pyrin PYD with the B-box (22). The R42W mutation also diminished the interaction between PYD and pyrinΔPYD (Fig. 9D). In contrast, the A89T mutation slightly enhanced this interaction.
Although the FMF-associated mutations had an effect on interactions of the pyrin PYD in isolation, it was not clear what effect these mutations would have in the context of full-length pyrin. This is critical to understanding how these mutations may lead to increased inflammation. The ability of full-length pyrin to interact with ASC in vivo is governed by the relative affinities of the intramolecular interaction between the PYD and B-box of pyrin, and the intermolecular interaction with ASC. To investigate the effect of FMF-associated mutations on interaction of full-length pyrin with ASC, HEK 293T cells were transiently transfected with wild-type or mutant pyrin and ASC. HEK 293T cells do not normally express pyrin, ASC, or PSTPIP1, which make them an ideal system to reconstitute these complexes (9, 22, 63). As shown in Fig. 9 (E and F), binding of ASC to the R42W and A89T pyrin mutants is similar to that of wild-type, in the presence or absence of PSTPIP1. Thus, when expressed in transfected cells, the FMF-associated mutations do not appear to affect pyrin interaction with ASC.
Biophysical Properties of Pyrin PYD and Effects of FMF-associated Mutations
Amino acid sequence alignment indicated that the residue corresponding to Arg42 of pyrin PYD is a tryptophan in several PYDs including NLRP4, NLRP6, NLRP7, NLRP12, and NLRP14 (Fig. 6). Furthermore, it has been postulated that the FMF-associated mutation, R42W, in pyrin PYD alters the helical propensity of helix α3 and stabilizes it via hydrophobic interactions (34, 41). This suggests that there may be a structural advantage to having a tryptophan at this position in the short helix α3. Moreover, although the FMF-associated R42W mutation reduced interaction with ASC and with the pyrin B-box domain, it was not clear whether this was due to structural effects. Therefore we tested the effect of the two mutations, R42W and A89T, on the structural and biophysical properties of pyrin PYD.
To examine the effects on structure, purified wild-type and mutant pyrin PYDs were initially analyzed using CD spectroscopy (Fig. 10A). As expected, the far-UV CD spectrum of pyrin PYD exhibits characteristic features of an α-helical protein, with a double minimum at 208 and 222 nm and a strong positive peak at 195 nm. Analysis of the CD spectrum using DichroWeb (55) predicted 86% α-helix, consistent with the high helical content of the pyrin PYD structure. The far-UV CD spectrum of the A89T mutant was identical to that of the wild-type pyrin PYD, indicating that the A89T mutation does not alter the secondary structure of the protein. A subtle but distinct difference in the CD spectrum of the R42W mutant was a change in the ratio of the 208/222 minima, with the 222 minimum becoming more shallow. Although tryptophan residues can make a significant contribution to the CD of proteins in the far-UV, they tend to increase absorption at 222 nm (64, 65), which is contrary to the observed effect. Thus, the change in ratio may be due to an alteration in helix packing, which is reported to reduce the 222/208 ratio (66).
FIGURE 10.

Effects of FMF-associated mutations on the structure and stability of pyrin PYD. A, far-UV CD spectra of WT (gray line), A89T mutant (dotted black line), and R42W mutant (solid black line) pyrin PYDs. B and C, purified WT and mutant pyrin PYDs were subjected to chemical denaturation with urea (B) or thermal denaturation (C). Filled circles, WT; filled triangles, A89T; filled squares, R42W; open diamonds, R42A. In B, the data were fitted to a two-state unfolding model (56), and the fraction of unfolded protein was plotted as a function of urea concentration. D and E, overlay of the two-dimensional 1H-15N HSQC spectra of wild-type (black) and mutant (red) pyrin PYD proteins A89T and R42W, respectively. All spectra were recorded in 50 mm sodium phosphate, pH 4, and 150 mm NaCl at 25 °C. Residues that show chemical shift changes are indicated with the one-letter amino acid code and sequence number. Horizontal lines connect peaks corresponding to side chain NH2 groups of Asn and Gln residues that exhibit changes in chemical shifts. In E, the peaks corresponding to the Trp42 backbone NH and side chain NH (W42ϵ) are labeled. F and H, histograms of the weighted backbone amide chemical shift changes (Δδav) versus residue number for the A89T and R42W pyrin PYD mutants, respectively. The dashed line indicates one standard deviation higher than the mean Δδav value (0.04 and 0.14 ppm, respectively). G and I, ribbon diagrams of the pyrin PYD structure showing residues with chemical shifts perturbed by the A89T and R42W mutations, respectively. Mutated residues are colored red, and residues with chemical shift changes greater than one standard deviation above the mean Δδav are colored orange, and their side chains are shown.
As a second approach to assess the effect of the R42W and A89T mutations on structural integrity, we measured the relative stability of the wild-type and mutant pyrin PYDs by monitoring unfolding in response to chemical and thermal denaturation (Fig. 10, B and C). Both the chemical and thermal denaturation data indicate that pyrin PYD undergoes a two-state unfolding transition. Surprisingly, the R42W mutation increased stability of the pyrin PYD to both chemical and thermal denaturation. The ΔGunfolding of the R42W mutant (21.1 kJ mol−1) is almost twice that of wild-type pyrin PYD (12.4 kJ mol−1). In contrast, the stability of the A89T mutant was slightly decreased (ΔGunfolding = 11.4 kJ mol−1) compared with wild-type pyrin PYD. Consistent with the observations from chemical denaturation, the thermal unfolding midpoint (Tm) of the R42W mutant was increased to 62.5 °C compared with wild-type pyrin PYD (Tm = 57 °C), whereas that of the A89T mutant was decreased to 53 °C (Fig. 10C).
To distinguish between the two effects of the R42W mutation, which are loss of the positively charged arginine side chain and introduction of a bulky hydrophobic tryptophan residue, we also determined the stability of the R42A mutant. Unfolding in response to chemical and thermal denaturation showed that the R42A mutant was only slightly more stable than wild-type pyrin PYD with ΔGunfolding of 15.0 kJ mol−1 and Tm of 59.5 °C (Fig. 10, B and C).
Finally, to assess the extent of conformational perturbations associated with the FMF mutations in greater detail, wild-type and mutant pyrin PYDs were compared using NMR spectroscopy (Fig. 10, D and E). Consistent with the CD results, the two-dimensional 1H-15N HSQC spectrum of the A89T mutant showed only a few altered resonances localized to adjacent residues, confirming that there were no major structural changes (Fig. 10, F and G). In contrast, the R42W mutant exhibited more extensive spectral perturbations, affecting not only adjacent residues in helix α3 but also affecting residues in helices α1 and α2, some of which are far removed in the sequence and structure (Fig. 10, H and I). Although the R42W mutation has a greater effect on structure than the R42A mutation, it stabilizes the pyrin PYD and retains interaction with ASC PYD (Fig. 9C). Stabilization of pyrin PYD by the presence of a tryptophan residue at position 42 is likely to be due to creation of additional hydrophobic contacts in the core of the structure. This may induce slight changes in helix packing and also stabilize the α2-α3 loop, which would be consistent with the changes seen in the NMR spectrum of R42W and the change in the 208/222 ratio in the CD.
DISCUSSION
Inflammasomes are macromolecular complexes that play a key role in mediating inflammatory or apoptotic responses to foreign or cellular danger signals (3, 10). Dysregulated inflammasome activation is associated with the progression of many common diseases (67) in addition to several inherited autoinflammatory diseases (3). Consequently, inflammasome components are targets for the development of therapeutic agents (68). However, the molecular details of inflammasome complexes are poorly understood. Inflammasome assembly is mediated by PYD and CARD domains, which are members of the death fold superfamily (19, 20). The adaptor protein ASC, which consists of a PYD and a CARD domain, plays a pivotal role in inflammasome assembly. One of the early events in inflammasome assembly is the recruitment of ASC via a homotypic PYD interaction to activated oligomerized pattern recognition receptors (10). To gain insights into recruitment of ASC to the pyrin inflammasome and to identify the binding modes of PYDs, we have examined the interaction between ASC PYD and pyrin PYD. Our data reveal a complex scenario whereby three binding sites on pyrin PYD and two binding sites on ASC PYD are important for interaction. These findings are consistent with assembly of an oligomeric complex stabilized by multiple interactions between PYDs.
Identification of the binding sites on pyrin and ASC PYDs was achieved using mutagenesis and in vitro binding studies. A direct role for residues implicated in binding was further validated by NMR spectroscopy to demonstrate that the mutations did not perturb protein structure. The residues important for interaction of pyrin PYD localized to three distinct sites, which we refer to as site 1 (including residue Glu14), site 2 (including residues Lys25 and Arg42), and site 3 (including residues Arg75 and Arg80). The residues on ASC PYD that are important for interaction with pyrin PYD overlap with residues that were previously implicated in ASC PYD self-association and interaction with NLRP3 and POP1 (30). These residues include Asp10, Glu13, Asp48, Asp51, and Asp54, which localize to an acidic surface patch, and residues Lys21, Arg38, and Arg41, which localize to a basic patch on ASC PYD. Interestingly, despite the sequence diversity between ASC and pyrin PYDs, a residue from each of the two interaction sites on ASC PYD, Glu13 and Arg41, is conserved in pyrin PYD (Glu14 and Arg42, respectively), and both residues are important for ASC binding. This is consistent with conservation of these two interaction sites in ASC and pyrin PYDs. Although a single residue of the third binding site in pyrin PYD (comprising residues Arg75 and Arg80) is conserved in ASC PYD (Arg74), this residue is not critical for any ASC PYD interactions tested (this study and Ref. 30).
A short helix α3 is a characteristic feature of PYD structures, and this region also appears to be functionally important. In particular, Arg42 of pyrin and Arg41 of ASC were shown to be important for their interaction (30). Our studies of the FMF-associated R42W mutation in pyrin indicated that Arg42 could be substituted by a tryptophan residue without disrupting ASC interaction and also showed that this mutation stabilizes the pyrin PYD. Although the R42W mutation decreased the interaction between ASC and pyrin PYD in isolation, oligomeric full-length pyrin can overcome the effect of this single mutation on interaction with ASC. This observation may also explain how the arginine to tryptophan substitution in this position is tolerated in other PYDs including NLRP6 and NLRP7, which are known to bind ASC and assemble into higher order oligomers (10). The corresponding tryptophan residue in helix α3 of NLRP7 (Trp43) has been shown to mediate hydrophobic contacts with helix α2, thus stabilizing helix α3 and the α2-α3 loop (37). Similarly, the R42W mutation in pyrin PYD is likely to increase stability via increased hydrophobic contacts between helices α3 and α2. This would account for the change in helix packing suggested by CD spectroscopy and further evidenced by NMR data showing chemical shift changes for residues in helices α1 and α2, as well as helix α3. As a result of an increased hydrophobic core, the stability of the R42W mutant to chemical and thermal denaturation was significantly increased. We infer that the presence of an arginine residue at the corresponding position in ASC PYD similarly allows increased dynamics of helix α3 and the α2-α3 loop and promotes interactions of ASC PYD. Interestingly, the R42W mutation reduced interaction of pyrin PYD with the B-box domain, suggesting that it may contribute to FMF by facilitating activation of the pyrin inflammasome.
Our molecular docking studies showing that the three binding sites on pyrin PYD and the two binding sites on ASC PYD can be engaged simultaneously without any steric hindrance (this study and Ref. 30) are consistent with the multifaceted binding modes that have been identified in the death fold superfamily (59–62). The death fold superfamily comprises the DD and death effector domain subfamilies in addition to the PYD and CARD. All members of this superfamily play a key role in assembly of apoptotic or inflammatory signaling complexes, to which effector proteins such as caspases or kinases are recruited to allow proximity-induced activation (19). The role of DDs is best characterized as the structures of several oligomeric complexes have been determined including the PIDDosome (where PIDD is a p53-induced protein with a death domain) (59); Myddosome (60); and Fas-FADD (Fas-associated death domain protein) complex (61). These structures revealed a multilayered helical arrangement of DDs such that each DD can engage in up to six interactions simultaneously. However, only three types of interactions have been observed in the complexes determined to date (59, 69). In this study, the binding modes of pyrin PYD and ASC PYD were elucidated based on molecular docking using the interaction sites identified on both PYDs. Sites 1 and 2 of pyrin PYD are predicted to mediate type I interactions, whereas site 3 can engage in a type II interaction. These data are consistent with conservation of the type I and type II interactions in the PYD subfamily.
Notably, although our approach to identifying binding sites has been unbiased, the spatial distribution of the three interaction sites on pyrin PYD (Figs. 8D and 11A) is similar to the distribution of corresponding type I and type II sites on a central RAIDD (RIP-associated ICH-1 homologous protein with a death domain) DD in the PIDDosome complex (Fig. 11B). Additional type II and type III interaction sites are present on RAIDD DD, raising the possibility that pyrin PYD could accommodate additional interactions. Furthermore, adjacent ASC PYDs that are bound to pyrin (such as ASC PYD2 and PYD3 shown in Fig. 11A) may be able to associate via a type III interaction as observed for the corresponding adjacent DDs in the PIDDosome (Fig. 11B). The importance of multiple binding sites for the interaction between ASC and pyrin PYDs indicates assembly of a complex higher order oligomer that is stabilized by multiple interactions. We can rationalize models for ASC recruitment to the pyrin inflammasome (Fig. 11, C–E) that take into account the three key findings from this study: (i) each pyrin PYD has three binding sites for ASC PYD; (ii) both ASC PYD binding sites are required for stable interaction with pyrin PYD; and (iii) ASC PYD can self-associate and interact with full-length pyrin. These data suggest that three ASC PYDs can be recruited to each PYD of a pyrin trimer. Two ASC PYDs that are bound to pyrin by type I interactions will have a second available binding site, allowing them to self-associate via another type I interaction when they are in close proximity (Fig. 11, C and D). The use of multiple binding sites on ASC PYD is likely to drive stable association and the ability of ASC to self-associate, whereas interacting with pyrin is critical for caspase-1 activation (9). The binding of self-associated ASC PYDs to adjacent pyrin PYDs of a trimer also explains the requirement for oligomerization of pattern recognition receptors prior to ASC recruitment (10). Furthermore, additional type II interactions between ASC and pyrin PYDs (Fig. 11E) could drive the clustering of multiple ASC-pyrin trimer complexes to form a single large inflammasome complex that is visible by microscopy (9). Self-association of the CARD domain of ASC (not illustrated) is also likely to drive compaction of the inflammasome into a speck, because multiple surfaces on ASC CARD have recently been shown to be important for the formation of ASC foci (31).
FIGURE 11.

Multiple interaction sites on ASC and pyrin PYDs can mediate assembly of an inflammasome complex. A, model of pyrin PYD interacting with three ASC PYDs via the interactions identified in this study. B, section of the PIDDosome complex comprised of DDs from PIDD and RAIDD. The numbering of RAIDD and PIDD DDs is as previously described (59). In A and B, the locations of type I, II, and III interactions are shown, and the helices and loops that mediate some of the interactions are indicated. C, model of ASC PYDs bound to a pyrin trimer. For simplicity, only the PYDs of pyrin are shown and are connected with a pink dashed line. Interaction of each pyrin PYD with ASC PYDs is as shown in A, and double headed orange arrows indicate ASC PYD self-association in the complex. ASC and pyrin PYDs are marked A and P, respectively. D, linear representation of the complex shown in C to further illustrate ASC self-association. E, the same complex shown in D, but illustrating how the terminal ASC PYD could interact with a pyrin PYD via a type II interaction (double headed pink arrow) to form a spiral rather than a circle.
In summary, this study gives new insights into the architecture of the inflammasome. Conservation of interaction modes and similarities in the spatial distribution of binding sites between PYDs and DDs suggest parallels between the assembly of PYD and DD complexes. Furthermore, our data reveal how ASC can be recruited to the inflammasome and allowed to cluster, which is pivotal for activation of procaspase-1 and procaspase-8. Consideration of these findings and the proposed models will allow strategic planning to produce larger complexes of PYDs for future structural studies that are needed to fully understand the mechanism of inflammasome assembly.
Acknowledgments
We thank Naohiro Inohara for the pcDNA-ASC expression plasmid, Deborah Gumucio for the pcDNA-Pyrin expression plasmid, Ruth Mirams for acquisition of selected NMR data, and the Queensland NMR Network for access to excellent NMR facilities.
This work was supported by the Australian National Health and Medical Research Council through Project Grant 511227 and R. D. Wright Biomedical Career Development Award 401748 (to J. M. H.).
The atomic coordinates and structure factors (code 2MPC) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- FMF
- familial Mediterranean fever
- CARD
- caspase recruitment domain
- DD
- death domain
- HSQC
- heteronuclear single-quantum coherence
- NLR
- NOD-like receptor
- PYD
- pyrin domain
- ASC
- apoptosis-associated speck-like protein containing a CARD.
REFERENCES
- 1. Almeida de Jesus A., Goldbach-Mansky R. (2013) Monogenic autoinflammatory diseases: concept and clinical manifestations. Clin. Immunol. 147, 155–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Park H., Bourla A. B., Kastner D. L., Colbert R. A., Siegel R. M. (2012) Lighting the fires within: the cell biology of autoinflammatory diseases. Nat. Rev. Immunol. 12, 570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lamkanfi M., Dixit V. M. (2012) Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 28, 137–161 [DOI] [PubMed] [Google Scholar]
- 4. Federici S., Martini A., Gattorno M. (2013) The central role of anti-IL-1 blockade in the treatment of monogenic and multi-factorial autoinflammatory diseases. Front. Immunol. 4, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dinarello C. A. (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. The International FMF Consortium (1997) Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 90, 797–807 [DOI] [PubMed] [Google Scholar]
- 7. French FMF Consortium (1997) A candidate gene for familial Mediterranean fever. Nat. Genet. 17, 25–31 [DOI] [PubMed] [Google Scholar]
- 8. Richards N., Schaner P., Diaz A., Stuckey J., Shelden E., Wadhwa A., Gumucio D. L. (2001) Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J. Biol. Chem. 276, 39320–39329 [DOI] [PubMed] [Google Scholar]
- 9. Yu J. W., Wu J., Zhang Z., Datta P., Ibrahimi I., Taniguchi S., Sagara J., Fernandes-Alnemri T., Alnemri E. S. (2006) Cryopyrin and pyrin activate caspase-1, but not NF-κB, via ASC oligomerization. Cell Death Differ. 13, 236–249 [DOI] [PubMed] [Google Scholar]
- 10. Latz E., Xiao T. S., Stutz A. (2013) Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 13, 397–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gavrilin M. A., Mitra S., Seshadri S., Nateri J., Berhe F., Hall M. W., Wewers M. D. (2009) Pyrin critical to macrophage IL-1β response to Francisella challenge. J. Immunol. 182, 7982–7989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gavrilin M. A., Abdelaziz D. H., Mostafa M., Abdulrahman B. A., Grandhi J., Akhter A., Abu Khweek A., Aubert D. F., Valvano M. A., Wewers M. D., Amer A. O. (2012) Activation of the pyrin inflammasome by intracellular Burkholderia cenocepacia. J. Immunol. 188, 3469–3477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yu J. W., Farias A., Hwang I., Fernandes-Alnemri T., Alnemri E. S. (2013) Ribotoxic stress through p38 mitogen-activated protein kinase activates in vitro the human pyrin inflammasome. J. Biol. Chem. 288, 11378–11383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chae J. J., Komarow H. D., Cheng J., Wood G., Raben N., Liu P. P., Kastner D. L. (2003) Targeted disruption of pyrin, the FMF protein, causes heightened sensitivity to endotoxin and a defect in macrophage apoptosis. Mol. Cell 11, 591–604 [DOI] [PubMed] [Google Scholar]
- 15. Chae J. J., Wood G., Masters S. L., Richard K., Park G., Smith B. J., Kastner D. L. (2006) The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1β production. Proc. Natl. Acad. Sci. U.S.A. 103, 9982–9987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hesker P. R., Nguyen M., Kovarova M., Ting J. P., Koller B. H. (2012) Genetic loss of murine pyrin, the Familial Mediterranean Fever protein, increases interleukin-1β levels. PLoS One 7, e51105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Centola M., Wood G., Frucht D. M., Galon J., Aringer M., Farrell C., Kingma D. W., Horwitz M. E., Mansfield E., Holland S. M., O'Shea J. J., Rosenberg H. F., Malech H. L., Kastner D. L. (2000) The gene for familial Mediterranean fever, MEFV, is expressed in early leukocyte development and is regulated in response to inflammatory mediators. Blood 95, 3223–3231 [PubMed] [Google Scholar]
- 18. Tidow N., Chen X., Müller C., Kawano S., Gombart A. F., Fischel-Ghodsian N., Koeffler H. P. (2000) Hematopoietic-specific expression of MEFV, the gene mutated in familial Mediterranean fever, and subcellular localization of its corresponding protein, pyrin. Blood 95, 1451–1455 [PubMed] [Google Scholar]
- 19. Park H. H., Lo Y. C., Lin S. C., Wang L., Yang J. K., Wu H. (2007) The death domain superfamily in intracellular signaling of apoptosis and inflammation. Annu. Rev. Immunol. 25, 561–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kersse K., Verspurten J., Vanden Berghe T., Vandenabeele P. (2011) The death-fold superfamily of homotypic interaction motifs. Trends Biochem. Sci. 36, 541–552 [DOI] [PubMed] [Google Scholar]
- 21. Pawlowski K., Pio F., Chu Z., Reed J. C., Godzik A. (2001) PAAD: a new protein domain associated with apoptosis, cancer and autoimmune diseases. Trends Biochem. Sci. 26, 85–87 [DOI] [PubMed] [Google Scholar]
- 22. Yu J. W., Fernandes-Alnemri T., Datta P., Wu J., Juliana C., Solorzano L., McCormick M., Zhang Z., Alnemri E. S. (2007) Pyrin activates the ASC pyroptosome in response to engagement by autoinflammatory PSTPIP1 mutants. Mol. Cell 28, 214–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wise C. A., Gillum J. D., Seidman C. E., Lindor N. M., Veile R., Bashiardes S., Lovett M. (2002) Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum. Mol. Genet. 11, 961–969 [DOI] [PubMed] [Google Scholar]
- 24. Touitou I. (2001) The spectrum of Familial Mediterranean Fever (FMF) mutations. Eur. J. Hum. Genet. 9, 473–483 [DOI] [PubMed] [Google Scholar]
- 25. Ben-Chetrit E., Touitou I. (2009) Familial Mediterranean fever in the world. Arthritis Rheum. 61, 1447–1453 [DOI] [PubMed] [Google Scholar]
- 26. Masumoto J., Taniguchi S., Ayukawa K., Sarvotham H., Kishino T., Niikawa N., Hidaka E., Katsuyama T., Higuchi T., Sagara J. (1999) ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 274, 33835–33838 [DOI] [PubMed] [Google Scholar]
- 27. Pierini R., Juruj C., Perret M., Jones C. L., Mangeot P., Weiss D. S., Henry T. (2012) AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 19, 1709–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sagulenko V., Thygesen S. J., Sester D. P., Idris A., Cridland J. A., Vajjhala P. R., Roberts T. L., Schroder K., Vince J. E., Hill J. M., Silke J., Stacey K. J. (2013) AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 20, 1149–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Masumoto J., Taniguchi S., Sagara J. (2001) Pyrin N-terminal homology domain- and caspase recruitment domain-dependent oligomerization of ASC. Biochem. Biophys. Res. Commun. 280, 652–655 [DOI] [PubMed] [Google Scholar]
- 30. Vajjhala P. R., Mirams R. E., Hill J. M. (2012) Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 287, 41732–41743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Proell M., Gerlic M., Mace P. D., Reed J. C., Riedl S. J. (2013) The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 449, 613–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liepinsh E., Barbals R., Dahl E., Sharipo A., Staub E., Otting G. (2003) The death-domain fold of the ASC PYRIN domain, presenting a basis for PYRIN/PYRIN recognition. J. Mol. Biol. 332, 1155–1163 [DOI] [PubMed] [Google Scholar]
- 33. Natarajan A., Ghose R., Hill J. M. (2006) Structure and dynamics of ASC2, a pyrin domain-only protein that regulates inflammatory signaling. J. Biol. Chem. 281, 31863–31875 [DOI] [PubMed] [Google Scholar]
- 34. Hiller S., Kohl A., Fiorito F., Herrmann T., Wider G., Tschopp J., Grütter M. G., Wüthrich K. (2003) NMR structure of the apoptosis- and inflammation-related NALP1 pyrin domain. Structure 11, 1199–1205 [DOI] [PubMed] [Google Scholar]
- 35. Bae J. Y., Park H. H. (2011) Crystal structure of NALP3 protein pyrin domain (PYD) and its implications in inflammasome assembly. J. Biol. Chem. 286, 39528–39536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eibl C., Grigoriu S., Hessenberger M., Wenger J., Puehringer S., Pinheiro A. S., Wagner R. N., Proell M., Reed J. C., Page R., Diederichs K., Peti W. (2012) Structural and functional analysis of the NLRP4 pyrin domain. Biochemistry 51, 7330–7341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pinheiro A. S., Proell M., Eibl C., Page R., Schwarzenbacher R., Peti W. (2010) Three-dimensional structure of the NLRP7 pyrin domain: insight into pyrin-pyrin-mediated effector domain signaling in innate immunity. J. Biol. Chem. 285, 27402–27410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pinheiro A. S., Eibl C., Ekman-Vural Z., Schwarzenbacher R., Peti W. (2011) The NLRP12 pyrin domain: structure, dynamics, and functional insights. J. Mol. Biol. 413, 790–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Su M. Y., Kuo C. I., Chang C. F., Chang C. I. (2013) Three-dimensional structure of human NLRP10/PYNOD pyrin domain reveals a homotypic interaction site distinct from its mouse homologue. PLoS One 8, e67843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jin T., Perry A., Smith P., Jiang J., Xiao T. S. (2013) Structure of the absent in melanoma 2 (AIM2) pyrin domain provides insights into the mechanisms of AIM2 autoinhibition and inflammasome assembly. J. Biol. Chem. 288, 13225–13235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Eliezer D. (2003) Folding pyrin into the family. Structure 11, 1190–1191 [DOI] [PubMed] [Google Scholar]
- 42. Hill J. M. (2008) NMR screening for rapid protein characterization in structural proteomics. Methods Mol. Biol. 426, 437–446 [DOI] [PubMed] [Google Scholar]
- 43. Delaglio F., Grzesiek S., Vuister G. W., Zhu G., Pfeifer J., Bax A. (1995) NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 [DOI] [PubMed] [Google Scholar]
- 44. Garrett D. S., Powers R., Gronenborn A. M., Clore G. M. (1991) A common sense approach to peak picking in two-, three-, and four-dimensional spectra using automatic computer analysis of contour diagrams. J. Magn. Reson. 95, 214–220 [DOI] [PubMed] [Google Scholar]
- 45. Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D. (2005) The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins 59, 687–696 [DOI] [PubMed] [Google Scholar]
- 46. Sattler M., Schleucher J., Griesinger C. (1999) Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog. NMR Spectrosc. 34, 93–158 [Google Scholar]
- 47. Pellecchia M., Sebbel P., Hermanns U., Wüthrich K., Glockshuber R. (1999) Pilus chaperone FimC-adhesin FimH interactions mapped by TROSY-NMR. Nat. Struct. Biol. 6, 336–339 [DOI] [PubMed] [Google Scholar]
- 48. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+: a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shen Y., Lange O., Delaglio F., Rossi P., Aramini J. M., Liu G., Eletsky A., Wu Y., Singarapu K. K., Lemak A., Ignatchenko A., Arrowsmith C. H., Szyperski T., Montelione G. T., Baker D., Bax A. (2008) Consistent blind protein structure generation from NMR chemical shift data. Proc. Natl. Acad. Sci. U.S.A. 105, 4685–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bhattacharya A., Tejero R., Montelione G. T. (2007) Evaluating protein structures determined by structural genomics consortia. Proteins 66, 778–795 [DOI] [PubMed] [Google Scholar]
- 51. de Vries S. J., van Dijk M., Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 52. Gill S. C., von Hippel P. H. (1989) Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 53. Schmid F. X. (1997) Optical spectroscopy to characterize protein conformation and conformational changes. In Protein Structure: A Practical Approach (Creighton T. E., ed) 2nd Ed., pp. 293–297, Oxford University Press, Oxford [Google Scholar]
- 54. Johnson W. C. (1999) Analyzing protein circular dichroism spectra for accurate secondary structures. Proteins 35, 307–312 [PubMed] [Google Scholar]
- 55. Whitmore L., Wallace B. A. (2004) DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 32, W668–W673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Santoro M. M., Bolen D. W. (1988) Unfolding free energy changes determined by the linear extrapolation method. 1. Unfolding of phenylmethanesulfonyl α-chymotrypsin using different denaturants. Biochemistry 27, 8063–8068 [DOI] [PubMed] [Google Scholar]
- 57. Steipe B., Schiller B., Plückthun A., Steinbacher S. (1994) Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 240, 188–192 [DOI] [PubMed] [Google Scholar]
- 58. Pace C. N., Scholtz J. M. (1997) Measuring the conformational stability of a protein. In Protein Structure: A Practical Approach (Creighton T. E., ed) 2nd Ed., pp. 253–259, Oxford University Press, Oxford [Google Scholar]
- 59. Park H. H., Logette E., Raunser S., Cuenin S., Walz T., Tschopp J., Wu H. (2007) Death domain assembly mechanism revealed by crystal structure of the oligomeric PIDDosome core complex. Cell 128, 533–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lin S. C., Lo Y. C., Wu H. (2010) Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 465, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang L., Yang J. K., Kabaleeswaran V., Rice A. J., Cruz A. C., Park A. Y., Yin Q., Damko E., Jang S. B., Raunser S., Robinson C. V., Siegel R. M., Walz T., Wu H. (2010) The Fas-FADD death domain complex structure reveals the basis of DISC assembly and disease mutations. Nat. Struct. Mol. Biol. 17, 1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Qin H., Srinivasula S. M., Wu G., Fernandes-Alnemri T., Alnemri E. S., Shi Y. (1999) Structural basis of procaspase-9 recruitment by the apoptotic protease-activating factor 1. Nature 399, 549–557 [DOI] [PubMed] [Google Scholar]
- 63. Agostini L., Martinon F., Burns K., McDermott M. F., Hawkins P. N., Tschopp J. (2004) NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20, 319–325 [DOI] [PubMed] [Google Scholar]
- 64. Arnold G. E., Day L. A., Dunker A. K. (1992) Tryptophan contributions to the unusual circular dichroism of fd bacteriophage. Biochemistry 31, 7948–7956 [DOI] [PubMed] [Google Scholar]
- 65. Woody R. W. (1994) Contributions of tryptophan side chains to the far-ultraviolet circular dichroism of proteins. Eur. Biophys. J. 23, 253–262 [DOI] [PubMed] [Google Scholar]
- 66. Lau S. Y., Taneja A. K., Hodges R. S. (1984) Synthesis of a model protein of defined secondary and quaternary structure. Effect of chain length on the stabilization and formation of two-stranded alpha-helical coiled-coils. J. Biol. Chem. 259, 13253–13261 [PubMed] [Google Scholar]
- 67. Masters S. L. (2013) Specific inflammasomes in complex diseases. Clin. Immunol. 147, 223–228 [DOI] [PubMed] [Google Scholar]
- 68. Di Virgilio F. (2013) The therapeutic potential of modifying inflammasomes and NOD-like receptors. Pharmacol. Rev. 65, 872–905 [DOI] [PubMed] [Google Scholar]
- 69. Weber C. H., Vincenz C. (2001) The death domain superfamily: a tale of two interfaces? Trends Biochem. Sci. 26, 475–481 [DOI] [PubMed] [Google Scholar]