

Background: Human proinsulin provides a model for studies of toxic protein misfolding.
Results: Folding efficiency of proinsulin is controlled by stereochemistry at position B8.
Conclusion: The sign of a single φ angle can facilitate or impede disulfide pairing.
Significance: A monogenic diabetes syndrome suggests that insulin has evolved to the edge of non-foldability.
Keywords: Chemical Biology, Diabetes, Mutant, Peptide Chemical Synthesis, Protein Folding, Proinsulin, Insulin Receptor
Abstract
Misfolding of proinsulin variants in the pancreatic β-cell, a monogenic cause of permanent neonatal-onset diabetes mellitus, provides a model for a disease of protein toxicity. A hot spot for such clinical mutations is found at position B8, conserved as glycine within the vertebrate insulin superfamily. We set out to investigate the molecular basis of the aberrant properties of a proinsulin clinical mutant in which residue GlyB8 is replaced by SerB8. Modular total chemical synthesis was used to prepare the wild-type [GlyB8]proinsulin molecule and three analogs: [d-AlaB8]proinsulin, [l-AlaB8]proinsulin, and the clinical mutant [l-SerB8]proinsulin. The protein diastereomer [d-AlaB8]proinsulin produced higher folding yields at all pH values compared with the wild-type proinsulin and the other two analogs, but showed only very weak binding to the insulin receptor. The clinical mutant [l-SerB8]proinsulin impaired folding at pH 7.5 even in the presence of protein-disulfide isomerase. Surprisingly, although [l-SerB8]proinsulin did not fold well under the physiological conditions investigated, once folded the [l-SerB8]proinsulin protein molecule bound to the insulin receptor more effectively than wild-type proinsulin. Such paradoxical gain of function (not pertinent in vivo due to impaired secretion of the mutant insulin) presumably reflects induced fit in the native mechanism of hormone-receptor engagement. This work provides insight into the molecular mechanism of a clinical mutation in the insulin gene associated with diabetes mellitus. These results dramatically illustrate the power of total protein synthesis, as enabled by modern chemical ligation methods, for the investigation of protein folding and misfolding.
Introduction
Impaired biosynthesis or function of the protein hormone insulin leads to diabetes mellitus (DM),3 which is a general name for a complex of metabolic diseases characterized by high blood glucose concentrations. A monogenic form of this disease is permanent neonatal-onset DM, which becomes apparent within 6 months of birth (1) and is caused in almost 50% of cases by single mutations in the insulin gene (2–5). This syndrome is the subject of a recent review (6). The clinical mutations generate variants of proinsulin that differ from wild-type proinsulin by only one amino acid. The amino acid substitutions (which can be at any of several positions) lie in regions of the proinsulin polypeptide critical to its normal folding in the endoplasmic reticulum (ER) and impair the proper secretion of both the variant and wild-type insulin (6). Misfolding of the variant proinsulins causes severe ER stress and death of β cells. To date, all mechanistic studies of proinsulin misfolding mutants have been at the cellular level.
Our goal was to develop molecular insight into the aberrant properties of the clinical mutant [SerB8]proinsulin, because this mutation in the prohormone leads to permanent neonatal DM (2–5). The wild-type residue GlyB8 (Fig. 1) is conserved among vertebrate insulins and is invariant among a vertebrate superfamily containing insulin-like growth factors (IGF-I and IGF-II), relaxin, and relaxin-related factors. GlyB8 plays a critical role in the process of proinsulin folding (7, 8). Studies of how GlyB8 contributes to proinsulin folding/misfolding and to the biological function of proinsulin may deepen our understanding of the pathogenesis of this syndrome and suggest novel avenues for therapeutic intervention. Uniquely, glycine can adopt the backbone conformation of a d-amino acid in the folded protein as the absence of a side chain enables glycine to employ both positive and negative φ dihedral angles without steric clash. In general, positive φ angles reside on the right side of the Ramachandran plot, a region unfavorable for l-amino acids but allowed for d-amino acids. Negative φ angles are characteristic of native l-amino acids in polypeptide chains.
FIGURE 1.
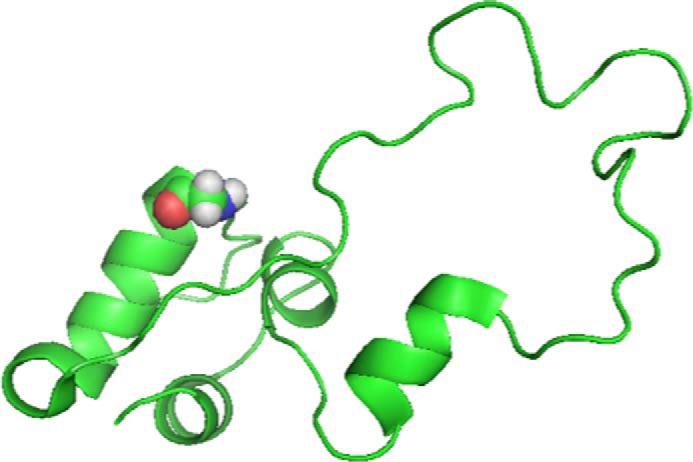
Structure of the proinsulin molecule as a schematic model with GlyB8 in the β-turn at amino acid residues B7-B10 (turn 1) shown with CPK (Corey-Pauling-Koltun) space-filling rendering. Image adapted from PDB code 2KQP.
In the mature two-chain insulin molecule, chiral amino acid substitutions at position B8 have been shown to have stereospecific effects on hormone activity: d-amino acid substitutions at B8 augment thermodynamic stability but markedly impair receptor binding, whereas l-amino acid substitutions have the opposite effects (7, 8). In mature insulin the folded zinc-insulin hexamer can appear in two different states: the T-state wherein residues B9–B19 are α-helical and residues B1–B8 adopt extended (B1–B6) and turn (B7–B10) conformations; and the R-state wherein the α-helix starts at or near the N terminus and continues to residue B19 (9, 10). In the native T state, which is the dominant conformation of the insulin monomer in solution (9, 11, 12), the glycine at B8 participates in the B7–B10 β turn (turn 1) and exhibits a positive φ dihedral angle as is typical of d-amino acids (7, 8, 13, 14).
Proinsulin contains a single polypeptide chain of 86 amino acid residues (Fig. 1). To probe how the sign of the φ angle at position B8 influences proinsulin folding, we sought to put both l- and d-amino acids in place of the natural GlyB8 (7). A proinsulin molecule containing a d-amino acid at this position can uniquely be prepared by chemistry. We thus undertook the total synthesis of three analogs: [d-AlaB8]proinsulin, [l-AlaB8]proinsulin, and the clinical mutation [l-SerB8]proinsulin. Wild-type [GlyB8]proinsulin was also synthesized as a control. By analogy with studies of insulin (7), we anticipated that substitution of l-Ala at position B8 of proinsulin would impair folding, whereas a d-Ala substitution would be competent for folding. In addition we anticipated that the clinical variant l-SerB8 would exhibit low folding yields. We also examined the effect of the enzyme protein-disulfide isomerase (PDI) on folding yields. PDI functions as a protein folding catalyst in the ER (15–18) and is involved in the formation of disulfide bonds in many proteins, including proinsulin (19). In addition, we measured binding of the synthetic proinsulin analogs to the insulin receptor (IR). Proinsulin binds to the IR; the affinity of wild-type proinsulin for the isolated IR is 1–2 orders of magnitude lower than that of the mature insulin protein molecule (20).
EXPERIMENTAL PROCEDURES
Reagents
N-tert-Butoxycarbonyl-(Nα-Boc) protected d- and l-amino acids (Peptide Institute, Osaka) and 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate were purchased from Peptides International Inc. Side chain protecting groups used were Arg(Tos), Asn(Xan), Asp(OcHex), Cys(4-CH3Bzl), Glu(OcHex), His(Dnp), Lys(2Cl-Z), Ser(Bzl), Thr(Bzl), and Tyr(2Br-Z). Aminomethyl resin was prepared from Bio-Beads S-X1 (Bio-Rad) (21). Boc-l-Asn-OCH2-phenylacetic acid and Boc-l-Ala-OCH2-phenylacetic acid were purchased from NeoMPS, Strasbourg, France. N,N-Diisopropylethylamine was obtained from Applied Biosystems Inc. (Foster City, CA). N,N-Dimethylformamide, dichloromethane, diethyl ether, HPLC-grade acetonitrile, and guanidine hydrochloride were purchased from Fisher. TFA was obtained from Halocarbon Products (NJ). HF was purchased from Matheson. All other reagents were purchased from Sigma or Fisher Scientific.
HPLC with On-line Electrospray Ionization Mass Spectrometry (LC-MS) Analysis and Preparative Reversed Phase HPLC
Analytical reverse phase high pressure liquid chromatography (HPLC) was performed on a Phenomenex C4 or C18 (3.6 μm, 300 Å) 2.1 × 50-mm silica column at a flow rate of 0.5 ml/min, or an Agilent C-8 (5 μm, 300 Å) 4.6 × 150-mm silica column at a flow rate of 1 ml/min using a linear gradient of 5–47% of buffer B in buffer A over 42 min or (9–53%) of buffer B in buffer A over 44 min at 40 °C (buffer A = 0.1% TFA in H2O; buffer B = 0.08% TFA in acetonitrile). The absorbance of the column eluate was monitored at 214 nm. The peptide masses were measured by on-line LC-MS using an Agilent 1100 LC/MSD ion trap. Calculated masses were based on average isotope composition. Preparative reverse phase HPLC of crude peptides was performed with an Agilent 1100 prep system Phenomenex C18 (5 μm, 300 Å) 10 × 250-mm column at 40 °C using an appropriate shallow gradient of increasing concentration of buffer B in buffer A at a flow rate of 5 ml/min. Fractions containing the purified target peptide were identified by analytical LC, and aliquots from each selected fraction were combined and checked by LC-MS. Selected fractions were then combined and lyophilized.
Peptide Synthesis
Peptide segments were synthesized using manual “in situ neutralization” Boc chemistry protocols for stepwise solid phase peptide synthesis (22). The C-terminal peptide segment Cys72-Asn86 (3) was synthesized on Boc-Asn-OCH2-phenyl-CH2CONH-resin (Boc-Asn-OCH2-Pam-resin) (21). The Phe1-Val18-thioester segment (1) and 1,3-thiazolidine-4-carboxo (Thz)-derivatized segment Thz19-Cys71-thioester (2) were synthesized on trityl-SCH2CH2CO-Ala-OCH2-Pam-resin (23). For peptide segment Phe1-Val18-thioester (1), prior to HF deprotection and cleavage, the side chain dinitrophenyl groups of residues His5 and His10 were removed from the resin-bound C-terminal peptide segment by treatment with a mixture of 2-mercaptoethanol/N,N-diisopropylethylamine/N,N-dimethylformamide, 2:1:7, twice for 15 min each. After removal of the last Nα-Boc group, peptides were cleaved from the resin and deprotected by treatment with anhydrous HF containing 5% (v/v) p-cresol as scavenger at 0 °C for 1 h. After removal of HF by evaporation under reduced pressure, the crude peptides were precipitated and washed with diethyl ether, then dissolved in 50% aqueous acetonitrile containing 0.1% TFA and lyophilized.
Preparative HPLC purification gave the following amounts of each peptide segment: 67 mg (21% yield based on 0.15 mmol of Boc-Ala-OCH2-Pam-resin) of pure peptide Phe1-Val18-αCOSCH2CH2CO-Ala-COOH, observed mass, 2200.8 ± 0.8 Da, calculated 2201.6 Da; 114 mg (17% yield based on 0.3 mmol of Boc-Ala-OCH2-Pam-resin) of pure peptide [d-AlaB8]Phe1-Val18-αCOSCH2CH2CO-Ala-COOH, observed mass, 2214.8 ± 0.8 Da, calculated 2215.6 Da; 130 mg (20% yield based on 0.3 mmol of Boc-Ala-OCH2-Pam-resin) of pure peptide [l-AlaB8]Phe1-Val18-αCOSCH2CH2CO-Ala-COOH, observed mass, 2215.0 ± 0.8 Da, calculated 2215.6 Da; 62 mg (14% yield based on 0.2 mmol of Boc-Ala-OCH2-Pam-resin) of pure peptide [l-SerB8]Phe1-Val18-αCOSCH2CH2CO-Ala-COOH, observed mass, 2231.4 ± 0.4 Da, calculated 2231.6 Da; 99 mg (11% yield based on 0.15 mmol of Boc-Ala-OCH2-Pam-resin) of pure peptide Thz19-Cys71-αCOSCH2CH2CO-Ala-COOH, observed mass, 5804.4 ± 0.5 Da, calculated 5805.5 Da; 228 mg (26% yield based on 0.5 mmol of Boc-Asn-OCH2-Pam-resin) of pure peptide Cys72-Asn86, observed mass, 1753.0 ± 0.8 Da, calculated 1753.9 Da.
“One Pot” Synthesis of the Proteins
Synthesis of the analog [d-AlaB8]proinsulin is representative and is described in detail. Based on the synthetic strategy shown in Fig. 2, the first native chemical ligation was performed between peptide segments Cys72-Asn86 (3, 7.4 mg, 4.2 μmol, 4.8 mm) and Thz19-Cys71-αCOSR (2, 22.2 mg, 3.8 μmol, 4.3 mm) under standard conditions (ligation buffer (880 μl) containing 6 m guanidine hydrochloride (GdnHCl), 0.1 m sodium phosphate buffer, pH 6.9, 4-mercaptophenylacetic acid (MPAA, 100 mm) (24), tris(2-carboxyethyl)phosphine·HCl (20 mm) to give the ligation product Thz19-Asn86 (4), not shown in Fig. 2B, within 4.5 h (Fig. 2, A–B). To ensure that the longer peptide segment (2) was completely consumed, we used an excess (1.1 eq) of the shorter peptide (3). On completion of the first ligation, Thz19 was converted to Cys19 by treatment with methoxylamine·HCl (430 mm) at pH 4.0 (25) to give Cys19-Asn86 (5, Fig. 3C). For the second native chemical ligation, the peptide-thioester segment [d-AlaB8]Phe1-Val18-αCOSR (1, 12.7 mg, 5.7 μmol, 6.5 mm) and another portion of tris(2-carboxyethyl)phosphine·HCl (50 mm total) and MPAA (200 mm total) were added to the same reaction mixture, and the solution was readjusted to pH 6.9 by addition of 5 n NaOH (Fig. 3D). This ligation is at a Val-Cys site, which is sterically hindered by the Val side chain, and therefore more MPAA catalyst was added to bring the concentration to 200 mm (26) and excess of the third peptide (1, 1.5 eq.) was used. The reaction was completed within 46 h (Fig. 3E), and the target full-length polypeptide [d-AlaB8]Phe1-Asn86 (6) was recovered by precipitation from the reaction mixture by 10-fold dilution with water, redissolved in 50% B and lyophilized; observed mass, 9408.4 ± 0.3 Da; calculated 9408.6 Da. Wild-type proinsulin and the l-AlaB8 and l-SerB8 analogs were made in the same way.
FIGURE 2.

Total chemical synthesis of proinsulin. A, amino acid sequence of wild-type human proinsulin. GlyB8 is in bold. Note that we use the amino acid residue numbering of the mature two-chain insulin molecule. B, synthetic strategy for the one pot total chemical synthesis of proinsulin and analogs. r = CH2CH2COAlaCOOH. All three analogs and wild-type human proinsulin were chemically synthesized by the same route. This modular synthetic strategy was particularly efficient for preparation of a related set of analogs. Instead of synthesizing afresh the full-length polypeptide for each target, only one new peptide segment was required per analog. Segments Thz19-Cys71-thioester (2) and Cys72-Asn86 (3) were common to all the analogs and were thus synthesized in larger amounts. Peptide segment Phe1-Val18-thioester (1) was resynthesized as necessary for each of the d- or l-AlaB8 and l-SerB8 analogs. The three peptide segments: Cys-peptide Cys72-Asn86 (3), Thz19-Cys71-αCOSR (2), and Phe1-Val18-αCOSR (1) (r = -CH2CH2CO-Ala-COOH) were covalently condensed in two native chemical-ligation steps starting from C-terminal segment Cys72-Asn86 (3) (see also Fig. 3).
FIGURE 3.

Analytical LC-MS data for one pot total chemical synthesis of [d-AlaB8]proinsulin. A, native chemical ligation of Cys72-Asn86 (3) and Thz19-Cys71-αCOSR (2) (r = -CH2CH2CO-Ala-COOH) to give Thz19-Asn86 (4) at t = 2 min. The asterisk (*) corresponds to the MPAA-exchanged thioester intermediate form of 2. B, the same ligation reaction upon completion after 4.5 h. C, crude reaction mixture after treatment with methoxylamine/HCl at pH 4.0 to convert Thz to Cys. D, native chemical ligation of Cys19-Asn86 (5) and [d-AlaB8]Phe1-Val18-αCOSR (1) to give the full-length polypeptide [l-AlaB8]Phe1-Asn86 (6) at t = 1 min. E, the same ligation reaction upon completion after 46 h. Because peptide segment 1 was used in excess (1.5 eq), several related peaks were observed at the end of the second native chemical ligation: (1) unreacted peptide segment; **, MPAA-exchanged thioester intermediate form of 1; ▴, hydrolysis of the thioester of peptide 1; ●, product of reaction of methoxylamine with thioester 1. Online ESI MS data (inset) is shown (observed mass, 9408.4 ± 0.3 Da; calculated mass (average isotope composition), 9408.6 Da). Analytical HPLC (λ = 214 nm) was performed using a linear gradient (5–47%) of buffer B in buffer A over 42 min (buffer A = 0.1% TFA in water; buffer B = 0.08% TFA in acetonitrile) on a C18 column.
Folding
After lyophilization, folding experiments were performed on a small scale beginning from the linear unfolded polypeptides. The following conditions were employed.
Folding at pH 10.5
The crude linear polypeptide(1–86) was folded in a buffer solution containing 50 mm glycine/NaOH (pH 10.5), 1 mm EDTA, 1 mm reduced glutathione (GSH), 1 mm oxidized glutathione (GSSG) at peptide concentration of 0.1 mg/ml (27). The folding solution was purged, sealed, and incubated overnight at 4 °C.
Folding at pH 7.5
The crude polypeptide was folded at room temperature at a concentration of 0.1 mg/ml in buffer solution containing 10 mm Tris, pH 7.5, 10 mm glycine, 1 mm EDTA, 1 mm GSH, 2 mm GSSG, 70 mm GdnHCl (19). The folding solution was purged, sealed, and incubated overnight.
Folding at pH 7.5 in the Presence of PDI
The same conditions as at pH 7.5 were used, plus PDI in a PDI/proinsulin molar ratio of 0.093. The folding solution was purged and sealed, and essentially complete after 1 h as evidenced by LC-MS.
Receptor Binding
Affinities of the purified synthetic proteins for the lectin-purified IR (isoform B) were measured by competitive displacement of 125I-TyrA14-labeled insulin (28, 29) (purchased from New England Nuclear); recombinant human insulin (purchased from Eli Lilly and Co.) was employed as a positive control.
RESULTS
Chemical Synthesis of the Polypeptide Chains of Human Proinsulin and Analogs
The wild-type and three analogs of human proinsulin were prepared using an improved form of our previously reported total chemical synthesis of human proinsulin (26). That synthesis had involved purification of each intermediate product. In an effort to make the synthesis more efficient, we developed a one pot series of reactions (30). We anticipated that the new strategy would minimize losses from purification steps and handling of intermediates, of special concern in the synthesis of analogs associated with misfolding. The 86-amino acid residue proinsulin polypeptide chain contains six cysteine residues (Fig. 2A), each of which is a potential site for native chemical ligation (31, 32). The synthetic strategy we used is shown in Fig. 2B. Three peptide segments were employed: Phe1-Val18-thioester (1), Thz19-Cys71-thioester (2), and Cys72-Asn86 (3). Each peptide was synthesized by manual stepwise Boc chemistry “in situ neutralization” solid phase peptide synthesis (22).
To avoid side reactions such as cyclization or oligomerization of peptide segment 3, the N-terminal Cys of this peptide segment was introduced as the R-Thz group (30), which was readily converted to an N-terminal L-Cys residue (Fig. 3C). The product full-length polypeptide was in each case isolated by precipitation from the reaction mixture by 10-fold dilution with water. In this way, multiple tens of milligram amounts of each target polypeptide chain were prepared for folding studies.
Folding Experiments
For each analog, folding was examined under three sets of conditions: first, the standard conditions used for folding recombinant proinsulin, pH 10.5 (50 mm glycine, pH 10.5, 1 mm EDTA, 1 mm reduced glutathione, 1 mm oxidized glutathione, at 4 °C and peptide concentration of 0.1 mg/ml (27)), second, at near physiological pH 7.5 (10 mm Tris, pH 7.5, 10 mm Gly, 1 mm EDTA, 1 mm reduced glutathione, 2 mm oxidized glutathione, 70 mm GdnHCl, at room temperature and peptide concentration of 0.1 mg/ml (19)), and third, at pH 7.5 in the presence of PDI. These three sets of conditions were chosen to give a better understanding of how the substitutions at B8 affect folding efficiency under commonly used folding conditions, and at physiological pH.
Recombinantly produced human proinsulin is routinely folded at pH 10.5, rather than at physiological pH, to minimize off-pathway aggregation (27, 33). It was of interest to test whether the B8 substitutions might impair folding at pH 7.5 but not at pH 10.5. A folding screen can in principle define two classes of mutations: a pH-independent class (i.e. mutations that impede folding at all pH values), and a pH-dependent class (mutations that impedes folding at pH 7.5 but permit efficient folding at pH 10.5). At pH 10.5 the Cys side chain thiols are ionized (to thiolates), which facilitates folding relative to the rate of off-pathway aggregation; the latter is prominent at lower pH (pH 6–8) (19). We employed the pH 10.5 folding conditions both for small scale experiments (to investigate and compare the results under other folding conditions), and for large scale folding (for the isolation of the folded product for receptor binding assays). The purpose of the folding efforts at pH 7.5 was to mimic physiological conditions. To further mimic those conditions, we decided to also perform studies at pH 7.5 in the presence of PDI, previously shown to improve proinsulin folding in vitro (19). According to Winter et al. (19), yields of proinsulin folding increased with increase of the molar ratio of PDI/proinsulin until it reached a plateau of 50% yield at the molar ratio of 0.033. In our experiments we used a molar ratio of PDI/proinsulin of 0.093 to obtain the highest possible yields.
All folding experiments were monitored by LC-MS. Relative to their respective unfolded polypeptides, the folded products each gave an earlier eluting, sharp peak with a mass lower by 6 Da, corresponding to the formation of three disulfide bonds. Folded globular proteins that contain disulfides typically have most of their hydrophobic residues buried in the core of the protein molecule and therefore have an earlier elution time in reverse-phase HPLC relative to the corresponding unfolded polypeptide.
Results of the folding trials are summarized in Table 1, and HPLC data for the folding of wild-type proinsulin, [l-SerB8]proinsulin, [d-AlaB8]proinsulin, and [l-AlaB8]proinsulin under each of the three conditions are shown in Figs. 4 and 5. As expected, the folding yields of wild-type and d-AlaB8 proinsulins were in general higher than the folding yields of l-AlaB8 and l-SerB8 analogs. Indeed, the wild-type and d-AlaB8 proinsulins folded very efficiently in near quantitative yields at pH 10.5 (Figs. 4D and 5D). Although yields were lower at pH 7.5 than at 10.5, the addition of PDI gave improved folding yields (Figs. 4, B and F, and 5B). Yields of the folded d-AlaB8 analog were higher than yields obtained for wild-type proinsulin. As shown in Table 1, the l-AlaB8 substitution impaired folding at both pH values (Fig. 4, F–H); at pH 10.5 a broad peak corresponding to a mixture of folded and misfolded products was obtained (Fig. 5H). l-SerB8 proinsulin folded at pH 10.5 but not at pH 7.5.
TABLE 1.
Folding yields of wild-type proinsulin and position B8 analogs under three conditions
| pH 7.5 + PDIa | pH 7.5b | pH 10.5c | |
|---|---|---|---|
| Wild-type hpid | 50% | 20% | 90% |
| l-SerB8 hpi (clinical mutation) | 15% | Does not fold | 90% |
| d-AlaB8 hpi | 60% | 40% | 90% |
| l-AlaB8 hpi | Does not fold | Does not fold | Broad peak of folded and misfolded products |
a 10 mm Tris, 10 mm glycine, 1 mm EDTA, 1 mm GSH, 2 mm GSSG, 70 mm GdnHCl, at room temperature with a PDI molar ratio of PDI/proinsulin = 0.093.
b Same as a without PDI.
c 50 mm glycine, 1 mm EDTA, 1 mm GSH, 1 mm GSSG, at 4 °C. The polypeptide concentration for all conditions was 0.1 mg/ml. Folded product was indentified by LC-MS. The yields were calculated according to the area under the identified peaks and are ±5%.
d hpi, human proinsulin.
FIGURE 4.

Folding of wild-type proinsulin (left column) and [l-SerB8]proinsulin (right column) in three different conditions: at pH 10.5 (50 mm glycine/NaOH, 1 mm EDTA, 1 mm reduced GSH, 1 mm GSSG, at 4 °C and peptide concentration of 0.1 mg/ml); at pH 7.5 (10 mm Tris, 10 mm glycine, 1 mm EDTA, 1 mm GSH, 2 mm GSSG, 70 mm GdnHCl, at room temperature and peptide concentration of 0.1 mg/ml); and at pH 7.5 in the presence of PDI. The top two chromatograms represent the unfolded polypeptide of wild-type proinsulin (A) and [l-SerB8]proinsulin (E). The six bottom chromatograms represent the reaction mixtures after overnight folding under the conditions shown on each chromatogram. The asterisk (*) corresponds to the PDI protein. All the reactions were monitored by LC-MS and the masses were confirmed by online ESI.
FIGURE 5.

Folding of [d-AlaB8]proinsulin (left column) and [l-AlaB8]proinsulin (right column) in three different conditions as described in the legend to Fig. 4. The top two chromatograms represent the unfolded polypeptide of [d-AlaB8]proinsulin (A) and [l-AlaB8]proinsulin (E). The six bottom chromatograms represent the reaction mixtures after overnight folding under the conditions shown on each chromatogram. The asterisk (*) corresponds to the PDI protein.
Isolation of the Proteins
For studies of the receptor binding activities of the various analog proinsulins, it was necessary to isolate each folded analog protein in purified form. We performed the folding reactions on a larger scale (∼10–15 mg of each polypeptide). For this purpose we employed conditions that gave the highest folding yields: 50 mm glycine/NaOH at pH 10.5, 1 mm EDTA, 1 mm reduced glutathione, 1 mm oxidized glutathione, at 4 °C and at a peptide concentration of 0.1 mg/ml (27). On completion of folding, the solution was acidified with dilute HCl to pH ∼2, and each folded protein was purified by reverse phase HPLC. The purified folded wild-type proinsulin and the three B8 analogs were characterized by LC-MS (Fig. 6). Observed masses were: wild-type proinsulin, 9388.6 ± 0.2 Da, calculated mass, 9388.6 Da (average isotope composition) (Fig. 6A); [l-SerB8]proinsulin, observed mass, 9418.8 ± 0.5 Da, calculated mass, 9418.6 Da (average isotope composition) (Fig. 6B); [d-AlaB8]proinsulin, 9402.5 ± 0.1 Da, calculated mass, 9402.6 Da (average isotope composition) (Fig. 6C); [l-AlaB8]proinsulin, observed mass, 9402.6 ± 0.8 Da, calculated mass, 9402.6 Da (average isotope composition) (Fig. 6D).
FIGURE 6.

Characterization of synthetic human proinsulin and its B8 analogs by LC-MS. The MS (ESI) taken across the entire main peak is shown in the inset. Note that distinct reverse phase packings and gradients were used, as follows: A, folded wild-type proinsulin. The chromatographic separations were performed on a C4 column using a linear gradient (5–47%) of buffer B in buffer A over 42 min. B, folded [l-SerB8] proinsulin. The chromatographic separations were performed on a C8 column using a linear gradient (5–47%) of buffer B in buffer A over 42 min. C, folded [d-AlaB8] proinsulin. The chromatographic separations were performed on a C4 column using a linear gradient (9–53%) of buffer B in buffer A over 44 min. D, folded [l-AlaB8] proinsulin. The chromatographic separations were performed on a C8 column using a linear gradient (5–47%) of buffer B in buffer A over 42 min.
Receptor-binding Assays
Affinities were measured by competitive displacement of labeled human proinsulin from lectin-purified IR (isoform B) (28, 29). Recombinant wild-type human insulin was used as positive control. The resulting data are shown in Fig. 7; dissociation constants are given in Table 2.
FIGURE 7.

Insulin receptor-binding assays for biosynthetic insulin (■), synthetic human proinsulin (hpi) (●), l-SerB8-hpi (♦), l-AlaB8-hpi (▾), d-AlaB8-hpi (▴). Vertical scale indicates fractional displacement of pre-bound 125I-TyrA14-insulin on addition of wild-type or variant proinsulins at successive protein concentrations (horizontal log scale). Inferred dissociation constants are given in Table 2.
TABLE 2.
Affinities of insulin, proinsulin, and analogs
| Insulin | Proinsulin (hpi)a | l-SerB8-hpi | d-AlaB8-hpi | l-AlaB8-hpib | |
|---|---|---|---|---|---|
| Kd (nm)c | 0.054 ± 0.008 | 4.1 ± 0.6 | 2.1 ± 0.3 | ∼1500 | 124 ± 27 |
a hpi, human proinsulin.
b The following findings together suggest that the [l-AlaB8]proinsulin is folded correctly: the relative potencies of l-AlaB8-DKP-insulin/l-SerB8-DKP-insulin and of l-AlaB8-proinsulin/l-SerB8-proinsulin are closely similar; for l-SerB8-DKP-insulin, the disulfide pairing in the folded structure has been authenticated by two-dimensioinal NMR structural analysis; the qualitative features of the NMR spectrum of l-AlaB8-DKP-insulin closely resemble those of the l-SerB8 analog.
DISCUSSION
The amino acid sequence of a protein specifies not only its native three-dimensional structure (34), but also enables an efficient conformational search en route to the native state (35, 36). Biological selection in general yields foldable protein sequences, a key property that may otherwise be unrepresentative of polypeptides as a chemical class of heteropolymers (37). Selection for folding efficiency thus provides a hidden constraint in the evolution of protein sequences (38).
The need to provide a productive protein-folding pathway has further focused attention on nucleation steps to form a transition-state ensemble stabilized by a critical subset of native-like contacts (39). The folding nucleus of a globular protein, as investigated by a variety of chemical, mutational, and biophysical probes (40, 41), typically contains a subset of native secondary structures related by long-range interactions that foreshadow in approximate (or distorted) form the ultimate state of the folded protein molecule (38). Interest in these fundamental mechanisms has deepened in the past two decades by direct links between these concepts and diverse diseases of toxic protein misfolding (42). Although such past studies have predominantly focused on extracellular amyloid (protein deposition diseases as exemplified by the mutant lysozyme and transthyretin syndromes (43, 44), recent attention has focused on the intracellular origins of ER stress arising from misfolding of nascent protein molecules (45–47). A paradigm for this class of genetic diseases is provided by the monogenic syndrome of permanent neonatal-onset DM due to the impaired foldability of proinsulin variants in the ER of pancreatic β-cells (48).
Insulin has long provided a favorable model for the study of protein stability, folding, and misfolding due to its small size, amenability to chemical synthesis (49, 50), and wealth of structural information (13). The present study extended the use of chemical synthesis to the human proinsulin protein molecule to investigate a DM-associated clinical mutation of the invariant GlyB8 residue. Mutation at this position affects the conformational properties of the polypeptide main chain rather than specific properties of the variant side chain. GlyB8 functions as a structural switch in the classical T state to R state conformational transition in the insulin molecule that is important for receptor binding. The GlyB8 residue has a positive backbone φ dihedral angle in the T state but a negative φ angle in the R state (10, 13, 51). Substitution of natural l-amino acids at this position restricts the backbone φ dihedral to negative values. In the present study, we used chemical synthesis of the proinsulin protein molecule to introduce both d- and l-amino acid substitutions in place of GlyB8 to probe both the right and left sides of the Ramachandran plot (7, 8).
The data reported here provide molecular insights into the aberrant folding properties of the clinical mutant [SerB8]human proinsulin and help explain why that mutation causes permanent neonatal DM. Distinct differences in folding efficiency were seen for the B8 analogs of human proinsulin. At physiological pH, [d-AlaB8]proinsulin folded better than the wild-type proinsulin and the l-amino acidB8 analogs. We attribute the efficient folding of [d-AlaB8]proinsulin to the fact that in the folded structure of the native protein GlyB8 adopts the backbone conformation of a d-amino acid. Thus, having a d-amino acid at position B8 favors the folded conformation (7). The d-AlaB8 substitution reduced receptor binding more than 300-fold (only an approximate lower limit was obtained for such weak binding). The profound block to receptor binding imposed by d-AlaB8 in proinsulin was also seen in studies of d-AlaB8-insulin (7, 52).
Significantly, the clinical mutation l-SerB8 impaired the folding of human proinsulin at pH 7.5 even in the presence of PDI, a resident ER chaperone and oxidoreductase. This poor folding under in vitro conditions that mimic physiological folding is in accord with data obtained for cellular studies of the SerB8 mutant insulin (6). Interestingly, [l-SerB8]proinsulin folded efficiently at pH 10.5, whereas [l-AlaB8]proinsulin did not (Table 1). Such pH-dependent rescue of folding may reflect accelerated formation of disulfide bridges and rapid disulfide exchange within the monomeric polypeptide leading to native disulfide pairing, relative to pH-dependent rates of competing off-pathway events (such as aggregation of the reduced polypeptide or one-disulfide partial folds). The absence of such rescue of folding for [l-SerB8]proinsulin in pancreatic β-cells, even following presumed activation of the ER stress pathways (53), suggests that there are limits to the chemical tactics ordinarily available to the oxidative folding machinery of the cell (17).
A surprising observation was that, although [l-SerB8]proinsulin did not fold well under the physiological conditions investigated, once it was folded the [l-SerB8]proinsulin protein molecule bound to the insulin receptor ∼2-fold more effectively than wild-type proinsulin. This remarkable increase suggests that, on receptor binding, the perturbation to the structure and stability observed in our studies of the free [l-SerB8]proinsulin protein molecule facilitates adoption of a novel receptor-bound conformation. The enhanced receptor binding of [l-SerB8]proinsulin may shed light on possible mechanisms of how insulin binds to the IR,4 but is at present without clinical significance due to the impaired folding, trafficking, processing, and secretion of the mutant proinsulin and mature hormone analog.
Might the underlying mechanisms of pH-dependent foldability of [l-SerB8]proinsulin be exploited in vivo? The findings reported here suggest the tantalizing possibility that therapeutic maneuvers to rescue the foldability of [l-SerB8]proinsulin would both preserve β-cell viability through alleviation of ER stress and enable regulated secretion of an active hormone variant (as well as co-secretion of wild-type insulin in this heterozygous syndrome). Such ER-based therapeutics would circumvent the complexity of insulin replacement therapy in this form of DM (53) and restore physiological regulation of metabolism, the key to avoiding the serious long term microvascular and macrovascular complications of DM (54, 55).
In conclusion, glycines may be conserved among protein sequences due to steric restrictions on side chain size (such as in the interior of the collagen triple helix), due to specific β-turn conformations (as a positive φ dihedral angle may optimize turn stability at a specific motif position), or due to general requirements for segmental flexibility (as in natively unfolded polypeptide segments (56)). We postulate that glycine is invariant at position B8 of the vertebrate insulin-related superfamily because of a requirement for a positive φ angle at a critical step in the mechanism of specific disulfide pairing. This requirement is transient as the compatibility of a negative B8 φ angle with a well organized ground state has rigorously been shown in classical crystal structures of zinc insulin hexamers (10, 13, 51). The enhanced receptor binding of [l-SerB8]proinsulin and decreased receptor binding of [d-AlaB8]proinsulin (relative to wild-type proinsulin) further demonstrate that a positive B8 φ angle is not necessary for, and indeed impedes, hormonal function. We therefore propose that GlyB8 is conserved as an achiral “ambidextrous” switch between folding-competent and functional conformations.
To the best of our knowledge, the present studies are the first of their kind for any misfolding proinsulin, and one of a small handful of such studies for any disease of protein misfolding. In addition to their implications for the evolution of the insulin-related superfamily of globular proteins, our results dramatically illustrate the power of a modular approach to total chemical synthesis (31, 57) in the preparation of analog protein molecules, including polypeptide diastereomers not readily accessible by conventional molecular biology. We foresee that continuing advances in synthetic technology will increasingly enable the application of chemistry to important biological questions.
Acknowledgments
We thank L. Whittaker for assistance with insulin receptor-binding assays, J. Racca and N. B. Phillips for pilot studies of circular dichroism, Prof. P. Arvan for advice regarding cellular folding mechanisms, M. C. Lawrence and B. Smith for discussion regarding the structure of the insulin receptor, and Q.-X. Hua, Z.-l Wan, M. Liu, and the late Prof. G. G. Dodson for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK069764 from the NIDDK (to M. A. W.) with a subcontract to S. B. Kent (University of Chicago). M. A. W. has equity in Thermalin Diabetes, LLC (Cleveland, OH) where he serves as Chief Scientific Officer and has also been a consultant to Merck Research Laboratories and DEKA Research and Development Corp.
A suite of four co-crystal structures has recently been reported depicting insulin bound to fragments of the IR ectodomain at 3.9-Å resolution (58). Although electron density was not observed for residues B1-B6, three of the four structures appear to exhibit a positive φ dihedral angle at GlyB8; the fourth exhibits a negative φ angle at B8 but would be precluded from adopting a positive value by a bound Fab fragment (M. C. Lawrence, personal communication). It is thus not known to what extent the bound conformation of insulin may resemble, in its N-terminal B-chain segment, the classical R state of insulin.
- DM
- diabetes mellitus
- Boc
- tert-butoxycarbonyl
- ER
- endoplasmic reticulum
- IR
- insulin receptor
- PDI
- protein-disulfide isomerase
- MPAA
- 4-mercaptophenylacetic acid
- Thz
- 1,3-thiazolidine-4-carboxo
- GdnHCl
- guanidine hydrochloride.
REFERENCES
- 1. Shield J. P., Gardner R. J., Wadsworth E. J., Whiteford M. L., James R. S., Robinson D. O., Baum J. D., Temple I. K. (1997) Aetiopathology and genetic basis of neonatal diabetes. Arch. Dis. Child. Fetal Neonatal Ed. 76, F39–F42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Støy J., Edghill E. L., Flanagan S. E., Ye H., Paz V. P., Pluzhnikov A., Below J. E., Hayes M. G., Cox N. J., Lipkind G. M., Lipton R. B., Greeley S. A., Patch A. M., Ellard S., Steiner D. F., Hattersley A. T., Philipson L. H., Bell G. I., and Neonatal Diabetes International Collaborative Group. (2007) Insulin gene mutations as a cause of permanent neonatal diabetes. Proc. Natl. Acad. Sci. U.S.A. 104, 15040–15044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Colombo C., Porzio O., Liu M., Massa O., Vasta M., Salardi S., Beccaria L., Monciotti C., Toni S., Pedersen O., Hansen T., Federici L., Pesavento R., Cadario F., Federici G., Ghirri P., Arvan P., Iafusco D., Barbetti F., and Early Onset Diabetes Study Group of the Italian Society of Pediatric Endocrinology and Diabetes (SIEDP) (2008) Seven mutations in the human insulin gene linked to permanent neonatal/infancy-onset diabetes mellitus. J. Clin. Investig. 118, 2148–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Edghill E. L., Flanagan S. E., Patch A. M., Boustred C., Parrish A., Shields B., Shepherd M. H., Hussain K., Kapoor R. R., Malecki M., MacDonald M. J., Støy J., Steiner D. F., Philipson L. H., Bell G. I., and Neonatal Diabetes International Collaborative Group, Hattersley A. T., Ellard S. (2008) Mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood. Diabetes 57, 1034–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Molven A., Ringdal M., Nordbø A. M., Raeder H., Støy J., Lipkind G. M., Steiner D. F., Philipson L. H., Bergmann I., Aarskog D., Undlien D. E., Joner G., Søvik O., Norwegian Childhood Diabetes Study Group, Bell G. I., Njølstad P. R. (2008) Mutations in the insulin gene can cause MODY and autoantibody-negative type 1 diabetes. Diabetes 57, 1131–1135 [DOI] [PubMed] [Google Scholar]
- 6. Weiss M. A. (2013) Diabetes mellitus due to the toxic misfolding of proinsulin variants. FEBS Lett. 587, 1942–1950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakagawa S. H., Zhao M., Hua Q. X., Hu S. Q., Wan Z. L., Jia W., Weiss M. A. (2005) Chiral mutagenesis of insulin: foldability and function are inversely regulated by a stereospecific switch in the B chain. Biochemistry 44, 4984–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hua Q. X., Nakagawa S., Hu S. Q., Jia W., Wang S., Weiss M. A. (2006) Toward the active conformation of insulin: stereospecific modulation of a structural switch in the B chain. J. Biol. Chem. 281, 24900–24909 [DOI] [PubMed] [Google Scholar]
- 9. Hua Q. X., Shoelson S. E., Kochoyan M., Weiss M. A. (1991) Receptor binding redefined by a structural switch in a mutant human insulin. Nature 354, 238–241 [DOI] [PubMed] [Google Scholar]
- 10. Derewenda U., Derewenda Z., Dodson E. J., Dodson G. G., Reynolds C. D., Smith G. D., Sparks C., Swenson D. (1989) Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature 338, 594–596 [DOI] [PubMed] [Google Scholar]
- 11. Hua Q. X., Hu S. Q., Frank B. H., Jia W., Chu Y. C., Wang S. H., Burke G. T., Katsoyannis P. G., Weiss M. A. (1996) Mapping the functional surface of insulin by design: structure and function of a novel A-chain analogue. J. Mol. Biol. 264, 390–403 [DOI] [PubMed] [Google Scholar]
- 12. Olsen H. B., Ludvigsen S., Kaarsholm N. C. (1996) Solution structure of an engineered insulin monomer at neutral pH. Biochemistry 35, 8836–8845 [DOI] [PubMed] [Google Scholar]
- 13. Baker E. N., Blundell T. L., Cutfield J. F., Cutfield S. M., Dodson E. J., Dodson G. G., Crowfoot Hodgkin D. M., Hubbard R. E., Isaacs N. W., Reynolds C. D., Sakabe K., Sakabe N., Vijayan N. M. (1988) The structure of 2Zn pig insulin crystals at 1.5-Å resolution. Philos. Trans. R. Soc. Lond. B Biol. Sci. 319, 369–456 [DOI] [PubMed] [Google Scholar]
- 14. Nakagawa S. H., Hua Q. X., Hu S. Q., Jia W., Wang S., Katsoyannis P. G., Weiss M. A. (2006) Chiral mutagenesis of insulin: contribution of the B20-B23 β-turn to activity and stability. J. Biol. Chem. 281, 22386–22396 [DOI] [PubMed] [Google Scholar]
- 15. Noiva R. (1999) Protein disulfide isomerase: the multifunctional redox chaperone of the endoplasmic reticulum. Semin. Cell Dev. Biol. 10, 481–493 [DOI] [PubMed] [Google Scholar]
- 16. Wang C. C. (1998) Isomerase and chaperone activities of protein disulfide isomerase are both required for its function as a foldase. Biochemistry 63, 407–412 [PubMed] [Google Scholar]
- 17. Frand A. R., Cuozzo J. W., Kaiser C. A. (2000) Pathways for protein disulphide bond formation. Trends Cell Biol. 10, 203–210 [DOI] [PubMed] [Google Scholar]
- 18. Tu B. P., Ho-Schleyer S. C., Travers K. J., Weissman J. S. (2000) Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science 290, 1571–1574 [DOI] [PubMed] [Google Scholar]
- 19. Winter J., Klappa P., Freedman R. B., Lilie H., Rudolph R. (2002) Catalytic activity and chaperone function of human protein-disulfide isomerase are required for the efficient refolding of proinsulin. J. Biol. Chem. 277, 310–317 [DOI] [PubMed] [Google Scholar]
- 20. Freychet P. (1974) The interactions of proinsulin with insulin receptors on the plasma membrane of the liver. J. Clin. Investig. 54, 1020–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mitchell A. R., Kent S. B. H., Engelhard M., Merrifield R. B. (1978) A new synthetic route to tert-butyloxycarbonylaminoacyl-4-(oxymethyl)phenylacetamidomethyl-resin, an improved support for solid-phase peptide synthesis. J. Org. Chem. 43, 2845–2852 [Google Scholar]
- 22. Schnölzer M., Alewood P., Jones A., Alewood D., Kent S. B. H. (2007) In situ neutralization in Boc-chemistry solid phase peptide synthesis. Int. J. Pept. Res. Ther. 13, 31–44 [DOI] [PubMed] [Google Scholar]
- 23. Hackeng T. M., Griffin J. H., Dawson P. E. (1999) Protein synthesis by native chemical ligation: expanded scope by using straightforward methodology. Proc. Natl. Acad. Sci. U.S.A. 96, 10068–10073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson E. C., Kent S. B. (2006) Insights into the mechanism and catalysis of the native chemical ligation reaction. J. Am. Chem. Soc. 128, 6640–6646 [DOI] [PubMed] [Google Scholar]
- 25. Villain M., Vizzavona J., Rose K. (2001) Covalent capture: a new tool for the purification of synthetic and recombinant polypeptides. Chem. Biol. 8, 673–679 [DOI] [PubMed] [Google Scholar]
- 26. Luisier S., Avital-Shmilovici M., Weiss M. A., Kent S. B. (2010) Total chemical synthesis of human proinsulin. Chem. Commun. (Camb). 46, 8177–8179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mackin R. B., Choquette M. H. (2003) Expression, purification, and PC1-mediated processing of (H10D, P28K, and K29P)-human proinsulin. Protein Expr. Purif. 27, 210–219 [DOI] [PubMed] [Google Scholar]
- 28. Whittaker J., Whittaker L. (2005) Characterization of the functional insulin binding epitopes of the full-length insulin receptor. J. Biol. Chem. 280, 20932–20936 [DOI] [PubMed] [Google Scholar]
- 29. Sohma Y., Hua Q. X., Liu M., Phillips N. B., Hu S. Q., Whittaker J., Whittaker L. J., Ng A., Roberts C. T., Jr., Arvan P., Kent S. B., Weiss M. A. (2010) Contribution of residue B5 to the folding and function of insulin and IGF-I: constraints and fine-tuning in the evolution of a protein family. J. Biol. Chem. 285, 5040–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bang D., Kent S. B. (2004) A one-pot total synthesis of crambin. Angew. Chem. Int. Ed. Engl. 43, 2534–2538 [DOI] [PubMed] [Google Scholar]
- 31. Dawson P. E., Muir T. W., Clark-Lewis I., Kent S. B. (1994) Synthesis of proteins by native chemical ligation. Science 266, 776–779 [DOI] [PubMed] [Google Scholar]
- 32. Kent S. B. (2009) Total chemical synthesis of proteins. Chem. Soc. Rev. 38, 338–351 [DOI] [PubMed] [Google Scholar]
- 33. Winter J., Lilie H., Rudolph R. (2002) Renaturation of human proinsulin: a study on refolding and conversion to insulin. Anal. Biochem. 310, 148–155 [DOI] [PubMed] [Google Scholar]
- 34. Anfinsen C. B. (1973) Principles the govern the folding of protein chains. Science 181, 223–230 [DOI] [PubMed] [Google Scholar]
- 35. Dill K. A., Chan H. S. (1997) From Levinthal to pathways to funnels. Nat. Struct. Biol. 4, 10–19 [DOI] [PubMed] [Google Scholar]
- 36. Karplus M. (1997) The Levinthal paradox: yesterday and today. Fold. Des. 2, S69–S75 [DOI] [PubMed] [Google Scholar]
- 37. Sali A., Shakhnovich E., Karplus M. (1994) How does a protein fold?. Nature 369, 248–251 [DOI] [PubMed] [Google Scholar]
- 38. Zeldovich K. B., Shakhnovich E. I. (2008) Understanding protein evolution: from protein physics to Darwinian selection. Annu. Rev. Phys. Chem. 59, 105–127 [DOI] [PubMed] [Google Scholar]
- 39. Alm E., Baker D. (1999) Prediction of protein-folding mechanisms from free-energy landscapes derived from native structures. Proc. Natl. Acad. Sci. U.S.A. 96, 11305–11310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Creighton T. E. (1992) What the papers say: protein folding pathways determined using disulphide bonds. Bioessays 14, 195–199 [DOI] [PubMed] [Google Scholar]
- 41. Schaeffer R. D., Fersht A., Daggett V. (2008) Combining experiment and simulation in protein folding: closing the gap for small model systems. Curr. Opin. Struct. Biol. 18, 4–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 43. Dobson C. M. (2003) Protein folding and misfolding. Nature 426, 884–890 [DOI] [PubMed] [Google Scholar]
- 44. Connelly S., Choi S., Johnson S. M., Kelly J. W., Wilson I. A. (2010) Structure-based design of kinetic stabilizers that ameliorate the transthyretin amyloidoses. Curr. Opin. Struct. Biol. 20, 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ron D. (2002) Proteotoxicity in the endoplasmic reticulum: lessons from the Akita diabetic mouse. J. Clin. Invest. 109, 443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao L., Ackerman S. L. (2006) Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 18, 444–452 [DOI] [PubMed] [Google Scholar]
- 47. Fonseca S. G., Gromada J., Urano F. (2011) Endoplasmic reticulum stress and pancreatic β-cell death. Trends Endocrinol. Metab. 22, 266–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu M., Wan Z. L., Chu Y. C., Aladdin H., Klaproth B., Choquette M., Hua Q. X., Mackin R. B., Rao J. S., De Meyts P., Katsoyannis P. G., Arvan P., Weiss M. A. (2009) Crystal structure of a “nonfoldable” insulin: impaired folding efficiency despite native activity. J. Biol. Chem. 284, 35259–35272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Katsoyannis P. G. (1966) Synthesis of insulin. Science 154, 1509–1514 [DOI] [PubMed] [Google Scholar]
- 50. Sohma Y., Kent S. B. (2009) Biomimetic synthesis of lispro insulin via a chemically synthesized “mini-proinsulin” prepared by oxime-forming ligation. J. Am. Chem. Soc. 131, 16313–16318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bentley G., Dodson E., Dodson G., Hodgkin D., Mercola D. (1976) Structure of insulin in 4-zinc insulin. Nature 261, 166–168 [DOI] [PubMed] [Google Scholar]
- 52. De Meyts P., Whittaker J. (2002) Structural biology of insulin and IGF1 receptors: implications for drug design. Nat. Rev. Drug Discov. 1, 769–783 [DOI] [PubMed] [Google Scholar]
- 53. Liu M., Hodish I., Haataja L., Lara-Lemus R., Rajpal G., Wright J., Arvan P. (2010) Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol. Metab. 21, 652–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. DCCT. (1993) The Effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 329, 977–986 [DOI] [PubMed] [Google Scholar]
- 55. Nathan D. M., Cleary P. A., Backlund J. Y., Genuth S. M., Lachin J. M., Orchard T. J., Raskin P., Zinman B. (2005) Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N. Engl. J. Med. 353, 2643–2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fink A. L. (2005) Natively unfolded proteins. Curr. Opin. Struct. Biol. 15, 35–41 [DOI] [PubMed] [Google Scholar]
- 57. Avital-Shmilovici M., Mandal K., Gates Z. P., Phillips N. B., Weiss M. A., Kent S. B. (2013) Fully convergent chemical synthesis of ester insulin: determination of the high resolution x-ray structure by racemic protein crystallography. J. Am. Chem. Soc. 135, 3173–3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Menting J. G., Whittaker J., Margetts M. B., Whittaker L. J., Kong G. K., Smith B. J., Watson C. J., Záková L., Kletvíková E., Jiráček J., Chan S. J., Steiner D. F., Dodson G. G., Brzozowski A. M., Weiss M. A., Ward C. W., Lawrence M. C. (2013) How insulin engages its primary binding site on the insulin receptor. Nature 493, 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]