

Abstract
Guided by recent experimental results suggesting that protein-folding rates and mechanisms are determined largely by native-state topology, we develop a simple model for protein folding free-energy landscapes based on native-state structures. The configurations considered by the model contain one or two contiguous stretches of residues ordered as in the native structure with all other residues completely disordered; the free energy of each configuration is the difference between the entropic cost of ordering the residues, which depends on the total number of residues ordered and the length of the loop between the two ordered segments, and the favorable attractive interactions, which are taken to be proportional to the total surface area buried by the ordered residues in the native structure. Folding kinetics are modeled by allowing only one residue to become ordered/disordered at a time, and a rigorous and exact method is used to identify free-energy maxima on the lowest free-energy paths connecting the fully disordered and fully ordered configurations. The distribution of structure in these free-energy maxima, which comprise the transition-state ensemble in the model, are reasonably consistent with experimental data on the folding transition state for five of seven proteins studied. Thus, the model appears to capture, at least in part, the basic physics underlying protein folding and the aspects of native-state topology that determine protein-folding mechanisms.
Recent experimental results suggest that protein-folding rates and mechanisms are largely determined by native-state topology (1). First, dramatic changes in sequence have been found to have little effect on protein-folding rates. Laboratory generated variants of the SH3 domain and IgG binding domain of protein L with large numbers of sequence changes were found to fold at rates similar to that of the naturally occurring proteins, suggesting that the sequences of small proteins are not extensively optimized for rapid folding (2, 3). Furthermore, naturally occurring proteins with similar folds but very different sequences generally have similar folding rates (4). Second, for two distinct protein folds, the SH3 domain and CspB, there is evidence that folding mechanisms and transition-state structures are conserved among homologs despite differences in amino acid sequence and stability (4–6). Third, the folding rates of small proteins were found to be strongly correlated with a property of the native-state topology: the average sequence separation between residues that make contacts in the three-dimensional structure (the contact order) (7). Recent theoretical work is also consistent with the idea that topology is an important determinant of protein-folding mechanisms (8). Taken together, these results suggest that native-state topology is a key determinant of protein-folding mechanisms and of the distribution of structure in the transition-state ensemble.
These findings imply that a model of protein folding need not take into account the complex details of the interactions between residues or consider nonnative interactions in order to reproduce the general features of the folding landscape. Rather, a simple treatment of the interactions in the native protein may suffice to account for most of the experimental data available on the folding of small protein domains. We describe a simple model based on surface area burial and chain conformational entropy that shows promise in reproducing some of the general features of the protein-folding landscapes of real proteins.
Basic Assumptions.
The goal of our approach is to explicitly map out the folding free-energy landscape using information from the native protein structure. From the free-energy landscape, all of the observable properties of the folding process can be inferred, including folding rates and transition-state structure. Two key approximations make mapping out the free-energy landscape possible. The first is that only native interactions contribute significantly to the folding process; configurations including nonnative interactions are ignored. The second is that in any given configuration, each residue is either fully ordered as in the native state or completely disordered. Because each residue can be in one of two states, ordered or disordered, the folding free energy landscape that follows from these assumptions consists of 2n possible configurations, where n is the number of residues in the protein considered.
Enumerating Configurations.
Although the second approximation reduces the continuum of possible configurations to a finite number, for most proteins there are still too many to evaluate. To reduce further the number of configurations that must be considered, all ordered residues can be required to occur within a single contiguous stretch in the linear protein sequence [the single sequence approximation of helix–coil theory (9, 10)]. A more general approach is to allow multiple ordered segments that are distant in linear sequence to interact. Increasing the number of allowed segments increases both the complexity of the energy landscape and the total number of configurations that must be considered. We have adopted a compromise between the generality of the multiple-segment model and the simplicity of smaller models, which we call the sequential binary collision model: only two contiguous segments of the chain are allowed to be ordered in any given configuration; however, if all of the intervening residues between two ordered segments become ordered, a new segment may be added, because the two initial segments can be described by a single larger segment. When one ordered segment has a length of zero, this model is identical to the contiguous sequence model.
METHODS
Free Energy Function.
Once a suitable set of configurations has been chosen to serve as the basis set for a folding landscape, the free energy of each configuration must be calculated. We use a simple free-energy function,
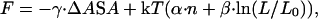 |
1 |
where the first term represents attractive native interactions, the second term, the entropic cost of ordering the residues in the ordered segments, and the third term is the entropic cost of closing the loop between the two ordered segments. The free-energy function for the contiguous sequence model contains only the first two terms, because only one ordered segment is allowed. A related free-energy function was developed by Finkelstein and Badretdinov (11).
The attractive native interactions [first term in Eq. 1] were taken to be proportional to the difference between the surface area buried by the ordered residues in the native state and the surface area buried by these residues in the denatured state. Total surface area burial (both hydrophobic and polar) was used; because only native interactions are considered, all burial is assumed to be favorable. The surface area buried by the ordered residues was computed from the positions of these residues in the native structures [1fmk (SH3), 1rnb (barnase), 3ci2 (CI-2), 2chf (Che Y,) residues 6-92 of chain 3 of 1lmb (λ repressor), residues 17–78 of 2ptl (protein L), and residues 10A through 81A from 1aye (procarboxypeptidase activation domain)] using the method of LeGrand et al. (12). The surface area buried in the unfolded state was taken to be the sum of the surface areas for each residue in its corresponding native tripeptide. γ in Eq. 1 was taken to be 16 cal/mol/Å2 (13). Specific hydrogen bonding patterns, van der Waals energy, and details of sidechain packing are not taken into account in this simple model. Such terms may not be relevant until late in the folding process when the protein structure is well determined, and thus may be more relevant to the analysis of protein unfolding and stability.
The conformational entropy lost on ordering a segment of consecutive residues was taken to be proportional to the number (n) of residues ordered (second term in Eq. 1). The model was tested on three of the proteins studied (the SH3 domain, barnase, and CI-2) over a range of values of α (corresponding to free energies of 1.6, 1.65, 1.7, 1.75, and 1.8 kcal/mol per residue at 298 K), in order to find the value that best characterized the experimentally observed features of the folding landscape. A fixed value equivalent to 1.75 kcal/mol per residue at 298 K was chosen on the basis of the position of the transition state ensemble on the reaction coordinate (compared to mf/m values) and on the performance of the model in reproducing experimentally observed φ-values for these three proteins. This is very close to the value of 1.8 kcal/mol per residue at 298 K estimated by Freire from calorimetric studies (14).
The decrease in entropy on loop closure in configurations in which there is more than one contiguous segment of ordered residues was estimated from off-lattice studies of loop closure frequencies in polypeptide chains (E.A. and D.B., unpublished results). The entropy of loop closure derived from these studies, S(loop closure)/k, was found to be well represented by β⋅ln(L/L0), where L is the length of the loop, β = 1.8 and L0 = 0.15. This expression is consistent with estimates from polymer theory and recent experimental results (11, 15–19).
Identifying Folding Pathways and Transition-State Configurations.
Folding kinetics are treated in the model by allowing only one residue to change state (from ordered to disordered or vice versa) at a time. Thus, a configuration with two ordered segments is kinetically connected only to configurations in which one of the two segments is unchanged and the other is one residue longer or shorter. With this constraint, folding pathways connecting the fully unfolded (all residues disordered) and folded (all residues ordered) configurations can be readily identified by using a standard recursive depth first-search algorithm. The kinetically relevant pathways are those that involve surmounting the lowest free-energy barriers; the lowest free-energy transition-state configurations are defined to be the highest free-energy configurations on the lowest free-energy folding pathways. A simple algorithm was used to identify these pathways and transition-state configurations:
0. Generate a list of all allowed configurations and their free energies.
1. Remove from the list the highest free-energy configuration
2. Determine whether there is still a path from the fully unfolded configuration to the fully folded configuration. If there is a path, go to step 1. If there is not a path, the configuration removed in step 1 is the lowest free-energy transition-state configuration.
The 100 lowest free-energy transition-state configurations were identified by repeatedly carrying out steps 0–2 and each time removing from the list the lowest free-energy transition-state configuration. The computational time needed to identify transition-state configurations was considerably reduced by replacing the linear search in step 1 with a bisection technique and by using a branch-and-bound strategy to determine if there is a path in step 2.
Calculation of φ-Values.
To compare with experimental data, φ-values were obtained by dividing the change in transition-state free energy accompanying a sidechain truncation by the change in native-state free energy. Because there were typically a collection of transition-state species of similar free energy, φ-values were computed for the 100 lowest free-energy transition-state configurations and were averaged.
Graphics.
All molecular structures shown were produced by using the rasmol and molscript software packages (20, 21). s-plus (Mathsoft, Seattle, WA) was used to generate images of the folding landscape for the two models described.
RESULTS AND DISCUSSION
Contiguous Sequence Model.
The major advantage of the contiguous sequence approximation is that the resulting folding landscapes can be represented directly on a two-dimensional plot. Fig. 1, column I, shows the contiguous sequence folding landscape for five proteins studied [several of these plots were shown for illustration in a recent review (1)]. The order parameter, Nf, used as the x axis in this and all subsequent plots, is the fraction of residues that are ordered; Nf = 0 (Left) corresponds to the fully unfolded state and Nf = 1 (Right) to the folded state (22). The y axis indicates the position of the ordered fragment, and the colors indicate free energy (the highest free-energy configurations are shown in yellow and the lowest, in blue). The folding landscapes for the SH3 domain, barnase, and Che Y each contain a distinct low free-energy pathway from the unfolded state to the native state. For the SH3 domain, this pathway is determined by a bottleneck on the folding landscape when about half of the residues in the protein are ordered: the 30-residue region centered at residue 35 is much lower in free energy than any other region of comparable size. This segment roughly corresponds to the region that has been shown experimentally to be important in stabilizing the folding transition state (5, 6). For Che Y, the stretch of low free-energy (blue) configurations that connects the unfolded and folded states along the N-terminal (Bottom) side of the landscape suggests that there is a low free-energy pathway that corresponds to ordering the protein from the N terminus to the C terminus. This is consistent with the experimental observation that the N terminus is ordered at the transition state (23). For barnase, the residues shown experimentally to be involved in the transition state are split between the C-terminal β-sheet and an N-terminal helix (24). Although, the contiguous sequence model correctly identifies one of these regions (the C-terminal beta sheet is the lowest free-energy structure with ∼40 residues ordered), the constraints of the model do not allow both regions to be identified. Thus, a major limitation of this simple model is the severe constraint that all configurations have only one stretch of ordered residues. CI-2 is another example of the limits of this simple model. The model correctly identifies the alpha helix (25) (the segment about 1/3 the length of the protein centered at residue 15) as a low free-energy configuration, but the helix cannot nucleate folding because of the large free-energy barrier to ordering the loop adjacent to the helix. In a more realistic model, the beta sheet could condense on the helix without fully ordering each intervening residue.
Figure 1.

Column I (free-energy landscape in the contiguous sequence model). Free-energy landscapes in the contiguous sequence model for the SH3 domain (row 1), CI-2 (row 2), barnase (row 3), Che Y (row 4), and λ repressor (row 5). In the contiguous sequence model, each configuration consists of a single stretch of ordered residues. Thus, each of the configurations on the free-energy landscape can be uniquely identified by two parameters, namely the length of the stretch of ordered residues or, equivalently, the fraction of residues ordered, Nf (x axis) and the location of the center of the ordered segment (y axis). Colors indicate free energy and are computed from Eq. 1 (without the loop entropy term); black indicates 0 kcal/mol, and white indicates 30 kcal/mol for SH3, 25 kcal/mol for CI-2 and λ repressor, 35 kcal/mol for Che Y, and 40 kcal/mol for barnase. The color scheme used to represent free energies between 0 kcal/mol and the upper bounds is black–blue (0–25% of upper bound), blue–magenta (25–50%), magenta–red (50–75%), red–yellow (75–88%) and yellow–white (88–100%). Column II (hierarchy of structure formation). The frequencies with which individual residues were ordered in a Boltzmann weighted ensemble of the configurations available under the sequential binary collision model are shown for each value of Nf (see text); the y axis is position along the sequence. Colors indicate the frequencies with which residues were ordered (black–blue, 0.–0.25; blue–magenta, 0.25–0.50; magenta–red, 0.50–.75; red–yellow, 0.75–0.88; and yellow–white, 0.88–1.00). Column III (structure in the transition-state ensemble). The number of times each residue was ordered in the top 100 transition states is shown as a function of Nf; the color scheme (with counts taking the place of frequencies) and axes are as in Column II. Column IV (free energy profile). The free energy, as a function of Nf, was computed from the partition function for each value of Nf as described in the text. The x axis is the reaction coordinate, Nf, and the y axis is the free energy in kcal/mol.
Free-Energy Profile of the Sequential Binary Collision Model.
The sequential binary collision model overcomes some of the limitations of the contiguous sequence model by allowing two segments of the protein distant in the linear sequence to come together in three dimensions. The full free-energy landscape for the sequential binary collision model cannot be represented easily in a two-dimensional graph as is the case for the contiguous sequence model. A free-energy profile, however, can be generated for this model with the caveat that the profile may include contributions from states that are not kinetically accessible. The free energy as a function of Nf,
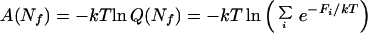 |
is shown in Fig. 1, column IV, where Fi is the free energy of configuration i (Eq. 1), and the sum is over all configurations with a particular Nf. For all of the proteins studied, the free energy of the folded state was found to be higher than that of the unfolded state by 5–15 kcal/mol. This lack of agreement with experimentally determined stabilities was not unexpected, because only contributions from surface area burial were included in our simple energy function (a more sophisticated energy function may not be appropriate because partially ordered conformations are probably not well ordered enough to receive the full stabilizing energies of most hydrogen bonds and van der Waals interactions). Of particular interest in Fig. 1, column IV, is the ruggedness of the free-energy profile for barnase and Che Y. Because of the limitations of our energy function in correctly estimating the stability of completely ordered proteins, the large dips in the profiles for these proteins may actually be lower than indicated if they involve the condensation of a fragment of the protein into a well structured subdomain; these local energy minima may correspond to the kinetic intermediates observed in the folding of these two proteins.
Hierarchy of Structure Formation.
To exhibit the hierarchy of structure formation in the sequential binary collision model, Fig. 1, column II, displays how often each residue would be ordered in the “thermodynamic limit” where each configuration is populated according to its Boltzmann weight, and kinetic barriers are ignored. All configurations were enumerated, and the Boltzmann weighted average frequency with which each residue was ordered
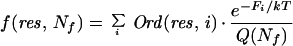 |
is shown as a function of Nf, where Ord(res, i) = 1 if residue res is ordered in the ith configuration and Ord(res, i) = 0 otherwise, and the sum is over all configurations at the specified value of Nf.
For SH3, it is clear that the first regions of structure to emerge (at Nf ∼ 0.2) are the distal loop (the blue/red region centered near residue 40) and the RT loop (the blue–red region centered near residue 10); however, when about half of the residues in the protein are ordered (at Nf ∼ 0.5), there is little structure in the RT loop, suggesting that the structure observed there early in the folding pathway does not serve to nucleate folding; in this view of the folding landscape, the early structure seen in the RT loop can be described as “off-pathway.” In contrast, the regions structured at Nf ∼ 0.5 remain structured throughout the rest of the folding pathway. Interestingly, the regions of the SH3 domain experimentally determined to be structured in the folding transition state are nearly identical to those structured in this representation of the folding landscape at Nf ∼ 0.5.
In the CI-2 free energy landscape, the alpha helix (centered at residue 15) exhibits some structure early but does not become completely ordered until residues in the C-terminal β-strands (near residue 50) are also ordered at Nf ∼ 0.6. The free-energy landscape for the λ repressor fragment does not suggest a well defined pathway in the thermodynamic limit; when about half the residues in the protein are ordered (Nf ∼ 0.5), there is a roughly uniform distribution of structure throughout the protein that gradually becomes more ordered until the protein reaches its native configuration (at Nf = 1). By contrast, the free-energy landscape for Che Y suggests an unambiguous folding mechanism in which the N-terminal region of the protein forms structure first and the C-terminal region becomes structured last.
In the barnase folding landscape, the beta sheet becomes ordered first, then the protein condenses to a low free-energy configuration (based on the one-dimensional free-energy profile in Fig. 1, column V) that includes some interactions between the first alpha-helix (centered at residue 10) and the C-terminal beta sheet. Interestingly, the distribution of structure in the configurations that correspond to the first local minimum on the free-energy profile is in qualitative agreement with the distribution of experimental φ-values for the barnase folding intermediate (26).
Distribution of Structure in the Transition-State Ensemble.
Although Fig. 1, column II, gives a rough idea of which regions of a protein might be ordered at a given point in the folding reaction, it is not completely rigorous because some low free-energy configurations may not be kinetically accessible. To overcome this limitation, individual transition-state configurations (defined as the highest free-energy configurations on low free-energy paths from the unfolded to folded states) were identified (see Methods). Fig. 1, column III, shows the occupancy by residue of the 100 lowest free-energy transition-state configurations. These configurations are distributed over a range of values of the reaction coordinate (Nf). For the SH3 domain and Che Y, all of the top transition states have the same general distribution of structure. In contrast, barnase appears to have two structurally distinct transition-state ensembles: one that corresponds to forming the β-sheet alone, and one that corresponds to bringing the N-terminal helix onto the sheet. The placement of these transition states on the reaction coordinate, Nf, agrees roughly with the position of the corresponding local maxima on the free-energy profile (Fig. 1, column IV) for barnase. The CI-2 and λ-repressor transition-state ensembles exhibit more structural diversity than those of SH3 and Che Y, but are not easily separable into distinct ensembles as is the case for barnase.
An important issue in computational studies of folding is whether purely thermodynamic approaches, which search for the maximum in the free energy along some suitably chosen reaction coordinate, can accurately identify the folding barrier (27, 28). Although there is a general correspondence between the peak in the free-energy profile and the location of the transition-state configurations, there are some transition-state configurations with values of Nf significantly different from that of the maximum in the free-energy profile, suggesting that in this system Nf is a good but not perfect reaction coordinate. For minimally frustrated systems, such as small proteins, with funnel-like free-energy landscapes, any order parameter that reflects the amount of native structure (the number of native contacts, the number of residues ordered, etc.) is likely to be a reasonable reaction coordinate (29).
The distributions of transition-state configurations (Fig. 2, column III) show that the breadth of the transition-state ensemble (30, 31) varies considerably among the five proteins shown. A complementary estimate of the diversity among the configurations sampled during folding is provided by Y(Nf), which measures the inverse number of thermally accessible states available to the system at a given value of Nf (32) (Y(Nf) = Σi(Pi2), where the sum is over all configurations at a particular value of Nf, and Pi is the Boltzmann weighted probability of observing the system in configuration i). Interestingly, for all of the proteins except λ repressor, a sharp reduction in the number of thermally accessible states was observed just after the transition barrier [the peak in Y(Nf) for the SH3 domain is evident near Nf ∼ 0.8 in Fig. 2 (solid line)]. For λ repressor (Fig. 2, dashed line), the peak in Y(Nf) was absent, and the average value of Y(Nf) was lower than for the other proteins. This result is intriguing given the relatively large variation in the position of the transition state in λ repressor mutants (33); the position of the transition state may be less rigidly constrained in λ repressor because the large number of thermally accessible states provides a number of alternative folding trajectories that can become populated in response to mutations [such behavior was observed in a recent diffusion-collision model of λ repressor folding (34)].
Figure 2.

Distribution of thermally accessible states. Y(Nf) = Σi(Pi2), which represents the inverse number of thermally accessible states available to the system, was computed for all values of Nf, and the result for SH3 and γ repressor is shown. The sharp peak near Nf ∼ 0.8 for the SH3 domain (solid line) indicates a drastic reduction in the number of accessible states late in folding that is also seen for CI-2, barnase, and Che Y. By contrast, λ-repressor (dotted line) does not exhibit such a drastic reduction in accessible states, suggesting a more plastic folding mechanism.
Comparison with Experiment.
There are two sets of experimental observables that can be used to test the sequential binary collision model: protein folding rates and φ-values. Folding rates can be approximated directly from the height of the free-energy barriers involved in folding. For the small data set studied under the computationally expensive sequential binary collision model, there is little correlation between folding rates and barrier height. A larger set of protein kinetic data was compared with barrier height under the contiguous sequence model, and although there was a correlation between barrier height and folding rates, the relative contact order (a simple measure of the balance between local and nonlocal contacts) was a better predictor of folding rates (7). Given the very large numbers being subtracted in each model to give the net free energy for individual configurations, it is not surprising that there is some error in calculating barrier height precisely.
φ-Values suggest which residues are important in stabilizing the folding transition state and provide a means to test whether the transition-state ensemble identified using the model involves the correct region of the protein. A φ-value of 0 indicates the residue probed does not contribute to the transition-state stability, whereas a φ-value of 1 indicates that the residue stabilizes the transition state to the same extent that it stabilizes the native state (35). Folding φ-values were computed directly from the top 100 transition-state structures (shown in Fig. 1, column III) and were averaged. Results did not change significantly when only 10 transition-state structures were used but did become less accurate when only the top transition state was used to compute φ-values. On a residue-by-residue basis, computed φ-values correlated reasonably well with those determined experimentally for five of the seven proteins we studied [the SH3 domain (r = .50), CI-2 (r = .58), barnase (r = .61), Che Y (r = .87), and λ-repressor (r = .71), where r is the correlation coefficient between the predicted and experimental φ-values; (Fig. 3)]. Of these, Che Y and λ repressor, which were not used to optimize the conformational entropy parameter (see Methods), demonstrated the best agreement between computed and experimental φ-values. The disagreement between computed and experimental φ-values for the SH3 domain is primarily because of the fact that the model predicts a mostly ordered transition-state configuration including the distal loop, diverging turn, and half of the RT loop, while experimental data indicate only the distal loop and diverging turn are ordered. This is precisely the region of the protein ordered at Nf ∼ 0.5 in the model (Fig. 2, column II).
Figure 3.

Comparison of computed and experimental φ values. (Left) Protein structures are shown colored by φ-values computed directly from the sequential binary collision model. Red indicates residues that are important in stabilizing the folding transition state (high φ-values), and blue indicates residues that do not stabilize the transition state. (Right) Structures colored by experimentally observed φ-values; residues with a phi-value of 0 are colored blue, residues with a φ-value of 1 are colored red; purple shades indicate residues with intermediate φ-values, and white regions indicate residues for which there are no experimental data.
The predictions for two of the seven proteins, the procarboxypeptidase activation domain and the IgG binding domain of protein L, did not agree with the experimentally determined transition-state structures. For protein L, the failure of the model probably results from the neglect of local sequence-structure propensities. Protein L possesses a high degree of structural symmetry: an N-terminal β hairpin is followed by a single helix and a C-terminal β hairpin. The two β-hairpins bury very similar amounts of surface area within themselves and against the helix, and thus are computed to have similar free energies of ordering in the model, yet only the N-terminal half is experimentally observed to be ordered in the folding transition-state ensemble (36) (D. Kim and D.B., unpublished observations). A constant value of conformational entropy per residue ignores sequence biases for specific local structures that may be responsible for the preferential formation of the first β hairpin (recent data suggest that the first hairpin is already populated to some extent in the denatured state; Q. Yi and D.B., unpublished observations). A more sophisticated approach might use database statistics to estimate the free-energy cost associated with formation of specific local structures.
CONCLUSIONS
The remarkable result of this study is that a very simple model for the folding free-energy landscape combined with a crude energy function is able to reproduce the qualitative features of the transition-state ensemble for five of the seven proteins studied. The success of the simple model combined with recent experimental results supports the idea that protein-folding landscapes are shaped primarily by gross topological features rather than by the details of interresidue interactions.
While completing this work, we became aware of very complementary studies being carried out by Munoz and Eaton at the National Institutes of Health and Galzitskaya and Finkelstein in Moscow (Russian Academy of Sciences). Munoz and Eaton (38) used the single-sequence approximation and a secondary structure-dependent free-energy cost for chain ordering and were successful in predicting protein-folding rates after setting the strength of interresidue interactions to reproduce native-state stabilities. Finkelstein and coworkers used an elegant dynamic programming method to identify transition states for multisegment models and succeeded in predicting φ-values distributions for several proteins (37). The common theme of all three approaches is a native-state centric description of the folding free-energy landscape (nonnative interactions are completely neglected); the success achieved by these and other simple models (8) suggests that the essential physics underlying the folding of small proteins may be simple indeed.
Acknowledgments
We thank Alexei Finkelstein for communicating results before publication and Peter Wolynes and members of the Baker group for helpful comments on the manuscript. This work was supported by a grant from the National Institutes of Health (NIH), and young investigator awards to D.B. from the National Science Foundation and the Packard Foundation, and a Molecular Biophysics predoctoral fellowship from the NIH to E.A.
Footnotes
This paper was submitted directly (Track II) to the Proceedings office.
References
- 1.Alm E, Baker D. Curr Opin Struct Biol. 1999;9:189–196. doi: 10.1016/S0959-440X(99)80027-X. [DOI] [PubMed] [Google Scholar]
- 2.Kim D E, Baker D. Proc Natl Acad Sci USA. 1998;95:4982–4986. doi: 10.1073/pnas.95.9.4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riddle D S, Santiago J V, Bray-Hall S T, Doshi N, Grantcharova V P, Yi Q, Baker D. Nat Struct Biol. 1997;4:805–809. doi: 10.1038/nsb1097-805. [DOI] [PubMed] [Google Scholar]
- 4.Perl D, Welker C, Schindler T, Schroder K, Marahiel M A, Jaenicke R, Schmid F X. Nat Struct Biol. 1998;5:229–235. doi: 10.1038/nsb0398-229. [DOI] [PubMed] [Google Scholar]
- 5.Grantcharova V P, Riddle D S, Santiago J V, Baker D. Nat Struct Biol. 1998;5:714–720. doi: 10.1038/1412. [DOI] [PubMed] [Google Scholar]
- 6.Martinez J C, Pisabarro M T, Serrano L. Nat Struct Biol. 1998;5:721–729. doi: 10.1038/1418. [DOI] [PubMed] [Google Scholar]
- 7.Plaxco K W, Simons K T, Baker D. J Mol Biol. 1998;277:985–994. doi: 10.1006/jmbi.1998.1645. [DOI] [PubMed] [Google Scholar]
- 8.Portman J J, Takada S, Wolynes P G. Phys Rev Lett. 1998;81:5237–5240. [Google Scholar]
- 9.Schellman J A. J Phys Chem. 1958;62:1485–1494. [Google Scholar]
- 10.Munoz V, Thompson P A, Hofrichter J, Eaton W A. Nature (London) 1997;390:196–1999. doi: 10.1038/36626. [DOI] [PubMed] [Google Scholar]
- 11.Finkelstein A V, Badretdinov A Y. Fold Des. 1997;2:115–121. doi: 10.1016/s1359-0278(97)00016-3. [DOI] [PubMed] [Google Scholar]
- 12.Le Grand S M, Merz K M., Jr J Comput Chem. 1993;14:349–352. [Google Scholar]
- 13.Eisenberg D, McLachlan A D. Nature (London) 1986;319:199–203. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 14.D’Aquino J A, Gomez J, Hilser V J, Lee K H, Amzel L M, Freire E. Proteins. 1996;25:143–156. doi: 10.1002/(SICI)1097-0134(199606)25:2<143::AID-PROT1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 15.Jacobson H, Stockmayer W H. J Chem Phys. 1950;18:1600–1606. [Google Scholar]
- 16.Chan H S, Dill K A. J Chem Phys. 1990;92:3118–3135. [Google Scholar]
- 17.Nagi A D, Regan L. Fold Des. 1997;2:67–75. doi: 10.1016/S1359-0278(97)00007-2. [DOI] [PubMed] [Google Scholar]
- 18.Ladurner A G, Fersht A R. J Mol Biol. 1997;273:330–337. doi: 10.1006/jmbi.1997.1304. [DOI] [PubMed] [Google Scholar]
- 19.Viguera A R, Serrano L. Nat Struct Biol. 1997;4:939–946. doi: 10.1038/nsb1197-939. [DOI] [PubMed] [Google Scholar]
- 20.Sayle R A, Milner-White E J. Trends Biochem Sci. 1995;20:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 21.Kraulis P J. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- 22.Wolynes P G. Proc Natl Acad Sci USA. 1997;94:6170–6175. doi: 10.1073/pnas.94.12.6170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.López-Hernández E, Serrano L. Fold Des. 1996;1:43–55. [PubMed] [Google Scholar]
- 24.Serrano L, Matouschek A, Fersht A R. J Mol Biol. 1992;224:805–818. doi: 10.1016/0022-2836(92)90563-y. [DOI] [PubMed] [Google Scholar]
- 25.Itzhaki L S, Otzen D E, Fersht A R. J Mol Biol. 1995;254:260–288. doi: 10.1006/jmbi.1995.0616. [DOI] [PubMed] [Google Scholar]
- 26.Matouschek A, Serrano L, Fersht A R. J Mol Biol. 1992;224:819–835. doi: 10.1016/0022-2836(92)90564-z. [DOI] [PubMed] [Google Scholar]
- 27.Socci N D, Onuchic J N, Wolynes P G. J Chem Phys. 1996;104:5860–5868. [Google Scholar]
- 28.Pande V S, Grosberg A Yu, Tanaka T, Rokhsar D S. Curr Opin Struct Biol. 1998;8:68–79. doi: 10.1016/s0959-440x(98)80012-2. [DOI] [PubMed] [Google Scholar]
- 29.Bryngelson J D, Onuchic J N, Socci N D, Wolynes P G. Proteins Struct Funct Genet. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 30.Thurmalai D, Klimov D K. Fold Des. 1998;13:R112–R118. doi: 10.1016/S1359-0278(98)00057-1. [DOI] [PubMed] [Google Scholar]
- 31.Shakhnovich E I. Fold Des. 1998;13:R108–R111. doi: 10.1016/s1359-0278(98)00056-x. [DOI] [PubMed] [Google Scholar]
- 32.Onuchic J N, Wolynes P G, Luthey-Schulten Z, Socci N D. Proc Natl Acad Sci USA. 1995;92:3626–3630. doi: 10.1073/pnas.92.8.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burton R E, Huang G S, Daugherty M A, Calderone T L, Oas T G. Nat Struct Biol. 1997;4:305–310. doi: 10.1038/nsb0497-305. [DOI] [PubMed] [Google Scholar]
- 34.Burton R E, Myers J K, Oas T G. Biochemistry. 1998;37:5337–5343. doi: 10.1021/bi980245c. [DOI] [PubMed] [Google Scholar]
- 35.Fersht A R. Curr Opin Struct Biol. 1994;5:79–84. doi: 10.1016/0959-440x(95)80012-p. [DOI] [PubMed] [Google Scholar]
- 36.Gu H, Kim D, Baker D. J Mol Biol. 1997;274:588–596. doi: 10.1006/jmbi.1997.1374. [DOI] [PubMed] [Google Scholar]
- 37.Galzitskaya O V, Finkelstein A V. Proc Natl Acad Sci USA. 1999;96:11299–11304. doi: 10.1073/pnas.96.20.11299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munoz V, Eaton W A. Proc Natl Acad Sci USA. 1999;96:11311–11316. doi: 10.1073/pnas.96.20.11311. [DOI] [PMC free article] [PubMed] [Google Scholar]