

Significance
Fermentation of dietary fiber in the lower gut of humans is a critical process for the function and integrity of both the bacterial community and host cells. Here we demonstrate that two human gut commensal Bacteroides are equipped with unique enzymes that allow degradation of xylan, a common hemicellulose in human diets. Furthermore, we identify a novel carbohydrate-binding module (CBM) family that disrupts the catalytic domain of a glycoside hydrolase 10 (GH10) endoxylanase and facilitates the hydrolytic activity of the enzyme. The conservation of the unique modular architecture of the GH10 endoxylanase in the genomes of diverse Bacteroidetes suggests a critical role in fiber digestion in this phylum.
Keywords: RNAseq, gut microbiota, xylanolytic bacteria, hemicellulose, human nutrition
Abstract
Enzymes that degrade dietary and host-derived glycans represent the most abundant functional activities encoded by genes unique to the human gut microbiome. However, the biochemical activities of a vast majority of the glycan-degrading enzymes are poorly understood. Here, we use transcriptome sequencing to understand the diversity of genes expressed by the human gut bacteria Bacteroides intestinalis and Bacteroides ovatus grown in monoculture with the abundant dietary polysaccharide xylan. The most highly induced carbohydrate active genes encode a unique glycoside hydrolase (GH) family 10 endoxylanase (BiXyn10A or BACINT_04215 and BACOVA_04390) that is highly conserved in the Bacteroidetes xylan utilization system. The BiXyn10A modular architecture consists of a GH10 catalytic module disrupted by a 250 amino acid sequence of unknown function. Biochemical analysis of BiXyn10A demonstrated that such insertion sequences encode a new family of carbohydrate-binding modules (CBMs) that binds to xylose-configured oligosaccharide/polysaccharide ligands, the substrate of the BiXyn10A enzymatic activity. The crystal structures of CBM1 from BiXyn10A (1.8 Å), a cocomplex of BiXyn10A CBM1 with xylohexaose (1.14 Å), and the CBM from its homolog in the Prevotella bryantii B14 Xyn10C (1.68 Å) reveal an unanticipated mode for ligand binding. A minimal enzyme mix, composed of the gene products of four of the most highly up-regulated genes during growth on wheat arabinoxylan, depolymerizes the polysaccharide into its component sugars. The combined biochemical and biophysical studies presented here provide a framework for understanding fiber metabolism by an important group within the commensal bacterial population known to influence human health.
The human lower gastrointestinal tract (GIT) is home to trillions of bacteria that equip humans with biological activities that are absent in human cells (1–3). The microbes within the human lower gut perform a wide variety of functions important for normal physiology, including promoting maturation of the immune system (4), development of the GIT (5), and improving energy capture from dietary components (6). Moreover, dysbiosis of the microbiota is associated with various diseases including inflammatory bowel disease (7), obesity (8), and atherosclerotic cardiovascular disease (9, 10). The importance of the microbial community in the GIT to human health has prompted analysis of gut bacteria on a massive scale. Using recent advances in DNA sequencing technologies, investigators have captured the diversity of organisms in these complex microbial assemblages (11, 12) and have generated enormous catalogs of microbial genes (1, 3, 13). However, the rapid accumulation of DNA sequences has far surpassed our ability to assign function to genes. Indeed, ∼75% of genes encoded by the gut microbiota have gene products that cluster into unknown orthologous groups or represent entirely novel protein families (3).
One of the most important functional roles specific to the lower gut microbiota is the degradation and fermentation of dietary fiber that is resistant to digestion by human enzymes. The enzymes involved in breaking down polysaccharides are members of carbohydrate active enzymes (CAZymes) and are categorized into families of carbohydrate-binding modules (CBMs), carbohydrate esterases (CEs), glycoside hydrolases (GHs), glycoside transferases (GTs), or polysaccharide lyases (PLs) (14). The CAZy gene catalog of the human genome pales in comparison with that of the gut microbiota, which exceeds the total number of human CAZymes by at least 600-fold (15). This observation suggests that humans rely heavily on their gut microbes to extract energy from dietary carbohydrates (16), especially in their lower GIT. Despite the importance of these genes in energy capture by the gut microbiota, very little is known of the function of their encoded polypeptides.
Xylan is the second most abundant structural polysaccharide in plant cell walls and is particularly high in cereal grains, a significant component of the human diet (17). Moreover, the digestibility rates of xylan in the human GIT are estimated at about 72% (18). Of the bacteria in the human gut, the genus Bacteroides possesses the most expanded glycolytic gene repertoires that target xylan degradation (19, 20). These organisms use a highly specialized system consisting of a core set of polysaccharide-binding proteins, outer membrane transporters, and glycolytic enzymes to cleave large polysaccharides into oligosaccharides that are then transported by TonB-dependent transporters into the periplasm. An arsenal of glycolytic enzymes then converts the oligosaccharides into fermentable monosaccharides (21). The xylan utilization system (XUS) appears to be conserved across xylan-degrading Bacteroidetes from different gastrointestinal systems (20, 22), but the roles of individual genes in the gene cluster have not been defined.
Xylan degradation is not universally shared by the Bacteroidetes in the human gut; rather, this capacity varies among different species and even between different strains. As recently reviewed (20), human gut Bacteroides that have the capacity to degrade xylan include strains of Bacteroides eggerthii, Bacteroides cellulosilyticus, Bacteroides intestinalis, Bacteroides ovatus, and Bacteroides xylanisolvens. Partial genome sequences for several strains of these bacteria are present in the publicly available databases, such as the National Center for Biotechnology Information GenBank. Among these xylanolytic bacteria, B. ovatus American Type Culture Collection (ATCC) 8483 and B. intestinalis Deutsche Sammlung von Mikroorganismen or German Culture Collection of Microorganisms (DSM) 17393 stand out from all other gut bacteria sequenced to date as having the most highly expanded complement of CAZymes (23). B. intestinalis DSM 17393 was isolated from fecal samples of Japanese volunteers using a selective medium for polyamine-producing bacteria (24). The organism has a large (∼60 kb) region of its genome dedicated to xylan degradation. B. ovatus ATCC 8483 is a human commensal bacterium known to ferment a wide variety of plant- and animal-derived glycans (19, 25). Although B. ovatus ATCC 8483 is genetically distinct from B. intestinalis DSM 17393, its genome sequence has revealed a similar abundance of xylan-degrading genes. A recent study demonstrated that a large number of B. ovatus xylanolytic genes are induced during growth with xylan (19). Furthermore, a hybrid two-component system (HTCS) regulator whose periplasmic-sensing domain binds xylooligosaccharides was identified adjacent to the highly induced xylanolytic genes, suggesting a role in the transcriptional response to xylan (19). Despite the vast diversity of xylan-degrading genes in these two organisms, very little is known about the specific roles of the individual genes in xylan utilization.
In the current study, we used whole-genome transcriptome sequencing to guide the study of genes with roles in xylan degradation by the two human gut commensal bacteria B. intestinalis DSM 17393 and B. ovatus ATCC 8483. The most highly up-regulated genes encode BiXyn10A and BACOVA_04390, two polypeptides with an unusual modular organization that is widespread, especially in the human lower gut Bacteroidetes. The transcriptome data from another gut Bacteroidetes (the ruminal bacterium Prevotella bryantii B14) showed that a gene encoding a polypeptide with similar architecture to BiXyn10A and BACOVA_04390 is the most highly up-regulated CAZy gene when the bacterium is metabolizing xylan (22). BiXyn10A and BACOVA_04390 are linked to a xylan utilization core cluster of genes that seem invariant in the genomes of xylan-degrading Bacteroidetes. Here, we present the structure/function analyses of BiXyn10A, a member of the Bacteroidetes cardinal xylan degradation enzymes, and show that the enzyme and its homologs harbor a novel group of CBMs that impact their capacity to degrade xylans. Our results provide an important framework for understanding xylan catabolism in the Bacteroidetes, a significant group of human gut commensal bacteria.
Results
Transcriptional Analysis of B. intestinalis DSM 17393 and B. ovatus ATCC 8483 Reveals an Extensive Repertoire of CAZymes.
Within the human gut bacterium B. intestinalis DSM 17393, 67 genes are highly up-regulated (>fivefold) during growth on the complex polysaccharide wheat arabinoxylan (WAX) (SI Appendix, Table S4) compared with growth on the monosaccharide xylose. Twenty-six of the up-regulated genes are predicted to encode CAZymes belonging to GH and CE families, and a number of the encoded proteins exhibited unusual modular arrangements (Fig. 1A). Most of the up-regulated CAZymes possess N-terminal signal peptides, suggesting that they are transported to the periplasm or outer membrane during xylan metabolism. The most highly induced CAZy gene (BACINT_04215) was up-regulated by 170-fold and encodes a modular enzyme defined by a GH10 polypeptide interrupted by an amino acid sequence with low similarity to hitherto characterized polypeptides. The next most highly up-regulated gene (135-fold) encodes a GH43 protein (BACINT_00570), followed by two genes encoding polypeptides with unusual arrangements of GH43 and GH10 modules. The arrangement of the catalytic modules between these two bimodular proteins has been flipped, with one having a GH43/GH10 arrangement (BACINT_00569) and the other a GH10/GH43 arrangement (BACINT_04202). An unexpected architectural arrangement common to the polypeptides encoded by several of the up-regulated genes consisted of GH modules inserted within or linked to CE modules, suggesting that the corresponding gene products attack both the ester-linked side chains and the sugar backbone. Thus, the transcriptome analysis revealed a great diversity in the modularity of the polysaccharides encoded by B. intestinalis genes, indicating a highly versatile capacity in xylan degradation.
Fig. 1.

CAZyme encoding genes up-regulated on soluble WAX compared with xylose. Each B. intestinalis DSM 17393 (A) or B. ovatus ATCC 8483 (B) gene that was overexpressed at least fivefold was used as a query in a BLASTp search of the nonredundant (nr) database at GenBank. Genes were assigned to CAZy (14) families if they exhibited significant similarity (E-value <1 × 10−5) to biochemically characterized proteins already cataloged in a CAZy family. The CAZy annotations were verified using the dbCAN server (47), and additional domains were predicted using both Pfam (48) and the conserved domain database (49). Signal peptides and lipoprotein signal sequences were predicted using SignalP v4.0 (50) and LipoP v1.0 (51), respectively.
A total of 63 genes were up-regulated more than fivefold by B. ovatus ATCC 8483 during growth on xylan relative to xylose (SI Appendix, Table S5). These genes included a similarly expansive complement of CAZy genes possessing unique domain arrangements to B. intestinalis DSM 17393 (Fig. 1B). Notable differences between the induced CAZy gene sets include the lack of GH8 enzyme induction by B. ovatus ATCC 8483, a reduced number of GH10 endoxylanases, and the presence of two GH115 α-glucuronidases not encoded by B. intestinalis DSM 17393. Importantly, the most highly induced CAZy gene for both B. ovatus ATCC 8483 and B. intestinalis DSM 17393 encodes an enzyme having the same predicted domain arrangement (BACINT_04215, 170-fold; BACOVA_04390, 335-fold). Both of these polypeptides are predicted to encode a GH10 catalytic module containing a 250 amino acid insertion sequence of unknown function. Investigating the biochemistry of BACINT_04215 and BACOVA_04390 should enhance our understanding of xylan hydrolysis and fermentation by the two gut bacteria, and in the ensuing analysis, we focused on biochemical characterization of the B. intestinalis homolog.
The Most Highly Up-Regulated CAZy Gene Encodes an Endoxylanase.
In B. intestinalis, the most highly up-regulated genes are contained within a single transcriptional unit comprising BACINT_04220-04215 (Fig. 2A). This gene cluster consists of two tandem repeats of susC and susD homologs, predicted to encode TonB-dependent outer membrane porins (XusA/C) and polysaccharide-binding proteins (XusB/D), respectively. Downstream of these genes is a gene encoding a hypothetical protein, followed by Bixyn10A. Given the unique domain architecture of BiXyn10A (Fig. 1A) and the fact that it is the most highly induced CAZy gene, we characterized the biochemical properties of this protein. The gene product of Bixyn10A was purified to near homogeneity and incubated with the WAX substrate used in the RNAseq experiment and also with the insoluble substrate oat spelt xylan (OSX). As shown in Fig. 2B, the polypeptide released reducing ends from both substrates. End product analysis, using TLC, revealed oligosaccharides (Fig. 2C), verifying that the protein possesses endoxylanase activity. A mutation in a glutamate (E654A) corresponding to the highly conserved catalytic nucleophile in GH10 proteins abolished hydrolytic activity on xylan substrates (Fig. 2 B–D), indicating that catalytic activity was restricted to the GH10 sequence and independent of the 250 amino acid insertion sequence.
Fig. 2.

Identification of BACINT_04125 as the most highly up-regulated CAZyme encoding gene and functional assignment as an endoxylanase (BiXyn10A). (A) Transcriptome map showing RNAseq coverage of the PUL containing BACINT_04215. (B and C) Demonstration of endoxylanase activity. BACINT_04215 WT and its E654A mutant were incubated with WAX or OSX for 16 h, and the products were analyzed using a reducing sugar assay (B) and by TLC (C). In D, the hydrolytic action on oligosaccharides was analyzed by TLC.
Delineation of Two Novel CBMs in the Unknown Region of BiXyn10A.
To identify the function of the amino acid sequence interrupting BiXyn10A, we expressed the coding sequence of the cryptic 250 amino acid polypeptide and purified the corresponding product for biochemical characterization (TM1; Fig. 3 A and B). Based on initial bioinformatics studies suggesting that the insertion sequence is a CBM, we carried out native gel analysis to evaluate potential carbohydrate-binding activity. An extensive library of soluble polysaccharides was used as substrates, as described in Experimental Procedures. As shown in Fig. 3C, addition of WAX to the gel retarded the migration of TM1, whereas the control protein BSA migrated to the same position in the two gels, irrespective of the presence of WAX (Fig. 3C, control and WAX). These results showed that the insertion sequence is a hitherto uncharacterized CBM. CBMs are usually about 90–120 amino acids in length. Therefore, the insertion sequence was unusually large for a single CBM. Bioinformatic analysis coupled with truncational and biochemical analyses demonstrated that the unknown region contains two CBMs, which we designated BiXyn10A CBM1 and BiXyn10A CBM2, and protein constructs encompassing each of these CBMs are designated as TM2 and TM3, respectively. The evidence for the two different CBMs is presented in Fig. 3C (control and WAX), where we demonstrate binding to the WAX-infused gel by TM2 and TM3 each. Lichenan and to a lesser extent laminarin retarded the migration of TM1 and TM3, however the migration of TM2 appeared unaffected (Fig. 3C). This suggested that CBM2 differs in its binding specificity compared with CBM1. Very minor changes in migration were seen for TM3 with xyloglucan and konjac glucomannan (Fig. 3C), whereas galactan, arabinogalactan, carboxymethyl cellulose (CMC), and linear or debranched arabinan did not alter the migration pattern of TM1, TM2, or TM3, suggesting that these polysaccharides are not recognized by the CBMs in BiXyn10A (SI Appendix, Fig. S2).
Fig. 3.

Truncational analysis of BiXyn10A reveals two CBMs in tandem in the polypeptide. (A) Domain organization of BiXyn10A (WT) and truncational mutants (TMs). (B) Purification of BiXyn10A WT and TMs. (C) Affinity gel electrophoresis of BiXyn10A tandem CBMs (TM1), CBM1 (TM2), and CBM2 (TM3). Native polyacrylamide gels were prepared with or without polysaccharides (0.1% wt/vol), and 2 μg of each truncational mutant or 1 μg of BSA was loaded and electrophoresed at 100 V for 6 h at 4 °C. The proteins were then visualized by staining with Coomassie Brilliant Blue G-250. Polysaccharides used in the assays were medium viscosity soluble WAX, konjac glucomannan (KGM), laminarin, lichenan, and xyloglucan.
Isothermal Titration Calorimetry Demonstrates Different Specificities of CBM1 and CBM2 for Oligosaccharides.
Isothermal titration calorimetry (ITC) was used to investigate the binding of the CBMs to xylose and xylooligosaccharides, as xylose constitutes the backbone of WAX. The CBM1 did not show detectable binding to xylose (×1), xylobiose (×2), xylotriose (×3), or xylotetraose (×4). However, the protein bound with increasing affinity as the chain length increased from xylopentaose (×5) to xylohexaose (×6) (Table 1 and SI Appendix, Fig. S3A). Additional binding experiments with mixed β-1,3/β-1,4 linked glucooligosaccharides derived from lichenan, and xyloglucanoheptaose derived from xyloglucan showed no detectable binding for CBM1 (SI Appendix, Fig. S4A). The CBM2, in contrast, exhibited binding activity to ×3, with the association constant increasing as the chain length of the oligosaccharide increased (Table 2 and SI Appendix, Fig. S3B). In addition, binding was detected for CBM2 with both lichenan-derived and xyloglucan-derived oligosaccharides, although binding to these oligosaccharides were weaker compared with binding to xylooligosaccharides (SI Appendix, Fig. S4B).
Table 1.
Affinity of BiXyn10A CBM1 for xylooligosaccharides
| Substrate | N | Ka × 103, M−1 | ΔG, kJ/mol | ΔH, kJ/mol | TΔS, kJ/mol |
| X1 | N.D. | N.D. | N.D. | N.D. | N.D. |
| X2 | N.D. | N.D. | N.D. | N.D. | N.D. |
| X3 | N.D. | N.D. | N.D. | N.D. | N.D. |
| X4 | N.D. | N.D. | N.D. | N.D. | N.D. |
| X5 | 0.78 ± 0.17 | 2.51 ± 0.01 | −19.4 | −68.9 ± 17.3 | −49.5 |
| X6 | 0.82 ± 0.15 | 5.00 ± 0.23 | −21.1 | −56.9 ± 11.4 | −35.8 |
Values are reported as means ± SEs from the mean for two or three independent experiments. N.D. indicates that no binding was detected.
Table 2.
Affinity of BiXyn10A CBM2 for xylooligosaccharides
| Substrate | N | Ka × 103, M−1 | ΔG, kJ/mol | ΔH, kJ/mol | TΔS, kJ/mol |
| X1 | N.D. | N.D. | N.D. | N.D. | N.D. |
| X2 | N.D. | N.D. | N.D. | N.D. | N.D. |
| X3 | 1.18 ± 0.28 | 2.11 ± 0.01 | −19.0 | −37.9 ± 8.46 | −18.8 |
| X4 | 0.91 ± 0.20 | 4.25 ± 0.23 | −20.8 | −51.8 ± 12.4 | −31.1 |
| X5 | 0.82 ± 0.08 | 10.3 ± 0.41 | −27.1 | −52.6 ± 5.1 | −25.4 |
| X6 | 1.23 ± 0.04 | 11.0 ± 0.23 | −23.1 | −33.9 ± 1.2 | −10.8 |
Values are reported as means ± SEs from the mean for two or three independent experiments. N.D. indicates that no binding was detected.
The CBMs in BiXyn10A Contribute to Hydrolysis of Xylan Substrates.
Scanning alanine mutagenesis of aromatic amino acid residues in the two CBMs identified W176 and W249 in CBM1 and W363 in CBM2 as amino acids in which alanine substitutions abolished binding to xylan (SI Appendix, Fig. S5) and xylooligosaccharides (SI Appendix, Fig. S6). To investigate the role of the CBMs in the enzymatic activity of BiXyn10A, the three single mutants and a triple mutant were created in the full-length polypeptide (Fig. 4A) and enzymatic activity examined through kinetic analysis, where we measured total release of reducing sugars as we varied concentration of the substrates. On OSX, single mutations led to less than half the estimated catalytic efficiency of the wild-type enzyme, and the triple mutant exhibited a value lower than a third of the estimate for the wild-type protein (SI Appendix, Table S6). Although a similar effect was observed with the mutants on WAX as substrate, in all cases the mutations reduced the estimated catalytic efficiency to only about half that of the wild-type enzyme (SI Appendix, Table S7). The changes in the estimated catalytic efficiencies mostly derived from reduced kcat due to the mutations.
Fig. 4.

Mutation of critical residues in CBM1 and CBM2 diminishes BiXyn10A-mediated hydrolysis of xylans. (A) Domain organization of BiXyn10A, with asterisks indicating locations within the gene where site-specific mutations were introduced. (B) BiXyn10A WT, the individual site-directed mutants, or the triple mutant (W176A/W249A/W363A) were incubated with WAX or OSX at 37 °C for 48 h, and the xylose released during hydrolysis was quantified by HPLC as indicated in Experimental Procedures. *P < 0.05, ***P < 0.001.
Endoxylanases cleave substrates in a random fashion. Therefore, components of the end products of hydrolysis were also examined for variations due to the presence of the mutations. Given that some of the end products may contain side chains, we focused our analysis on the most reliable estimate—that is, determination of xylose concentrations in the mixture by HPLC analysis. Other than the triple mutant, which reduced xylose release, the mutations had no significant effect on the amount of xylose produced by BiXyn10A on the soluble substrate WAX (Fig. 4B, WAX). Although the W249A in CBM1 and W363A in CBM2 led to statistically significant increases in xylose release from OSX, the triple mutant led to a highly significant decrease in xylose in the end products (Fig. 4B, OSX). Thus, mutations that likely abolished the substrate-binding activity of the CBMs (triple mutant) decreased the hydrolytic activity of the enzyme on xylan substrates and also affected the composition of the end products. Importantly, the mutations did not lead to gross changes in the secondary structure of the protein as revealed by circular dichroism scans (SI Appendix, SI Materials and Methods and SI Results, and Tables S8–S10).
Phylogenetic Distribution of the Novel CBM and Functional Characterization of Homologs.
A search of the nonredundant GenBank database with the novel CBM sequence as the query retrieved nearly 100 homologs. Phylogenetic analysis revealed broad distribution of this CBM in the Bacteroidetes largely derived from the gut microbial community (Fig. 5A). The novel CBM was invariably found in association with GH10 proteins and is inserted either as a single CBM module (e.g., the ruminal bacterium P. bryantii B14 Xyn10C, EFI72438.1|136–303|) or tandem CBM repeats as seen for BiXyn10A. The level of amino acid sequence homology among the CBMs varied dramatically, with some proteins sharing 99% identity and others exhibiting only 12% identity. Remarkably, this pattern of divergence was noted even between two CBMs within a single polypeptide. For example, the two CBMs in BACINT_04197 (EDV05054.1) share 71% amino acid sequence identity, whereas the two CBM repeats in BiXyn10A (EDV05072.1) share just 12% identity.
Fig. 5.

Phylogenetic tree of insertion sequences detected in GH10 polypeptides and xylan-binding activities of representatives. (A) A phylogenetic tree based on amino acid sequences aligned using ClustalW (52) was constructed by the maximum likelihood algorithm in MEGA6 (53). Bootstraps were performed with 1,000 replicates. The sequence IDs are color coded according to whether they represent a single CBM sequence or are part of a pair of repeated CBM sequences, in which case a designation of CBM1 for N-terminal CBM and CBM2 for C-terminal CBM is used. The proteins indicated by arrows were cloned, expressed, purified, and subsequently assessed for xylan-binding activity. The GenBank accession numbers or locus IDs in this tree are as follows: EJP31444.1 (GH family 10, Prevotella sp. MSX73), EFC76216.1 (GH family 10, Prevotella buccae D17), EFU31470.1 (GH family 10, P. buccae ATCC 33574), EEF88961.1 (putative GH family 10, B. cellulosilyticus DSM 14838), EFI38462.1 (GH family 10, Bacteroides sp. 3_1_23), CBK67953.1 (Beta-1,4-xylanase, Bacteroides xylanisolvens XB1A), EGK05990.1 (hypothetical protein HMPREF9456_02254, Dysgonomonas mossii DSM 22836), EHO73033.1 (hypothetical protein HMPREF9944_00626, Prevotella maculosa OT 289), EFI72438.1 (GH family 10, P. bryantii B14, PBR_0377), EEF88944.1 (GH family 10, B. cellulosilyticus DSM 14838), EDV05054.1 (GH family 10, B. intestinalis DSM 17393, BACINT_04197), EDO10010.1 (GH family 10, B. ovatus ATCC 8483, BACOVA_04390), AFL84731.1 (beta-1,4-xylanase, Belliella baltica DSM 15883), WP_010528479.1 (carbohydrate-binding CenC domain protein, Thermophagus xiamenensis), ACX30647.1 (XynB19 precursor, Sphingobacterium sp. TN19), EEC54455.1 (putative GH family 10, Bacteroides eggerthii DSM 20697, BACEGG_01298), EFB34367.1 (putative GH family 10, Prevotella copri DSM 18205), EFA43871.1 (GH family 10, Prevotella bergensis DSM 17361), EFI48293.1 (carbohydrate binding domain-containing protein, Prevotella oris C735), EDV05072.1 (putative GH family 10, B. intestinalis DSM 17393, BACINT_04215), EIY38981.1 (hypothetical protein HMPREF1062_00490, B. cellulosilyticus CL02T12C19), EDY95689.1 (putative GH family 10, Bacteroides plebeius DSM 17135), ADF51103.1 (family 10 GH, Zunongwangia profunda SM-A87), EGK02428.1 (hypothetical protein HMPREF9455_01698, Dysgonomonas gadei ATCC BAA-286), ADY53105.1 (carbohydrate-binding CenC domain protein, Pedobacter saltans DSM 12145), EJL65303.1 (beta-1,4-xylanase, Flavobacterium sp. CF136), WP_010417124.1 (carbohydrate-binding CenC domain protein, Anaerophaga thermohalophila), AEV98992.1 (endo-1,4-beta-xylanase, Niastella koreensis GR20-10), CCH01345.1 (endo-1,4-beta-xylanase, Fiberlla aestuarina BUZ 2), ADB38141.1 (GH family 10, Spirosoma linguale DSM 74), CCH00356.1 (GH family 10, F. aestuarina BUZ 2), ADE81777.1 (endo-1,4-beta-xylanase, P. ruminicola 23, PRU_2739), EDO14052.1 (putative GH family 10, B. ovatus ATCC 8483, BACOVA_00247), EEZ05210.1 (hypothetical protein HMPREF0102_00209, Bacteroides sp. 2_1_22), EIY59304.1 (hypothetical protein HMPREF1069_04286, B. ovatus CL02T12C04), EDO10798.1 (hypothetical protein BACOVA_03431, B. ovatus ATCC 8483), EFS31249.1 (hypothetical protein BSGG_1949, Bacteroides sp. D2), EEF86710.1 (hypothetical protein BACCELL_05690, B. cellulosilyticus DSM 14838), EDV03684.1 (putative GH family 10, B. intestinalis DSM 17393, BACINT_02810), EDO14247.1 (putative GH family 10, B. ovatus ATCC 8483), and EIY28996.1 (hypothetical protein HMPREF1062_03304, B. cellulosilyticus CL02T12C19). (B) Affinity gel electrophoresis of selected proteins. Native polyacrylamide gels were prepared with or without 0.1% wt/vol WAX, and 2 μg of each protein or 1 μg of BSA was loaded and electrophoresed at 100 V for 6 h at 4 °C. The proteins were then visualized by staining with Coomassie Brilliant Blue G-250.
The broad diversity at the primary sequence level, confirmed by the low bootstrap values in the phylogenetic tree (Fig. 5A), suggested that these CBMs have diverged to perform different functions. To evaluate this possibility, representative CBMs (indicated by arrows, Fig. 5A) from the different lineages were selected for analyses. Each selected CBM was expressed in Escherichia coli and purified (SI Appendix, Fig. S7A) for electrophoretic mobility analysis. The electrophoretic mobility of five out of seven newly studied proteins were dramatically retarded in the presence of soluble WAX (Fig. 5B) but not CMC (SI Appendix, Fig. S7B), indicating that these proteins bind to xylan. The two proteins that did not exhibit binding (EDV03684.1 and EDV014052.1) were present at the top of both gels, suggesting that the native gel conditions did not support migration of the proteins. The ITC analysis revealed that of the five proteins that bound to xylan, four bind to xylopentaose (EDV05054.1, EDO10010.1, EEC54455.1, and ADE81777.1) (SI Appendix, Fig. S7C). Interestingly, the CBM from P. bryantii B14 Xyn10C (Pbr_0377) bound to soluble WAX (Fig. 5B, EFI72438.1) but not to xylopentaose (SI Appendix, Fig. S7C, EFI72438.1), indicating that its binding preference was different to the remaining CBMs that bound both polysaccharide and oligosaccharides. Thus, despite the diversity in amino acid sequence, the new CBM family likely binds mostly to xylans.
Structural Characterization of BiXyn10A CBM1 and P. bryantii B14 Xyn10C CBM.
Crystallization attempts were carried out on each of the nine recombinant CBMs, with only BiXyn10A CBM1 and PbXyn10C CBM yielding diffraction quality crystals. Crystallographic phases were independently determined using single-wavelength anomalous diffraction for each apo CBM, and the structures were subsequently refined to 1.8 Å (apo BiXyn10A CBM1) and 1.68 Å (apo PbXyn10C CBM). In addition, the cocrystal structure of BiXyn10A CBM1 bound to xylohexaose was determined to 1.14 Å resolution (SI Appendix, Table S11).
Despite limited conservation in primary amino acid sequence (15% identity), the structures of the two CBMs are architecturally similar and consisted of a characteristic β-sandwich fold (Fig. 6 A and B). A structure-based search of the Protein Data Bank (PDB) using the Dali Server (26) with BiXyn10A CBM1 identified the closest homolog as a CBM4 engineered to bind xylans from Rhodothermus marinus (X-2; 18% sequence identity; rmsd of 2.4 Å over 138 aligned Cα atoms, PDB ID code 2Y6L) (Fig. 7B).
Fig. 6.

Overall structures of BiXyn10A CBM1 and PbXyn10C CBM. The overall structures of (A) BiXyn10A CBM1 and (B) PbXyn10C CBM contain the typical β-sandwich fold. (C) Stereoview of xylotriose bound to BiXyn10A CBM1.
Fig. 7.
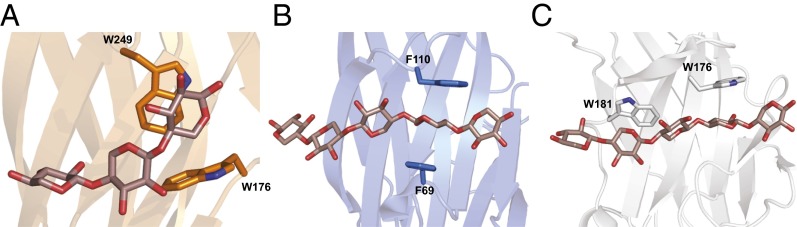
BiXyn10A CBM1 binds xylotriose in a novel conformation. (A–C) The BiXyn10A CBM1, R. marinus X-2 CBM4, and CjXyn10C CBM15, respectively, with bound xylooligosaccharide. BiXyn10A CBM1 binds xylotriose in a bent conformation, whereas the R. marinus X-2 CBM4 (B) and C. japonicus Xyn10C CBM15 (C) bind xylopentaose in a linear manner.
The cocrystal structure of BiXyn10A CBM1 with xylohexaose reveals continuous density for only three of the six sugars of the oligosaccharide (Fig. 6C), suggesting that the remaining three sugars are dynamically disordered. Binding of the oligosaccharide by CBM1 is facilitated by three π-stacking interactions—namely, sandwiching of the reducing end between W176 and W249 and stacking of the nonreducing end with H210. These observations are in agreement with the mutagenesis studies described above that indicate the importance of the two tryptophan residues in engaging the xylooligosaccharide ligand. Other important hydrogen bonding interactions are provided by E178, which interacts with the hydroxyls of the center xylose, and Q282 and Q213, which form hydrogen bonds with the nonreducing end sugar (Fig. 6C). An additional, water-mediated hydrogen bond occurs between R252 and the xylose at the nonreducing end. Although PbXyn10C CBM was crystallized in the absence of substrate, a structure-based alignment with BiXyn10A CBM1 shows conservation of the binding site. Notably, the amino acids predicted to be responsible for the π-stacking interactions, Y209 and W201 in PbXyn10C CBM, are located on the opposite side of the binding pocket as W176 and W249 from BiXyn10A CBM1.
Compared with the few available CBM structures with a xylan substrate bound, BiXyn10A CBM1 engages the oligosaccharide in a novel manner; in particular, the polypeptide induced a bend in the bound oligosaccharide (Fig. 7A). A loop consisting of residues 247–252 and an α-helix encompassing residues 209–211 pinch the ligand to create the bend. The binding mode for the sugar is in contrast with that observed for other iso-functional CBMs, such as the engineered X-2 (R. marinus CBM4) and CjXyn10C CBM15 of Cellvibrio japonicus (PDB ID code 1GNY). In each of these structures, the oligosaccharide binds in an extended fashion, orthogonal to the direction of the β-strands of the CBM (Fig. 7 B and C).
BiXyn10A Functions Synergistically with Gene Products of Other Up-Regulated Genes to Release Fermentable Sugars.
To determine the hydrolytic potential of the enzymes encoded by the most highly up-regulated genes during WAX fermentation, the top three most highly up-regulated genes were expressed in their recombinant forms (SI Appendix, Fig. S8) and examined as an enzyme mix for their hydrolytic products on WAX and OSX. In each case, several oligosaccharides and also the monosaccharides xylose and arabinose were released. Addition of BiXyl3A (Fig. 1A, BACINT_00926), a GH3 β-xylosidase, to the enzyme mixture led to detection of largely monosaccharides (xylose and arabinose) from WAX and OSX (Fig. 8 A and B).
Fig. 8.

BiXyn10A functions synergistically with the most highly up-regulated gene products to degrade xylans. (A) WAX and (B) OSX hydrolysis in the presence of the indicated combinations of enzymes were carried out in citrate buffer at 37 °C for 24 h. The end products of the reactions were resolved by HPAEC by comparison of peaks with retention times of arabinose (A1) and xylose (×1). Control reactions were composed of the same constituents in the absence of enzymes. These experiments were performed in triplicate, and representative results are shown.
Discussion
Members of the phylum Bacteroidetes are among the most abundant organisms in the distal human GIT (15). These bacteria possess a remarkable repertoire of genes targeted at the utilization of dietary and host-derived polysaccharides (27), which allow for successful competition in the gut microbial community. The polysaccharide-targeting genes are arranged in clusters termed polysaccharide utilization loci (PULs), which characteristically contain genes encoding outer membrane porins, polysaccharide-binding proteins, and hydrolytic enzymes in addition to transcriptional regulators that govern expression of genes in the corresponding PUL (21). The Bacteroidetes PULs target specific substrates (19), confer competitive advantages in the presence of the substrate in the diet (28), and are often within integrative and conjugative elements, permitting their transfer among Bacteroidetes in the GIT (29).
Xylan degradation is increasingly being recognized as an important process in the gut and is the subject of intense research as a prebiotic dietary supplement (30). The genome sequences of gut bacteria have revealed a large diversity of xylan-degrading enzymes, with the Bacteroidetes clearly equipped with the most extensive enzyme systems (20). In particular, the genome of B. intestinalis DSM 17393 encodes the greatest abundance of CAZy gene families (23), and our study suggests that B. intestinalis has the most elaborate xylan-specific gene repertoire among the bacteria with genomes sequenced to date. The xylan-specific transcriptome of B. intestinalis DSM 17393 consists of nearly 70 genes. Twenty-two of these genes map to a ∼60 kb contiguous region on the genome comprised of two large PULs each containing an individual HTCS regulator (SI Appendix, Fig. S9). The proteins encoded by the genes in this cluster are highly modular in their polypeptide architecture, and many of these structures are exceedingly rare.
The genes that make up the core XUS in human gut and ruminal Bacteroidetes consist of two tandem repeats of SusC/SusD homologs (XusA–XusD) followed by a hypothetical protein (XusE) and finally an endoxylanase gene (homologous to Xyn10C from P. bryantii B14) (reviewed in refs. 22 and 31). Recently it has been demonstrated that the core xylan utilization genes in the human gut symbiont B. cellulosilyticus WH2 are critical to fitness in the gut of mice fed a polysaccharide-rich diet (32). Furthermore, in B. intestinalis DSM 17393 (current study), B. ovatus ATCC 8683 (ref. 19 and the current study), B. cellulosilyticus WH2 (32), and P. bryantii B14 (22), these genes are the most highly up-regulated when the bacteria are grown on xylan. However, the biochemical and functional properties of proteins encoded by the core genes are poorly understood.
The modular GH10 proteins within these core clusters are 180–325 amino acids longer than other GH10 proteins, indicating the presence of additional domains within these enzymes. A previous study of the homolog from P. bryantii B14 (Xyn10C) demonstrated that the protein contains a catalytic endoxylanase module that is disrupted by a 160 amino acid sequence of unknown function (33). Another GH10 endoxylanase from a strain of Prevotella ruminicola was disrupted by a segment of 280 amino acids consisting of an imperfect tandem of 130 amino acids (33). We demonstrate that these proteins contain either single or tandem repeats of a CBM that binds to xylans. Instead of the usual N- or C-terminal location to the GH domain, the CBMs are integrated into a loop present in the GH10 catalytic module. The binding activity of the CBMs is partially mediated by tryptophan residues, and mutation of these residues diminishes enzymatic activity on the two xylans used as substrates in the present study. The reduction in activity, especially for the triple mutant, was more pronounced on OSX than on WAX (Fig. 4B and SI Appendix, Tables S6 and S7), suggesting that the CBM improves enzymatic activity by targeting the enzyme to insoluble xylan. Analogous to BiXyn10A, the alpha-amylase (SusG) from the prototypical starch utilization system in Bacteroides thetaiotaomicron also possesses a catalytic domain (GH13) interrupted by a CBM (CBM58) (34). Although the biological significance of having a CBM emanating from a loop within the catalytic domain is not clear, the existence of two such models within the Bacteroides spp. suggests there may be an evolutionary benefit. This may be confirmed by future studies that seek to address functional interactions within the outer membrane complexes that consist of transporters, polysaccharide-binding proteins, and hydrolytic enzymes.
Although structural alignments with other CBMs demonstrate a common fold, the BiXyn10A CBM1-xylohexaose cocrystal structure reveals a distinct strategy for engaging oligosaccharide substrates. Specifically, the aromatic residues that typically mediate stacking interactions to engage oligosaccharide ligands are located nearly orthogonal to each other, resulting in the inducement of a bend in the bound sugar. Several lines of evidence support the assignment of the CBMs to a new family: (i) The primary amino acid sequences show low conservation with defined CBM families, (ii) structural comparisons of CBMs with other characterized CBMs reveal that the xylan substrate binds in a novel orientation with unique amino acids, and (iii) the CAZy database currently groups these proteins into a “nonclassified” category of CBMs.
The current study provides important molecular insights into the glycolytic capacity of gut bacteria. Beyond the sheer number of enzymes produced by B. intestinalis, a salient point is the degree to which these polypeptides are assembled into multifunctional proteins. This is most notable for the GH10 proteins, where all six proteins display some degree of modularity. The GH10 protein architectures include C-terminal (GH10-CE1, GH10-GH43) or N-terminal (GH43-GH10) appendages as well as two genes with CBM insertions within the GH10 catalytic domain (Fig. 1A). The GH10 and GH43 fusion proteins in the Bacteroidetes with the GH43/GH10 arrangement (BACINT_00569, Fig. 1A) are currently found only in strains of B. intestinalis and B. cellulosilyticus (GenBank accession nos. EEF89781.1 and CDB72001.1), whereas genes encoding GH10/GH43 polypeptides (BACINT_04202) are found in strains of B. intestinalis, B. cellulosilyticus, B. oleiciplenus, B. clarus (human isolates), and B. gallinarum (chicken cecum). The most similar protein to BACINT_04202 is a protein from B. intestinalis CAG:564 (GenBank accession no. CCY87565.1), which shares >99% amino acid sequence identity over the GH10 and GH43 domains, but just 30% identity over the region joining the two domains (SI Appendix, Fig. S10). Analysis of the nucleotide sequence identified two 16 bp inverted repeats in the BACINT_04202 gene corresponding to the amino acid region joining the two domains (SI Appendix, Fig. S11). Although further studies are needed, this observation suggests that integrative and conjugative elements serve as a driving force in the evolution of multidomain enzymes in these organisms.
The other genes that were up-regulated over 100-fold (BACINT_00570 and BACINT_00569, Fig. 1A) were expressed and the proteins biochemically analyzed for xylan hydrolysis. BACINT_00570 hydrolyzed xylooligosaccharides, mostly into xylobiose and xylotriose, and on WAX cleaved only the side-chain arabinose, suggesting an enzyme with both β-xylosidase and arabinofuranosidase activities in a single catalytic domain. The recombinant BACINT_00569 released xylooligosaccharides and arabinose from WAX, and site-directed mutagenesis demonstrated that the endoxylanase and arabinofuranosidase activities reside in different catalytic domains—that is, in the GH10 and GH43 catalytic domains, respectively. Although an enzyme mixture from the top three highly up-regulated genes (Fig. 1A) released a mixture of oligosaccharides and monosaccharides, addition of the enzyme encoded by BACINT_00926, a protein determined in our previous report to function as a β-xylosidase (35), led to depolymerization of both the soluble and insoluble xylans to their unit monosaccharides (xylose and arabinose). Both sugars are fermentable by the bacterium to generate energy and building blocks. Thus, the gene products of only four of the highly up-regulated genes completely depolymerized the complex substrate into fermentable sugars.
The gut microbial community is a complex environment where the interplay of diet and host factors and microbial community dynamics converge to define the specific niche of gut microbes. Although the true physiological contributions of the xylan degradation genes in the context of the human gut microbial community remain to be completely unraveled, our data show that a human colonic commensal bacterium has evolved an elaborate mechanism to depolymerize one of the recalcitrant polysaccharides present in the diet. Furthermore, our results demonstrate that fiber digestion in the human lower gut microbial community represents another paradigm, other than the ruminant and termite gut environments, in the search for plant cell wall-degrading enzymes.
Experimental Procedures
Materials.
The human colonic bacterial isolate B. intestinalis DSM 17393 was obtained from the Deutsche Sammlung von Mikroorganismen und Zellkulturen or German Collection of Microorganisms and Cell Cultures, and B. ovatus ATCC 8483 was obtained from the ATCC. E. coli XL10 and BL21-CodonPlus(DE3) RIPL competent cells and the PicoMaxx high-fidelity DNA polymerase were purchased from Stratagene. The pET-46 Ek/LIC kit was obtained from Novagen. The QIAprep Spin Miniprep, RNAprotect, RNase-free DNase, and RNeasy kits were obtained from Qiagen. The Talon Metal Affinity resin was obtained from Clontech Laboratories, Inc. The QuikChange Lightening Multi-Site Directed Mutagenesis kit was obtained from Agilent. The xylooligosaccharides, glucotetraose A-C, xyloglucanoheptaose, arabinan (sugar beet), debranched arabinan (sugar beet), konjac glucomannan, lichenan (icelandic moss), galactan (potato), arabinogalactan (larch), xyloglucan (tamarind seed) 1,4-β-d-mannan, and medium viscosity WAX were purchased from Megazyme. Carboxymethyl cellulose was purchased from Acros Organic. Laminarin, OSX, and all other reagents were of the highest possible purity and were purchased from Sigma-Aldrich.
Growth of B. intestinalis DSM 17393, B. ovatus ATCC 8483, and Isolation of RNA.
B. intestinalis or B. ovatus cells were cultured anaerobically at 37 °C in a modified basal cellulolytic (BC) medium (36) (SI Appendix, Table S1) with 0.5% wt/vol glucose. At an optical density of ∼1, 100 μL of culture was inoculated into the modified BC medium in duplicate with either 0.5% wt/vol WAX or xylose as the main carbohydrate source. The optical density at 600 nm wavelength was monitored using a Spectronic 20D (ThermoFisher Scientific) over a period of 27 h. At midlog phase of growth (OD600nm = 0.4), 50 mL of each culture was removed, combined with two volumes of RNAprotect, and RNA was extracted using the RNeasy mini kit based on the manufacturer’s protocol. The optional on-column DNase treatment was performed to ensure total removal of DNA. The RNA was then quantitated using a Qubit RNA assay kit (Invitrogen) and stored at −80 °C until RNA sequencing as described in our earlier report (22).
Library Preparation, RNA Sequencing, and Expression Analysis.
Ribosomal RNA was removed from 1 μg of total RNA using the Ribo-Zero rRNA Removal Metabacteria kit (Epicentre Biotechnologies). The mRNA-enriched fraction was converted to indexed RNAseq libraries with the TruSeq RNA Sample Prep kit (Illumina), and the final libraries were pooled in equimolar concentrations and analyzed by quantitative PCR (Library Quantification kit, Kapa Biosystems).
Four libraries were prepared from two biological replicates of each culture medium (WAX1, WAX2, Xyl1, and Xyl2), and RNAseq data were analyzed using CLC Genomics Workbench v5.0 (CLC Bio) as outlined in our previous report (22). The RNAseq output files were then analyzed for statistical significance by using the proportion-based test of Baggerly et al. (37). A summary of the read mapping statistics is provided in SI Appendix, Table S2.
Gene Cloning, Site-Directed Mutagenesis, Gene Expression, and Protein Purification.
All genes cloned for expression were amplified from B. intestinalis DSM 17393 genomic DNA by PCR using the primers listed in SI Appendix, Table S3 (constructs listed in SI Appendix, Fig. S1). The cloning of the correct PCR amplicons into pET-46b vector, overexpression of the genes, and site-directed mutagenesis were as described in our earlier report (22). The recombinant proteins in the cell lysates were purified with Talon metal affinity resin (22), and the purity was assessed by SDS/PAGE. The protein concentrations were determined by the method of Gill and von Hippel based on the molecular mass and computed extinction coefficients (38).
Hydrolysis of Polysaccharides.
Hydrolysis of xylooligosaccharides and the natural substrates WAX and OSX by BiXyn10A was performed at 37 °C for 16 h in citrate buffer (50 mM citrate, 150 mM NaCl, pH 5.5). Hydrolysis of the natural substrates WAX and OSX by the BiXyn10A WT and its mutants was performed at 37 °C for 48 h in citrate buffer (50 mM citrate, 150 mM NaCl, pH 5.5) to determine the influence of residues essential for binding of the CBMs in BiXyn10A on hydrolysis of xylan. The end products of hydrolysis were analyzed by TLC, reducing sugar assays, and high-performance anion exchange chromatography (HPAEC), all as described previously (39).
Affinity Gel Electrophoretic Mobility Assays.
Assays to monitor the binding activity of the proteins were performed according to Tomme et al. (40) with minor modifications. The stacking gel contained 3% wt/vol polyacrylamide in 1.5M Tris⋅HCl buffer (pH 8.3). Each protein (1 μg) was loaded in a native gel (12% wt/vol polyacrylamide) containing soluble polysaccharides (0.1% wt/vol) or a control gel without polysaccharides, and the gels were electrophoresed simultaneously at 4 °C at 100 V for 6 h. The polysaccharides used for affinity gel electrophoretic assays include WAX, konjac glucomannan, laminarin, lichenan, xyloglucan, CMC, galactan, linear arabinan, debranched arabinan, and arabinogalactan. The gels were then stained with Coomassie Brilliant Blue G-250 for protein visualization.
ITC.
The ITC measurements were performed using a VP-ITC microcalorimeter with a 1.4 mL cell volume from MicroCal, Inc. The proteins were exchanged into phosphate buffer (50 mM sodium phosphate, 150 mM NaCl, pH 7.0) by dialysis, and the oligosaccharide ligands were dissolved in the same buffer. The proteins (50 µM) were then injected with 28 successive 10 µL aliquots of ligand (2 mM) at 300-s intervals. The data were fitted to a nonlinear regression model using a single binding site (MicroCal Origin software). The thermodynamic parameters were calculated using the Gibbs free energy equation (ΔG = ΔH – TΔS), and the relationship ΔG = –RTlnKa.
Crystallography Studies.
Both BiXyn10A CBM1 and PbXyn10C CBM were crystallized by screening with sparse matrix conditions followed by optimization. Specifically, BiXyn10A CBM1 crystals were optimized to 42 mg/mL at 23 °C using a precipitant consisting of 2.5 M ammonium sulfate and 0.1 M bicine pH 9.0. Crystals were soaked in mother liquor containing 30% (wt/vol) threitol before vitrification by direct immersion into liquid nitrogen. Cocrystals of BiXyn10A CBM1 with xylohexaose were grown at 23 °C with 30 mg/mL protein and 5 mM xylohexaose using a precipitant of 1.6 M sodium citrate tribasic dehydrate pH 6.5. Crystals were vitrified by direct immersion into liquid nitrogen. PbXyn10C CBM was optimized to yield crystals at 4 °C and 5 mg/mL protein using a precipitant solution containing 2.0 M ammonium sulfate, 0.1 M sodium chloride, and 0.1 M sodium cacodylate pH 6.5. Briefly before vitrification, crystals were soaked in 30% (wt/vol) trehalose to serve as a cyroprotectant. X-ray diffraction data were collected at the Life Science Collaborative Access Team, Sector 21, Argonne National Laboratory for crystals of both BiXyn10A CBM1 and PbXyn10C CBM.
Structure Determination and Refinement.
Crystallographic phases for BiXyn10A CBM1 were determined by anomalous diffraction data collected from crystals soaked with 1 mM 4-chloromercuribenzoic acid for 4 h at 23 °C. After soaking, 4-chloromercuribenzoic acid-derived crystals were treated identically as native crystals. PbXyn10C CBM was phased using selenomethionine containing protein crystals grown at 4 °C with 30 mg/mL protein containing a precipitating solution of 1.2 M sodium phosphate monobasic, 0.8 M potassium phosphate dibasic, 0.2 M lithium sulfate, and 0.1 M 3-(cyclohexylamino)-1-propanesulfonic acid pH 10.5, and vitrified in the same solution supplemented with 20% (wt/vol) threitol. Data were indexed and scaled using HKL2000 or autoPROC (41). Phasing was carried out using the AutoSol package from the software PHENIX suite (42). Solutions from AutoSol were automatically built using ARP/wARP (43, 44). The structures were further refined using a combination of COOT (45), REFMAC5 (46), and PHENIX refine (42).
Synergy of BiXyn10A with Other Up-Regulated Enzymes.
The capacity to depolymerize xylan polysaccharides by the gene products of the three most highly up-regulated genes was examined with WAX and OSX as substrates by measuring release of reducing ends at 37 °C for 24 h. Reactions were initiated by addition of enzymes (50 nM each, final concentration), and reaction end products were subjected to HPAEC as described in our earlier report (22). Arabinose, xylose, xylobiose, and xylotriose were injected as standards.
Supplementary Material
Acknowledgments
We thank Alvaro Hernandez and staff of the W.M. Keck Center for Comparative and Functional Genomics for assistance with Illumina sequencing and Eoghan M. Smyth of the Energy Biosciences Institute for assistance with phylogenetic analysis. We thank Keith Brister and Joseph Brunzelle for facilitating data collection at Life Sciences Collaborative Access Team (21 ID) at the Advanced Photon Source. This research was funded by the Energy Biosciences Institute and in part by Agriculture and Food Research Initiative Competitive Grant 2012-67015-19451 from the US Department of Agriculture National Institute of Food and Agriculture.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4MGS, 4QPW, and 4MGQ).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1406156111/-/DCSupplemental.
References
- 1.Consortium HMP. Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gill SR, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006;312(5778):1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin J, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van de Pavert SA, Mebius RE. New insights into the development of lymphoid tissues. Nat Rev Immunol. 2010;10(9):664–674. doi: 10.1038/nri2832. [DOI] [PubMed] [Google Scholar]
- 5.Sommer F, Bäckhed F. The gut microbiota—Masters of host development and physiology. Nat Rev Microbiol. 2013;11(4):227–238. doi: 10.1038/nrmicro2974. [DOI] [PubMed] [Google Scholar]
- 6.Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9(10):577–589. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 7.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448(7152):427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 8.Ley RE, et al. Obesity alters gut microbial ecology. Proc Natl Acad Sci USA. 2005;102(31):11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karlsson FH, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun. 2012;3:1245. doi: 10.1038/ncomms2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, et al. Organismal, genetic, and transcriptional variation in the deeply sequenced gut microbiomes of identical twins. Proc Natl Acad Sci USA. 2010;107(16):7503–7508. doi: 10.1073/pnas.1002355107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science. 2005;307(5717):1915–1920. doi: 10.1126/science.1104816. [DOI] [PubMed] [Google Scholar]
- 13.Nelson KE, et al. Human Microbiome Jumpstart Reference Strains Consortium A catalog of reference genomes from the human microbiome. Science. 2010;328(5981):994–999. doi: 10.1126/science.1183605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009;37(Database issue):D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turnbaugh PJ, Henrissat B, Gordon JI. Viewing the human microbiome through three-dimensional glasses: Integrating structural and functional studies to better define the properties of myriad carbohydrate-active enzymes. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(Pt 10):1261–1264. doi: 10.1107/S1744309110029088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Selvendran RR. The plant cell wall as a source of dietary fiber: Chemistry and structure. Am J Clin Nutr. 1984;39(2):320–337. doi: 10.1093/ajcn/39.2.320. [DOI] [PubMed] [Google Scholar]
- 18.Slavin JL, Brauer PM, Marlett JA. Neutral detergent fiber, hemicellulose and cellulose digestibility in human subjects. J Nutr. 1981;111(2):287–297. doi: 10.1093/jn/111.2.287. [DOI] [PubMed] [Google Scholar]
- 19.Martens EC, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9(12):e1001221. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dodd D, Mackie RI, Cann IK. Xylan degradation, a metabolic property shared by rumen and human colonic Bacteroidetes. Mol Microbiol. 2011;79(2):292–304. doi: 10.1111/j.1365-2958.2010.07473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: The Bacteroidetes Sus-like paradigm. J Biol Chem. 2009;284(37):24673–24677. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dodd D, Moon YH, Swaminathan K, Mackie RI, Cann IK. Transcriptomic analyses of xylan degradation by Prevotella bryantii and insights into energy acquisition by xylanolytic bacteroidetes. J Biol Chem. 2010;285(39):30261–30273. doi: 10.1074/jbc.M110.141788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat Rev Microbiol. 2013;11(7):497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- 24.Bakir MA, Kitahara M, Sakamoto M, Matsumoto M, Benno Y. Bacteroides intestinalis sp. nov., isolated from human faeces. Int J Syst Evol Microbiol. 2006;56(Pt 1):151–154. doi: 10.1099/ijs.0.63914-0. [DOI] [PubMed] [Google Scholar]
- 25.Salyers AA, Vercellotti JR, West SE, Wilkins TD. Fermentation of mucin and plant polysaccharides by strains of Bacteroides from the human colon. Appl Environ Microbiol. 1977;33(2):319–322. doi: 10.1128/aem.33.2.319-322.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holm L, Rosenström P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38(Web Server issue):W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu J, et al. A genomic view of the human-Bacteroides thetaiotaomicron symbiosis. Science. 2003;299(5615):2074–2076. doi: 10.1126/science.1080029. [DOI] [PubMed] [Google Scholar]
- 28.Sonnenburg ED, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141(7):1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hehemann JH, Kelly AG, Pudlo NA, Martens EC, Boraston AB. Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc Natl Acad Sci USA. 2012;109(48):19786–19791. doi: 10.1073/pnas.1211002109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aachary AA, Prapulla SG. Xylooligosaccharides (XOS) as an emerging prebiotic: Microbial synthesis, utilization, structural characterization, bioactive properties, and applications. Compr Rev Food Sci Food Safety. 2011;10(1):2–16. [Google Scholar]
- 31.White BA, Lamed R, Bayer EA, Flint HJ. Biomass utilization by gut microbiomes. Annu Rev Microbiol. 2014;68:279–296. doi: 10.1146/annurev-micro-092412-155618. [DOI] [PubMed] [Google Scholar]
- 32.McNulty NP, et al. Effects of diet on resource utilization by a model human gut microbiota containing Bacteroides cellulosilyticus WH2, a symbiont with an extensive glycobiome. PLoS Biol. 2013;11(8):e1001637. doi: 10.1371/journal.pbio.1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flint HJ, Whitehead TR, Martin JC, Gasparic A. Interrupted catalytic domain structures in xylanases from two distantly related strains of Prevotella ruminicola. Biochim Biophys Acta. 1997;1337(2):161–165. doi: 10.1016/s0167-4838(96)00213-0. [DOI] [PubMed] [Google Scholar]
- 34.Koropatkin NM, Smith TJ. SusG: A unique cell-membrane-associated alpha-amylase from a prominent human gut symbiont targets complex starch molecules. Structure. 2010;18(2):200–215. doi: 10.1016/j.str.2009.12.010. [DOI] [PubMed] [Google Scholar]
- 35.Hong PY, et al. Two new xylanases with different substrate specificities from the human gut bacterium Bacteroides intestinalis DSM 17393. Appl Environ Microbiol. 2014;80(7):2084–2093. doi: 10.1128/AEM.03176-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robert C, Bernalier-Donadille A. The cellulolytic microflora of the human colon: Evidence of microcrystalline cellulose-degrading bacteria in methane-excreting subjects. FEMS Microbiol Ecol. 2003;46(1):81–89. doi: 10.1016/S0168-6496(03)00207-1. [DOI] [PubMed] [Google Scholar]
- 37.Baggerly KA, Deng L, Morris JS, Aldaz CM. Differential expression in SAGE: Accounting for normal between-library variation. Bioinformatics. 2003;19(12):1477–1483. doi: 10.1093/bioinformatics/btg173. [DOI] [PubMed] [Google Scholar]
- 38.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182(2):319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 39.Han Y, et al. Comparative analyses of two thermophilic enzymes exhibiting both beta-1,4 mannosidic and beta-1,4 glucosidic cleavage activities from Caldanaerobius polysaccharolyticus. J Bacteriol. 2010;192(16):4111–4121. doi: 10.1128/JB.00257-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tomme P, Boraston A, Kormos JM, Warren RA, Kilburn DG. Affinity electrophoresis for the identification and characterization of soluble sugar binding by carbohydrate-binding modules. Enzyme Microb Technol. 2000;27(7):453–458. doi: 10.1016/s0141-0229(00)00246-5. [DOI] [PubMed] [Google Scholar]
- 41.Vonrhein C, et al. Data processing and analysis with the autoPROC toolbox. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):293–302. doi: 10.1107/S0907444911007773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perrakis A, Morris R, Lamzin VS. Automated protein model building combined with iterative structure refinement. Nat Struct Biol. 1999;6(5):458–463. doi: 10.1038/8263. [DOI] [PubMed] [Google Scholar]
- 44.Murshudov GN, et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67(Pt 4):355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 4):486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vagin AA, et al. REFMAC5 dictionary: Organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 47.Yin Y, et al. dbCAN: A web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res. 2012;40(Web Server issue):W445–W451. doi: 10.1093/nar/gks479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Punta M, et al. The Pfam protein families database. Nucleic Acids Res. 2012;40(Database issue):D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marchler-Bauer A, et al. CDD: Conserved domains and protein three-dimensional structure. Nucleic Acids Res. 2013;41(Database issue):D348–D352. doi: 10.1093/nar/gks1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: Discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 51.Juncker AS, et al. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12(8):1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22(22):4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.