

Abstract
Recent and rapid human population growth has led to an excess of rare genetic variants that are expected to contribute to an individual’s genetic burden of disease risk. To date, much of the focus has been on rare protein-coding variants, for which potential impact can be estimated from the genetic code, but determining the impact of rare noncoding variants has been more challenging. To improve our understanding of such variants, we combined high-quality genome sequencing and RNA sequencing data from a 17-individual, three-generation family to contrast expression quantitative trait loci (eQTLs) and splicing quantitative trait loci (sQTLs) within this family to eQTLs and sQTLs within a population sample. Using this design, we found that eQTLs and sQTLs with large effects in the family were enriched with rare regulatory and splicing variants (minor allele frequency < 0.01). They were also more likely to influence essential genes and genes involved in complex disease. In addition, we tested the capacity of diverse noncoding annotation to predict the impact of rare noncoding variants. We found that distance to the transcription start site, evolutionary constraint, and epigenetic annotation were considerably more informative for predicting the impact of rare variants than for predicting the impact of common variants. These results highlight that rare noncoding variants are important contributors to individual gene-expression profiles and further demonstrate a significant capability for genomic annotation to predict the impact of rare noncoding variants.
Introduction
Studies using deep and population-scale sequencing have reported large numbers of rare variants (minor allele frequency [MAF] < 1%) present as a consequence of recent and rapid human population expansion.1–6 However, interpreting the impact of rare variation remains an ongoing challenge. Several exome sequencing studies have suggested that rare variants are of broad importance with the finding that they represent the majority of potentially deleterious and damaging protein-coding alleles2 and can contribute to complex disease risk.7–11 In contrast, population-genetic models have indicated that rare alleles are unlikely to be large overall contributors to heritable variation for many complex diseases.12 Indeed, large population studies of rare variants in autoimmune disorders have so far found negligible impact,13 and analyses of personal genomes have reported multiple rare and protein-code-disrupting sites in presumably healthy individuals.14,15 Further compounding the challenge of understanding the impact of rare variation has been that most studies have focused on only protein-coding alleles whose interpretation is facilitated by the genetic code. For rare variants in noncoding regions, there is no analogous code to aid in the prediction of their impact even though these regions harbor considerable complex-disease-associated variation16,17 and most likely contain an abundance of important rare alleles.
Currently, genetic studies of gene expression provide a systematic means of identifying functional noncoding variants; such studies have identified noncoding variants associated with gene expression, splicing, and allele-specific expression (ASE).18–20 However, insight into the impact of rare noncoding variants has been limited. Few studies have had the advantage of full genome sequencing data and, even when these data are available, they have only assayed unrelated individuals, providing limited power to describe rare-variant effects.18,21,22 To overcome this challenge and provide more systematic insight into the impact of rare noncoding variants, we coupled high-quality genomes with transcriptomes within a large family (n = 17 individuals). The advantage of this design is that the large number of children (n = 11) provides high-confidence rare variants established through both deep sequencing and Mendelian segregation as well as sufficient power to test for cis-expression quantitative trait loci (eQTLs) present within a single human family. Furthermore, eQTLs from the family can be compared to eQTLs from a cell-type- and ethnicity-matched population sample recently reported by the Geuvadis Consortium,18 providing the unique ability to identify large genetic effects specific to the family and test their relationship to rare variants.23 Indeed, we report that rare regulatory variants are enriched near genes that exhibit large-effect cis-eQTLs for gene expression, splicing, and ASE within the family. Furthermore, the family eQTL genes are more evolutionarily constrained than comparable eQTL genes in the population, and several of the genes have established relationships with complex disease, indicating a potential for rare variants to further influence genetic risk.
In addition, as genome-interpretation approaches are becoming increasingly informed by diverse noncoding genome annotation,24–26 genome and transcriptome analysis within a single large family provides unique insight into the predictive power of diverse noncoding annotation for rare variants. In our study, we demonstrated that the combination of variant location, epigenomic information, and evolutionary constraint is considerably more informative for predicting the impact of rare noncoding variants than for predicting the impact of common variants. Likewise, we observed equivalent increases in predictive strength for rare splicing variants. This suggests that many rare noncoding variants are likely to be interpretable via existing noncoding annotation and supports their more routine integration in rare-variant association studies.
Material and Methods
Cell Culture and RNA Sequencing
Epstein-Barr-virus-transformed peripheral blood B lymphocytes (catalog no. XC01463) from families from the CEU population (Utah residents with ancestry from northern and western Europe from the CEPH collection) were purchased from the Coriell Institute and grown in RPMI 1640 supplemented with 10% fetal calf serum and penicillin and streptomycin in humidified 5% CO2 at a concentration of ∼1 × 106 cells/ml. Total RNA was isolated with Trizol. RNA quality was assessed with the Agilent Bioanalyzer 2100, and RNA integrity numbers above 9 were used for cDNA production. One microgram of total RNA was used for isolating polyA-purified mRNA and subsequently used for cDNA-library construction with the Illumina TruSeq RNA Preparation Kit. Strand specificity was performed with 2'-deoxyuridine 5'-triphosphate during second-strand synthesis.27 All samples were indexed with Illumina adapters and sequenced with an Illumina HiScanSQ. We subsequently sequenced each cDNA library on an Illumina HiSeq to obtain 30 million 75 bp paired-end reads per individual. We performed RNA sequencing (RNA-seq) for all 17 individuals (all three generations); however, for eQTL association, we only used the 11 children, and for ASE analysis, we used the two parents and 11 children. All RNA-seq data for all 17 individuals are freely available at the Gene Expression Omnibus under accession number GSE56961.
Quantification of Gene Expression, Splicing, and ASE
We used Tophat and Cufflinks to obtain gene-expression levels from RNA-seq. We used Tophat to map RNA reads to the human reference genome (UCSC Genome Browser, hg19) and Cufflinks to quantify transcript-expression levels. Gene-expression levels were the sum of transcript-expression levels. Gencode28 v.12 was used as the input annotation for Cufflinks. We calculated transcript ratios to quantify alternative splicing patterns. Gene-expression and transcript-ratio data for Geuvadis samples were downloaded from the Geuvadis website; we used quantified gene-level reads per kilobase per million both before (for assessing effect sizes) and after (for eQTL mapping) normalization via probabilistic estimation of expression residuals.29 We assessed ASE by counting RNA read depth at heterozygous sites. We performed multiple quality-control steps to reduce known technical artifacts (see Figure S2). We obtained read counts at each heterozygous site by using SAMtools30 mpileup and our own ASE pipeline based on a binomial test modified for reference-mapping bias with a filter for observing at least five reads for each allele and a minimum read depth of 20× per site.21,31 To assess the quality of ASE estimates, we compared ASE correlation between double-IBD (identical-by-descent) siblings, half-IBD siblings, and non-IBD siblings. Indeed, we observed an expected increase in correlation between degree of IBD and allelic ratio measured across all sites (Figures S28–S30).
Whole-Genome Sequencing Data
Whole-genome sequencing data for the family were downloaded from the Complete Genomics website. Family members were originally sequenced to an average genome-wide coverage of 80×. We used variants called by the Complete Genomics Analysis Pipeline (v.2.0.0). We performed an additional filtering step testing for Mendelian inconsistency to obtain a high-confidence set of variants, and we eventually retained 5,546,682 out of the original 6,181,281 SNPs. We further compared our selected variants to those assessed by long-fragment read (LFR) technology (N50s 400–1,500 kb).32 LFR has a claimed error rate of 1 in 10 Mb. Our comparison showed that variant concordance between the 80× shotgun-sequencing approach and LFR technology was 99.91% to 99.95% (Table S2). In addition, the same family was also sequenced to 50× by Illumina Platinum Genomes, and the genotyping concordance with Complete Genomics was found to be 99.62% to 99.83% (Table S4).
Haplotyping and Verification by Long-Fragment Sequencing
We inferred recombination positions and haplotypes of the family by using our software tool Ped-IBD.33 Haplotype blocks are defined by recombination positions. We identified a total of 813 recombination positions over 22 chromosomes. Haplotype blocks range in size from 0.02 to 12 Mb (90% interval) and have a median length of 1.65 Mb. We further confirmed haplotyping results with molecular haplotypes from the LFR technology in three individuals (NA12877, NA12885, and NA12886; Table S3). The comparison showed that phasing was 99.84% to 99.92% concordant between inferred and molecular haplotypes.
Linkage Mapping of cis-eQTLs in the Family
We used linear regression to evaluate correlation of gene-expression levels within local haplotype blocks. We measured effect size by using the regression slope, β, and the coefficient of determination, R2. The linear model we used considers additive effects of two haplotype blocks. More specifically, for each block, the two parental haplotypes of each child are encoded with two covariates, p and m. The maternal haplotype mi of child i, for example, is either 0 or 1, depending on which of the two possible maternal alleles is present. Then, an expression trait is regressed as the summation of effects of two parental haplotypes, Ti ∼ μ + βjpi + βkmi, where Ti is the trait of individual i, the effects of two parental alleles k and j are expressed by βj and βk, and μ is the intercept. Each sibling has two choices of parental haplotypes on each side—p,m∈{0,1}—to yield four total combinations. Gene expression Ti uses log2(FPKM [fragments per kilobase per million] values). For splicing quantifications, we used relative transcript abundances, which we calculated by dividing the FPKM of each isoform by the FPKM of the whole gene (see Table S5). For cis-eQTLs, we only tested the local haplotypes containing the genes, which is sufficient for including most cis-eQTL signals (Figures S3–S5). Furthermore, we confirmed gene-expression levels and eQTL effect sizes with existing microarray data on the same family (Figures S6 and S7).
Comparison of cis-eQTL Effect Sizes between Population and Family
To compare cis-eQTL effect sizes, β, between the population and family, we sought to first correct for the overestimation of effect sizes (such discoveries exhibit characteristic regression to the mean). To address this in the population eQTLs, we divided the European-descended Geuvadis samples (n = 373) in half and partitioned them into discovery (n = 180) and replication (n = 193) panels. Within the discovery panel, we identified the strongest cis-associated variant per gene (by p value and within the same interval tested in the family). This allowed us to use the replication panel to more accurately measure the effect size of each cis-eQTL variant. However, to account for the difference in sample sizes between the replication panel (n = 193) and the family (n = 11), we further sought to estimate how much variance in effect-size measurements (β) could be obtained from sampling 11 people in the population at random. In this way we controlled for chance observations of larger effect sizes for some genes in the family. To achieve this, we repeatedly subsampled (100 times) 11 individuals from the replication panel while maintaining the exact same genotypes of the best associated variant between the subsample and the family. Figure S8 illustrates this subsample scheme. Effect sizes were then measured with the same regression formula, Ti ∼ μ + βjp + βkm, for both the family and the subsample; note that two regressors, p and m, match segregating patterns of both the haplotypes of the family and the best SNP of the population subsample. We note that estimation of β in the population was highly correlated independently of the use of a one- or two-regressor model (Figure S9). This allowed us to create a distribution of measured effect sizes that would be expected from randomly measuring the same number of individuals and genotypes in both the family and the population. Using this approach, we identified empirical p values representing how often measured effect sizes in the family were greater than that of the best associated SNP in the population. We also repeated this analysis by using fit (R2) given that we observed differences in the distribution of raw β values between the family and population and also observed higher variance in gene expression in Geuvadis overall (see Figure S17).
We analyzed several features that could result in over- or underestimation of effect-size measurements between the family and population (see Figures S17–S19). First, because effect-size measurements can be influenced by differences in quantification pipelines, we repeated the experiment by using different quantification approaches (Tophat + Cufflinks and GEM34 + Flux Capacitor;31 Figures S13 and S14). Second, effect sizes in the population could potentially be underestimated if the best associated SNP in the discovery panel is not causal given that subsequent effect-size measurements, in the replication panel, might not accurately measure the largest effect. To address this, we examined different discovery-panel sizes (Table S6 and Figure S15) and different criteria (Figure S16) for selecting the best SNP from the population. In addition, we observed through permutation that levels of noise in measurements of effect size (β) were different between the family and the population (Figure S17). To better gauge confidence intervals (CIs) of family effect sizes, we estimated the degree of inflation through permutation and adjusted effect-size CIs by scaling. These adjusted CIs were only applied to comparisons of β values and are denoted by CIadjusted (see Figure S17–S19). For the main manuscript, we report only unadjusted CIs. Furthermore, without using subsampling or permutation, we also directly compared effect sizes with Welch’s t test by applying analytic estimation of SEs of β. As a correctness check of the subsampling method, we compared and verified that analytic p values by Welch’s t test and empirical p values by subsampling were concordant (Figure S19).
We applied the same subsampling method to identify large-effect splicing quantitative trait loci (sQTLs) and ASE. To compare ASE between the family and population, we focused on a subset of genes that had substantial data for the measurement and comparison of allelic ratios (n = 1,777 genes). For a gene to be included, allelic ratios at a single site had to be measurable for at least five siblings and at least 30 population samples. We tested each gene once and excluded genes that were not tested for eQTLs, such as pseudogenes or genes within high-complexity regions (human leukocyte antigen and immunoglobulin loci). For a site to be considered measurable, it needed to be covered by a minimum of 20 reads with at least five reads for each allele. We then took the maximum allelic ratio in the family and compared it with the maximum allelic ratio found in 1,000 subsamples of the Geuvadis; each subsample was matched to the number of heterozygous individuals found in the family for that site. This approach generated an empirical p value that we used to assess whether an ASE effect in the family was greater than that in the population. To account for ASE biases caused by differing read depths between the family and population, we downsampled (hypergeometric) Geuvadis reads by a factor of 1.97—we calculated this scaling factor by measuring the average level of read-depth differences between Geuvadis and family samples at those selected heterozygous sites for each gene. To exclude the possibility that large-effect ASE was due to technical artifacts such as mapping biases or sequencing errors, we also looked at ASE for the second-largest-effect siblings and IBD siblings (Figure S25).
Variant Annotation
We obtained annotations (missense, synonymous, regulatory, and splice region) by using the Variant Effect Predictor tool,35 which queries annotation from the Ensembl website. ENCODE transcription factor (TF) binding and DNase I hypersensitivity peaks were obtained from RegulomeDB.24 Conservation scores obtained from PhyloP36 (phyloP100way) software were downloaded from the UCSC Genome Browser. Motif-disrupting sites were downloaded from HaploReg (v.2).37 Variant allele frequency was based on phase 1 of the 1000 Genomes Project38 as calculated across European populations.
Conservation and Network Annotation
We examined the conservation of family eQTL genes between humans and chimpanzees (Pan Troglodytes) by using the dN/dS ratios; dN measures the rate of amino acid substitutions, and dS measures the background rate of neutral DNA subsitutions.39 The dN and dS values were obtained from BioMart40 (Ensembl v.70), and the dN/dS ratios were computed. dN/dS is negatively correlated with the conservation status of a gene, so higher dN/dS ratios indicate lower conservation of a gene. We also compared centrality of eQTL genes by using the protein-protein interaction (PPI) network as another indication of the biological importance of the affected genes.19 We computed connectivity of family and population eQTL genes in the PPI network. The PPI network was integrated from BioGRID,41 the Molecular Interaction database,42 the Human Protein Reference Database,43 and IntAct,44 all data obtained from the GeneMANIA45 data repository (downloaded on January 4, 2012).
Rare-Variant Enrichment Analyses
To control for site discovery and genotyping differences between the population (1000 Genomes Project) and family (Complete Genomics) genomes, we performed enrichment analyses only for variants in the family genomes. Using these data, we calculated enrichment of rare variants at large-effect-size genes by dividing the proportion of large-effect-size genes with a rare variant by the proportion of all tested genes with a rare variant.
Results
We set out to develop an improved understanding of the impact and interpretability of rare noncoding variants. Our approach involved combining high-quality genomes and transcriptomes within a single large family to identify cis-eQTLs and compare these to cis-eQTLs discovered in a large population sample. Through the use of RNA-seq data, we were also able to conduct comparable analyses for alternative splicing and ASE. Our analyses focused on the enrichment of rare and potentially regulatory variants in large-effect eQTLs and sQTLs in the family, and we sought to identify the properties of genes that exhibit such effects. Furthermore, we investigated the degree to which family transcriptome data enable the detection of noncoding annotation relevant to interpreting rare noncoding variants genome-wide.
Family Transcriptome Sequencing Identifies Large-Effect cis-eQTLs
We hypothesized that rare variants acting either alone or in combination with common variants can cause an eQTL to exhibit a larger effect size in the family than in the population. To identify such cases, we applied a ranking scheme in which we compared gene-expression cis-eQTLs between the family and the population to find genes that exhibited larger effect sizes within the family (see Material and Methods). At CI > 0.95 (or empirical p value < 0.05), we found that 319 (including both paternal and maternal β measurements) of the 7,341 genes we tested had effect sizes exceeding that of the best population cis-eQTL SNP (false-discovery rate [FDR] = 7,341 × 0.05 × 2 / 319 > 1; Figure 1A). Using comparisons of β, we did not find more relatively large-effect eQTLs than we would expect by chance; however, we identified that this FDR is likely overconservative primarily because of differences in noise between the family and population (see Figures S17–S19), and we therefore also discuss less conservative estimates of FDR (see Figures S17–S19). It is important to note that FDR here measures whether there are more large effects in the family than in the population; however, ranking relative effect sizes by empirical p values is biologically meaningful whether there is an excess or a depletion. Such relative effects overlap (to a degree) genes measured only by absolute effect size in the family; for instance, when comparing genes at the 95% percentile for absolute β versus relative β, we observed an overlap of 52% (Figure S12). However, we chose to use in all subsequent analyses the ranking of genes according to their relative effect sizes instead of absolute effect sizes because we hypothesized that the former might better inform family-specific effects. By instead measuring fit (R2), we identified 577 cis-eQTLs that had a better fit in the family than the best population-level cis-eQTL variant (CI > 0.95; FDR = 7,341 × 0.05 / 577 = 63%; Figure S10). Among those genes that exhibited the largest effect sizes and fits in the family (both at a CI > 0.95), there was a significant overlap of 36.4% (Figure S11). To exclude the possibility of technical factors underlying effect-size differences, we repeated the analysis by using different quantification pipelines (Figures S13 and S14), population discovery-panel sizes (Table S6), and alternative methods for choosing the best SNP (Figure S16); we observed no significant difference in the discovery set of large-effect genes or on further downstream analyses (see Material and Methods).
Figure 1.
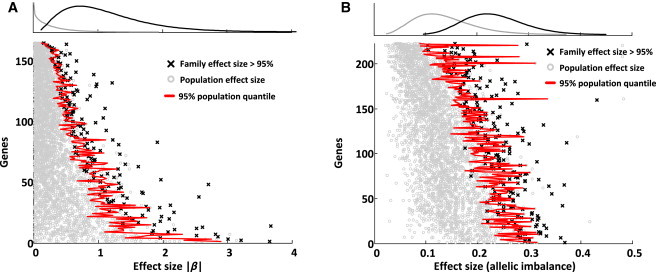
Large-Effect eQTLs and ASE in the Family
(A) Large-effect cis-eQTLs. Effect sizes are shown as β, the regression slope. The distribution of family effect sizes (black) is compared to the distribution of population effect sizes (gray). We show cis-eQTL genes for which family effect sizes are greater than 95% of population effect sizes. Here, we only plot the distribution of paternal effect sizes (maternal effects have a very similar distribution).
(B) Large-effect ASE genes. ASE effect sizes were assessed by allelic imbalance (0 is balanced, and 0.5 is monoallelic expression). We picked the maximum ASE effect out of 11 siblings and compared it to the maximum ASE effect out of the subsampled population. Plotted are family ASE effects greater than 95% of population ASE effects. To exclude outlier effects, we further tested this for the second-strongest ASE effect in the siblings (Figure S22).
We also identified genes that exhibited larger ASE effects in the family than in the population. We found that 223 of the 1,777 genes we tested had larger ASE effect sizes in the family (CI > 0.95, FDR = 1,777 × 0.05 / 223 = 40%; Figure 1B; Figure S25). We expected that on an individual basis, the family and population would actually have the same distribution of ASE effect sizes (no excess of large effects, FDR = 1). We controlled for some initially observed excess in the family by matching read depths via downsampling; however, this did not address all the excess in the family, and unknown factors still remained. We expected that any excess, however, would only add noise to subsequent rare-variant enrichment analyses, and we further validated large ASE effects by using evidence from IBD siblings (Figure S25). In addition, we applied ASE to support discoveries of cis-eQTLs in the family; by stratifying their degree of effect size relative to those in the population, we detected a proportionally increased enrichment of detectable ASE (significant ASE sites defined as allelic imbalance > 0.05, binomial test p value < 0.05; Figure S21). This relationship supports a potential regulatory role of rare variants because it indicates that large-effect cis-eQTLs in the family might be the consequence of heterozygous variants that manifest in ASE. This idea is further supported by our observation of a direct and simple linear relationship between cis-eQTL effect size among children and ASE effect size among parents (Figure S20).
Large-Effect cis-eQTLs in the Family Are Enriched with Rare Variants
We hypothesized that rare noncoding variants might be responsible for a considerable proportion of the large-effect-size cis-eQTLs in the family. Taking advantage of full genome data in the family, we assessed the enrichment of rare and potentially regulatory variants near the transcription start site (TSS) of genes with different magnitudes of relative effect sizes between the family and the population. Here, we used PhyloP to define potentially regulatory variants on the basis of ENCODE TF peaks, DNase I hypersensitivity peaks, and evolutionarily constrained regions across 99 vertebrate genomes; we will later further explore the relative importance of each of these annotations. We observed enrichment of rare and potentially regulatory noncoding variants in genes that had the largest effect sizes (CI > 0.95 and CI > 0.80; Figure 2A). This relationship was most pronounced within the first 5 kb close to the TSS and decayed as a function of distance. It was also related to the degree to which the family effect was larger than that detected in the population across the full distribution of measured effects (Figure 2B). Likewise, we tested both large-effect cis-eQTLs by fit (R2) and large-effect ASE genes and observed similar strong enrichment of rare and potentially regulatory variants (Figures S23 and S25C).
Figure 2.
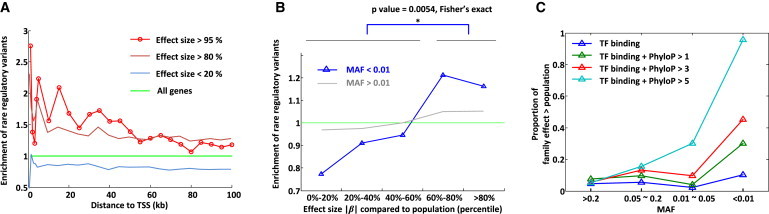
Enrichment of Rare Variants in Large-Effect eQTLs
(A) Enrichment of rare and potentially regulatory variants near the TSS of large-effect (β) cis-eQTL genes. Variants are restricted to those with a MAF < 0.01, within ENCODE TF binding and DNase I hypersensitivity peaks, and with a PhyloP score > 1. We observed increased enrichment of rare regulatory variants near the TSS of larger-effect-size genes in the family.
(B) Enrichment of potentially regulatory variants depends on allele frequency and relative effect sizes. We ranked genes (x axis) on the basis of how often their effect sizes in the family were greater than their effect sizes in the population subsamples, which is also 1 − their empirical p values (see Material and Methods). Variants are restricted to those within <100 kb of the TSS, within ENCODE TF binding and DNase I hypersensitivity peaks, and with a PhyloP score > 1. We observed that variant enrichment was dependent on whether the variant was rare (blue) or not (gray). We calculated enrichment by dividing the proportion of genes with such an annotated rare variant in each effect-size bin by the proportion of genes with an annotated rare variant across all effect-size bins.
(C) Conservation scores and allele frequency predict genes with a larger effect in the family than in the population. We restricted to variants within <100 kb of the TSS, within ENCODE TF binding and DNase I hypersensitivity peaks, and with different PhyloP thresholds. Proportions were computed by π1 statistics on permutation-based p values of family effect larger than population effect. We observed that rare and highly conserved variants overlapping epigenomic data (light blue) were highly predictive of a larger effect in the family than in the population.
We also evaluated the utility of known regulatory annotations in predicting eQTLs for rare variants. Comparing annotated rare variants with all rare variants, we observed strong enrichment (up to a 2-fold increase near the TSS) of annotated variants, indicating that annotation is highly informative in predicting eQTLs (Figure S22). Furthermore, we observed that the enrichment was higher in family-based eQTLs than in population eQTLs as a function of effect size (Figure S24). To test the contribution of different annotations to a large effect in the family, we further stratified by MAF and strength of evolutionary constraint. We observed that variants with lower MAF and with increasing degree of evolutionary constraint were the most informative factors indicative of large cis-eQTL effects in the family (Figure 2C).
Large-Effect cis-eQTLs in the Family Influence Essential Genes
It has been previously reported that cis-eQTLs based on population studies are depleted among essential genes.19 We hypothesized that if rare variation was indeed responsible for large-effect cis-eQTLs in the family, reduced impact of purifying selection on rare variants would result in family eQTLs disproportionately affecting essential genes. We tested this hypothesis in two ways: defining gene essentiality by (1) its degree of evolutionary constraint and (2) its centrality within a PPI network. To assess evolutionary constraint, we used dN/dS ratios between humans and chimps to compare large-effect cis-eQTL genes in the family to cis-eQTL genes in the population. We observed that large-effect cis-eQTL genes in the family had significantly higher conservation status than population cis-eQTL genes (Figure 3A). This was even more pronounced for genes with a rare and potentially regulatory variant within 5 kb of the TSS. By contrast, cis-eQTL genes in the population were less constrained for increasingly stringent p values.
Figure 3.
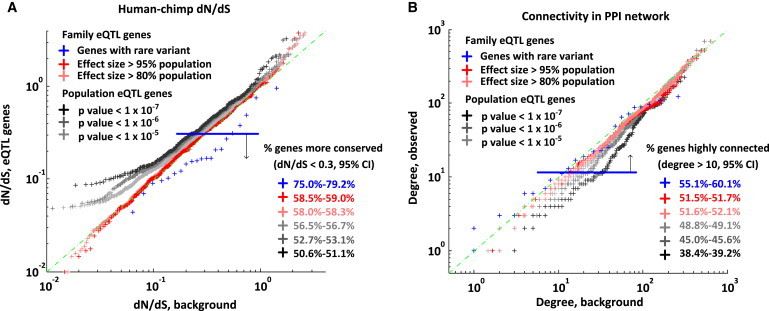
Large-Effect eQTLs Influence Essential Genes
(A) dN/dS ratio comparing large-effect family cis-eQTLs to population cis-eQTLs. We selected family eQTLs on the basis of their effect sizes relative to population eQTL effect sizes and plotted the distributions of dN/dS ratios. As a comparison, we show the distribution of dN/dS ratios for the most significant cis-eQTL genes identified only in the population (373 unrelated European individuals from the Geuvadis study) given different p value cutoffs. This is further compared to family-level genes that have rare and potentially regulatory variants (within 5 kb of the TSS, within ENCODE TF binding and DNase I hypersensitivity peaks, and with a PhyloP score > 1). We observed that for large-effect cis-eQTLs and family-level genes with a rare variant, a higher proportion were more conserved (described as the percentage of genes with a dN/dS < 0.3; lower dN/dS ratios indicate higher conservation).
(B) Comparison of centrality in the PPI network between large-effect cis-eQTLs in the family and population cis-eQTLs. Centrality is measured by the number of interacting proteins (degrees). Different groups of genes are defined in the same way as in (A). We show proportions of high-connectivity (hub) genes (degree > 10; higher degrees indicate more essential genes) among these groups. We observed that the proportion of high-connectivity genes was greatest for large-effect cis-eQTLs and family-level genes with a rare variant. This suggests that common regulatory variants are less likely to occur at conserved genes. In contrast, family-specific eQTL effects, because they arise from rare variants, can affect conserved genes.
We next applied PPI networks with the premise that genes that are more central in the network or have more connections to other genes are more essential than less connected ones. We found significantly higher connectivity for large-effect cis-eQTL genes in the family than for cis-eQTL genes in the population (Figure 3B). Furthermore, this contrast became stronger when we focused only on those genes that also contained a proximal rare and potentially regulatory variant (Figure 3B).
Family Transcriptome Sequencing Identifies Large-Effect sQTLs
By comparing cis-sQTLs between the family and the population, we also ranked genes with larger relative effect sizes (measured as R2) in the family than in the population (n = 726, >95% population, n = 5,622 genes; FDR = 39%). Differences in isoform-quantification pipelines probably overestimate the excess number of large-effect sQTLs because there is also more noise in isoform quantification in the population. However, as for large-effect eQTLs, we also observed enrichment of rare and potentially functional variants for large-effect sQTL genes in the family (Figure 4A). Furthermore, by stratifying on allele frequency, distance to splice sites, and evolutionary-constraint thresholds, we found that large-effect sQTLs in the family were much better predicted by rare variants than by common variants, especially for conserved regions near splice sites (Figure 4B). In addition to observing large effect sizes, we also found that sQTLs could exhibit very high heritabilities, nearly as high as those for Mendelian traits (examples in Figures S26 and S27).
Figure 4.
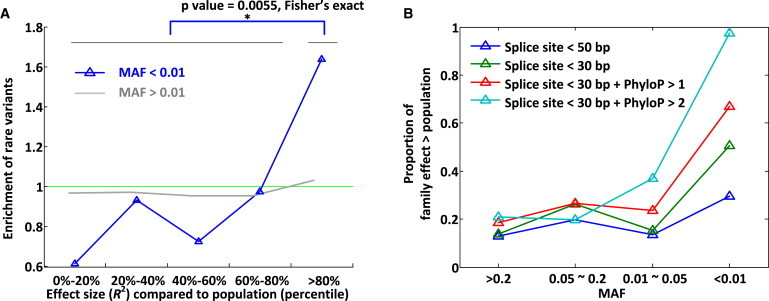
Large-Effect sQTLs in the Family
(A) Enrichment of rare variants at large-effect sQTL genes. We ranked genes (x axis) on the basis of how often their effect sizes in the family were greater than their effect sizes in the population subsamples (see Material and Methods). We restricted to variants within 30 bp of splice sites and with a PhyloP score > 1. As for cis-eQTLs (in Figure 2B), we observed that enrichment was dependent on allele frequency. We calculated enrichment by dividing the proportion of genes with such an annotated rare variant in each effect-size bin by the proportion of genes with an annotated rare variant across all effect-size bins.
(B) Conservation scores, the distance to splice site, and allele frequency predict genes with a larger effect in the family than in the population. We observed that rare and conserved variants near splice sites (light blue) were highly predictive of a larger splicing effect in the family than in the population.
Large-Effect cis-eQTLs in the Family Might Further Modify Complex-Disease-Associated Genes
There has been considerable interest in whether rare variants modify risk of complex disease.46,47 Although we were unable to directly test disease associations within this family because of the anonymity of the individuals, we sought to quantify the number of genome-wide association study (GWAS) genes in which cis-eQTLs exhibited larger effects in the family than in the population. We identified 315 GWAS genes in which the known GWAS variant was an eQTL in the population (at an FDR of 5%), suggesting a regulatory basis to disease pathogenesis. Of these genes, we identified 65 with a larger-effect cis-eQTL in the family (>80th percentile). Of those, 17 (Table S9) were not polymorphic for the known GWAS SNP in the family, and two had a rare and potentially regulatory variant (within <100 kb of the TSS, within an ENCODE TF binding and DNase I hypersensitivity peak, and with a PhyloP score > 0) influencing genes implicated in body mass index, hypertension, and obesity. In addition, regardless of relative effect sizes of eQTLs between the family and population, we identified four GWAS genes (Table S10) in which the known GWAS SNP was an eQTL in the population and that had a rare and potentially regulatory variant (within <100 kb of the TSS, within an ENCODE TF binding and DNase I hypersensitivity peak, and with a PhyloP score > 3) in the family according to strong predictor variables. Although increased risk in this family is not known, the presence of rare and potentially regulatory variants in complex-disease-associated genes whose expression is implicated in disease pathogenesis suggests that complex traits and genes should be further studied with rare-variant association tests.
Functional Noncoding Annotations Are Informative of the Impact of Rare Noncoding Variants
Genome and transcriptome data from a single large family allowed us to test the utility of various noncoding annotations for predicting the impact of noncoding variants on expression. Here, our goal was to identify those annotations that could inform a functional variant from genome sequence alone. We chose to include the following as potentially informative annotations: ENCODE TF binding, DNase I hypersensitivity peaks, evolutionary constraint, motif disruption as computed by HaploReg, and distance to the TSS. We identified that each noncoding annotation was more informative for predicting the impact of rare variants than the impact of common variants on expression (Figure 5A; Table S7). We observed that evolutionary constraint and distance to the TSS were the most informative for rare variants, and they further increased their utility with increasing strength of constraint and shorter distances, respectively. One potential concern we identified is that we might be only predicting a gene’s ability to harbor an eQTL such that having a rare variant possessing specific annotation might indirectly inform genes tolerant of arbitrary functional variants (both common and rare). However, when assessing whether genes containing different annotations for rare variants were also more likely to have common eQTLs in the population, we saw no significant difference (Figure 5A, right panel). This demonstrates that particular species of rare noncoding variants might be interpretable from genome sequence data alone provided that there is sufficiently high-confidence genotyping of those rare variants. Furthermore, provided increasing availability of genome-interpretation methods, this method offers a means of determining and calibrating the efficacy of different approaches.
Figure 5.
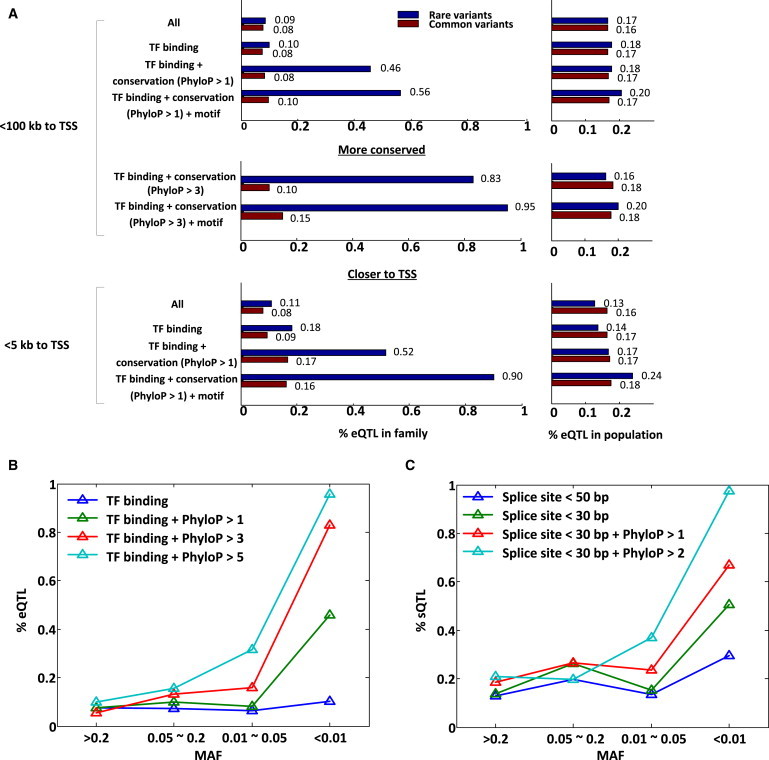
Predicting Rare and Common eQTLs
(A) Utility of diverse noncoding annotation for predicting rare and common eQTLs. We considered the enrichment in eQTLs (measured with the π1 statistic) for rare (MAF < 0.01) and common (MAF > 0.01) variants overlapping the following different functional annotations: ENCODE TF binding and DNase I hypersensitivity peaks, distance to TSS, PhyloP conservation scores, and motif disruption (score change > 10); annotations were added one at a time. We found that these functional annotations were significantly more powerful for detecting an eQTL when intersecting rare variants rather than common variants. Furthermore, on the right, we demonstrate that none of the genes possessing rare variants overlapping the different categories of annotation were disproportionally enriched in their ability to also be eQTLs in the population. A full matrix summarizing intersections of these annotations is provided in Table S7.
(B) Conservation scores and allele frequency predict genes with an eQTL. We restricted to variants within 100 kb of the TSS, within ENCODE TF binding and DNase I hypersensitivity peaks, and with different PhyloP scores and allele frequencies to assess each variant class’s enrichment in eQTLs. We observed that highly conserved and rare variants were strongly predictive of an eQTL.
(C) Conservation scores, the distance to splice site, and allele frequency predict genes with a sQTL. We considered different thresholds on distance to splice sites, PhyloP conservation scores, and allele frequencies. We observed that rare and conserved variants near splice sites (light blue) were highly predictive of a sQTL.
Through finer stratification of allele frequency, we were able to observe the degree to which genome annotation influenced predictions of cis-eQTLs. We observed that predictions of eQTLs were most informative for potentially regulatory variants when those variants were rare (Figure 5B). This was also the case for sQTLs: predictor variables such as evolutionary constraint and distance to splice sites were the most informative factors for predicting a sQTL when a variant was rare (Figure 5C).
Discussion
Our study combined high-quality genome sequencing and RNA-seq data for a 17 member, three-generation family, enabling us to investigate the role and interpretability of rare noncoding variants. In contrast to low-pass approaches, high-quality full-coverage genome sequencing and patterns of Mendelian segregation provided the ability to more confidently identify and genotype rare variants within the family. More importantly, the large number of children provided us with the ability to detect eQTLs caused by rare variants specific to the family. In contrast, the power of a design that includes many small families or trios would be reduced by the overall heterogeneity of causal rare variants in each family. A further advantage is that with matched cell type and population, we were able to compare family eQTLs to population eQTLs reported by the Geuvadis Consortium.18 We identified genes that exhibit larger eQTL effect sizes in the family than in the population and demonstrated that these family-specific eQTLs are enriched with rare regulatory variants, influence more evolutionarily constrained and central genes, and are potential contributors to risk of complex disease.
One limitation of the study is that we did not observe many more large-effect eQTLs in the family than expected by chance; high FDRs were observed for all categories of large-effect eQTLs. This could suggest that there is not an overabundance of large-effect eQTLs specific to the family. It might also simply reflect limited power or imperfect comparison of effect sizes between cohorts, given that we explored by varying quantification pipelines, discovery-panel sizes, and methodologies for selecting testable variants. However, the enriched properties we identified for large-effect family eQTLs appear to be robust to such limitations, and we highlight that although there might not be a strong excess of large-effect eQTLs, the relative degree of effect between the family and population conveys meaningful properties of family eQTLs. For instance, as the degree of effect size increased in the family relative to the population, we observed an increasing enrichment of rare and potentially regulatory variants. Furthermore, such large-effect eQTLs in the family exhibited increasing enrichment in ASE, implicating a heterozygous causal variant. Additionally, the enrichment of family eQTLs among constrained and central genes was most extreme for the subset of genes in which a rare and potentially regulatory variant could be identified. These observations fit with population-genetic expectation given that rare variants can influence more essential genes because of a reduced impact of purifying selection. Furthermore, this is in contrast to the general properties of population eQTL genes; for increasing effect sizes, they have previously been shown to be less constrained and less central.19 Taken together, these results implicate an important role of rare regulatory variants in large-effect eQTLs in the family.
We compared, in addition to gene expression, ASE and alternative splicing between the family and the population. As with gene expression, we observed enrichment in rare variants for large-effect ASE and sQTLs in the family. Furthermore, we observed that evolutionary constraint and distance to splice sites for rare splicing variants was significantly informative of large splicing effects in the family. With both large-effect eQTLs and large-effect sQTLs predicted by rare variants, this study highlights existing potential for routine integration of these variants in rare-variant association tests.
Ultimately, a principal goal in genome interpretation is to develop the ability to predict the impact of all variants, including those that are rare or novel. In our study, we were able to test the importance of diverse noncoding annotations for predicting the impact of noncoding variants on gene expression. For rare variants, we identified that evolutionary constraint coupled with distance to the TSS and epigenomic information was highly informative in predicting eQTLs. For common variants, such annotations did not provide comparable predictive power. The likely reason for this difference is that common variants, regardless of genomic annotation, are very likely to be neutral, whereas rare variants have a higher prior likelihood of functional impact that can be further informed by genomic annotation. Given that no previous analyses have had access to high-quality genomes and transcriptomes in a single large human family, this study provides data to support a much-needed framework for frequency-independent evaluation of genome interpretation for noncoding variants and suggests that the impact of many rare and causal noncoding variants might be easier to predict than expected.
Acknowledgments
We would like to thank Tomas Babak, Christopher Brown, Hunter Fraser, Arend Sidow, and members of the S.B.M. lab for critical review of this work and manuscript. This work was supported by the Edward Mallinckrodt, Jr. Foundation and the Li Ka Shing Foundation.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Contributor Information
Xin Li, Email: xxli@stanford.edu.
Stephen B. Montgomery, Email: smontgom@stanford.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Phase 1 Analysis Results, ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/phase1/analysis_results/
Complete Genomics, 69 Genomes Data, http://www.completegenomics.com/public-data/69-Genomes/
Ensembl Genome Browser, http://www.ensembl.org
Ensembl Variant Effect Predictor, http://www.ensembl.org/info/docs/tools/vep/
GeneMANIA, http://www.genemania.org/
Geuvadis Data Browser, http://www.ebi.ac.uk/Tools/geuvadis-das/
Geuvadis RNA sequencing project, http://www.geuvadis.org/web/geuvadis/RNAseq-project
GWAS catalog, http://www.genome.gov/admin/gwascatalog.txt
Illumina Platinum Genomes, whole-genome sequencing data, http://www.illumina.com/platinumgenomes/
LFR data for family members, ftp://ftp2.completegenomics.com/
PhyloP conservation scoring, http://hgdownload.cse.ucsc.edu/goldenpath/hg19/phyloP100way/
RegulomeDB, http://regulomedb.org/
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The Gene Expression Omnibus accession number for the RNA-seq data of all 17 individuals reported in this paper is GSE56961.
References
- 1.Nelson M.R., Wegmann D., Ehm M.G., Kessner D., St Jean P., Verzilli C., Shen J., Tang Z., Bacanu S.A., Fraser D. An abundance of rare functional variants in 202 drug target genes sequenced in 14,002 people. Science. 2012;337:100–104. doi: 10.1126/science.1217876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tennessen J.A., Bigham A.W., O’Connor T.D., Fu W., Kenny E.E., Gravel S., McGee S., Do R., Liu X., Jun G., Broad GO. Seattle GO. NHLBI Exome Sequencing Project Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;337:64–69. doi: 10.1126/science.1219240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keinan A., Clark A.G. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science. 2012;336:740–743. doi: 10.1126/science.1217283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coventry A., Bull-Otterson L.M., Liu X., Clark A.G., Maxwell T.J., Crosby J., Hixson J.E., Rea T.J., Muzny D.M., Lewis L.R. Deep resequencing reveals excess rare recent variants consistent with explosive population growth. Nat. Commun. 2010;1:131. doi: 10.1038/ncomms1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marth G.T., Yu F., Indap A.R., Garimella K., Gravel S., Leong W.F., Tyler-Smith C., Bainbridge M., Blackwell T., Zheng-Bradley X., 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu W., O’Connor T.D., Jun G., Kang H.M., Abecasis G., Leal S.M., Gabriel S., Rieder M.J., Altshuler D., Shendure J., NHLBI Exome Sequencing Project Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature. 2013;493:216–220. doi: 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Flannick J., Thorleifsson G., Beer N.L., Jacobs S.B., Grarup N., Burtt N.P., Mahajan A., Fuchsberger C., Atzmon G., Benediktsson R., Go-T2D Consortium. T2D-GENES Consortium Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat. Genet. 2014;46:357–363. doi: 10.1038/ng.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanna S., Li B., Mulas A., Sidore C., Kang H.M., Jackson A.U., Piras M.G., Usala G., Maninchedda G., Sassu A. Fine mapping of five loci associated with low-density lipoprotein cholesterol detects variants that double the explained heritability. PLoS Genet. 2011;7:e1002198. doi: 10.1371/journal.pgen.1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Momozawa Y., Mni M., Nakamura K., Coppieters W., Almer S., Amininejad L., Cleynen I., Colombel J.F., de Rijk P., Dewit O. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat. Genet. 2011;43:43–47. doi: 10.1038/ng.733. [DOI] [PubMed] [Google Scholar]
- 10.Raychaudhuri S., Iartchouk O., Chin K., Tan P.L., Tai A.K., Ripke S., Gowrisankar S., Vemuri S., Montgomery K., Yu Y. A rare penetrant mutation in CFH confers high risk of age-related macular degeneration. Nat. Genet. 2011;43:1232–1236. doi: 10.1038/ng.976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Panoutsopoulou K., Tachmazidou I., Zeggini E. In search of low-frequency and rare variants affecting complex traits. Hum. Mol. Genet. 2013;22(R1):R16–R21. doi: 10.1093/hmg/ddt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simons Y.B., Turchin M.C., Pritchard J.K., Sella G. The deleterious mutation load is insensitive to recent population history. Nat. Genet. 2014;46:220–224. doi: 10.1038/ng.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunt K.A., Mistry V., Bockett N.A., Ahmad T., Ban M., Barker J.N., Barrett J.C., Blackburn H., Brand O., Burren O. Negligible impact of rare autoimmune-locus coding-region variants on missing heritability. Nature. 2013;498:232–235. doi: 10.1038/nature12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacArthur D.G., Tyler-Smith C. Loss-of-function variants in the genomes of healthy humans. Hum. Mol. Genet. 2010;19(R2):R125–R130. doi: 10.1093/hmg/ddq365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicolae D.L., Gamazon E., Zhang W., Duan S., Dolan M.E., Cox N.J. Trait-associated SNPs are more likely to be eQTLs: annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., Geuvadis Consortium Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Battle A., Mostafavi S., Zhu X., Potash J.B., Weissman M.M., McCormick C., Haudenschild C.D., Beckman K.B., Shi J., Mei R. Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Res. 2014;24:14–24. doi: 10.1101/gr.155192.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kwan T., Benovoy D., Dias C., Gurd S., Provencher C., Beaulieu P., Hudson T.J., Sladek R., Majewski J. Genome-wide analysis of transcript isoform variation in humans. Nat. Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- 21.Montgomery S.B., Lappalainen T., Gutierrez-Arcelus M., Dermitzakis E.T. Rare and common regulatory variation in population-scale sequenced human genomes. PLoS Genet. 2011;7:e1002144. doi: 10.1371/journal.pgen.1002144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaffney D.J., Veyrieras J.B., Degner J.F., Pique-Regi R., Pai A.A., Crawford G.E., Stephens M., Gilad Y., Pritchard J.K. Dissecting the regulatory architecture of gene expression QTLs. Genome Biol. 2012;13:R7. doi: 10.1186/gb-2012-13-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lupski J.R., Belmont J.W., Boerwinkle E., Gibbs R.A. Clan genomics and the complex architecture of human disease. Cell. 2011;147:32–43. doi: 10.1016/j.cell.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boyle A.P., Hong E.L., Hariharan M., Cheng Y., Schaub M.A., Kasowski M., Karczewski K.J., Park J., Hitz B.C., Weng S. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–1797. doi: 10.1101/gr.137323.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ritchie G.R., Dunham I., Zeggini E., Flicek P. Functional annotation of noncoding sequence variants. Nat. Methods. 2014;11:294–296. doi: 10.1038/nmeth.2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parkhomchuk D., Borodina T., Amstislavskiy V., Banaru M., Hallen L., Krobitsch S., Lehrach H., Soldatov A. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stegle O., Parts L., Piipari M., Winn J., Durbin R. Using probabilistic estimation of expression residuals (PEER) to obtain increased power and interpretability of gene expression analyses. Nat. Protoc. 2012;7:500–507. doi: 10.1038/nprot.2011.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montgomery S.B., Sammeth M., Gutierrez-Arcelus M., Lach R.P., Ingle C., Nisbett J., Guigo R., Dermitzakis E.T. Transcriptome genetics using second generation sequencing in a Caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peters B.A., Kermani B.G., Sparks A.B., Alferov O., Hong P., Alexeev A., Jiang Y., Dahl F., Tang Y.T., Haas J. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature. 2012;487:190–195. doi: 10.1038/nature11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X., Yin X., Li J. Efficient identification of identical-by-descent status in pedigrees with many untyped individuals. Bioinformatics. 2010;26:i191–i198. doi: 10.1093/bioinformatics/btq222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marco-Sola S., Sammeth M., Guigó R., Ribeca P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods. 2012;9:1185–1188. doi: 10.1038/nmeth.2221. [DOI] [PubMed] [Google Scholar]
- 35.McLaren W., Pritchard B., Rios D., Chen Y., Flicek P., Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–2070. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward L.D., Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40(Database issue):D930–D934. doi: 10.1093/nar/gkr917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abecasis G.R., Auton A., Brooks L.D., DePristo M.A., Durbin R.M., Handsaker R.E., Kang H.M., Marth G.T., McVean G.A., 1000 Genomes Project Consortium An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurst L.D. The Ka/Ks ratio: diagnosing the form of sequence evolution. Trends Genet. 2002;18:486. doi: 10.1016/s0168-9525(02)02722-1. [DOI] [PubMed] [Google Scholar]
- 40.Flicek P., Amode M.R., Barrell D., Beal K., Brent S., Carvalho-Silva D., Clapham P., Coates G., Fairley S., Fitzgerald S. Ensembl 2012. Nucleic Acids Res. 2012;40(Database issue):D84–D90. doi: 10.1093/nar/gkr991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stark C., Breitkreutz B.J., Reguly T., Boucher L., Breitkreutz A., Tyers M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34(Database issue):D535–D539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chatr-aryamontri A., Ceol A., Palazzi L.M., Nardelli G., Schneider M.V., Castagnoli L., Cesareni G. MINT: the Molecular INTeraction database. Nucleic Acids Res. 2007;35(Database issue):D572–D574. doi: 10.1093/nar/gkl950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goel R., Harsha H.C., Pandey A., Prasad T.S.K. Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 2012;8:453–463. doi: 10.1039/c1mb05340j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aranda B., Achuthan P., Alam-Faruque Y., Armean I., Bridge A., Derow C., Feuermann M., Ghanbarian A.T., Kerrien S., Khadake J. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38(Database issue):D525–D531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Warde-Farley D., Donaldson S.L., Comes O., Zuberi K., Badrawi R., Chao P., Franz M., Grouios C., Kazi F., Lopes C.T. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gibson G. Rare and common variants: twenty arguments. Nat. Rev. Genet. 2011;13:135–145. doi: 10.1038/nrg3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cirulli E.T., Goldstein D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.