

Abstract
The retinogeniculate synapse, the connection between retinal ganglion cells (RGC) and thalamic relay neurons, undergoes robust changes in connectivity over development. This process of synapse elimination and strengthening of remaining inputs is thought to require synapse specificity. Here we show that glutamate spillover and asynchronous release are prominent features of retinogeniculate synaptic transmission during this period. The immature excitatory postsynaptic currents exhibit a slow decay time course that is sensitive to low-affinity glutamate receptor antagonists and extracellular calcium concentrations, consistent with glutamate spillover. Furthermore, we uncover and characterize a novel, purely spillover-mediated AMPA receptor current from immature relay neurons. The isolation of this current strongly supports the presence of spillover between boutons of different RGCs. In addition, fluorescence measurements of presynaptic calcium transients suggest that prolonged residual calcium contributes to both glutamate spillover and asynchronous release. These data indicate that, during development, far more RGCs contribute to relay neuron firing than would be expected based on predictions from anatomy alone.
Keywords: glutamate spillover, asynchronous release, development, retinogeniculate synapse, visual system
even before eye opening, information encoded in retinal activity is relayed to the visual cortex and plays a critical role in the proper formation of retinotopic maps in the visual cortex (Akerman et al. 2002; Huttenlocher 1967; Kirkby et al. 2013). Remarkably, this information transfer via the retinogeniculate synapse is maintained, despite rapidly changing properties of the immature connection (Hanganu et al. 2006; Mooney et al. 1996). Between postnatal day (p) 8 and p30, the average strength of a retinal ganglion cell (RGC) input increases 20-fold, while the number of retinal inputs that innervate a given relay neuron is pruned from many to few (Chen and Regehr 2000; Jaubert-Miazza et al. 2005). The synaptic mechanisms that allow immature weak retinal inputs to drive relay neuron firing and maintain information transfer are still incompletely understood.
One feature of the immature synapse that has been proposed to aid in the transmission of visual information is the prolonged waveform of the retinogeniculate excitatory postsynaptic current (EPSC) (Chen and Regehr 2000; Hooks and Chen 2006; Liu and Chen 2008; Ramoa and McCormick 1994; Ziburkus and Guido 2006). The decay time course of both α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) and N-methyl-d-aspartate receptor (NMDAR) EPSCs is slower in the young than the old (Chen and Regehr 2000; Liu and Chen 2008; Ramoa and Prusky 1997). Although receptor subunit changes contribute to part of the developmental acceleration of the EPSC, they cannot account for all of the observed changes in kinetics. Notably, the AMPAR quantal event does not exhibit similar slow decay kinetics to the evoked response, nor does the miniature EPSC waveform significantly accelerate with age (Liu and Chen 2008).
These findings suggest that other synaptic mechanisms contribute to the slow time course of the immature EPSC. For example, the slow component could be due to glutamate pooling within the synaptic cleft from impeded diffusion. Alternatively, it could be delayed (asynchronous) release, where the release of additional vesicles persists for up to hundreds of milliseconds following depolarization of the presynaptic terminal (Atluri and Regehr 1998; Barrett and Stevens 1972; Goda and Stevens 1994; Kaeser and Regehr 2014; Rahamimoff and Yaari 1973). Asynchronous release is driven by presynaptic residual calcium (Cares) and prolongs the decay time course of the postsynaptic response, thus enhancing synaptic charge transfer (Cummings et al. 1996; Vanderkloot and Molgo 1993; Zengel and Magleby 1981; Zucker and Lara-Estrella 1983). Finally, the slow EPSC decay could be due to glutamate spillover between release sites located within the same bouton or in neighboring boutons (Barbour et al. 1994; DiGregorio et al. 2002; Otis et al. 1996; Takahashi et al. 1995; Trussell et al. 1993). At the immature retinogeniculate synapse, glutamate can spill over to extrasynaptic metabotropic glutamate receptors (mGluRs), but whether glutamate also reaches neighboring release sites is not known (Hauser et al. 2013).
Here, we distinguish between these different explanations for the prolonged current observed at the immature retinogeniculate synapse. We find that both spillover between synaptic release sites and asynchronous release contribute to the extended glutamate time course. Moreover, we characterize purely spillover-mediated currents from relay neurons, indicating that neighboring RGCs can influence relay neuron firing without making direct contacts onto the cell. This prolonged glutamate transient is essential for the propagation of retinal activity to the developing cortex during a dynamic period of robust synaptic maturation and refinement.
METHODS
Slice preparation.
All experimental procedures were performed in accordance with federal guidelines and protocols approved by Boston Children's Hospital. Parasagittal brain slices containing both the optic tract and the dorsal lateral geniculate nucleus (LGN) were obtained as previously described (Chen and Regehr 2000; Liu and Chen 2008) from p8–34 C57BL/6 mice [C57BL/6J from Jackson Laboratory or C57BL/6NTac (B6) from Taconic]. Briefly, the brain was quickly removed and immersed into an oxygenated 4°C choline-based cutting solution containing the following (in mM): 87 NaCl, 25 NaHCO3, 37.5 choline chloride, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 7 MgCl2, and 0.5 CaCl2 or 130 choline chloride, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7.0 MgCl2, 0.5 CaCl2 and 25 glucose. Both cutting solutions yielded healthy slices at all ages. The brain tissue was then mounted on the cutting stage of a vibratome (Leica VT1000S) and submerged into oxygenated 4°C cutting solution. Cut 250-μm slices were allowed to recover for 15–20 min at 30°C in the oxygenated cutting solution and then for another 10–20 min at 30°C in oxygenated artificial cerebral spinal fluid (aCSF) containing the following (in mM): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 2 CaCl2 and 25 glucose. This aCSF solution was used also for recording.
Electrophysiology.
Whole cell voltage-clamp synaptic recordings from geniculate neurons were obtained using glass pipettes (1–2.0 MΩ) filled with an internal solution consisting of the following (in mM): 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, and the L-type calcium channel antagonist, 0.1 methoxyverapamil (Sigma, St. Louis, MO). This solution was designed to minimize the contributions from postsynaptic intrinsic membrane conductances and second-messenger systems. In experiments examining AMPAR rectification, 100 μM spermine were included in the internal solution. EPSCs were evoked with stimulus intensities that ranged from 10 to 150 μA. Series resistance was <10 MΩ (average 5 MΩ). Cells were excluded if series resistance changed more than 10% during an experiment.
For current clamp experiments, the internal solution contained the following (in mM): 116 KMeSO4, 6 KCl, 2 NaCl, 20 HEPES, 0.5 EGTA, 4 MgATP, 0.3 NaGTP, 10 Na phosphocreatine, pH 7.25 with KOH. To elicit successful spikes, the amplitude of first EPSC waveform injected were 300 and 1,500 pA for immature and mature neurons, respectively (Liu and Chen 2008). The bath saline solution for all electrophysiological experiments contained the GABAA receptor antagonist bicuculline (20 μM) or picrotoxin (Sigma, 50 μM), and the GABAB receptor antagonist N-[1-(S)-3,4-dichlorophenyl ethyl]amino-2-(S)-hydroxypropyl-P-benzyl-phosphinic acid (CGP55845, 2 μM) and the A1 adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (10 μM). Five micromoles of 2,3-dihydro-6-nitro-7-sulphamoyl-benzo(f)quinoxaline (NBQX), 4-(8-methyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-5-yl)-benzenamine dihydrochloride (GYKI 52466, 50 μM), 20–100 μM 3-(R)-2-carboxypiperazin-4-yl-propyl-1-phosphonic acid [(R)-CPP], and 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl)propanoic acid (LY341495 50 μM) were used to block AMPAR, NMDAR, and mGluRs, respectively. All of the experiments were performed at 35 ± 1°C, unless otherwise indicated.
EGTA-acetoxymethyl ester (AM) experiments were performed as previously described (Atluri and Regehr 1998; Chen and Regehr 1999). Briefly, EGTA-AM was dissolved in dimethysulfoxide, diluted to a final concentration of 50 μM in aCSF, and continuously bath applied to slices for 15 min to allow the chelator to be taken up by all cells in the slice. Excess EGTA-AM was then washed out with aCSF. Measurements of the EPSC or relative change in fluorescence (ΔF/F) waveforms for the EGTA-AM condition were obtained 15 min after bath washout of EGTA-AM.
Calcium measurements.
RGCs were labeled with calcium indicator dyes, as previously described (Chen and Regehr 2003). Briefly, the mouse was placed in an enclosed chamber, and isoflurane was delivered via a vaporizer with proper scavenging of fumes. Two to four percent isoflurane with a mixture of oxygen was used for induction, and 1–3% for maintenance during the procedure. Once properly anesthetized, as tested by foot pad stimulation, a sharp glass electrode (2- to 5-μm tip diameter) filled with a solution that consisted of a 1:1 mixture of 20% wt/vol Calcium Green-1 dextran [molecular weight (MW) 3,000] and 20% wt/vol Texas Red dextran (MW 10,000) in 0.1% Triton X-100 was inserted into the retina (Invitrogen, OR). One microliter of the solution was pressure injected into the nasal and temporal regions of the retina (30 psi, 10- to 15-ms duration; Parker Instruments). Five to ten days after injection, the animal was killed for experiments. Examination of the slices under ×60 objective (Olympus) revealed that retinogeniculate fibers, but not postsynaptic cells, were labeled.
All fluorescence recordings were performed in the presence of the glutamate receptor antagonists NBQX (5 μM) and CPP (20 μM), bicuculline and CGP55845 (20 μm, 2 μm) using a 450–490 excitation/FT510 dichroic/510WB40 emission and 580DF15 excitation/600 dichroic long pass/610 long pass emission filter sets for Calcium Green-1 and Texan Red signals, respectively. Illumination was provided by a 150-W Xenon lamp (Optiquip) and gated with a transistor logic pulse to an electromechanical shutter (Vincent Associates). Collected light from the labeled slice was digitized, and the ΔF/F was calculated as described previously (Kreitzer et al. 2000). Calcium measurements were performed at 25°C.
Stock solutions of pharmacological agents were stored at −20°C and diluted according to the final concentrations immediately prior to experiments. Constant bath flow during application of drugs was ensured by a perfusion pump (Gilson, MA). Dead space in the perfusion tubing was reduced to 1 ml, allowing rapid bath exchange of pharmacological agents. All pharmacological agents, including l-(+)-2-amino-5-phosphonopentanoic acid (l-AP5, 1 mM), γ-d-glutamylgycine (γ-DGG, 1–3 mM), and 6-chloro-3,4-dihydro-3-(5-norbornen-2-yl)-2H-1,2,4-benzothiazidiazine-7-sulfonamide-1,1-dioxide (cyclothiazide, CTZ, 50 μM), were purchased from Tocris, unless otherwise indicated. EGTA, tetra(AM) (EGTA, AM), calcium green-1 dextran and Texas Red 10,000 MW dextran were obtained from Invitrogen.
Data acquisition and analysis.
Current and voltage-clamp recordings were acquired with a Multiclamp 700A amplifier (Axon Instruments, CA) filtered at 1 kHz and digitized at 10–20 kHz with an ITC-16 interface (Instrutech). Data analysis was performed using Igor software (Wavemetrics), Excel (Microsoft) and Prism (GraphPad Software). EPSCs were analyzed as the average of 5–10 waves. The decay time constant (τ) of both AMPA and NMDA receptor-mediated EPSCs at immature retinogeniculate synapse were best fitted with a double exponential, f(x) = y0 + A1e(−x/τfast) + A2e(−x/τslow) and was quantified as the weighted τ = [τfast × A1/(A1 + A2)] +[τslow × A2/(A1 + A2)] (Liu and Chen 2008).
Studies examining short-term synaptic plasticity involved evoking synaptic responses to pairs of retinal input stimuli. Randomized interstimulus intervals (ISIs) ranging from 10 to 8,000 ms were interleaved with a single conditioning pulse, as previously described (Chen and Regehr 2000). For each cell recorded, averages of three to five trials were used to measure the paired pulse ratio (EPSC2/EPSC1 × 100) for each ISI.
Spike probability was calculated by summing the number of action potentials (APs) that occur over the period following current injection (∼10 trials) and dividing the value by number of trials. The criterion for an AP was a depolarizing change in membrane potential ≥ 10 V/s. Synaptic charge was calculated by integrating the evoked synaptic current. All data are summarized as means ± SE, using the two-tailed paired t-test, unless otherwise indicated.
RESULTS
Acceleration of the retinogeniculate EPSC waveform over development.
Changes in the EPSC waveform over development are evident when comparing the decay kinetics of the AMPAR current of immature (p9–11, red trace) and mature (p26–32, black trace) synapses (Fig. 1A, left). The synaptic currents, evoked by optic nerve stimulation, were recorded in whole cell voltage clamp mode [holding potential (Vh) = −70 mV] and in the presence of the NMDAR selective antagonist, 20 μM (R)-CPP. The immature EPSC waveform exhibited slower decay kinetics compared with the mature synapse, and this difference persisted even in the presence of CTZ (50 μM), an inhibitor of AMPAR desensitization (Fig. 1A, right). The AMPAR decay time course, in control and CTZ conditions, could be approximated by a double-exponential τ, where the average weighted τ is two to three times greater for immature synapses compared with the mature synapses (Fig. 1B). Therefore, developmental changes in the AMPAR EPSC waveform cannot be simply explained by differences in receptor desensitization.
Fig. 1.

Properties of the retinogeniculate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) excitatory postsynaptic current (EPSC) change over development. Comparison is shown of the decay time course of the immature [postnatal days (p) 9–11, red traces] and mature (p26–32, black traces) retinogeniculate AMPAR EPSCs [holding potential (Vh) = −70 mV] in the absence (A, left) and presence (A, right) of 50 μM cyclothiazide (CTZ). Representative traces are averages from 10–15 trials and normalized to the peak. B: summary graph of the time course of the AMPAR EPSC decay [weighted time constant (τ)] from the two age groups in control (white) and in CTZ (gray); n = 4. *P < 0.05. **P < 0.01. Recordings were made in the presence of (R)-2-carboxypiperazin-4-yl-propyl-1-phosphonic acid [(R)-CPP] to block N-methyl-d-aspartate receptor (NMDAR)-mediated EPSCs. Bath temperature: 35 ± 1°C. Ci: representative examples of AMPAR EPSC traces evoked at different Vh (−60 to 60 mV in 20-mV increments). Cii: average current (I)-voltage (Vm) relationship for immature (red, n = 5 cells) and mature (black, n = 6 cells) synapses. Synaptic currents were recorded with intracellular spermine of p9–11 significantly different from p26–32 (P << 0.001, 2-way ANOVA). Recording was at room temperature.
Our results involving CTZ suggested that the immature EPSC is more sensitive to the inhibitor of desensitization (6.37 ± 0.93-fold increase in weighted τ vs 3.3 ± 0.43-fold) and raised the possibility that the subunit composition of AMPARs may change with age. One means of testing for a change in AMPAR subunit composition is to assess whether the contribution of calcium-permeable AMPAR subunits change with age. In the presence of intracellular polyamines such as spermine, the current-voltage relationship of Ca2+-permeable AMPARs is known to rectify, whereas that of Ca2+-impermeable AMPARs is linear (Blaschke et al. 1993; Hollmann et al. 1991). Therefore, we compared the AMPAR current-voltage relationship of immature to mature synapses using an intracellular recording solution containing 100 μM spermine. Figure 1C shows increased AMPAR rectification with age. The rectification index, calculated as the peak EPSC current measured at +60 mV/−60 mV, was significantly different at p9–11 compared with p26–32 (0.74 ± 0.06 vs. 0.34 ± 0.05, P < 0.01, n = 5, 6, Student t-test). This developmental change in AMPAR composition is opposite that described in hippocampus, but similar to that in another thalamic synapse, the leminscal input onto relay neurons in the somatosensory thalamus (Takeuchi et al. 2012). However, the increased contribution of calcium-permeable AMPARs at older ages cannot fully explain the observed change in the EPSC waveform over development. Some calcium-permeable AMPAR subunits have been shown to have slower, not faster, decay kinetics (Partin et al. 1996). Moreover, our previous studies showed no significant difference in the quantal waveform of the immature and mature synapses (Liu and Chen 2008). Taken together, these data suggest that a change in subunit composition is not responsible for the slow component of the immature AMPAR EPSC.
γ-DGG accelerates the decay of immature AMPAR EPSCs.
As postsynaptic mechanisms cannot account for the acceleration of the synaptic waveform, we next asked whether the slow decay of the immature EPSC is influenced by an extended time course of glutamate. For these studies, we took advantage of low-affinity glutamate receptor antagonists (Olverman et al. 1988). These agents actively compete with glutamate for binding to receptors, and their efficacy of inhibition is dependent on the relative concentration of glutamate. Previous studies have shown that receptors directly across from the site of release experience a peak glutamate concentration much greater than receptors that are located further away from this site (Clements et al. 1992; Diamond 2001; DiGregorio et al. 2002). We examined the effects of these antagonists on the immature retinogeniculate waveforms.
We recorded isolated AMPAR-mediated currents in the presence of 50 μM CTZ and NMDAR antagonists to gain an accurate measurement of the glutamate transient. Bath application of the low-affinity AMPAR antagonist, γ-DGG (3 mM), reduced the peak AMPAR EPSC amplitude to 51 ± 2.8% of control (P < 0.01, n = 4) and significantly accelerated the decay of the current to 48.8 ± 3.6% of control (P < 0.01, n = 4; weighted τ control: 21.7 ± 3 ms, γ-DGG: 10.6 ± 1.8 ms, Fig. 2C). Notably, γ-DGG preferentially inhibited the slow component of the EPSC decay (control vs. γ-DGG: τslow = 18.6 ± 3.6 ms vs. 8.4 ± 2 ms, n = 4, P = 0.01; τfast = 3.1 ± 1.2 vs. 2.2 ± 0.4 ms, n = 4, P = 0.28). To ensure the acceleration of the current was not due to voltage-clamp errors, we performed parallel experiments with a low concentration of NBQX (200 nM), a high-affinity antagonist that dissociates from the receptor slowly. Unlike γ-DGG, NBQX inhibition of AMPAR currents is independent of glutamate concentration. NBQX (200 nM) reduced the AMPAR EPSC amplitude to a similar extent as γ-DGG (48.1 ± 5% of control, n = 5; P > 0.6 NBQX vs. γ-DGG), but the two antagonists differed in their effects on the time course of the decay kinetics. In Fig. 2C, the average AMPAR current waveform, normalized to the peak of the EPSC, is shown before and during bath application of γ-DGG (gray trace) or NBQX (dashed trace). γ-DGG inhibits a greater fraction of the decaying phase of the EPSC, compared with the peak of the current. In contrast, NBQX did not have a significant effect on the time course of the EPSC (Fig. 2C, weighted τ = 84.1 ± 9.5% of control, n = 5, P > 0.2, Fig. 2C; γ-DGG vs. NBQX, P < 0.02). These data suggest that AMPARs that contribute to the tail of the immature EPSC experience a lower peak concentration of glutamate than those receptors open during the peak of the current.
Fig. 2.
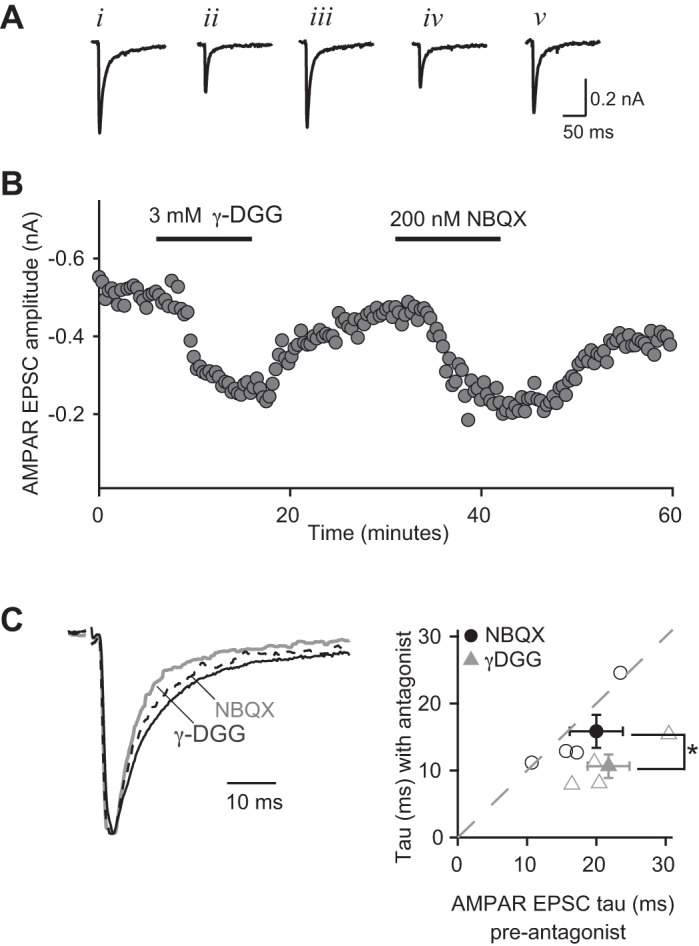
γ-d-Glutamylgycine (γ-DGG) accelerates the AMPAR EPSC τ at the immature synapse. A: AMPAR EPSCs (Vh = −70 mV) recorded in the presence of CTZ (50 μM) and NMDAR antagonists [50 μM d-(+)-2-amino-5-phosphonopentanoic acid (d-AP5), 20 μM CPP] under control conditions (i, iii, and v) and in the presence of 3 mM γ-DGG (ii) and 200 nM 2,3-dihydro-6-nitro-7-sulphamoyl-benzo(f)quinoxaline (NBQX) (iv). Traces are the average of 5–10 consecutive responses and correspond to the peak amplitudes in the time course plotted in B. C, left: representative normalized AMPAR EPSCs before (black trace) and during (gray trace) application of γ-DGG or NBQX (dashed trace). C, right: scatterplot of AMPAR EPSC weighted τ shown as means ± SE (closed circles or triangles) before (X-axis) and in the presence of antagonist (Y-axis) with individual experiments plotted (open circles or triangles). γ-DGG (gray triangle): before τ, 21.8 ± 3.0 ms; in antagonist, 10.6 ± 1.8 ms; n = 4. NBQX: before τ, 20.0 ± 3.8 ms; in antagonist, 15.9 ± 2.5 ms; n = 5. Bath temperature: 35 ± 1°C. *P < 0.05.
l-AP5 accelerates the decay of the immature retinogeniculate NMDAR EPSC.
If the waveform retinogeniculate EPSC does indeed reflect a gradient of peak glutamate concentrations, then we would predict that low-affinity antagonists of NMDARs would also accelerate the decay of the EPSC. To test this hypothesis, we compared the effects of bath application of the low-affinity NMDAR antagonist l-AP5 (1 mM) to that of low concentration of the high-affinity antagonist, R-CPP (1–1.25 μM) in a similar approach as our AMPAR experiments. Figure 3, A and B, shows that, at these concentrations, both antagonists inhibited the peak amplitude of the NMDAR EPSC to comparable levels (l-AP5 to 34.4 ± 2.3% and CPP to 40.9 ± 4.3% of control, n = 7 each, P > 0.2). However, the antagonists have distinct effects on the decay of the NMDAR current (Fig. 3C). On average, l-AP5 significantly accelerated the EPSC decay τ to 65.0 ± 3.3% of control (P < 0.01, n = 7, Fig. 3C). R-CPP had a significantly smaller effect on the time course of the EPSC than l-AP5 (CPP decay τ: 82.1 ± 2% of control, n = 7, P < 0.001; l-AP5 vs. CPP, P < 0.01, Fig. 3C). These data provide further support for the presence of a gradient in peak concentration of glutamate at the immature synapse.
Fig. 3.
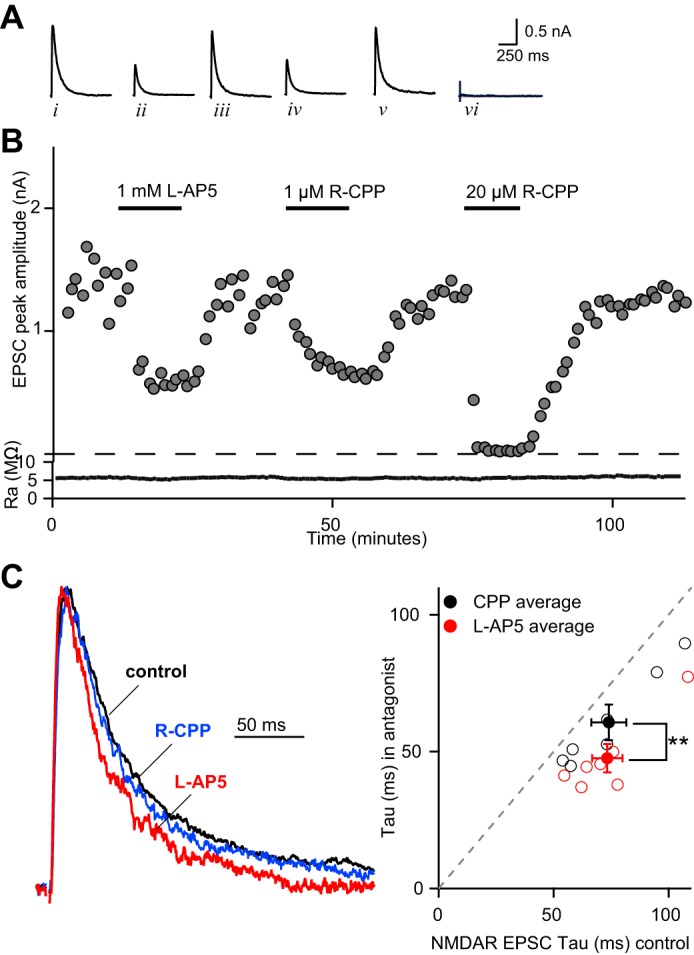
l-AP5 accelerates the NMDAR EPSC τ at the immature synapse. A: NMDAR EPSCs (Vh = +40 mV) recorded under control conditions (i, iii, and v) and in the presence of 1 mM l-AP5 (ii), 1 μM (R)-CPP (iv) or 20 μM (R)-CPP (vi). Traces are the average of 5 consecutive responses and correspond to the amplitudes plotted against time in minutes (B, top). Bottom: access resistance (Ra) is plotted. Recordings were performed in the presence of the AMPAR antagonist NBQX (5 μM). Bath temperature: 35 ± 1°C. C, left: representative normalized NMDAR EPSCs from before (black trace) and during application of l-AP5 (red trace) or low concentration of (R)-CPP (blue trace). C, right: scatterplot of NMDAR EPSC weighted τ before (X-axis) and in the presence of antagonist (Y-axis). Average values are represented by solid circles shown as means ± SE. CPP (black circle), control τ: 74.0 ± 7.7 ms, CPP τ: 60.7 ± 6.5 ms; l-AP5 (red circle), control τ: 73.3 ± 6.6 ms; l-AP5 τ: 47.6 ± 5.2 ms. Individual experiments are shown as open circles. **P = 0.01.
EPSC decay time course is dependent on extracellular calcium.
Compared with NMDARs, AMPARs are much less likely to be located extrasynaptically and have a significantly lower affinity for glutamate (Dingledine et al. 1999; Tarusawa et al. 2009). Therefore, our low-affinity AMPAR data suggest that glutamate may diffuse between release sites (aka, “spillover”) at the immature synapse. If this were true, then the EPSC decay time course could be sensitive to changes in probability of release (PR) (Mennerick and Zorumski 1995; Trussell et al. 1993). To alter PR, we changed the extracellular calcium concentrations (0.5 mM [Ca2+]o vs. 2.0 mM [Ca2+]o) and compared the normalized AMPAR EPSC waveform (Fig. 4A). In the presence of 2.0 mM [Ca2+]o/1.0 mM [Mg2+]o, the average weighted τ of the AMPAR EPSC decay was 4.1 ± 0.7 ms. Bath exchange to an external solution containing 0.5 mM [Ca2+]o/2.5 mM [Mg2+]o reduced the peak EPSC to 25.4 ± 2.6% of that in 2.0 mM [Ca2+]o/1.0 mM [Mg2+]o (n = 5, P < 0.001) and significantly accelerated the weighted τ to 1.7 ± 0.2 ms (n = 5, P < 0.05, Fig. 4Aiii). These data provide support for the existence of glutamate spillover at the developing retinogeniculate synapse.
Fig. 4.

Experimental evidence in support of glutamate spillover. Ai: overlaid AMPAR EPSC traces normalized to peak amplitude in 2.0 mM (black and dashed traces) and 0.5 mM (gray) external Ca2+, recorded at Vh = −70 mV in R-CPP (20 μM) and d-AP5 (50 μM) to block NMDAR-mediated EPSCs. To facilitate comparison of decay, normalized EPSCs are aligned by peak amplitude. ii: Peak amplitude of AMPAR ESPSCs plotted over the time course of an experiment. iii: Bar graph of AMPAR weighted τ in 2.0 and 0.5 mM external Ca2+ (n = 5, P < 0.05); individual experiments are shown in gray. CTZ was not included in the bath solution. [Ca2+]o, extracellular Ca2+ concentration. *P < 0.05. B: superimposed AMPAR EPSC2 traces evoked at different interstimulus intervals (ISIs) before (black) and during (gray) bath application of 50 μM CTZ. Traces are the average of 3–5 trials. The EPSC2 waveforms were calculated by subtracting the average single EPSC from the average EPSC response to a pair of stimuli. The single EPSC (EPSC1) waveform is shown in bold. C: the average paired pulse ratio (PPR) calculated as %EPSC2/EPSC1 is plotted against ISIs for control (black; n = 5 cells) and CTZ (gray; n = 5 cells). Error bars indicate SE (P << 0.001, 2-way ANOVA). Inset: initial phase of the recovery from depression is shown on an expanded time scale. Recordings were made in the presence of (R)-CPP (20 μM) to block NMDAR-mediated EPSCs. Bath temperature: 35 ± 1°C.
Cyclothiazide relieves paired-pulse depression at the immature retinogeniculate synapse.
If spillover exists at the immature retinogeniculate synapse, then glutamate released from one site can bind, activate and desensitize receptors at more distant sites. Therefore a population of AMPARs across from a quiescent release site can become desensitized when bound to glutamate released from a neighboring site. AMPAR desensitization that occurs this way would transiently reduce the number of available AMPARs at neighboring quiescent release sites. If glutamate is subsequently released at these neighboring sites within a few miliseconds after the initial release event, the synaptic response will be reduced. At the mature retinogeniculate synapse, postsynaptic AMPAR desensitization is known to contribute to short-term synaptic plasticity because paired-pulse depression (PPD) is relieved in the presence of CTZ. These observations have been used to describe the presence of glutamate spillover between release sites at the mature synapse (Budisantoso et al. 2012; Chen et al. 2002). We asked whether short-term depression in response to pairs of optic tract stimuli is also relieved by CTZ at the immature synapse.
Figure 4B shows superimposed traces of the second EPSCs (EPSC2, thin traces) evoked at different ISIs, compared with the first EPSC (EPSC1, bold trace). Bath application of CTZ slowed the AMPAR EPSC decay and increased the paired pulse ratio (EPSC2/EPSC1, Fig. 4C). These data are consistent with the findings at the mature synapse and suggest glutamate can diffuse between release sites at the immature retinogeniculate contact (Budisantoso et al. 2012; Chen et al. 2002). However, our results do not distinguish whether the observed glutamate gradient is intrabouton (within the confines of a single bouton), or interbouton (where glutamate escapes from the bouton to spill over to neighboring quiescent sites located in other boutons). To differentiate between these possibilities, we next examined the glutamate transient of single retinal inputs.
Two populations of single-fiber AMPAR EPSCs.
We hypothesized that, if release of glutamate from distant release sites contributes to the slow component of the immature EPSC, then we may be able to detect single retinal fiber synaptic responses that are mediated purely by spillover. These currents should have distinctly slower kinetics than conventional synaptic responses. Therefore, we recorded isolated AMPAR-mediated single-fiber EPSC in the presence of 50 μM CTZ. Single-fiber EPSCs were identified as the synaptic response to minimal stimulation (for details see Noutel et al. 2011; Fig. 5A). Figure 5B shows two examples of single-fiber currents recorded from the same relay neuron. Although the EPSCs were evoked with similar stimulation intensities from two nearby sites in the optic nerve, the activation and decay kinetics were very different. Analysis of 63 single-fiber synaptic responses recorded from 50 relay neurons revealed at least two types of EPSCs based on their kinetics. In addition to the familiar fast rise/fast decay single-fiber currents, we observed a previously unrecognized population of slow-rising currents with small amplitudes and slow decay kinetics. While the time course of the EPSC decay was highly variable among the single-fiber responses, there was a strong correlation between the rise time and decay time of the synaptic current (Fig. 5C). Fast-rising currents could be distinguished from the slow-rising currents by their 10–90% rise times: a histogram of the rise times shows two distinct peaks, one at 0.67 ms and the other at 5.2 ms (Fig. 5D, n = 63 single-fiber currents, n = 43 mice).
Fig. 5.

Identification of slow- and fast-rising single-fiber AMPAR EPSCs. Ai: example of a fast-rising single-fiber current. Pairs of stimuli separated by 50 ms (arrows indicate the time of stimulus) are increased from subthreshold until a response emerges from noise (Vh = −70 mV). A response on the second, but not first, pulse indicates the single-fiber response is near threshold. ii: The same technique is used to identify slow-rising single-fiber currents. B: comparison of two types of single-fiber AMPAR EPSC responses recorded from the same thalamic relay neuron. The stimulus electrode position in the optic tract differed for the two types of synaptic currents. Overlay of fast- (left) and slow-rising (right) single-fiber currents evoked from 7–10 consecutive trials is shown. The average waveform is shown in black. Currents were recorded at Vh = −70 mV in the presence of NMDAR antagonists and CTZ. C: scatterplot of half decay time (Thalf) vs. rise time for all single fibers (n = 63); dotted line represents linear regression fit (Pearson correlation coefficient r2 = 0.47; P << 0.0001). D: histogram of the 10–90% rise times of all of the observed single-fiber AMPAR EPSCs. Ei: representative averaged traces showing paired-pulse depression occurs with both fast-rising (top) and slow-rising EPSCs (bottom). ii: Summary graph of average PPR of fast rise (<1 ms, n = 5) and slow rise (>1 ms, n = 5) EPSCs. F: average traces (i) and time course of the peak amplitude (ii) of slow-rising EPSC before and during bath application of an AMPAR-specific antagonist 50 μM GYKI52466. iii: Summary plot of the effects of GYKI52466 on the peak amplitude of slow-rising EPSCs (n = 3). **P < 0.01. Bath temperature: 35 ± 1°C. ns, Nonsignificant.
One possible explanation for these slow-rising currents is that they are evoked by inadvertent stimulation of the corticothalamic tract. Relay neurons in the LGN receive excitatory inputs from both retina and cortex, and corticothalamic EPSCs are known to innervate the distal dendrites of relay neurons and to exhibit slower kinetics than retinogeniculate currents (Jurgens et al. 2012; Kielland et al. 2006; Turner and Salt 1998). However, corticothalamic innervation of the LGN is not complete until after eye-opening in mouse (Seabrook et al. 2013), making this explanation less likely. Nevertheless, we tested whether the slow single-fiber EPSCs could arise from corticothalamic inputs by taking advantage of the known fact that retinogeniculate and corticothalamic inputs exhibit distinct forms of short-term synaptic plasticity. In response to a pairs of stimulation, retinogeniculate synapses exhibit PPD because of their high PR, while corticothalamic connections display paired-pulse facilitation due to their low PR (Turner and Salt 1998). We found that, in response to pairs of stimulation, the slow-rising EPSC displayed a similar degree of PPD as the fast-rising single-fiber EPSCs (Fig. 5E). These results demonstrate that the slow-rising EPSCs are evoked by stimulating retinal fibers.
We next asked whether the slow-rising single-fiber EPSCs were mediated by glutamatergic receptors. We found that bath application of the non-NMDA receptor antagonist NBQX (5 μM) completely abolished the slow-rising current (data not shown). However, NBQX inhibits both AMPAR and kainate receptors, and the latter are known to give rise to currents with slow kinetics (Bureau et al. 2000). To distinguish between AMPAR and kainate receptors, we took advantage of a selective AMPAR antagonist, GYKI 52466 [IC50 ∼10–20 μM for AMPA and ∼450 μM for kainate receptors; (Tarnawa et al. 1989)]. Figure 5F illustrates the effects of bath application of 50 μM GYKI 52466 on the peak of the slow-rising EPSC. On average, the AMPAR antagonist inhibited the slow-rising current to 18.0 ± 1.0% of control (n = 3, P < 0.01, Fig. 5Fiii). Therefore, the slow-rising retinogeniculate EPSCs result from activation of AMPARs and not other glutamatergic receptors.
The effects of γ-DGG on slow- and fast-rising AMPAR EPSCs.
If the slow-rising AMPAR EPSCs represent a population of receptors activated by glutamate spillover from distant release sites, we would predict that the receptors would experience a lower peak concentration of glutamate than that of fast-rising EPSCs. Therefore, we compared the effects of 2 mM γ-DGG on the peak amplitude of the two types of single-fiber AMPAR EPSCs. Figure 6, A and B, shows representative examples of γ-DGG inhibition of a fast- and slow-rising EPSC, respectively. A plot of the degree of blockade by the low-affinity antagonist as a function of all single-fiber EPSC rise times showed that synaptic currents with slower rise times were more sensitive to inhibition by γ-DGG than those with faster rise times (Fig. 6C). We sorted the EPSCs into two groups based on their 10–90% rise times (fast <1.0 ms, slow >1.0 ms) and compared the average block by γ-DGG. The low-affinity antagonist blocked a significantly greater percentage of the peak amplitude of the slow-rising currents than of the fast-rising currents (76.2 ± 4.2% vs 35.7 ± 2.7% of baseline; n = 6 for each group, P << 0.0001, Fig. 6C). Therefore, the slow-rising EPSCs experience a lower peak concentration of glutamate than fast-rising EPSCs, consistent with glutamate diffusing a significant distance after its release. These results suggest the presence of interbouton spillover at the immature developing retinogeniculate synapse.
Fig. 6.

AMPA receptors of fast- and slow-rising EPSCs are activated by different glutamate concentrations. The effects are shown of 2 mM γ-DGG (gray) on fast-rising (A) and slow-rising (B) AMPAR EPSCs. C: plot of the percentage peak amplitude blocked by γ-DGG vs. the EPSC rise time (10–90% rise time, ms). The data are fit by an exponential equation (dashed line). D: summary graph of the average percent inhibition of the peak amplitude by γ-DGG (“fast” n = 6; “slow” n = 6). Ei: time course of peak slow-rising AMPAR EPSC before and in the presence of the glutamate transport blocker dl-threo-β-benzyloxyaspartic acid (TBOA; 10 μM) followed by the application of 50 μM GYKI52466. ii: Average EPSCs from the same experiment before (black line), during bath application of TBOA (gray line) and in the presence of TBOA+GYKI (dashed line). Traces are the average of 15–20 trials. F: summary graph of the effects of TBOA on the slow-rising EPSCs (n = 8), quantifying the average change in peak amplitude, rise time and normalized charge transfer. Q, synaptic charge. Individual experiments are shown as gray circles. All recordings were performed in the presence of (R)-CPP and d-AP5 to block NMDAR-mediated EPSCs and CTZ to prevent AMPAR desensitization. Bath temperature: 35 ± 1°C. *P < 0.05. ***P < 0.001.
The amplitude of slow-rising AMPAR EPSCs is potentiated by inhibition of glutamate transporters.
To further strengthen the argument that the slow-rising EPSC represents a spillover-mediated current, we asked whether it was sensitive to glutamate transporter activity. We reasoned that the degree of interbouton glutamate spillover would increase with the inhibition of glutamate uptake. Figure 6E illustrates the effects of bath application of 10 μM dl-threo-β-benzyloxyaspartic acid (TBOA) on the slow-rising single-fiber EPSC (Fig. 6, E and F). The peak amplitude of the current increased to 226.0 ± 32.2% of control (average peak amplitude control: 76.3 ± 15.1 pA to 160.2 ± 27.7 pA in TBOA, n = 8, P = 0.01). These changes led in an increase in the normalized charge transfer of the EPSC to 214.1 ± 17.9% of control (n = 6, P < 0.01, Fig. 6G). Therefore, glutamate transporters can regulate the amplitude and time course of the slow-rising EPSCs, consistent with a current driven by a prolonged glutamate transient.
Inhibiting glutamate uptake does not influence the peak amplitude of fast AMPAR EPSCs.
Previous studies have shown that purely spillover-mediated AMPAR currents display a robust increase in peak amplitude in the presence of TBOA, whereas direct synaptic contacts do not (Szapiro and Barbour 2007). We asked whether this was also true at the retinogeniculate synapse. We found that the amplitude of fast-rising AMPAR EPSCs evoked by stimulating multiple retinogeniculate fibers did not change with bath application of 10 μM TBOA (peak amplitude: 696.1 ± 154.8 pA to 658.2 ± 140.3 pA in TBOA, n = 6 P > 0.3; Fig. 7, A–C). Similar to our previous finding, the decay of the EPSC was prolonged (Hauser et al. 2013; Fig. 7, A and D).
Fig. 7.

Inhibiting glutamate uptake does not influence the peak amplitude of evoked AMPAR EPSCs at the immature synapse. A: example traces of AMPAR-EPSCs in the presence of 50 μM CTZ, NMDAR antagonists, and metabotropic glutamate receptor antagonist LY341495 (Vh = −70) before (black trace) and during (gray trace) bath application of 10 μM TBOA. B: time course of an experiment showing that bath application of TBOA does not alter the peak amplitude of the AMPAR-EPSC. C: summary plots show no significant change in the peak amplitude of the AMPAR EPSC during application of TBOA (average: black trace, individual experiments: gray traces, n = 6, P > 0.3). D: example traces of AMPAR-EPSCs recorded in the presence of 1 mM γ-DGG to prevent receptor saturation, LY341495, CTZ and NMDAR antagonists. Traces are shown before (black), during (gray) and after washing out (dashed trace) bath application of 10 μM TBOA. E: time course of an experiment performed in 1 mM γ-DGG. The current is abolished in the presence of 5 μM NBQX. F: summary plots show no significant change in peak amplitude of the AMPAR EPSC during application of TBOA in the presence of γ-DGG (n = 3, P > 0.9; average: black trace, individual experiments: gray traces). Bath temperature: 35 ± 1°C.
The absence of an effect of TBOA on the peak AMPAR EPSC could potentially be influenced by receptor saturation. To control for this possibility, we repeated experiments in the presence of the low-affinity antagonist γ-DGG (1 mM). Even in the presence of γ-DGG, the peak amplitudes of the evoked AMPAR EPSCs were unaltered with bath application of TBOA (1090.2 pA to 1083.3 pA in TBOA, n = 3, P = 0.9; Fig. 7, D–F). Therefore, the decay, but not the peak, of the AMPAR EPSC is influenced by glutamate released from a distant source. These results further support the presence of direct and indirect glutamatergic signaling between RGCs and relay neurons in the developing retinogeniculate synapse.
The effects of EGTA-AM on EPSC waveform over age.
Our results strongly suggest that glutamate spillover between boutons contributes to the slow decay of the evoked retinogeniculate EPSC. This would be consistent with our previous findings that retinogeniculate quantal events did not exhibit the slow decay kinetics seen in the EPSCs evoked by suprathreshold stimulation. Previously, we proposed that delayed release of glutamate could contribute to the observed differences in the decay of the quantal and evoked EPSC waveform at the immature synapse (Liu and Chen 2008). In light of our current findings that glutamate spillover contributes to the slow component of the AMPAR EPSC, we asked whether delayed release and spillover coexist at the immature retinogeniculate synapse.
To test for the presence of delayed release, we used a calcium chelator with a slow binding rate, EGTA. Because of the slow binding rate, EGTA has little effect on the high calcium concentration transient near open calcium channels (local calcium). However, at later time points, after the channels have closed, this high-affinity chelator is effective in binding to calcium as it equilibrates within the presynaptic bouton and decays to lower concentrations; this calcium signal is referred to as Cares. Previous studies at other central nervous system (CNS) synapses have demonstrated that introduction of relatively low concentrations of EGTA into presynaptic terminals affects Cares much more than the local calcium (Borst and Sakmann 1996). Thus EGTA has been used as a tool to preferentially reduce delayed release over synchronous release (Atluri and Regehr 1998; Cummings et al. 1996; Delaney et al. 1989; Feller et al. 1996; Ravin et al. 1997; Vanderkloot and Molgo 1993).
We introduced EGTA into RGC terminals by bath, applying the membrane-permeable form of EGTA, EGTA-AM (50 μM), to LGN brain slices (see methods). For voltage-clamp recordings, we used an internal solution that contained 10 mM EGTA; therefore, any effect of EGTA-AM on the EPSC decay time course could be attributed to alterations in presynaptic calcium. Figure 8A shows representative traces of EPSCs recorded at −70 mV in the absence (black trace) and 15 min following bath application (red trace) of 50 μM EGTA-AM for young (left) and old (right) synapses. EGTA-AM reduced the peak amplitude of the immature EPSC to 85.1 ± 3% of control (P < 0.01, n = 8, Fig. 8A, left). Normalization of the currents to the peak EPSC amplitude before and after exposure to EGTA-AM showed an acceleration of the decay time course of the immature EPSC. The weighted τ of the EPSC decay decreased to 65.1 ± 5.9% of control in the presence of EGTA-AM (P < 0.05, n = 8, Fig. 8, B and C). Notably, the portion of the immature EPSC most sensitive to the EGTA-AM was the slow component of the EPSC decay time course (Fig. 8D, red bars, P < 0.05, n = 8, paired t-test). In contrast to the immature synapse, EGTA-AM did not significantly alter the peak EPSC amplitude or the decay time course (P > 0.7, n = 5–6). These results are consistent with a greater contribution of delayed release to the immature compared with the mature EPSC.
Fig. 8.
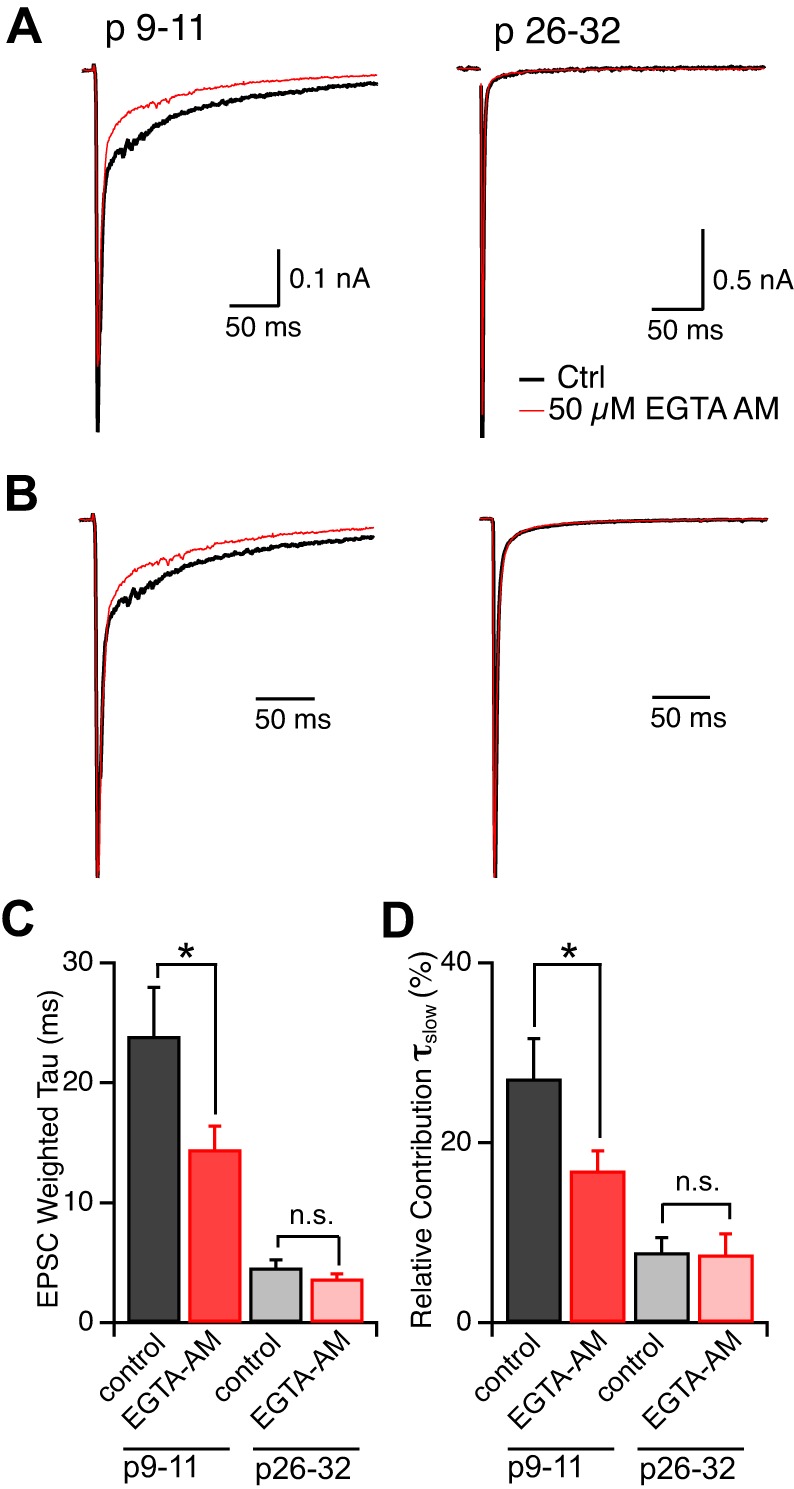
EGTA-acetoxymethyl ester (AM) accelerates the decay kinetics of the immature EPSC. A: representative traces of EPSCs before (black) and 15 min after (red) bath application of 50 μM EGTA-AM recorded from immature (left) and mature (right) synapses. B: traces from A, normalized to the peak. C: summary of average weighted τ, before and after bath application of EGTA-AM (for immature: 23.9 ± 4.1 ms to 14.4 ± 1.9 ms; n = 8, P < 0.09; for mature: 4.6 ± 0.6 ms to 3.7 ± 0.4 ms; P > 0.09, n = 6, paired t-test). EPSC decay was fit to a double-exponential relationship: y0 + A1e(−x/τfast) + A2e(−x/τslow), and weighted τ = [τfast × A1/(A1 + A2)] +[τslow × A2/(A1 + A2)]. D: the relative contribution of τslow to the weighted τ, [%A2/(A1 + A2)], is summarized before (black) and after bath application of EGTA-AM (red). Immature: control, 27.2 ± 4.4% to EGTA-AM, 16.9 ± 2.2%; mature: 7.8 ± 1.6% to 7.6 ± 2.3% of τtotal. *P < 0.05.
The immature presynaptic calcium transient is sensitive to EGTA.
As delayed release is driven by presynaptic Cares, we next examined the effect of EGTA-AM on presynaptic Cares transient (Cummings et al. 1996; Vanderkloot and Molgo 1993; Zengel and Magleby 1981; Zucker and Lara-Estrella 1983). We introduced calcium green-1 dextran into RGC in vivo, as previously described (Chen and Regehr 2003). A mixture of calcium green-1 dextran (Kd = 540 nM) and Texas red dextran was introduced into the eyes of anaesthetized mice. Mice were allowed to recover as the dye was taken up by retinal cells. Only axons of RGCs project to the LGN; thus fluorometric calcium measurements obtained from acute LGN slices cut 3–7 days after eye injections represent presynaptic calcium signals from the retinogeniculate synapse. All experiments were performed with inhibitors of GABAA and GABAB (bicuculline, CGP55845A) and glutamatergic receptors [(R)-CPP and NBQX] to eliminate both inhibitory and excitatory synaptic transmission.
Figure 9, A and B (right) shows the calcium green-1 ΔF/F transient evoked by optic tract stimulation (black trace) and after (gray trace) bath application of EGTA-AM for a p11 and p19 mouse. Under control conditions, the decay time course of ΔF/F at the immature synapse is significantly slower compared with that at a more mature synapse (p9–11: 349.6 ± 70 ms vs. p18–23: 111.7 ± 24 ms, P < 0.05, n = 4 and 5, respectively). At the immature synapse, EGTA-AM reduced the peak of the ΔF/F to 60.6 ± 6% of control, while the time course of the calcium transient accelerated from 349.6 ± 70 to 24.4 ± 5 ms (to 9.2 ± 4% of baseline) (P < 0.05, n = 4). In contrast, EGTA-AM did not significantly alter the peak of the ΔF/F signal in older synapses (101.7 ± 6% of baseline; Fig. 9C). Although the calcium chelator accelerated the decay time course of the ΔF/F transient at all ages tested, the effects were much less pronounced at older ages (in mature synapses accelerated decay τ from 93.6 ± 20 ms to 38.8 ± 5.7 ms; n = 4, P < 0.05, 43.3 ± 3.5% of baseline).
Fig. 9.
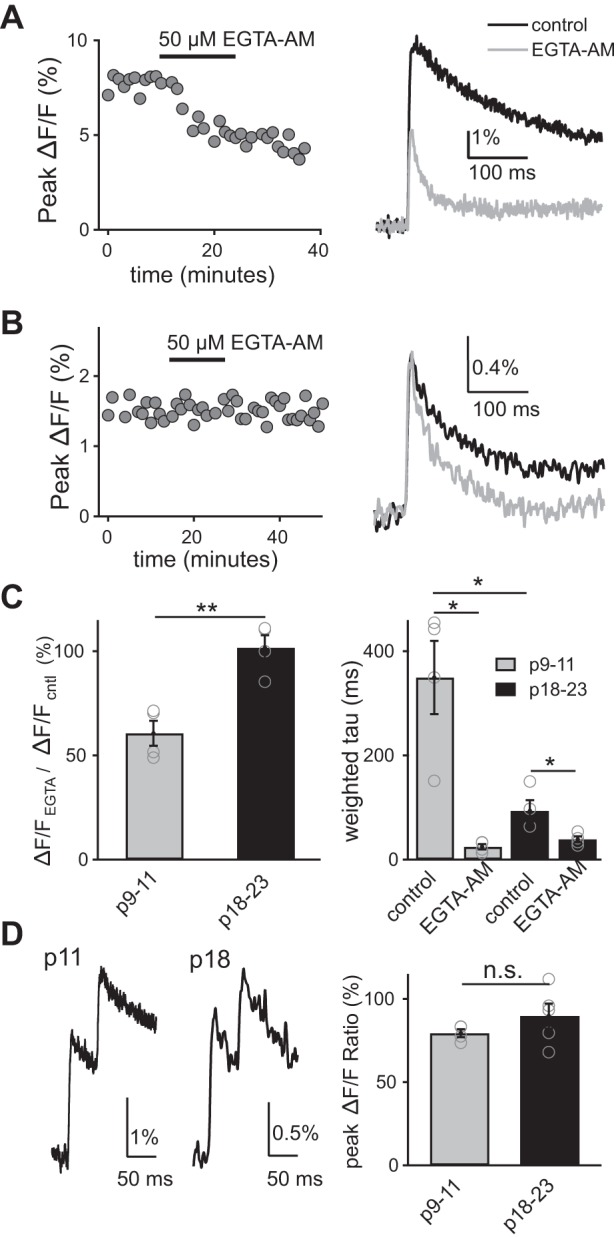
The presynaptic calcium transient at the immature synapse is more sensitive to EGTA-AM than that of the mature. A and B, left: representative time course is shown of peak relative change in fluorescence (ΔF/F) signal before, during and 15 min following application of 50 μM EGTA-AM from p9–11 (A) and p18–23 (B) synapses. A and B, right: representative ΔF/F signals evoked by optic tract stimulation before (black) and 15 min after (gray) EGTA-AM application in immature (A) and p18–23 (B) mice. Traces are the average of 3–5 trials. C: summary of the average change in the peak amplitude (left) and weighted τ (right) of the ΔF/F in the presence of EGTA-AM relative to baseline in p9–11 (gray) and p18–23 (black). *P < 0.05. **P < 0.01. D: averaged representative traces of ΔF/F signal in response to a pair of optic tract stimuli (ISI = 50 ms) shown for immature (left) and p18–23 (middle) synapses. D, right: summary of peak ΔF/F ratio from immature and p18–23 (n = 4 and 5, respectively, P > 0.2).
The slower time course of the presynaptic calcium transient at the immature synapse could be explained by less dye uptake in young vs. in old RGCs, leading to saturation of the calcium fluorophore. If this were true, one would predict that the response of the presynaptic fluorescence transient to pairs of stimuli separated by short interpulse intervals would be different over development. With saturation, fewer fluorophores are available to bind to calcium after the first stimulus, leading to a reduced second response. To test this possibility, we compared the ΔF/F signal in response to a pair of optic tract stimuli (ISI = 50 ms) at two different age ranges. We found no significant difference in the paired pulse response of peak ΔF/F signals over development (Fig. 9D). Therefore, differences in the time course of the presynaptic calcium transient cannot be attributed to a change in the saturation of the fluorophore. These data suggest that a prolonged presynaptic Ca2+ transient could be responsible, in part, for the slow EPSC decay at the immature synapse.
EGTA-AM decreases charge transfer in train of optic tract stimulation.
Since EGTA-AM inhibits the peak of the calcium transient at the immature synapse, it will likely affect both synchronous and delayed release (Borst and Sakmann 1996). As glutamate spillover is known to decrease with a reduction in PR, EGTA-AM cannot be used to distinguish between delayed release and glutamate spillover. However, our results show that EGTA-AM can accelerate the decay time course of the immature EPSC. Thus it is a useful tool for assessing the physiological contributions of the slow component of the EPSC waveform in the developing synapse.
Visual information is encoded in trains of RGC APs (Demas et al. 2003; Kerschensteiner and Wong 2008; Torborg and Feller 2005). We next asked how bath application of EGTA-AM influences the synaptic response to trains of optic nerve stimuli at different frequencies (Fig. 10). The synaptic current evoked by trains of optic tract stimuli were compared before and after bath application of 50 μM EGTA-AM. To compare the charge transfer contributed by the slow component of the EPSC waveform, the synaptic response in each condition was normalized to the first EPSC of each train, and total charge was calculated. We found that the relative charge transfer was significantly reduced by a 15-min EGTA-AM bath application for the immature synapse (Fig. 10, A and B, P < 0.01, n = 8–10) across all three frequencies tested. In contrast, at the mature synapse, EGTA-AM had a much smaller effect, albeit significant, on relative charge transfer at 100 and 1 Hz (P < 0.05, n = 7) and no effect at 10 Hz (P > 0.2, n = 7, Fig. 10, C and D). Therefore, the contribution of the slow component of the synaptic waveform to the total synaptic charge transfer in response to trains of stimuli is much greater at immature compared with mature retinogeniculate synapses.
Fig. 10.

The slow component of the EPSC contributes significantly to the synaptic response to trains of stimuli. Representative EPSCs are normalized to the peak of the first EPSC. EPSCs are recorded from immature (A) and mature (C) synapses in response to trains of 15 stimuli given at 100-, 10- and 1-Hz frequencies. Traces are the average of 3–5 trials and are shown before (black) and 15 min following bath application of 50 μM EGTA-AM (red). Summary is shown of the normalized charge transfer in response to different stimulus frequencies before (black) and following application of EGTA-AM (red) for immature (B) and mature (D) synapses. Vh = −70 mV. *P < 0.05. **P < 0.01.
Prolonged synaptic currents contribute to relay neuron firing.
To investigate how the slow decay component of the immature EPSC contributes to the relay of information at the retinogeniculate synapse, we examined how relay neuron spiking changed when the slow component of the current was reduced. The internal solution used for current clamp recordings contains a low concentration of EGTA, unlike that for recording in voltage clamp mode. Because EGTA-AM would be taken up in both the pre- and postsynaptic neurons, we could not simply bath apply the calcium chelator while recording relay neuron spikes. An alternative approach to examining the relationship between the slow component of the EPSC and postsynaptic firing is to compare the spike response to the injection of different synaptic current waveforms. For immature and mature mice, representative synaptic responses at different stimulation frequencies obtained from our voltage clamp experiments before and after 50 μM EGTA-AM application (Fig. 10) were converted into waveforms (Fig. 11, A and B, top) and injected into the soma of relay neurons. Waveforms were scaled such that the amplitude of the first EPSC could reliably drive AP firing (300 and 1,500 pA for immature and mature neurons, respectively) at a holding potential of −55 mV in control conditions.
Fig. 11.

Spike pattern of immature relay neurons is influenced by the EPSC decay kinetics. Current clamp recordings are shown of immature (A) and mature (B) synapses in response to injection of their respective current waveforms. Waveforms were obtained from representative average synaptic responses to 15 stimuli at different frequencies (from left to right: 100, 10 and 1 Hz) before (black traces) and after bath application of 50 μM EGTA-AM (red traces). Relay neuron spiking was assessed at both Vh of −70 mV (middle) and −55 mV (bottom). i: Control. ii: EGTA-AM. C and D: summary of spike probability following current injection waveform at a Vh of −70 mV (C) and −55 mV (D) at immature (i) and mature (ii) synapse. Statistical data for 1 Hz for immature neurons are not available because neurons did not fire action potentials in response to 1-Hz stimulation/injected current while holding at −70 mV. *P < 0.05.
Figure 11, A and B, shows the relay neuron firing response to the injection of current waveforms representing control conditions (left) and after (right) EGTA-AM application for immature and mature synapses. Relay neuron spike probability was analyzed at two holding potentials: one at −70 mV (middle) and one at −55 mV (bottom). These two holding potentials were chosen to mimic the burst and tonic firing of thalamic relay neurons, respectively. Previous studies have shown that immature relay neurons rest at a more depolarized potential than those of mature neurons, consistent with immature neurons firing predominantly in the tonic mode (MacLeod et al. 1997; Pirchio et al. 1997; Ramoa and McCormick 1994).
Spike probability of the immature relay neuron was dramatically reduced in response to the EGTA-AM waveforms, mimicking the currents evoked by 10- and 100-Hz optic tract stimulation. In contrast, the difference between relay neuron spike probability in response to control and EGTA-AM waveform injections was much smaller at the mature synapse. In the burst mode of firing, the EGTA-AM waveform did not decrease spike probability of mature relay neurons. In fact, there was a significant increase of firing at 100 Hz with EGTA-AM (Fig. 11Cii), suggesting that prolonged synaptic currents in mature relay neurons could depolarize the membrane enough to result in inactivation of Na channels and, in turn, reduced AP firing. These data are consistent with a major role for the prolonged component of the EPSC decay in driving relay neuron firing at the immature retinogeniculate synapse.
DISCUSSION
Here we provide evidence that the additional current generated by glutamate spillover and by asynchronous release increases the probability of immature relay neuron firing. The prolonged synaptic current improves the efficiency of RGC transmission to the developing cortex during a period when individual synaptic inputs are weak. By examining single-fiber synaptic responses from immature RGC inputs, we uncovered a novel, purely spillover-mediated current. Our results suggest that a much larger number of RGCs than those that make direct synaptic contacts influence immature relay neuron spiking: this relay of information is critical for proper wiring of the visual cortex during development.
Asynchronous release.
Given the slow decay of the presynaptic calcium transient at the immature synapse, it is difficult to distinguish contributions of asynchronous release vs. spillover to the EPSC waveform. Reducing [Ca2+]o will decrease Cares (Fig. 4A; Chen and Regehr 2003). In addition, EGTA-AM reduced the amplitude of the ΔF/F signal and significantly decreased the peak of the immature EPSC (Figs. 8 and 9A). At the Calyx of Held, asynchronous release can be clearly separated from spillover using fluctuation analysis (Neher and Sakaba 2001). However, this approach requires voltage control over the presynaptic bouton, which has not been established at visual synapses. Asychronous release is often shown as the individual quantal events that can be resolved following the synchronous evoked response (Chen and Regehr 1999; Isaacson and Walmsley 1995). At the immature retinogeniculate synapse, we were unable to reliably isolate such events for several reasons: 1) the peak quantal amplitude at young ages is small (7 pA); 2) both retinogeniculate and corticothalamic contacts contribute to a relatively high frequency of spontaneous miniature EPSCs; and 3) spillover leads to extensive AMPAR desensitization that may mask individual single asynchronous events (Chen et al. 2002; Chen and Regehr 2000; Liu and Chen 2008). However, we have shown that the immature EPSC decay accelerates in the presence of the slow calcium buffer EGTA-AM. This suggests that release at the immature synapse is sensitive to Cares, consistent with asynchronous glutamate release.
Asynchronous release of glutamate has been described at both excitatory and inhibitory synapses in the CNS (Atluri and Regehr 1998; Diamond and Jahr 1995; Hefft and Jonas 2005; Hjelmstad 2006; Isaacson and Walmsley 1995; Lu and Trussell 2000) and has been shown to influence synaptic integration and postsynaptic firing patterns (Crowley et al. 2009; Iremonger and Bains 2007; Rudolph et al. 2011). We find that the developmental decrease in EPSC time course corresponds to the acceleration of the presynaptic calcium transient. The slow kinetics of the immature transient cannot simply be explained by saturation of the calcium indicator, because ΔF/F response to pairs of stimuli does not change with age (see Fig. 9).
The substantial developmental acceleration of the calcium transient (τ= ∼350 ms vs. ∼100 ms) may be due to changes in the concentration and composition of endogenous calcium buffers and/or calcium reuptake mechanisms (Sabatini and Regehr 1996; Vyleta and Jonas 2014). Other factors, such as shortening of the presynaptic AP width, changes in presynaptic ion channel expression levels and kinetics or decrease in AP temporal jitter could also contribute to age-dependent acceleration in the presynaptic calcium transient (Taschenberger and von Gersdorff 2000; Wang et al. 1997).
In our experiments, EGTA significantly accelerated the decay and decreased the peak of the EPSC at the immature but not the mature retinogeniculate synapse. At other CNS synapses, where the exact concentration of calcium buffer can be controlled, reduction of the peak EPSC amplitude by presynaptic EGTA suggests loose coupling between calcium channels and release machinery (Eggermann et al. 2012; Fedchyshyn and Wang 2005; Vyleta and Jonas 2014; Yang et al. 2010). A caveat of using membrane-permeable EGTA-AM is that the exact intracellular concentration of the calcium chelator is unknown; thus we cannot assess how components of the release machinery change with age. Despite these limitations, we used EGTA-AM to selectively reduce the prolonged component of the EPSC waveform and investigate its contribution to synaptic transmission.
Glutamate spillover at the immature synapse.
We show that excitatory currents at the developing retinogeniculate synapse are by both direct and spillover-mediated inputs from RGC axons. We provide multiple lines of evidence involving low-affinity antagonists for NMDARs and AMPARs, AMPAR desensitization, manipulations of PR, and characterization of slow- and fast-rising single-fiber currents. Taken together, our data strongly suggest that the prolonged decay of the immature EPSC is influenced by spillover of glutamate.
Few central excitatory synapses display such extensive spillover at near physiological temperature and with glutamate transporter activity intact. One example is the mossy fiber to granule cell (GC) synapse in the cerebellar cortex (DiGregorio et al. 2002). Like the retinogeniculate synapse, individual slow-rising spillover-mediated AMPAR currents have also been recorded from mature GCs (DiGregorio et al. 2002; Nielsen et al. 2004). However, the morphology of the two synapses is very different. At the cerebellar synapse, the absence of intervening glia promotes glutamate spillover to non-postsynaptic neurons within a glomerular glial sheath (DiGregorio et al. 2002). In contrast, glomeruli do not encompass retinogeniculate contacts in the immature LGN (Aggelopoulos et al. 1989; Bickford et al. 2010). The two synapses also respond differently to glutamate transporter inhibition. TBOA did not change the peak amplitude of AMPAR currents at the mossy fiber-GC synapse, whereas our results show that TBOA potentiated the peak of the slow-rising current at the immature retinogeniculate synapse (DiGregorio et al. 2002; Fig. 5). This result suggests that, despite the presence of glutamate transporters, there is substantial interaction between retinogeniculate release sites. TBOA's effects on the slow-rising retinogeniculate single-fiber currents are more similar to those of the purely spillover-mediated connection between climbing fibers and molecular layer interneurons of the cerebellum (Coddington et al. 2013; Szapiro and Barbour 2007). Consistent with the studies in the cerebellum, our results suggest that anatomy alone underestimates the influence of presynaptic neurons on postsynaptic firing.
Functional role of a prolonged glutamate transient and synaptic currents.
Significant glutamate spillover at the immature synapse is unexpected; the classic view of activity-dependent refinement at the retinogeniculate synapse assumes synapse specificity (Butts et al. 2007; Mooney et al. 1993). It is generally assumed that cross talk between synapses would degrade information signaling in the CNS (Barbour 2001). This may be the case at synapses in the auditory system, where information is coded in the precise timing of high-frequency transmission (Oertel 1997). However, in the visual system, relay of information relies heavily on temporal summation of synaptic currents (Sincich et al. 2007, 2009). Evidence from other central synapses has shown that the consequences of glutamate spillover can be desirable: spillover can increase reliability of synaptic signals, synchronize neural output and influence processing of local circuits (Arnth-Jensen et al. 2002; Coddington et al. 2013; Crowley et al. 2009; DiGregorio et al. 2002; Isaacson 1999). Therefore, glutamate spillover at the retinogeniculate connection could ensure reliable transmission of retinal activity to the visual cortex.
There are many potential ways that prolonged synaptic currents could influence activity-dependent synaptic strengthening and weakening. Previously, we demonstrated the presence of high-affinity group II/III mGluRs that, when activated, reduce the probability of glutamate release (Hauser et al. 2013). Our studies show that the spillover-mediated component of the EPSC is sensitive to PR. Therefore, heterosynaptic glutamate spillover and/or differential expression of mGluRs on RGC axon terminals could play a major role in activity-dependent refinement. Notably, the mGluR-mediated response downregulates with age (Hauser et al. 2013).
Alternatively, early in the development of a synaptic circuit, specificity may be less important than establishing connections onto a postsynaptic cell. In this case, spillover could aid in coordinating postsynaptic firing driven by neighboring RGCs. It is feasible that, before competition occurs between RGCs innervating the same relay neuron, all synapses strengthen to a level at which each RGC is strong enough to drive relay neuron spiking. The idea that neighboring synapses strengthen and weaken together has been proposed before in other developmental systems. Studies in the hippocampus, as well as the retinotectal synapse in Xenopus, have demonstrated that heterosynaptic plasticity exists (Engert and Bonhoeffer 1999; Harvey and Svoboda 2007; Tao et al. 2001). At the retinogeniculate synapse, neighboring release sites are more likely to be from the same RGC or from a neighboring RGC (Wong 1999). A prolonged glutamate transient may help establish and stabilize the initial retinotopy in the developing visual system.
GRANTS
This work was supported by National Eye Institute (NEI) Grant T32 EY-007110 to J. L. Hauser, the Childrens' Hospital Postdoctoral Career Development Fellowship to X. Liu, “PhD Training in Neuroscience” National Institute of Mental Health Grant 5T32-MH-020017 to E. Y. Litvina, NEI Grant RO1-EY-013613 to C. Chen, and the Children's Hospital Boston Intellectual and Developmental Disabilities Research Center National Institute of Child Health and Human Development Grant P01-HD-18655.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.L.H., X.L., and C.C. conception and design of research; J.L.H., X.L., and E.Y.L. performed experiments; J.L.H., X.L., and E.Y.L. analyzed data; J.L.H., X.L., and C.C. interpreted results of experiments; J.L.H. and X.L. prepared figures; J.L.H., X.L., and C.C. drafted manuscript; J.L.H., E.Y.L., and C.C. edited and revised manuscript; J.L.H., E.Y.L., and C.C. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank P. A. Rosenberg, A. Thompson, S. Park, and J. Leffler for helpful discussions of the experiments and manuscript.
REFERENCES
- Aggelopoulos N, Parnavelas JG, Edmunds S. Synaptogenesis in the dorsal lateral geniculate nucleus of the rat. Anat Embryol (Berl) 180: 243–257, 1989 [DOI] [PubMed] [Google Scholar]
- Akerman CJ, Smyth D, Thompson ID. Visual experience before eye-opening and the development of the retinogeniculate pathway. Neuron 36: 869–879, 2002 [DOI] [PubMed] [Google Scholar]
- Arnth-Jensen N, Jabaudon D, Scanziani M. Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci 5: 325–331, 2002 [DOI] [PubMed] [Google Scholar]
- Atluri PP, Regehr WG. Delayed release of neurotransmitter from cerebellar granule cells. J Neurosci 18: 8214–8227, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B. An evaluation of synapse independence. J Neurosci 21: 7969–7984, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron 12: 1331–1343, 1994 [DOI] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol 227: 691–708, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bickford ME, Slusarczyk A, Dilger EK, Krahe TE, Kucuk C, Guido W. Synaptic development of the mouse dorsal lateral geniculate nucleus. J Comp Neurol 518: 622–635, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschke M, Keller BU, Rivosecchi R, Hollmann M, Heinemann S, Konnerth A. A single amino acid determines the subunit-specific spider toxin block of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor channels. Proc Natl Acad Sci U S A 90: 6528–6532, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature 383: 431–434, 1996 [DOI] [PubMed] [Google Scholar]
- Budisantoso T, Matsui K, Kamasawa N, Fukazawa Y, Shigemoto R. Mechanisms underlying signal filtering at a multisynapse contact. J Neurosci 32: 2357–2376, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, Dieudonne S, Coussen F, Mulle C. Kainate receptor-mediated synaptic currents in cerebellar Golgi cells are not shaped by diffusion of glutamate. Proc Natl Acad Sci U S A 97: 6838–6843, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butts DA, Kanold PO, Shatz CJ. A burst-based “Hebbian” learning rule at retinogeniculate synapses links retinal waves to activity-dependent refinement. PLoS Biol 5: e61, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Contributions of residual calcium to fast synaptic transmission. J Neurosci 19: 6257–6266, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Developmental remodeling of the retinogeniculate synapse. Neuron 28: 955–966, 2000 [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG. Presynaptic modulation of the retinogeniculate synapse. J Neurosci 23: 3130–3135, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Blitz DM, Regehr WG. Contributions of receptor desensitization and saturation to plasticity at the retinogeniculate synapse. Neuron 33: 779–788, 2002 [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science 258: 1498–1501, 1992 [DOI] [PubMed] [Google Scholar]
- Coddington LT, Rudolph S, Vande Lune P, Overstreet-Wadiche L, Wadiche JI. Spillover-mediated feedforward inhibition functionally segregates interneuron activity. Neuron 78: 1050–1062, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Fioravante D, Regehr WG. Dynamics of fast and slow inhibition from cerebellar golgi cells allow flexible control of synaptic integration. Neuron 63: 843–853, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings DD, Wilcox KS, Dichter MA. Calcium-dependent paired-pulse facilitation of miniature EPSC frequency accompanies depression of EPSCs at hippocampal synapses in culture. J Neurosci 16: 5312–5323, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney KR, Zucker RS, Tank DW. Calcium in motor nerve terminals associated with posttetanic potentiation. J Neurosci 9: 3558–3567, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demas J, Eglen SJ, Wong RO. Developmental loss of synchronous spontaneous activity in the mouse retina is independent of visual experience. J Neurosci 23: 2851–2860, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS. Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci 21: 8328–8338, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Asynchronous release of synaptic vesicles determines the time course of the AMPA receptor-mediated EPSC. Neuron 15: 1097–1107, 1995 [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron 35: 521–533, 2002 [DOI] [PubMed] [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev 51: 7–61, 1999 [PubMed] [Google Scholar]
- Eggermann E, Bucurenciu I, Goswami SP, Jonas P. Nanodomain coupling between Ca2+ channels and sensors of exocytosis at fast mammalian synapses. Nat Rev Neurosci 13: 7–21, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399: 66–70, 1999 [DOI] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci 25: 4131–4140, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller MB, Delaney KR, Tank DW. Presynaptic calcium dynamics at the frog retinotectal synapse. J Neurophysiol 76: 381–400, 1996 [DOI] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Two components of transmitter release at a central synapse. Proc Natl Acad Sci U S A 91: 12942–12946, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanganu IL, Ben-Ari Y, Khazipov R. Retinal waves trigger spindle bursts in the neonatal rat visual cortex. J Neurosci 26: 6728–6736, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CD, Svoboda K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 450: 1195–1200, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser JL, Edson EB, Hooks BM, Chen C. Metabotropic glutamate receptors and glutamate transporters shape transmission at the developing retinogeniculate synapse. J Neurophysiol 109: 113–123, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefft S, Jonas P. Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8: 1319–1328, 2005 [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO. Interactions between asynchronous release and short-term plasticity in the nucleus accumbens slice. J Neurophysiol 95: 2020–2023, 2006 [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA–gated glutamate receptor channels depends on subunit composition. Science 252: 851–853, 1991 [DOI] [PubMed] [Google Scholar]
- Hooks BM, Chen C. Distinct roles for spontaneous and visual activity in remodeling of the retinogeniculate synapse. Neuron 52: 281–291, 2006 [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR. Development of cortical neuronal activity in the neonatal cat. Exp Neurol 17: 247–262, 1967 [DOI] [PubMed] [Google Scholar]
- Iremonger KJ, Bains JS. Integration of asynchronously released quanta prolongs the postsynaptic spike window. J Neurosci 27: 6684–6691, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS. Glutamate spillover mediates excitatory transmission in the rat olfactory bulb. Neuron 23: 377–384, 1999 [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Counting quanta: direct measurements of transmitter release at a central synapse. Neuron 15: 875–884, 1995 [DOI] [PubMed] [Google Scholar]
- Jaubert-Miazza L, Green E, Lo FS, Bui K, Mills J, Guido W. Structural and functional composition of the developing retinogeniculate pathway in the mouse. Vis Neurosci 22: 661–676, 2005 [DOI] [PubMed] [Google Scholar]
- Jurgens CW, Bell KA, McQuiston AR, Guido W. Optogenetic stimulation of the corticothalamic pathway affects relay cells and GABAergic neurons differently in the mouse visual thalamus. PLos One 7: e45717, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG. Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu Rev Physiol 76: 333–363, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerschensteiner D, Wong RO. A precisely timed asynchronous pattern of ON and OFF retinal ganglion cell activity during propagation of retinal waves. Neuron 58: 851–858, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kielland A, Erisir A, Walaas SI, Heggelund P. Synapsin utilization differs among functional classes of synapses on thalamocortical cells. J Neurosci 26: 5786–5793, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkby LA, Sack GS, Firl A, Feller MB. A role for correlated spontaneous activity in the assembly of neural circuits. Neuron 80: 1129–1144, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Gee KR, Archer EA, Regehr WG. Monitoring presynaptic calcium dynamics in projection fibers by in vivo loading of a novel calcium indicator. Neuron 27: 25–32, 2000 [DOI] [PubMed] [Google Scholar]
- Liu X, Chen C. Different roles for AMPA and NMDA receptors in transmission at the immature retinogeniculate synapse. J Neurophysiol 99: 629–643, 2008 [DOI] [PubMed] [Google Scholar]
- Lu T, Trussell LO. Inhibitory transmission mediated by asynchronous transmitter release. Neuron 26: 683–694, 2000 [DOI] [PubMed] [Google Scholar]
- MacLeod N, Turner C, Edgar J. Properties of developing lateral geniculate neurones in the mouse. Int J Dev Neurosci 15: 205–224, 1997 [DOI] [PubMed] [Google Scholar]
- Mennerick S, Zorumski CF. Presynaptic influence on the time course of fast excitatory synaptic currents in cultured hippocampal cells. J Neurosci 15: 3178–3192, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney R, Madison DV, Shatz CJ. Enhancement of transmission at the developing retinogeniculate synapse. Neuron 10: 815–825, 1993 [DOI] [PubMed] [Google Scholar]
- Mooney R, Penn AA, Gallego R, Shatz CJ. Thalamic relay of spontaneous retinal activity prior to vision. Neuron 17: 863–874, 1996 [DOI] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Estimating transmitter release rates from postsynaptic current fluctuations. J Neurosci 21: 9638–9654, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen TA, DiGregorio DA, Silver RA. Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron 42: 757–771, 2004 [DOI] [PubMed] [Google Scholar]
- Noutel J, Hong YK, Leu B, Kang E, Chen C. Experience-dependent retinogeniculate synapse remodeling is abnormal in MeCP2-deficient mice. Neuron 70: 35–42, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel D. Encoding of timing in the brain stem auditory nuclei of vertebrates. Neuron 19: 959–962, 1997 [DOI] [PubMed] [Google Scholar]
- Olverman HJ, Jones AW, Watkins JC. [3H]d-2-amino-5-phosphonopentanoate as a ligand for N-methyl-d-aspartate receptors in the mammalian central nervous system. Neuroscience 26: 1–15, 1988 [DOI] [PubMed] [Google Scholar]
- Otis TS, Wu YC, Trussell LO. Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci 16: 1634–1644, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci 16: 6634–6647, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirchio M, Turner JP, Williams SR, Asprodini E, Crunelli V. Postnatal development of membrane properties and delta oscillations in thalamocortical neurons of the cat dorsal lateral geniculate nucleus. J Neurosci 17: 5428–5444, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahamimoff R, Yaari Y. Delayed release of transmitter at the frog neuromuscular junction. J Physiol 228: 241–257, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoa AS, McCormick DA. Developmental changes in electrophysiological properties of LGNd neurons during reorganization of retinogeniculate connections. J Neurosci 14: 2089–2097, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramoa AS, Prusky G. Retinal activity regulates developmental switches in functional properties and ifenprodil sensitivity of NMDA receptors in the lateral geniculate nucleus. Brain Res Dev Brain Res 101: 165–175, 1997 [DOI] [PubMed] [Google Scholar]
- Ravin R, Spira ME, Parnas H, Parnas I. Simultaneous measurement of intracellular Ca2+ and asynchronous transmitter release from the same crayfish bouton. J Physiol 501: 251–262, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph S, Overstreet-Wadiche L, Wadiche JI. Desynchronization of multivesicular release enhances Purkinje cell output. Neuron 70: 991–1004, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384: 170–172, 1996 [DOI] [PubMed] [Google Scholar]
- Seabrook TA, El-Danaf RN, Krahe TE, Fox MA, Guido W. Retinal input regulates the timing of corticogeniculate innervation. J Neurosci 33: 10085–10097, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sincich LC, Horton JC, Sharpee TO. Preserving information in neural transmission. J Neurosci 29: 6207–6216, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sincich LC, Adams DL, Economides JR, Horton JC. Transmission of spike trains at the retinogeniculate synapse. J Neurosci 27: 2683–2692, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szapiro G, Barbour B. Multiple climbing fibers signal to molecular layer interneurons exclusively via glutamate spillover. Nat Neurosci 10: 735–742, 2007 [DOI] [PubMed] [Google Scholar]
- Takahashi M, Kovalchuk Y, Attwell D. Pre- and postsynaptic determinants of EPSC waveform at cerebellar climbing fiber and parallel fiber to Purkinje cell synapses. J Neurosci 15: 5693–5702, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, Yamasaki M, Nagumo Y, Imoto K, Watanabe M, Miyata M. Rewiring of afferent fibers in the somatosensory thalamus of mice caused by peripheral sensory nerve transection. J Neurosci 32: 6917–6930, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao HW, Zhang LI, Engert F, Poo M. Emergence of input specificity of LTP during development of retinotectal connections in vivo. Neuron 31: 569–580, 2001 [DOI] [PubMed] [Google Scholar]
- Tarnawa I, Farkas S, Berzsenyi P, Pataki A, Andrasi F. Electrophysiological studies with a 2,3-benzodiazepine muscle relaxant: GYKI 52466. Eur J Pharmacol 167: 193–199, 1989 [DOI] [PubMed] [Google Scholar]
- Tarusawa E, Matsui K, Budisantoso T, Molnar E, Watanabe M, Matsui M, Fukazawa Y, Shigemoto R. Input-specific intrasynaptic arrangements of ionotropic glutamate receptors and their impact on postsynaptic responses. J Neurosci 29: 12896–12908, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H. Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci 20: 9162–9173, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torborg CL, Feller MB. Spontaneous patterned retinal activity and the refinement of retinal projections. Prog Neurobiol 76: 213–235, 2005 [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron 10: 1185–1196, 1993 [DOI] [PubMed] [Google Scholar]
- Turner JP, Salt TE. Characterization of sensory and corticothalamic excitatory inputs to rat thalamocortical neurones in vitro. J Physiol 510: 829–843, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderkloot W, Molgo J. Facilitation and delayed release at about 0°C at the frog neuromuscular-junction–effects of calcium chelators, calcium-transport inhibitors, and okadaic acid. J Neurophysiol 69: 717–729, 1993 [DOI] [PubMed] [Google Scholar]
- Vyleta NP, Jonas P. Loose coupling between Ca2+ channels and release sensors at a plastic hippocampal synapse. Science 343: 665–670, 2014 [DOI] [PubMed] [Google Scholar]
- Wang GY, Ratto G, Bisti S, Chalupa LM. Functional development of intrinsic properties in ganglion cells of the mammalian retina. J Neurophysiol 78: 2895–2903, 1997 [DOI] [PubMed] [Google Scholar]
- Wong RO. Retinal waves and visual system development. Annu Rev Neurosci 22: 29–47, 1999 [DOI] [PubMed] [Google Scholar]
- Yang YM, Fedchyshyn MJ, Grande G, Aitoubah J, Tsang CW, Xie H, Ackerley CA, Trimble WS, Wang LY. Septins regulate developmental switching from microdomain to nanodomain coupling of Ca2+ influx to neurotransmitter release at a central synapse. Neuron 67: 100–115, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengel JE, Magleby KL. Changes in miniature endplate potential frequency during repetitive nerve stimulation in the presence of Ca2+, Ba2+, and Sr2+ at the frog neuromuscular junction. J Gen Physiol 77: 503–529, 1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziburkus J, Guido W. Loss of binocular responses and reduced retinal convergence during the period of retinogeniculate axon segregation. J Neurophysiol 96: 2775–2784, 2006 [DOI] [PubMed] [Google Scholar]
- Zucker RS, Lara-Estrella LO. Post-tetanic decay of evoked and spontaneous transmitter release and a residual-calcium model of synaptic facilitation at crayfish neuromuscular junctions. J Gen Physiol 81: 355–372, 1983 [DOI] [PMC free article] [PubMed] [Google Scholar]