

Abstract
The ubiquitous nature of C-H bonds in organic molecules makes them attractive as a target for rapid complexity generation, but brings with it the problem of achieving selective reactions. In developing new methodology for C-H functionalization, alkenes are an attractive starting material due to their abundance and low cost. Here we describe the conversion of 1-alkenes into 1,4-diols. The method involves the installation of a Si,N-type chelating auxiliary group on the alkene followed by iridium catalyzed C-H silylation of an unactivated δ-C(sp3)-H bond to produce an silolane intermediate. Oxidation of the C-Si bonds affords a 1,4-diol. The method is demonstrated to have broad scope and a good functional group compatibility by application to the selective 1,4-oxygenation of several natural products and derivatives.
Transition metal catalyzed C–H activation reactions have emerged as a powerful tool in organic chemistry.1–6 However, aliphatic C–H bonds, which are ubiquitous in organic molecules, are the most challenging targets for selective functionalization due to the lack of the active frontier orbitals, which could interact with a transition metal center. 7,8 Among a variety of aliphatic C–H functionalizations,9,10 the C–H oxygenation is one of the most attractive transformations, since a number of important biochemical processes involve this step.11–13 Although, a number of transition metal-catalyzed aliphatic C–H oxygenation reactions have been reported, they are mostly limited to functionalization of activated C–H bonds. On the other hand, the development of selective oxygenation of unactivated sp3 C–H bond is still in its infancy.14–22 Therefore, the design of new methods, which can be applied for selective oxygenation of unactivated aliphatic C–H bonds, is highly warranted.
Results and discussion
The alkene fragment is widely found in feedstock materials, as well as in a variety of organic building blocks and in natural products. Although, oxygenations of the double bond and the allylic C–H bonds of alkenes are well developed, the oxygenation of homoallylic position of 1-alkenes has not been disclosed. Herein, we report an unprecedented double 1,4-functionalization of 1-alkenes, which includes a C–H activation of the homoallylic position, as well as a formal anti-Markovnikov hydration of the double bond. Our approach is based on the introduction of hydrosilane functionality, followed by an intramolecular dehydrogenative silylation and a subsequent oxidation step. The overall transformation represents the conversion of abundant 1-alkenes 1 into valuable 1,4-diols 4 (Fig. 1a).
Figure 1. Synthesis of 1,4-diols from 1-alkenes and alkyl halides.

a, General concept for formal 1,4-oxygenation of 1-alkenes 1 into 1,4 diols 4 via activation of homoallylic C–H bond. b, Conversion of 1-alkenes 1 (or 1-haloalkanes 1′) into 1,4-diols 4 via installation of the TBPicSi to form 2, followed by its iridium catalyzed C-H silylation of an unactivated C(sp3)-H bond (to produce the silolane 3), and subsequent oxidation. We designed tert-butylpicolylsilyl (TBPicSi), a new Si,N-type chelating directing group which can be easily installed on alkenes (and alternatively on alkyl halides). Notably, its picolyl moiety enables an efficient Si–H/C–H activation step (via iridacycle A) and, being easily removable from silicon, ensures a successful subsequent oxidation of silolane 3 into the final diol 4. The bulky tert-butyl substituent at silicon is requisite for stability of 2. cod, cyclooctadiene; nbe, norbornene.
As a key step (2→3) in the formal homoallylic C–H oxygenation of alkenes, we chose the Ir-catalyzed method developed by Hartwig for dehydrogehative Si–H/C–H coupling in alkoxysilanes.23 However, in order for this approach to be synthetically useful for a fully hydrocarbon chain of 2, a silicon group should possess at least one non-alkyl substituent (R1 or R2 ≠ alkyl; for rare examples on synthesis of dialkylsilolanes via the transition metal-catalyzed Si–H/C–H coupling see references 24, 25), which would ensure a successful subsequent oxidation of Si–C bonds26,27 of 3 into 4 (Fig. 1b). However, screening a few different removable groups at silicon, including siloxy-, aryl-, and benzyl groups indicated either no reaction (2→3) or instability of 2 under the reaction conditions (Supplementary Fig. S1).
Next, we proposed that a new Si,N-type chelation-assisted auxiliary could facilitate the dehydrogehative Si–H/C–H coupling reaction. This idea was inspired by Daugulis’ N,N-chelation concept, which was proven efficient for a remote Pd-catalyzed aliphatic C–H activation reactions.28 To this end, we screened a number of potential directing groups and reaction conditions (Supplementary Fig. 1–3, Supplementary Tables 1–5). Gratifyingly, we found that a new Si,N-type chelating group, tert-butylpicolylsilicon hydride (TBPicSi), is highly efficient for the dehydrogenative intramolecular silylation of δ-C(sp3)–H bond (Fig. 1b). A control experiment revealed the importance of the picolyl group, as the benzyl analog of TBPicSi was not efficient in the C-H activation reaction (see Supplementary Information for details). Moreover, its picolyl moiety has a double duty; it not only enables an efficient Si–H/C–H activation step via the six membered chelation stabilized iridacycle A, but also, being easily removable from silicon, ensures a successful subsequent oxidation of 3. The bulky tert-butyl substituent at silicon, in turn, proved vital for stability of 2. Next, we developed an efficient method for installation of TBPicSi directing group onto alkenes 1. Thus, transition metal-catalyzed hydrosilylation of alkenes with dichlorosilane,29 followed by a sequential substitution of two chlorine atoms at silicon with tert-butyl- and picolyl groups furnished the hydrosilane 2. Alternatively, synthesis of 2 can be easily achieved via alkylation of tBuSiHCl2 with organolithium/organomagnesium reagents, routinely available from the corresponding alkyl halides 1′. Mostly, these 3-step procedures allow to obtain starting hydrosilanes 2 in about 50% overall yields.
Next, the scope of this intramolecular dehydrogenative silylation reaction has been investigated. Thus, unsubstituted n-butyl silane 2a underwent the dehydrogenative Si–C coupling reaction affording silolane 3a in high yield (Table 1, Entry 1). The silanes 2b-f bearing α-, β-, and γ-alkyl substituents were efficiently converted into silolanes 3b-f as well (Entries 2–6). Cyclic cyclohexane-containing substrates can also be converted into bicyclic products 3g, h (Entries 7, 8). The substrates containing benzene ring 2i, as well as protected phenol 2j and catechol 2k fragments, were perfectly tolerated. Importantly, C(sp2)–Hal (Hal = F, Cl, Br) bonds, as well as amine and CF3 functionalities, remained intact under these reaction conditions (2l-p). Notably, silylation of δ-C–H bond of CH3 groups in hydrosilanes 2 is highly preferable over other competitive CH2, CH and benzylic CH2 groups. As an exception, a highly active secondary δ-C(sp3) H bond of a cyclopropane ring can also be silylated under these conditions (Entry 17).
Table 1.
Ir-catalyzed δ-C–H dehydrogenative silylation reaction.
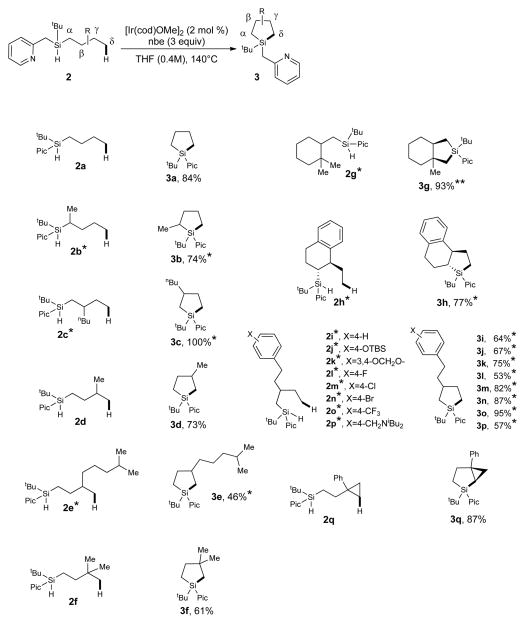
|
Mixture of stereoisomers (see Supplementary Information for details);
Major diastereomer is drawn (see Fig. 3 for details); nbe, norbornene; Pic, picolyl; TBS, tert-butyldimethylsilyl.
The obtained silolanes 3 could be efficiently converted into 1,4-diols using Woerpel’s oxidation procedure (Fig. 2).30 For convenience, the diols were isolated as diacetates 4. We have demonstrated that the oxidation procedure successfully affords aliphatic acyclic or cyclic primary diols (4c, 4g), as well as primary/secondary diol 4h, in which the alcohol moieties could be routinely differentiated. Bicyclic silolane 3g, upon oxidation, yields 4g as a mixture of cis/trans isomers in a 3:1 ratio. Obviously, this ratio is a result of activation of both methyl groups in hydrosilane 2g during the C–H activation step (Table 1, Entry 7).
Figure 2. Conversion of silacycle intermediates 3 into 1,4-diol derivatives 4.

The reaction was performed under Woerpel’s oxidation conditions.30 TBHP, tert-butyl hydroperoxide; TBAF, tetrabutylammonium fluoride; NMP, N-methylpyrrolidone; RT, room temperature; Ac, acetyl; DMAP, 4-dimethylaminopyridine; DCM, dichloromethane.
C–H functionalization is the most promising method for the late stage modification of natural products and drugs, since it eliminates prefunctionalization steps.31 Accordingly, our new method has been tested for modification of natural products and derivatives (Fig. 3). To our delight, camphene, 2-methylenebornane and the derivative of lithocholic acid 1t were successfully converted into the corresponding 1,4-diols 4r-t. In the case of camphene, the method resulted in endo-diol 4r, whereas 2-methylenebornane furnished exo-diol 4s, which can be explained by the preferable hydrosilylation of camphene and 2- methylenebornane from the less sterically hindered face of the double bond.
Figure 3. 1,4-Oxygenation of alkene-containing natural products and derivatives.

Application of the developed protocol for conversion of alkene-containing natural products and derivatives into the corresponding 1,4-diols. a, Conversion of Camphene into 1,4-diol 4r; b, Conversion of 2-Methylenebornane (derived from Bornane) into 1,4-diol 4s. c, Conversion of alkene 1t (derived form Lithoholic acid) into 1,4-diol 4t. *Mixture of stereoisomers endo/exo 8:1, major isomer is drawn (for details, see Supplementary Fig. S12). **Mixture of stereoisomers endo/exo 1:3, major isomer is drawn (for details, see Supplementary Fig. S13). THF, tetrahydrofuran; Pic, 2-picolyl; cod, cyclooctadiene; nbe, norbornene; TBAF, tetrabutylammonium fluoride; NMP, N-methylpyrrolidone; RT, room temperature; Ac, acetyl; DMAP, 4-dimethylaminopyridine; DCM, dichloromethane.
Conclusion
In summary, we developed an unprecedented conversion of 1-alkenes into 1,4-diols. This was accomplished by using a new Si,N-type tert-butylpicolylsilicon hydride (TBPicSi) directing group, which can be easily installed onto alkenes. TBPicSi group allows for intramolecular dehydrogenative silylation of unactivated δ-C(sp3)–H bonds, as well as for efficient oxygenation of C–Si bonds. The developed method was successfully applied for the 1,4-dioxygenation of several alkene-containing natural products and derivatives.
Methods
Detailed description of experiments as well as analytical data are provided in Supplementary Information.
Supplementary Material
Acknowledgments
The support of the National Institute of Health (GM-64444) and the National Science Foundation (CHE-1112055) is gratefully acknowledged.
Footnotes
Author contributions
N.G., F.S.M. and A.V.G. contributed equally to this work. N.G., F.S.M. and A.V.G. designed and performed the experiments and wrote the manuscript. C.H. performed the experiments at early stage of the project. All authors participated in the discussion of the results. V.G. conceived and guided the research.
Additional information
Supplementary Information and chemical compound information are available in the online version of the paper. Reprints and permissions information is available online at www.nature.com/reprints.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Yu J-Q, Shi Z, editors. C–H activation. Topics in Current Chemistry. 2010;292 [PubMed] [Google Scholar]
- 2.Engle KM, Mei TS, Wasa M, Yu JQ. Weak coordination as a powerful means for developing broadly useful C–H functionalization reactions. Acc Chem Res. 2011;45:788–802. doi: 10.1021/ar200185g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wencel-Delord J, Droge T, Liu F, Glorius F. Towards mild metal-catalyzed C-H bond activation. Chem Soc Rev. 2011;40:4740–4761. doi: 10.1039/c1cs15083a. [DOI] [PubMed] [Google Scholar]
- 4.Ackermann L. Carboxylate-assisted transition-metal-catalyzed C–H bond functionalizations: Mechanism and Scope. Chem Rev. 2011;111:1315–1345. doi: 10.1021/cr100412j. [DOI] [PubMed] [Google Scholar]
- 5.Lyons TW, Sanford MS. Palladium-catalyzed ligand-directed C–H functionalization reactions. Chem Rev. 2010;110:1147–1169. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Labinger JA, Bercaw JE. Understanding and exploiting C–H bond activation. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- 7.Li H, Li BJ, Shi ZJ. Challenge and progress: palladium-catalyzed sp3 C–H activation. Catal Sci Technol. 2011;1:191–206. [Google Scholar]
- 8.Jazzar R, Hitce J, Renaudat A, Sofack-Kreutzer J, Baudoin O. Functionalization of organic molecules by transition-metal-catalyzed C(sp3) H activation. Chem Eur J. 2010;16:2654–2672. doi: 10.1002/chem.200902374. [DOI] [PubMed] [Google Scholar]
- 9.He G, Zhao Y, Zhang S, Lu C, Chen G. Highly efficient syntheses of azetidines, pyrrolidines, and indolines via palladium catalyzed intramolecular amination of C(sp3)-H and C(sp2)-H bonds at gamma and delta positions. J Am Chem Soc. 2012;134:3–6. doi: 10.1021/ja210660g. [DOI] [PubMed] [Google Scholar]
- 10.Nadres ET, Daugulis O. Heterocycle synthesis via direct C-H/N-H coupling. J Am Chem Soc. 2012;134:7–10. doi: 10.1021/ja210959p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.White MC. Adding aliphatic C–H bond oxidations to synthesis. Science. 2012;335:807–809. doi: 10.1126/science.1207661. [DOI] [PubMed] [Google Scholar]
- 12.Newhouse T, Baran PS. If C–H bonds could talk: selective C–H bond oxidation. Angew Chem Int Ed. 2011;50:3362–3374. doi: 10.1002/anie.201006368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ortiz de Montellano PR. Hydrocarbon hydroxylation by Cytochrome P450 Enzymes. Chem Rev. 2009;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McNeill E, Du Bois J. Catalytic C–H oxidation by a triazamacrocyclic ruthenium complex. Chem Sci. 2012;3:1810–1813. [Google Scholar]
- 15.Bigi MA, Reed SA, White MC. Diverting non-haem Iron catalysed aliphatic C–H hydroxylations towards desaturations. Nature Chem. 2011;3:216–222. doi: 10.1038/nchem.967. [DOI] [PubMed] [Google Scholar]
- 16.Prat I, et al. Observation of Fe(V)=O using variable-temperature mass spectrometry and its enzyme-like C–H and C=C oxidation reactions. Nature Chem. 2011;3:788–793. doi: 10.1038/nchem.1132. [DOI] [PubMed] [Google Scholar]
- 17.Chen MS, White MC. Combined effects on selectivity in Fe-catalyzed methylene oxidation. Science. 2010;327:566–571. doi: 10.1126/science.1183602. [DOI] [PubMed] [Google Scholar]
- 18.Kamata K, Yonehara K, Nakagawa Y, Uehara K, Mizuno N. Efficient stereo- and regioselective hydroxylation of alkanes catalysed by a bulky polyoxometalate. Nature Chem. 2010;2:478–483. doi: 10.1038/nchem.648. [DOI] [PubMed] [Google Scholar]
- 19.Chen MS, White MC. A Predictably selective aliphatic C–H oxidation reaction for complex molecule synthesis. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 20.Das S, Incarvito CD, Crabtree RH, Brudvig GW. Molecular recognition in the selective oxygenation of saturated C–H bonds by a dimanganese catalyst. Science. 2006;312:1941–1943. doi: 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]
- 21.Giri R, et al. Pd-Catalyzed stereoselective oxidation of methyl groups by inexpensive oxidants under mild conditions: a dual role for carboxylic anhydrides in catalytic C–H bond oxidation. Angew Chem Int Ed. 2005;44:7420–7424. doi: 10.1002/anie.200502767. [DOI] [PubMed] [Google Scholar]
- 22.Desai LV, Hull KL, Sanford MS. Palladium-catalyzed oxygenation of unactivated sp3 C–H bonds. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 23.Simmons EM, Hartwig JF. Catalytic functionalization of unactivated primary C-H bonds directed by an alcohol. Nature. 2012;483:70–73. doi: 10.1038/nature10785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsukada N, Hartwig JF. Intermolecular and intramolecular, platinum-catalyzed, acceptorless dehydrogenative coupling of hydrosilanes with aryl and aliphatic methyl C-H bonds. J Am Chem Soc. 2005;127:5022–5023. doi: 10.1021/ja050612p. [DOI] [PubMed] [Google Scholar]
- 25.Kuninobu Y, Nakahara T, Takeshima H, Takai K. Rhodium-catalyzed intramolecular silylation of unactivated C(sp3)–H bonds. Org Lett. 2013;15:426–428. doi: 10.1021/ol303353m. [DOI] [PubMed] [Google Scholar]
- 26.Jones GR, Landais Y. The oxidation of the carbon-silicon bond. Tetrahedron. 1996;52:7599–7662. [Google Scholar]
- 27.Kuznetsov A, Gevorgyan V. General and practical one-Pot synthesis of dihydrobenzosiloles from styrenes. Org Lett. 2012;14:914–917. doi: 10.1021/ol203428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zaitsev VG, Shabashov D, Daugulis O. Highly regioselective arylation of sp3 C–H bonds catalyzed by palladium acetate. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 29.Watanabe H, Aoki M, Sakurai N, Watanabe K-i, Nagai Y. Selective synthesis of mono-alkyldichlorosilanes via the reaction of olefins with dichlorosilane catalyzed by group VIII metal phosphine complexes. J Organomet Chem. 1978;160:C1–C7. [Google Scholar]
- 30.Smitrovich JH, Woerpel KA. Oxidation of Sterically Hindered Alkoxysilanes and Phenylsilanes under Basic Conditions. J Org Chem. 1996;61:6044–6046. [Google Scholar]
- 31.Wencel-Delord J, Glorius F. C-H bond activation enables the rapid construction and late-stage diversification of functional molecules. Nature Chem. 2013;5:369–375. doi: 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.