

Abstract
Recurrent or sustained inflammation plays a causal role in the development and progression of left ventricular hypertrophy (LVH) and its transition to failure. Interleukin (IL)-18 is a potent pro-hypertrophic inflammatory cytokine. We report that induction of pressure overload in the rabbit, by constriction of the descending thoracic aorta induces compensatory hypertrophy at 4 weeks (mass/volume ratio: 1.7±0.11) and ventricular dilatation indicative of heart failure at 6 weeks (mass/volume ratio: 0.7±0.04). In concordance with this, fractional shortening was preserved at 4 weeks, but markedly attenuated at 6 weeks. We cloned rabbit IL-18, IL-18Rα, IL-18Rβ, and IL-18 binding protein (IL-18BP) cDNA, and show that pressure overload, while enhancing IL-18 and IL-18R expression in hypertrophied and failing hearts, markedly attenuated the level of expression of the endogenous IL-18 antagonist IL-18BP. Cyclical mechanical stretch (10% cyclic equibiaxial stretch, 1 Hz) induced hypertrophy of primary rabbit cardiomyocytes in vitro and enhanced ANP, IL-18, and IL-18Rα expression. Further, treatment with rhIL-18 induced its own expression and that of IL-18Rα via AP-1 activation, and induced cardiomyocyte hypertrophy in part via PI3K/Akt-dependent GATA4 activation. In contrast, IL-18 potentiated TNF-α-induced cardiomyocyte death, and induced cardiac endothelial cell death. These results demonstrate that pressure overload is associated with enhanced IL-18 and its receptor expression in hypertrophied and failing myocardium in rabbits. Since IL-18BP expression is markedly inhibited, our results indicate a positive amplification in IL-18 pro-inflammatory signaling during pressure overload, and suggest IL-18 as a potential therapeutic target in pathological hypertrophy and cardiac failure.
Keywords: Myocardial hypertrophy, Cardiac failure, Cyclical stretch, Interleukins, Inflammation
Introduction
Left ventricular hypertrophy (LVH) is an adaptation of the heart to chronic pressure or volume overload. Although initially beneficial in normalizing wall stress, prolonged LVH significantly increases the risk of heart failure and sudden death. Recurrent or sustained inflammation plays a causal role in the development of LVH and its transition to failure.
Interleukin (IL)-18 is a proinflammatory cytokine, and exerts cell type-specific effects in the heart; it induces (i) cardiomyocyte hypertrophy [1], (ii) fibroblast proliferation and migration [2–4], and (iii) death of endothelial cells via activation of both intrinsic and extrinsic cell death pathways [5]. Similar to its effects on fibroblasts, IL-18 induces SMC migration and proliferation [6, 7]. Together, hypertrophy of cardiomyocyte, migration and proliferation of fibroblasts, and endothelial cell death contribute causally to myocardial hypertrophy, fibrosis, adverse remodeling, and failure. While we have demonstrated IL-18’s pro-growth effects in vitro, its continuous infusion for 7 days has been shown to induce myocardial hypertrophy in vivo in mouse models [8, 9]. Further, Il18 gene deletion blunts pressure overload-induced myocardial hypertrophy in the mouse [10]. Though systemic IL-18 levels are found elevated in human heart failure patients [11] and in hypertrophied heart following pressure overload in a mouse model [12], it is not known whether pressure overload persistently elevates IL-18 in failing hearts.
IL-18 signals via IL-18 receptor, a heterodimer comprising of the ligand-binding α subunit (IL-18Rα) and the signal-traducing β-subunit (IL-18Rβ). Both receptor subunits are necessary for IL-18 to exert its biological effects. Importantly, IL-18 and IL-18Rα expressions are increased in explanted failed human hearts [11], and IL-18BP levels are decreased in human heart failure. IL-18 binding protein (IL-18BP), a natural IL-18 antagonist, is expressed at high levels in normal human serum [13]. IL-18BP is encoded by a separate gene, is transcriptionally regulated, and does not bind the extracellular domain of IL-18Rα, but binds IL-18 at a 1:1 ratio and with high affinity (400 pM) [13], thus acting as a decoy receptor. Importantly, by regulating both Th1 and Th2 responses, it reduces overall inflammatory response. Recently, we demonstrated that the β2-adrenergic receptor agonist induces IL-18 expression in cardiac endothelial cells via NF-κB and AP-1-dependent manner, and IL-18BP expression in CREB/C/EBP-mediated signaling [14, 15]. Since heart failure is characterized by sustained β-AR activation, and as β-AR activation induces both IL-18 and IL-18BP expression [14, 15], it is possible that a fine balance between the ligand and its endogenous inhibitor in the progression of heart failure. However, to date no systematic studies have been performed describing the expression levels of IL-18 and its regulators (IL-18Rα, IL-18Rβ and IL-18BP) during pressure overload hypertrophy and cardiac failure in a translationally important preclinical model.
Here we have investigated the expression of IL-18, IL-18R and IL-18BP in a rabbit model of pressure overload-induced myocardial hypertrophy and heart failure. Our results demonstrate that IL-18, IL-18Rα, and IL-18Rβ are increased in both hypertrophied and failing rabbit hearts, suggesting amplification in IL-18 proinflammatory signaling. Further, in studies in vitro, cyclical stretch, which mimics the effects of pressure overload, induced cardiomyocyte hypertrophy in part via IL-18. However, although IL-18 alone could induce cardiomyocyte hypertrophy, it potentiated TNF-α-induced cardiomyocyte death and induced endothelial cell death. Together, these in vivo and in vitro results suggest a causal role for IL-18 in myocardial hypertrophy and failure in a translationally important animal model.
Materials and methods
Animals and pressure-overload hypertrophy
The investigation conforms to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. All protocols were approved by the Institutional Animal Care and Use Committees of the Harvard Medical School and the University of Texas Health Science Center in San Antonio. Here we used 10-day-old New Zealand White rabbits as a hypertrophy/heart failure model. This model mimics progression of the human disease found in children with congenital heart defects. All animals underwent the same process of physiologic growth (i.e., physiologic hypertrophy) since littermates were used as sham-operated controls. Thus, the results reported here are due to imposing pressure-overload resulting in pathological hypertrophy signaling.
The animals were sedated with ketamine (40–60 mg/kg im) and xylazine (2–5 mg/kg im). The surgical sites were shaved and prepped with Betadine and 70% isopropyl alcohol (×3). The entire animal, except the surgical site, was draped with sterile towels. Bupivacaine (0.25%; <3 mg/kg) was administered into the wound at the time of surgery. Anesthesia was maintained with 0.5–2% isoflurane (Forane; Abbott Laboratories, Chicago, IL) under spontaneous respiration. Following left thoracotomy in the 4th intercostal space, pressure overload was achieved by placing a 2-0 silk suture around the descending aorta just distal to the ligamentum arteriosum as previously described [16–18]. Care was taken to make the banding snug without causing stenosis of the descending aorta as previously described [17].
Implanting a fixed constriction in an immature animal and allowing it to grow, induces pressure-overload hypertrophy by 2 to 3 weeks of age. The progression of LV hypertrophy was determined by weekly transthoracic echocardiography (Hewlett-Packard Sonos 1500 Cardiac Imager) with a 7.5-MHz transducer. During these procedures, the animals remained unsedated to avoid the influence of anesthetics on the results. Pressure-overload results in compensatory increase of muscle mass to keep wall stress normal by 4 weeks (concentric hypertrophy) which is followed by ventricular dilatation, indicative of cardiac failure by 6 weeks [16–18]. Sham-operated animals (Sham) and animals that did not undergo any procedure (Naïve) served as controls. For isolation of cardiomyocytes, naïve rabbits were used.
Echocardiography
Two-dimensional cross-sectional images and M-modes of the left ventricle were obtained by echocardiography [16–18]. Simultaneous measurements of ECG and LV short-axis dimensions by M-mode were recorded on hard copy at a paper speed of 100 mm/s. These examinations were started at 3 weeks of age and performed at 1-week intervals. LV epicardial and endocardial surfaces were traced by computer-aided offline analysis on a bit-mapped digitizing tablet. End-diastolic (maximum dimension) and end-systolic (minimum dimension) LV wall thicknesses were determined using previously described methods [16–18]. A representative cross-sectional view of the LV is shown in Fig. 1A. A representative echocardiogram for M-mode for calculation of M/V ratio is shown in Fig. 1B. Left ventricular mass/volume (M/V) ratio was calculated and was used as an index for progression of hypertrophy. As a measure of cardiac performance, fractional shortening (FS) was determined with the formula (systolic diameter-diastolic diameter/diastolic diameter) and is expressed as percentage.
Fig. 1. Pressure overload induces myocardial hypertrophy and transition to failure in a rabbit model.

A, Male 10 day-old NZW rabbits underwent pressure overload by constriction of the descending thoracic aorta. Myocardial function was analyzed after 4 (hypertrophy) and 6 weeks (heart failure) by transthoracic echocardiography. Rabbits that underwent sham surgery served as controls. While representative cross-sectional views are shown in the upper panels, M-mode of LVs used in the calculation of M/V ratio is shown in the bottom panels. ECG, electrocardiogram; EDD, end-diastolic diameter; ESD, end-systolic diameter; PWD, posterior wall thickness in diastole. B, As a measure of cardiac performance, fractional shortening (FS, %) was determined with the formula (systolic diameter-diastolic diameter/diastolic diameter) and expressed as percentage. P < 0.001 vs. respective sham-operated control (n=5–6/group). S, sham; H, hypertrophy; HF, heart failure. C, Pressure overload induced myocardial hypertrophy. Rabbits that underwent pressure overload were analyzed for body and heart weights after 4 and 6 weeks. Both sham-operated and naïve (N) rabbits served as controls. Myocardial hypertrophy was analyzed by the heart to body weight ratios. D, Pressure overload induces ANP expression. Left ventricular (LV) tissues at 4 and 6 weeks post-constriction were analyzed for ANP mRNA expression by RT-qPCR. ANP protein levels were analyzed by immunoblotting in a subset of animals (n=3/group selected at random) as shown in the inset. E, Pressure overload induces MMP9 mRNA expression. LV tissues described in D were analyzed for MMP9 mRNA expression by RT-qPCR. D, E, *P < 0.01 vs. Naïve or Sham (n=5–6 animals/group).
Cloning of IL-18, IL-18Rα, IL-18Rβ, and IL-18BP
To analyze rabbit IL-18, IL-18Rα, IL-18Rβ, and IL-18BP mRNA expression during hypertrophy and failure, we cloned the respective full-length cDNAs using DNA-free total RNA isolated from rabbit heart or spleen, and the oligonucleotide primers shown in Table 1. These primers were designed based on their predicted coding regions in genomic DNA using the GenBank database.
Table 1. Primers used to amplify coding sequences of rabbit IL-18, IL-18Rα, IL-18Rβ and IL-18BP.
Primers were designed against predicted rabbit IL-18, IL-18Rα, IL-18Rβ and IL-18BP coding regions in genomic DNA using the GenBank database. Coding region of each gene was predicted based on the comparison between different species (human, mouse and rat). RT-PCR was performed on cDNA prepared from rabbit heart, and amplified DNA fragments were cloned into pGEM-T vector (Promega) for DNA sequencing.
| IL-18 | Sense, 5’- TCATTAGGAATAAAGATGGCTGC-3’ Antisense, 5’- CTAATTCTTGTTTTGGACACTG-3’ |
| IL-18Rα | Sense, 5’-ATGCGTCAGAGAGAATTACCC-3’ Antisense, 5’- CTTGAGTTATAAGAAAGAGACTTATC-3’ |
| IL-18Rβ | Sense, 5’-ATGTGCTGCTTGGGCTGGATGTTTCTTTGGC-3’ Antisense, 5’-TCATGACATAGTTTTGAATTTGTTGCATGTG-3’ |
| IL-18BP | Sense, 5’-ATGGCCGTGAGACAGAACTGGACTCCAG-3’ Antisense, 5’-TCAAGGCTGTGCTGTTGCTGTGCTGAG-3’ |
Northern blot analysis
DNA-free total RNA was prepared using the RNAqueous®-4PCR kit (Ambion). Expression of IL-18, IL-18Rα, IL-18Rβ and IL-18BP was analyzed by Northern blot analysis using 20 µg of DNA-free total RNA. RNA quality was assessed by capillary electrophoresis using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). All RNA samples used for quantitative PCR had RNA integrity numbers greater than 8.9 (scale = 1–10) as assigned by default parameters of the Expert 2100 Bioanalyzer software package (v2.02). A 300–400 nt cDNA was amplified for labeling and probing using the primers described in Table 2. ANP and BNP mRNA expression was also analyzed by Northern blot analysis using cDNAs described in Supplementary Table S1. 18S rRNA served as a loading control.
Table 2.
Primers used to amplify cDNA for Northern blot analysis
| cDNA | GenBank accession # |
Primers |
|---|---|---|
| IL-18 | EU419127.1 | Sense, 5’-ACCTGGAATCAGATTACTTTGGC-3’ Antisense, 5’-ATCTTATCTTTCTGTCCTGCGAGAT-3’ Size: 352 nt |
| IL-18Rα | EU731009.1 | Sense, 5’-TGCACTGCGGTCAGCGAAGG-3’ Antisense, 5’-TCAACAACAGCTCCTCCAGGCA-3’ Size: 388 nt |
| IL-18Rβ | JF271681.1, JF271682.1, and JF271683.1 | Sense, 5’-AACAAGGATGAGACGCTCGGAGATG-3’ Antisense, 5’-CTGCCTGCAGTTCAAAGACACTGGG-3 Size: 300 nt |
| IL-18BP | JQ657816.1 and JQ657817.1 | Sense, 5’-GACGGCAGCGCAGGAGCAGAAGCAC-3’ Antisense, 5’-TCAAGGCTGTGCTGTTGCTGTGC-3’ Size: 247 nt |
RT-qPCR
Expression levels of ANP and MMP9 were analyzed by real-time quantitative RT-PCR (RT-qPCR) using the primers described in Supplementary Table S1. No-template controls were also performed for each assay, and samples processed without the reverse transcriptase step served as negative controls. Each cDNA sample was run in triplicate, and the amplification efficiencies of all primer pairs were determined by serial dilutions of input template. Data were analyzed using the 2−ΔΔCt method. GAPDH served as the endogenous invariant control gene, and all data were normalized to corresponding GAPDH levels.
Western blot analysis
Extraction of protein homogenates, Western blot analysis, autoradiography, and densitometry were performed as described previously [1, 3–7]. Briefly, myocardial tissue was homogenized in a hypotonic lysis buffer containing 10 mM Tris (pH 7.5), 1 mM EDTA, 1 mM Na3VO4, 10 mM sodium pyrophosphate, 10 mM β-glycerophosphate, 10 mM NaF, and 1% protease inhibitor cocktail (Sigma, St. Louis, MO). Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL) and extracts boiled in SDS sample buffer. Electrophoresis was carried out 10% polyacrylamide gels, proteins transferred to PVDF membrane, and probed with primary antibodies diluted in 2% nonfat dry milk in Tris-buffered saline containing 0.05% Tween 20 (TTBS). After incubation overnight at 4°C, blots were incubated in horseradish peroxidase-conjugated secondary antibodies in 5% milk in TTBS for 1 h, and the signal detected using a chemiluminescent substrate (SuperSignal Pico West; Pierce) supplemented with 5% SuperSignal Femto (Pierce) and exposure to film. The antibodies used in Western blotting are detailed in Supplementary Table S1. To determine the signal transduction pathways involved in IL-18-mediated Akt activation in vitro, cardiomyocytes were incubated with the PI3K inhibitor wortmannin (50 nM in DMSO for 1 h) or LY294002 (20 µM in DMSO for 1 h), or the Akt inhibitor SH-5 (1 µM in water for 1 h) prior to IL-18 addition. To determine the role of JNK in IL-18-mediated c-Jun phosphorylation, cardiomyocytes were incubated with the JNK inhibitor SP600125 (20 µM for 30 min) or transduced with lentiviral GATA4 shRNA (moi 0.5 for 24 h; Sigma-Aldrich) prior to IL-18 addition.
GATA DNA binding activity by EMSA and ELISA
Nuclear extracts were prepared as previously described [3–7] using the Panomics Nuclear Extraction kit (no. AY2002, Panomics/Affymetrix, Freemont, CA). GATA DNA binding activity in nuclear protein extracts was analyzed by electrophoretic mobility shift assay (EMSA) as previously described [1] using double-stranded consensus GATA-specific oligonucleotides (sense, 5′-TCGCTGGACTGATAACTTTAAAAG-3′) from the ANP promoter. Double-stranded mutant oligonucleotides (sense, 5′-TCGCTGGACTGGTAACTTTAAAAG-3′) served as controls. Lamin A/C (nuclear) served as a loading control. The formation of GATA4 protein-DNA complexes was also analyzed by ELISA (TransAM® TF ELISA kits, #46966; Active Motif, Carlsbad, CA) using equal amounts of nuclear protein extracts.
Isolation of adult rabbit cardiomyocytes
Cardiomyocytes were isolated using collagenase as detailed previously by Blaauw et al [19]. In brief, hearts excised from naïve rabbits were rinsed in ice-cold modified Krebs-Henseleit (KH) buffer A (pH 7.4) containing (in mM) 133 NaCl, 5 KCl, 2 MgCl2, 1.2 NaH2PO4, 10 glucose, 10 HEPES, 6 taurine, and a low Ca2+ concentration (10 µM CaCl2). After the removal of non-cardiac tissue, the aorta was cannulated, and the heart perfused in retrograde flow with oxygenated KH buffer A (25 ml/min) at 37°C for 10 min. The perfusion buffer was then changed to a collagenase-containing digestion buffer consisting of 150 ml fresh KH buffer A supplemented with BSA [1% (wt/vol), fraction V, and 290 U/ml collagenase type II (Worthington). The digestion buffer (pH 7.4, 37°C) was oxygenated and recirculated for 8–10 min. Atrial and right ventricular tissue were then removed, and the left ventricle (LV) tissue was incubated in KH buffer B (pH 7.4), containing (in mM) 120 NaCl, 5 KCl, 2 MgCl2, 1.2 NaH2PO4, 10 glucose, 10 HEPES, and 6 taurine supplemented with 10 µM CaCl2 and 1% (wt/vol) BSA (fraction V) at 22°C for 10 min. The tissue was then diced, and completely dissociated with gentle shaking. Dissociated cells were filtered through nylon gauze and allowed to recover at room temperature for 20 min. Thereafter, Ca2+ concentration was increased stepwise from 10 to 50, 75, 100, 150, 200, 400, 800, and 1,200 µM, with each step separated by 3 min. After mild centrifugation at 50 g for 3 min at 22°C, the supernatant was discarded, and the cardiomyocytes resuspended in 25 ml of medium 199 supplemented with 5% FBS, and L-carnitine (final concentration: 2 mM), taurine (5 mM), penicillin (100 U/ml), streptomycin (100 mg/ml), and gentamicin (0.05 mg/ml). Cell viability was assessed by trypan blue exclusion.
Stretch-induced cardiomyocyte hypertrophy
Cyclical mechanical serves an in vitro model for load, and exerts hypertrophic response [20]. Cardiomyocytes on a UniFlex®-untreated flexible-bottomed tissue culture plates (UF-4001U; Flexcell International Corporation, Hillsborough, NC) underwent pulsatile stretch (10% extension, 30 cycles/min) as previously described [20, 21] in a Flexcell FX-4000 (V4.0) strain unit in an incubator at 37°C and 5% CO2 atmosphere. Non-stretched cells under identical conditions served as controls. After 24 h, 100 cells from each experiment were randomly selected and digitally photographed using an Olympus CKX41 inverted microscope equipped with Olympus digital camera (C5050 Zoom) at ×20 magnification. Cell surface area was quantified using the computerized digital microscopic software Image-Pro® Plus 4. Results are expressed as fold increase from control cells. IL-18 mRNA expression was analyzed by RT-qPCR and protein level by immunoblotting. To determine if IL-18 mediates stretch-induced hypertrophy, cells were incubated with IL-18 neutralizing antibodies (10 µg/ml) or recombinant human IL-18 BPa-Fc chimera (Cat. #119-BP, R & D Systems; 10 µg/ml) one hour prior to initiation of stretch. Normal IgG or Fc served as controls. Rabbit and human IL-18BP show 62.3% homology (Table 1).
IL-18-induced cardiomyocyte hypertrophy
To investigate the direct effects of IL-18 on cardiomyocyte hypertrophy, cells were incubated with recombinant human (rh) IL-18 (10 ng/ml) for 24 h. Human IL-18 is 73% identical to rabbit IL-18 (Table 3), and has been previously used to induce sleep in a rabbit model [22]. Cell growth was analyzed as previously described [23] using two independent methods, increased cell surface area and enhanced protein synthesis. Surface area was analyzed as described above using the Image-Pro® Plus 4 software, and the results expressed as fold increase from PBS-treated cells. The rate of protein synthesis was determined by the incorporation of [3H]leucine. Briefly, after incubation for 18 h with rhIL-18, 0.5 µCi of [3H]leucine was added to the culture medium, and the incubation continued for an additional 6 h. The radioactivity incorporated into the trichloroacetic acid-precipitable material was determined in a liquid scintillation counter. Total DNA levels were analyzed in duplicate samples using the DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA). The [3H]leucine incorporation was normalized to DNA, and the ratio of [3H]leucine incorporation/DNA from untreated cells was considered as 1, and the results are expressed as fold increase from untreated controls. Further, to investigate the signal transduction pathway involved in IL-18-mediated cardiomyocyte hypertrophy, cells were treated with either the PI3K or Akt inhibitors or traduced with lentiviral GATA4 shRNA prior to IL-18 addition.
Table 3.
Comparison of amino acid identity between rabbit vs. human, mouse and rat coding sequences
| Amino acid identity (%) | ||||
|---|---|---|---|---|
| Rabbit vs. | Human | Mouse | Rat | |
| IL-18 | 73.1 | 61.6 | 61.3 | |
| IL-18Rα | 74 | 61.6 | 62.1 | |
| IL-18Rβ | 71.4 | 65.3 | 57.7 | |
| IL-18BP | 62.3 | 59 | 52.5 | |
IL-18 + TNF-α induces cardiomyocyte death
To determine whether co-treatment IL-18 potentiates TNF-α-induced cardiomyocyte death, cardiomyocytes were incubated with IL-18 (10 ng/ml), rhTNF-α (10 ng/ml) or IL-18 + TNF-α added simultaneously (10 ng each/ml). Cell death was analyzed after 12 h using the Cell Death Detection ELISAPLUS kit (Roche Applied Science). The assay quantifies mono- and oligonucleosomes released into the cytoplasm of cells undergoing apoptosis. In addition, cell death was analyzed by DNA laddering using the Apoptotic DNA Ladder Kit (11 835 246 001; Roche Applied Science) and by the release of cytochrome c from mitochondria into the cytoplasm. Cleaved caspase-3 levels were analyzed by immunoblotting using cleared whole cell lysates. Mitochondrial and cytoplasmic fractions were prepared using the Mitochondrial Fractionation Kit (Active Motif, Carlsbad, CA, USA). Purity of extracts was analyzed by immunoblotting using cytoplasmic (GAPDH) and mitochondrial (V-β) markers.
Isolation of adult rabbit cardiac endothelial cells
Endothelial cells were isolated from rabbit heart using a collagenase digestion method described by Widmann et al [24]. In brief, following removal of coronary arteries, the left ventricular tissue was minced into 1–2 mm3 pieces in sterile DMEM at room temperature. After centrifugation at 1000 ×g for 5 min, the pellet was incubated for 1 h at 37°C in 0.2% Type II collagenase (Worthington Biochemical, Freehold, NJ) in 0.1% bovine serum albumin. The preparation was passed through a sterile 110 µm-nylon mesh and plated onto a T-75 culture flask containing DMEM supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml streptomycin, 0.05% porcine heparin, 10 mM glutamine, and human acidic fibroblast growth factor. The media was replaced with fresh medium every other day until confluent. The cultures were passaged with 0.25% trypsin-EDTA, and used between passages 3–6. Twelve hours prior to IL-18 treatment, the media with 10% serum was replaced with media containing 0.5% BSA.
Statistical analysis
Comparisons between controls and various treatments were performed by analysis of variance with post hoc Dunnett's t test. All assays were performed at least three times, and the error bars in the figures indicate the S.E. Densitometric results were shown as ratios at the bottom of the panels whenever the results are less clear.
Results and Discussion
We and others have previously reported that IL-18 is a potent pro-growth factor inducing cardiomyocyte hypertrophy in vitro [1] and myocardial hypertrophy in vivo [8, 9]. We have also demonstrated that pressure overload-induced hypertrophy is markedly attenuated in IL-18-null mice [10]. However, it is also possible that expression levels of IL-18 receptors and IL-18BP that modulate IL-18 binding and signaling may also influence cardiac hypertrophy. In addition, their expression levels in pressure overload-induced cardiac failure are not known. Therefore, we investigated IL-18, IL-18R and IL-18BP expression in a translationally important rabbit model of pressure overload-induced myocardial hypertrophy and failure.
Pressure-overload in the rabbit model results in myocardial hypertrophy by 4 weeks and cardiac failure by 6 weeks [16–18]. In fact, transthoracic echocardiography (Fig. 1A and 1B) revealed compensated hypertrophy at 4 weeks (mass/volume ratio: 1.7±0.11) and ventricular dilatation indicative of failure at 6 weeks (mass/volume ratio: 0.7±0.04). Further, while contractile function (fractional shortening) was preserved during the hypertrophic phase (4 weeks), it was significantly reduced as the LV started to dilate at 6 weeks (Fig. 1B). In addition, the heart to body weight ratios showed a significant increase at 4 weeks and remained at this level after 6 weeks (Fig. 1C). The levels of expression of ANP (Fig. 1D) and MMP9 (Fig. 1E) were elevated in both hypertrophied and failing hearts.
Since transcripts encoding the rabbit IL-18, IL-18Rα, IL-18Rβ and IL-18BP have not been characterized, we amplified, cloned, and sequenced the corresponding cDNAs. The nucleotide sequence data are deposited with GenBank under accession numbers EU419127.1 (IL-18), EU731009.1 (IL-18Rα), JF271681, JF271682 and JF271683 (IL-18Rβ), and JQ657816 and JQ657817 (IL-18BP). We also compared their homology to human, mouse and rat. The ORF (open reading frame) of rabbit IL-18 cDNA contains 579 nucleotides and 192 amino acids (Table 3). Mutational studies have shown that Asp17, Asp35, and Asp132 of human IL-18 play a role in IL-18 binding to IL-18Rα [25], and Lys79, Lys84 and Asp98 are important in forming the ternary complex of IL-18, IL-18Rα, and IL-18Rβ. The corresponding residues in rabbit IL-18 are all conserved (Supplementary Fig. S1). The alignment of rabbit, human, mouse and rat IL-18Rα is shown in the Supplementary Fig. S2. The amino acid sequence of rabbit IL-18Rα also showed higher homology with human (74.0%), compared to mouse (61.6%) or rat (62.1%) (Table 3). We identified three different isoforms of IL-18Rβ (JF271681, JF271682 and JF271683). JF271681 is comprised of 10 exons and encodes a predicted protein with 575 amino acids. In JF271682, part of the exon 2 is skipped and the length of translated protein is 549 amino acids. In JF271683, the entire exon 2 is skipped, which results in a premature stop codon and is unlikely to encode a functional IL-18Rβ protein. The amino acid sequence alignment of rabbit, human, rat and mouse IL-18Rβ is summarized in Supplementary Fig. S3. We identified two different isoforms of rabbit IL-18BP (JQ657816 and JQ657817), which showed the highest amino acid identity to human isoform a (NM_173042) and isoform c (NM_005699), respectively (Supplementary Fig. S4 and Table 3). However, rabbit IL-18BP showed relatively lower homology to rat and mouse IL-18BP (Supplementary Fig. S4).
IL-18 is a pro-hypertrophic cytokine [1] and is elevated in explanted failed human hearts [11]. Therefore we next analyzed its expression levels in both hypertrophied and failing rabbit hearts. Since rabbit IL-18, IL-18 receptors and IL-18BP showed higher homology with human than other species (Table 3), we tested anti-human polyclonal antibodies against each target protein from three or more vendors. After confirming specificity and reproducibility (data not shown), we used the antibodies indicated in Supplementary Table S1. Our results showed low levels of IL-18 mRNA and mature IL-18 (18 kDa) protein levels in both naïve and sham-operated rabbit LV protein homogenates, but markedly elevated levels in both hypertrophied and failing hearts, indicating sustained upregulation of IL-18 in response to transaortic constriction (Fig. 2A, 2B, 3A and 3B). Similarly, IL-18Rα levels were low in naïve hearts, but were increased by at least 3-fold in both hypertrophied and failing hearts (Fig. 2A, 2C, 3A and 3B), indicating amplification in IL-18 pro-growth and proinflammatory signaling. Since we identified three isoforms of IL-18Rβ cDNA, a probe that recognizes all three isoforms was designed (Table 2). Our results show that while IL-18Rβ levels were low and similar in both naïve and hypertrophied hearts, its expression levels were significantly increased in the failing heart (Fig. 2A, 2C, 3A and 3B), again indicating the potential amplification of IL-18 signaling.
Fig. 2. Differential regulation of IL-18, IL-18R and IL-18BP mRNA expression following pressure overload.

A, Sham surgery fails to modulate basal IL-18, IL-18R and IL-18BP expression. Left ventricular tissue from rabbits that underwent sham surgery was analyzed for IL-18 (left hand panel), IL-18R subunits (IL-18Rα, IL-18Rβ; middle panel), and IL-18BP (right hand panel) expression by Northern blotting. LV tissue from naïve rabbits served as controls (n=5/group). A representative blot containing n=2/group is shown. Densitometric analysis of the autoradiographic signals for all samples is summarized in the bottom panels. N, naïve; S, Sham-operated. B, C, D, Pressure overload enhances IL-18 and its receptor expression, but markedly suppresses IL-18BP expression. Rabbits that underwent pressure overload as in Fig. 1A were analyzed for IL-18 (B), IL-18Rα and IL-18Rβ (C) and IL-18BP (D) mRNA expression by Northern blotting. A representative blot containing n=2/group is shown. Densitometric analysis of the autoradiographic signals is summarized in the bottom or on the right side of respective panels. B–D, *P < at least 0.05 vs. sham, † P < 0.05 vs. sham (n=5–6 animals/group). S, sham; H, hypertrophy; HF: heart failure.
Fig. 3. Pressure differentially regulates IL-18, IL-18R and IL-18BP protein expression in vivo.

A, Sham surgery fails to modulate basal IL-18, IL-18R and IL-18BP protein expression. Left ventricular tissue from 10 day-old male NZW rabbits that underwent sham surgery was analyzed for IL-18, IL-18R heterodimer, and IL-18BP expression by immunoblotting using cleared whole tissue homogenates. LV tissue from naïve rabbits served as controls (n=3/group). B, Pressure overload enhances IL-18 and its receptor expression, but markedly suppresses IL-18BP protein expression. Rabbits that underwent pressure overload as in Fig. 1A were analyzed for IL-18, IL-18Rα, IL-18Rβ, and IL-18BP protein expression by immunoblotting. A representative blot containing n=3/group is shown. Densitometric analysis of immunoreactive bands is summarized on the right. *P < at least 0.05 vs. sham, † P < 0.05 vs. sham (n=5–6 animals/group). C, Sham surgery fails to modulate basal active caspase-3 levels. LV tissue from rabbits that underwent sham surgery was analyzed for the activation of caspase-3 by immunoblotting using cleared whole tissue homogenates and antibodies that detect the 17 kDa active form of caspase-3 (cleaved caspase-3, n=3/group). D, Pressure overload markedly upregulates active caspase-3 levels in failing rabbit hearts. LV tissue from rabbits that underwent pressure overload as in Fig. 1A were analyzed for active form of caspase-3 by immunoblotting. A representative blot containing n=3/group is shown. Densitometric analysis of the immunoreactive band from all samples is summarized on the right. *P < at least 0.05 vs. sham, † P < 0.05 vs. sham (n=5–6 animals/group). S, sham; H, hypertrophy; HF: heart failure.
Importantly, expression levels of IL-18BP were high in naïve and sham-operated hearts, and in hypertrophied hearts, but its levels were markedly downregulated in the failing hearts (Fig. 2A, 2D, 3A and 3B), suggesting that the initial increase in hypertrophied hearts may be a compensatory mechanism to blunt IL-18 signaling. However, its decreased levels in failing hearts suggest uncontrolled IL-18 proinflammatory signaling. In fact, we previously reported a rapid and robust increase in IL-18 and IL-18Rα expression by TNF-α and hydrogen peroxide in adult rat cardiomyocytes, but a delayed and marked attenuation in IL-18BP expression [26]. TNF-α upregulation and enhanced ROS generation were reported in both hypertrophied and failing hearts [27–29]. The mechanisms involved in IL-18BP downregulation during heart failure are not known.
Both hypertrophy and failure are characterized by enhanced MMP expression [30, 31]. MMP9 was increased in the rabbit hypertrophied hearts, and remained elevated in the failing hearts (Fig. 1F). MMP9 is a NF-κB and AP-1 responsive gene, and IL-18 a potent inducer of MMP9 transcription in cardiomyocytes [32]. Further, the active form of caspase-3 (cleaved caspase-3, p17) was similar and low in both naïve and sham-operated hearts (Fig. 3C), and its expression did not change significantly in the hypertrophied hearts (Fig. 3D). However, a marked increase in active caspase-3 expression was detected in the failing hearts (Fig. 3D), suggestive of cell death. Cell death contributes to progression of heart failure, and in fact has been reported in failing human hearts [33, 34].
Cyclical mechanical stretch, an in vitro model of load, has been shown to exert hypertrophic response in cardiomyocytes [20]. Therefore, we next investigated whether stretch-induced rabbit cardiomyocyte growth is IL-18 dependent. Indeed, stretch induced both cardiomyocyte hypertrophy and the robust expression of IL-18 and IL-18Rα (Fig. 4A, B). Stretch-induced cardiomyocyte hypertrophy was markedly attenuated by pre-incubation with IL-18 neutralizing antibodies or its endogenous antagonist IL-18BP (Fig. 4C). Consistent with this, IL-18 neutralization or pre-incubation with IL-18BP blunted stretch-induced expression of the prohypertrophic marker ANP and the pro-survival factor Akt (Fig. 4D, E). Together, these results indicate that load modulates the expression of IL-18 and its receptor both in vivo and in vitro, and suggests that IL-18 signaling is an essential mediator of load-induced hypertrophic response.
Fig. 4. Cyclical mechanical stretch induces cardiomyocyte hypertrophy in part via IL-18.

A, Stretch induces cardiomyocyte hypertrophy. Isolated rabbit cardiomyocytes underwent pulsatile stretch (10% extension, 30 cycles/min) in a Flexcell FX-4000 (V4.0) strain unit at 37°C and 5% CO2 atmosphere. Non-stretched cells under identical conditions served as controls. After 24 h, 100 cells from each experiment were randomly selected and digitally photographed using an Olympus CKX41 inverted microscope equipped with Olympus digital camera (C5050 Zoom) at ×20 magnification. Cell surface area was quantified using the computerized digital microscopic software Image-Pro® Plus 4. The surface area of non-stretched control cells was considered one, and the results are expressed as fold increase from control cells. *P < 0.001 vs. unstretched controls. B, Stretch upregulates IL-18 and IL-18Rα expression. Cardiomyocytes that underwent stretch as in A were analyzed for IL-18 and IL-18Rα expression by immunoblotting. Densitometric analysis of the immunoreactive band is summarized on the right. *P < 0.05 vs. unstretched control. C, Neutralization or blocking IL-18 effects blunts stretch-induced cardiomyocyte size. Cardiomyocytes were incubated with IL-18 neutralizing antibodies or IL-18BP (10 µg/ml for 1 h) prior to stretch. Cell size was quantified as in A. *P < 0.001 vs. unstretched controls, †P < 0.01 vs. control IgG or Fc. D, E, Blocking or neutralization of IL-18 blunts stretch-induced hypertrophic marker ANP and the pro-survival factor Akt. Cardiomyocytes incubated as in C were analyzed for ANP mRNA expression by Northern blotting and phospho-Akt levels by immunoblotting. A representative of three independent experiments is shown.
We next investigated the direct effects of IL-18 on cardiomyocyte hypertrophy. Confirming our earlier report in rat cardiomyocytes [1], IL-18 induced a marked increase in cardiomyocyte size, as evidenced by an increase in cell surface area, an effect markedly attenuated by pre-treatment with IL-18 neutralizing antibodies or IL-18BP (Fig. 5A). ([3H]leucine incorporation confirmed these results (data not shown). Further, IL-18, but not neutralized IL-18, induced the hypertrophic markers ANP and BNP (Fig. 5B). Notably, IL-18 induced its own expression and that of IL-18Rα in an AP-1-dependent manner (Fig. 5C), indicating that IL-18 autoregulation is a contributing factor in myocardial hypertrophy and possibly to failure. IL-18 also induced a time-dependent increase in the activation of Akt (Fig. 6A), a pro-survival and pro-hypertrophic factor, in a PI3K-dependent manner (Fig. 6B). Further, pharmacological inhibition of PI3K and Akt attenuated IL-18 induced c-Jun phosphorylation (Fig. 6C, D) and GATA4 DNA binding activity (Fig. 6E). Notably, inhibition of PI3K and Akt, and knockdown of GATA4 each attenuated IL-18 induced cardiomyocyte hypertrophy (Fig. 6F). Similar results were obtained when IL-18-treated cardiomyocytes were analyzed for cell growth using [3H]leucine incorporation (Fig. 6G). Together, these results indicate that IL-18 is a potent pro-growth factor for rabbit cardiomyocytes, and induces hypertrophy in part via a PI3K/Akt/GATA4-dependent pathway.
Fig. 5. IL-18 is a potent inducer of cardiomyocyte hypertrophy in vitro.
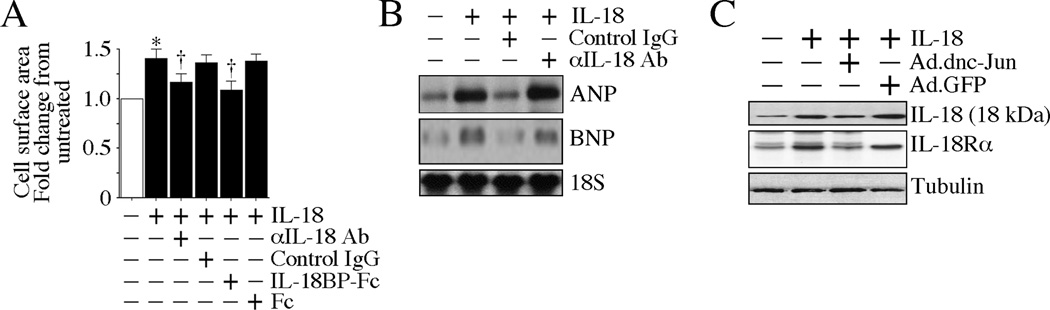
A, IL-18 induces cardiomyocyte growth. Rabbit cardiomyocytes were incubated with IL-18 neutralizing antibodies or IL-18BP-Fc (10 µg/ml) for 1 h prior to IL-18 addition (10 ng/ml). Cell size was analyzed after 24 h as in Fig. 4A. *P < 0.001 vs. untreated controls, †P < 0.01 vs. control IgG or Fc. B, IL-18 enhances pro-hypertrophic markers. Rabbit cardiomyocytes incubated as A were analyzed for ANP and BNP mRNA expression by Northern blotting. A representative of three independent experiments is shown. C, Forced expression of a dominant negative mutant of c-Jun blocks IL-18-induced IL-18 and IL-18Rα expression. Rabbit cardiomyocytes transduced with Ad.dnc-Jun (100 moi for 24 h) were incubated with IL-18 (10 ng/ml for 2 h) were analyzed for mature IL-18 and IL-18Rα expression by immunoblotting (n=3). Ad.GFP served as a control.
Fig. 6. IL-18 induces cardiomyocyte hypertrophy via PI3K, Akt and GATA4.

A, IL-18 induces Akt activation. Rabbit cardiomyocytes were incubated with rhIL-18 (10 ng/ml). Phospho-Akt levels were analyzed by immunoblotting using cleared whole cell lysates and activation-specific antibodies (n=3). B, IL-18 induces Akt activation via PI3K. Rabbit cardiomyocytes were incubated with the PI3K inhibitors wortmannin (50 nM in DMSO) or LY294002 (20 µM in DMSO) for 1 h prior to IL-18 addition (10 ng/ml for 1 h). Total and phospho-Akt levels were analyzed as in A (n=3). C, SH-5 inhibits IL-18-induced Akt activation. Rabbit cardiomyocytes were incubated with the Akt inhibitor SH-5 (1 µM in DMSO for 1 h) prior to IL-18 addition (10 ng/ml for 1 h). Total and phospho-Akt levels were analyzed as in A (n=3). D, IL-18 induces c-Jun activation via Akt and JNK. Rabbit cardiomyocytes incubated with SH-5 (1 µM in DMSO for 1 h) or the SP600125 (20 µM for 30 min) prior to IL-18 addition. Phospho-c-Jun levels were analyzed by immunoblotting using nuclear protein extracts and activation-specific (Ser73) antibodies (n=3). Lamin A/C served as a loading control, and is shown in the bottom panel. E, IL-18 induces GATA4 DNA binding activity via PI3K and Akt. Rabbit cardiomyocytes treated with the PI3K inhibitor wortmannin (50 nM in DMSO) or the Akt inhibitor SH-5 (1 µM in DMSO for 1 h) prior to IL-18 addition (10 ng/ml for 1 h). GATA4 DNA binding activity was analyzed by EMSA (left hand panel) or ELISA (right hand panel) using the nuclear protein extracts and a labeled consensus GATA probe (n=3). Lamin A/C served as a loading control, and is shown in the bottom panel. For competition, 50-fold molar excess cold consensus (Wild type or WT) or mutant oligonucleotides were used with the nuclear extracts before the addition of labeled consensus GATA4 probe. The arrow in the left panel indicates specific DNA-protein complexes. F, IL-18 induces cardiomyocyte hypertrophy via PI3K, Akt and GATA4. Rabbit cardiomyocytes incubated with wortmannin (50 nM in DMSO) or SH-5 (1 µM in DMSO for 1 h), or infected with lentiviral GATA4 shRNA (moi 0.5 for 24 h) were treated with IL-18 (10 ng/ml for 24 h). Cardiomyocyte size was analyzed as in Fig. 4A. Knockdown of GATA4 was confirmed by immunoblotting and is shown on the right (n=3). *P < 0.001 vs. untreated controls, †P < 0.01 vs. IL-18 ± DMSO or GFP shRNA.
In addition to IL-18, several other proinflammatory cytokines with negative inotropic effects are expressed during heart failure. For example, TNF-α is expressed, and is known to depress myocardial function in vivo and cardiomyocyte contractility in vitro, and to induce cardiomyocyte death. Since cytokines act in concert, we next examined the effect of IL-18 on TNF-α-induced cardiomyocyte death. While TNF-α alone stimulated translocation of the proapoptotic bax from cytoplasm to mitochondria, co-treatment with IL-18 further increased its levels in the mitochondrial compartment (Fig. 7A, B). Co-treatment with TNF-α + IL-18 also increased mitochondrial release of cytochrome c into cytoplasm (Fig. 7A, B). Moreover, while IL-18 failed to affect basal levels of cleaved caspase-3 (Fig. 7C), it potentiated TNF-α-induced caspase-3 activation (Fig. 7D) and cell death (Fig. 7E). Cell death was analyzed by quantifying mono- and oligonucleosomal fragmented DNA in cytoplasm (Fig. 7E) and fragmented nucleosomal DNA (DNA laddering; Fig. 7E, inset). Thus, while treatment with IL-18 alone elicits a pro-hypertrophic response, in concert with proinflammatory cytokines that are known to induce cardiomyocyte death, it may enhance the progression to heart failure by potentiating cell death.
Fig. 7. IL-18 potentiates and TNF-a-induced cardiomyocyte death in vitro.

A, B, Co-treatment with IL-18 and TNF-α induces bax translocation from cytoplasm to mitochondria. Rabbit cardiomyocytes were treated with IL-18 and TNF-α simultaneously (10 ng each/ml) for 12 h. Bax and cytochrome c levels in the cytoplasmic and mitochondrial fractions were analyzed by immunoblotting (n=3). GAPDH (cytoplasmic) and V-β (mitochondria) served as loading controls. C, IL-18 alone fails to induce caspase-3 activation. Cardiomyocytes were incubated with IL-18 (1 or 10 ng/ml) for 12 h. Cleaved caspase-3 (17 kDa) levels were analyzed by immunoblotting using cleared whole cell homogenates (n=3). D, IL-18 potentiates TNF-a-induced caspase-3 activation. Cardiomyocytes treated as in A were analyzed for cleaved caspase-3 levels (17 kDa) by immunoblotting using cleared whole cell homogenates (n=3). The intensity of immunoreactive bands was analyzed by densitometry, and results from three independent experiments are summarized on the right. *P < 0.05 vs. untreated controls, †P < 0.01 vs. untreated or IL-18 alone. E, IL-18 potentiates TNF-α-induced cardiomyocyte death. Cardiomyocytes treated as in A were analyzed by ELISA for mono- and oligonucleosomal fragmented DNA in cytoplasmatic extracts. Cell death was also analyzed by nucleosomal DNA fragmentation (DNA laddering). Genomic DNA was isolated and equal amounts were electrophoresed in a 1% agarose gel, visualized by ethidium bromide staining, and photographed. A representative of three independent experiments is shown (inset). Ladder, 100 bp.
Heart failure is characterized by endothelial dysfunction. Serum from heart failure patients has been shown to induce endothelial cell death [35]. Since IL-18 levels are increased in heart failure patients [11], we investigated whether IL-18 induces cardiac endothelial cell death. Confirming our earlier results in rat cardiac endothelial cells [5], treatment with IL-18 alone induced rabbit cardiac endothelial cell death (Fig. 8A). IL-18 enhanced bax levels in mitochondria and cytochrome c levels in cytoplasm (Fig. 8B). Further, IL-18 induced caspase-3 activation (Fig. 8C) and accumulation of nucleosomal fragmented DNA in cytoplasm (Fig. 8D). These results demonstrate that IL-18 is a pleiotropic proinflammatory cytokine, and exerts pro-hypertrophic and pro-apoptotic effects in a cell type-specific manner.
Fig. 8. IL-18 induces cardiac endothelial cell death.

A, B, IL-18 stimulates bax translocation from cytoplasm to mitochondria. Rabbit cardiac endothelial cells (A) were serum starved for 12 h, and incubated with IL-18 (10 ng/ml) for 12 h. Bax and cytochrome c levels in the cytoplasmic and mitochondrial fractions were analyzed by immunoblotting (n=3). GAPDH (cytoplasmic) and V-β (mitochondria) served as loading controls. C, IL-18 stimulates caspase-3 activation. Cardiomyocytes treated as in B were analyzed for cleaved caspase-3 (17 kDa) levels by immunoblotting using cleared whole cell homogenates (n=3). D, IL-18 stimulates cardiomyocyte death. Cardiomyocytes treated as in B were analyzed by ELISA for mono- and oligonucleosomal fragmented DNA in cytoplasmic extracts. Cell death was also analyzed by nucleosomal DNA fragmentation (DNA laddering) as described in Fig. 7E. A representative of three independent experiments is shown (inset). *P < 0.05 vs. untreated controls.
The acknowledged role that inflammatory cytokines play in the pathogenesis of myocardial hypertrophy and heart failure has led to the development of various anti-cytokine therapies. However, while showing early promise, some studies have had to be stopped due to adverse effects. Thus while blocking TNF-α showed promising results initially, in a large-scale clinical trial, blocking TNF-α by infliximab was unsuccessful due to increased mortality rate in the heart failure subjects [36]. Nevertheless, the strategy as a whole may still be effective and depend on the specific cytokine blocked. Thus targeting IL-1β, whose expression is increased during heart failure, has shown some promising results. In two small clinical trials, blockade of IL-1 with Anakinra, a recombinant, nonglycosylated form of the human interleukin-1 receptor antagonist, markedly attenuated LV remodeling in patients with ST-segment elevation AMI [37, 38]. Of note, AMI is the leading cause of heart failure. In another large-scale study, neutralization of IL-1β is being investigated to reduce cardiovascular events in high-risk patients [39].
IL-18 belongs to the IL-1 family, and has significant structural similarities to IL-1β [40]. Interestingly, in a recent report, plasma from human heart failure patients or infusion of IL-1β was shown to induce LV systolic dysfunction in mice, and this effect was abrogated by Il18 gene deletion or neutralization [41]. These results suggest that IL-18 is downstream of IL-1b, and targeting IL-18 not only blocks IL-18’s deleterious signaling, but may also blunt IL-1β proinflammatory signaling and overall inflammation. Consistent with our studies, these authors concluded that IL-18 blockade could represent a novel and more targeted therapeutic approach to the treatment of HF [41].
Supplementary Material
Highlights.
-
►
Expression of IL-18 and its receptor are increased during cardiac hypertrophy and failure
-
►
IL-18BP expression is enhanced during hypertrophy, but suppressed during failure
-
►
Cyclical mechanical stretch induces IL-18 and its receptor expression in cardiomyocytes
-
►
Co-treatment with IL-18 and TNF-α induces cardiomyocyte death
-
►
IL-18 may be a potential therapeutic target in hypertrophy and failure
Acknowledgements
This work was supported by Veterans Affairs Office of Research and Development Biomedical Laboratory Research and Development (ORD-BLRD) Service Award 1IO1BX000246 and the NIH/NHLBI Grant HL-86787 (to BC), HL-075430 (IF), and HL-70241 and HL-80682 (PD). SM is supported by Veterans Affairs ORD-BLRD Service Award I01BX000975. TY is supported by a NIH/NIGMS Tulane COBRE Pilot Project (P20GM103629). The contents of this report do not represent the views of the Department of Veterans Affairs or the United States Government.
Abbreviations
- AMI
acute myocardial infarction
- ANP
atrial natriuretic protein
- AP-1
activator protein 1
- BNP
Brain natriuretic peptide
- ECG
echocardiography
- EDD
end-diastolic diameter
- ESD
end-systolic diameter
- EMSA
electrophoretic gel mobility shift assay
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- H
hypertrophy
- HF
heart failure
- IL
Interleukin
- IL-18BP
IL-18 binding protein
- LVH
left ventricular hypertrophy
- MMP
matrix metalloproteinase
- N
naïve
- NF-κB
nuclear factor kappa B
- ORF
open reading frame
- PI3K
Phosphatidylinositol-3-kinase
- PWD
posterior wall thickness in diastole
- rh
recombinant human
- ROS
reactive oxygen species
- S
sham
- SMC
smooth muscle cells
- TRAF
TNF receptor-associated factor
- TNF
Tumor necrosis factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures: None
References
- 1.Chandrasekar B, Mummidi S, Claycomb WC, Mestril R, Nemer M. Interleukin-18 is a pro-hypertrophic cytokine that acts through a phosphatidylinositol 3-kinase-phosphoinositide-dependent kinase-1-Akt-GATA4 signaling pathway in cardiomyocytes. J Biol Chem. 2005 Feb 11;280(6):4553–4567. doi: 10.1074/jbc.M411787200. [DOI] [PubMed] [Google Scholar]
- 2.Fix C, Bingham K, Carver W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine. 2011 Jan;53(1):19–28. doi: 10.1016/j.cyto.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siddesha JM, Valente AJ, Sakamuri SS, Gardner JD, Delafontaine P, Noda M, et al. Acetylsalicylic Acid Inhibits IL-18-Induced Cardiac Fibroblast Migration through the Induction of RECK. J Cell Physiol. 2013 Nov 22; doi: 10.1002/jcp.24511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valente AJ, Sakamuri SS, Siddesha JM, Yoshida T, Gardner JD, Prabhu R, et al. TRAF3IP2 mediates interleukin-18-induced cardiac fibroblast migration and differentiation. Cell Signal. 2013 Nov;25(11):2176–2184. doi: 10.1016/j.cellsig.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chandrasekar B, Vemula K, Surabhi RM, Li-Weber M, Owen-Schaub LB, Jensen LE, et al. Activation of intrinsic and extrinsic proapoptotic signaling pathways in interleukin-18-mediated human cardiac endothelial cell death. J Biol Chem. 2004 May 7;279(19):20221–20233. doi: 10.1074/jbc.M313980200. [DOI] [PubMed] [Google Scholar]
- 6.Chandrasekar B, Mummidi S, Mahimainathan L, Patel DN, Bailey SR, Imam SZ, et al. Interleukin-18-induced human coronary artery smooth muscle cell migration is dependent on NF-kappaB- and AP-1-mediated matrix metalloproteinase-9 expression and is inhibited by atorvastatin. J Biol Chem. 2006 Jun 2;281(22):15099–15109. doi: 10.1074/jbc.M600200200. [DOI] [PubMed] [Google Scholar]
- 7.Reddy VS, Valente AJ, Delafontaine P, Chandrasekar B. Interleukin-18/WNT1-inducible signaling pathway protein-1 signaling mediates human saphenous vein smooth muscle cell proliferation. J Cell Physiol. 2011 Dec;226(12):3303–3315. doi: 10.1002/jcp.22676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Woldbaek PR, Sande JB, Stromme TA, Lunde PK, Djurovic S, Lyberg T, et al. Daily administration of interleukin-18 causes myocardial dysfunction in healthy mice. Am J Physiol Heart Circ Physiol. 2005 Aug;289(2):H708–H714. doi: 10.1152/ajpheart.01179.2004. [DOI] [PubMed] [Google Scholar]
- 9.Platis A, Yu Q, Moore D, Khojeini E, Tsau P, Larson D. The effect of daily administration of IL-18 on cardiac structure and function. Perfusion. 2008 Jul;23(4):237–242. doi: 10.1177/0267659108101511. [DOI] [PubMed] [Google Scholar]
- 10.Colston JT, Boylston WH, Feldman MD, Jenkinson CP, de la Rosa SD, Barton A, et al. Interleukin-18 knockout mice display maladaptive cardiac hypertrophy in response to pressure overload. Biochem Biophys Res Commun. 2007 Mar 9;354(2):552–558. doi: 10.1016/j.bbrc.2007.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mallat Z, Heymes C, Corbaz A, Logeart D, Alouani S, Cohen-Solal A, et al. Evidence for altered interleukin 18 (IL)-18 pathway in human heart failure. FASEB J. 2004 Nov;18(14):1752–1754. doi: 10.1096/fj.04-2426fje. [DOI] [PubMed] [Google Scholar]
- 12.Yu Q, Vazquez R, Khojeini EV, Patel C, Venkataramani R, Larson DF. IL-18 induction of osteopontin mediates cardiac fibrosis and diastolic dysfunction in mice. Am J Physiol Heart Circ Physiol. 2009 Jul;297(1):H76–H85. doi: 10.1152/ajpheart.01285.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim SH, Eisenstein M, Reznikov L, Fantuzzi G, Novick D, Rubinstein M, et al. Structural requirements of six naturally occurring isoforms of the IL-18 binding protein to inhibit IL-18. Proc Natl Acad Sci U S A. 2000 Feb 1;97(3):1190–1195. doi: 10.1073/pnas.97.3.1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandrasekar B, Marelli-Berg FM, Tone M, Bysani S, Prabhu SD, Murray DR. Betaadrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3K, Akt, IKK, and NF-kappaB. Biochem Biophys Res Commun. 2004 Jun 25;319(2):304–311. doi: 10.1016/j.bbrc.2004.04.185. [DOI] [PubMed] [Google Scholar]
- 15.Murray DR, Mummidi S, Valente AJ, Yoshida T, Somanna NK, Delafontaine P, et al. beta2 adrenergic activation induces the expression of IL-18 binding protein, a potent inhibitor of isoproterenol induced cardiomyocyte hypertrophy in vitro and myocardial hypertrophy in vivo. J Mol Cell Cardiol. 2012 Jan;52(1):206–218. doi: 10.1016/j.yjmcc.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friehs I, Moran AM, Stamm C, Colan SD, Takeuchi K, Cao-Danh H, et al. Impaired glucose transporter activity in pressure-overload hypertrophy is an early indicator of progression to failure. Circulation. 1999 Nov 9;100(19 Suppl):II187–II193. doi: 10.1161/01.cir.100.suppl_2.ii-187. [DOI] [PubMed] [Google Scholar]
- 17.Moran AM, Friehs I, Takeuchi K, Stamm C, Hammer PE, McGowan FX, et al. Noninvasive serial evaluation of myocardial mechanics in pressure overload hypertrophy of rabbit myocardium. Herz. 2003 Feb;28(1):52–62. doi: 10.1007/s00059-003-2392-0. [DOI] [PubMed] [Google Scholar]
- 18.Friehs I, Margossian RE, Moran AM, Cao-Danh H, Moses MA, del Nido PJ. Vascular endothelial growth factor delays onset of failure in pressure-overload hypertrophy through matrix metalloproteinase activation and angiogenesis. Basic Res Cardiol. 2006 May;101(3):204–213. doi: 10.1007/s00395-005-0581-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blaauw E, van Nieuwenhoven FA, Willemsen P, Delhaas T, Prinzen FW, Snoeckx LH, et al. Stretch-induced hypertrophy of isolated adult rabbit cardiomyocytes. Am J Physiol. Heart Circ Physiol. 2010 Sep;299(3):H780–H787. doi: 10.1152/ajpheart.00822.2009. [DOI] [PubMed] [Google Scholar]
- 20.Sadoshima J, Izumo S. Mechanotransduction in stretch-induced hypertrophy of cardiac myocytes. J Recept Res. 1993;13(1–4):777–794. doi: 10.3109/10799899309073692. [DOI] [PubMed] [Google Scholar]
- 21.Leychenko A, Konorev E, Jijiwa M, Matter ML. Stretch-induced hypertrophy activates NFkB-mediated VEGF secretion in adult cardiomyocytes. PLoS One. 2011;6(12):e29055. doi: 10.1371/journal.pone.0029055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kubota T, Fang J, Brown RA, Krueger JM. Interleukin-18 promotes sleep in rabbits and rats. Am J Physiol Regul Integr Comp Physiol. 2001 Sep;281(3):R828–R838. doi: 10.1152/ajpregu.2001.281.3.R828. [DOI] [PubMed] [Google Scholar]
- 23.Valente AJ, Clark RA, Siddesha JM, Siebenlist U, Chandrasekar B. CIKS (Act1 or TRAF3IP2) mediates Angiotensin-II-induced Interleukin-18 expression, and Nox2-dependent cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2012 Jul;53(1):113–124. doi: 10.1016/j.yjmcc.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Widmann MD, Letsou GV, Phan S, Baldwin JC, Sumpio BE. Isolation and characterization of rabbit cardiac endothelial cells: response to cyclic strain and growth factors in vitro. J Surg Res. 1992 Oct;53(4):331–334. doi: 10.1016/0022-4804(92)90057-7. [DOI] [PubMed] [Google Scholar]
- 25.Kato Z, Jee J, Shikano H, Mishima M, Ohki I, Ohnishi H, et al. The structure and binding mode of interleukin-18. Nat Struct Biol. 2003 Nov;10(11):966–971. doi: 10.1038/nsb993. [DOI] [PubMed] [Google Scholar]
- 26.Chandrasekar B, Colston JT, de la Rosa SD, Rao PP, Freeman GL. TNF-alpha and H2O2 induce IL-18 and IL-18R beta expression in cardiomyocytes via NF-kappa B activation. Biochem Biophys Res Commun. 2003 Apr 18;303(4):1152–1158. doi: 10.1016/s0006-291x(03)00496-0. [DOI] [PubMed] [Google Scholar]
- 27.Diwan A, Tran T, Misra A, Mann DL. Inflammatory mediators and the failing heart: a translational approach. Curr Mol Med. 2003 Mar;3(2):161–182. doi: 10.2174/1566524033361537. [DOI] [PubMed] [Google Scholar]
- 28.Dhalla NS, Temsah RM, Netticadan T. Role of oxidative stress in cardiovascular diseases. J Hypertens. 2000 Jun;18(6):655–673. doi: 10.1097/00004872-200018060-00002. [DOI] [PubMed] [Google Scholar]
- 29.Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail. 2002 May-Jun;8(3):132–140. doi: 10.1111/j.1527-5299.2002.00717.x. [DOI] [PubMed] [Google Scholar]
- 30.Deschamps AM, Spinale FG. Pathways of matrix metalloproteinase induction in heart failure: bioactive molecules and transcriptional regulation. Cardiovasc Res. 2006 Feb 15;69(3):666–676. doi: 10.1016/j.cardiores.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 31.Mishra PK, Givvimani S, Chavali V, Tyagi SC. Cardiac matrix: a clue for future therapy. Biochim Biophys Acta. 2013 Dec;1832(12):2271–2276. doi: 10.1016/j.bbadis.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddy VS, Prabhu SD, Mummidi S, Valente AJ, Venkatesan B, Shanmugam P, et al. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-kappaB activation. Am J Physiol Heart Circ Physiol. 2010 Oct;299(4):H1242–H1254. doi: 10.1152/ajpheart.00451.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kang PM, Izumo S. Apoptosis and heart failure: A critical review of the literature. Circ Res. 2000 Jun 9;86(11):1107–1113. doi: 10.1161/01.res.86.11.1107. [DOI] [PubMed] [Google Scholar]
- 34.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, et al. Apoptosis in the failing human heart. N Engl J Med. 1997 Apr 17;336(16):1131–1141. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 35.Rossig L, Haendeler J, Mallat Z, Hugel B, Freyssinet JM, Tedgui A, et al. Congestive heart failure induces endothelial cell apoptosis: protective role of carvedilol. J Am Coll Cardiol. 2000 Dec;36(7):2081–2089. doi: 10.1016/s0735-1097(00)01002-0. [DOI] [PubMed] [Google Scholar]
- 36.Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL) Circulation. 2004 Apr 6;109(13):1594–1602. doi: 10.1161/01.CIR.0000124490.27666.B2. [DOI] [PubMed] [Google Scholar]
- 37.Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, et al. Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study) Am J Cardiol. 2010 May 15;105(10):1371–1377. doi: 10.1016/j.amjcard.2009.12.059. e1. [DOI] [PubMed] [Google Scholar]
- 38.Abbate A, Van Tassell BW, Biondi-Zoccai G, Kontos MC, Grizzard JD, Spillman DW, et al. Effects of interleukin-1 blockade with anakinra on adverse cardiac remodeling and heart failure after acute myocardial infarction [from the Virginia Commonwealth University-Anakinra Remodeling Trial (2) (VCU-ART2) pilot study] Am J Cardiol. 2013 May 15;111(10):1394–1400. doi: 10.1016/j.amjcard.2013.01.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1beta inhibition and the prevention of recurrent cardiovascular events: rationale and design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) Am Heart J. 2011 Oct;162(4):597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 40.Dinarello CA, Novick D, Kim S, Kaplanski G. Interleukin-18 and IL-18 Binding Protein. Front Immunol. 2013;4:289. doi: 10.3389/fimmu.2013.00289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toldo S, Mezzaroma E, O'Brien L, Marchetti C, Seropian IM, Voelkel NF, et al. Interleukin-18 mediates interleukin-1-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol. 2014 Apr 1;306(7):H1025–H1031. doi: 10.1152/ajpheart.00795.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.