

Abstract
Background
The suborder Anoplura contains 540 species of blood-sucking lice that parasitize over 840 species of eutherian mammals. Fragmented mitochondrial (mt) genomes have been found in the lice of humans, pigs, horses and rats from four families: Pediculidae, Pthiridae, Haematopinidae and Polyplacidae. These lice, eight species in total, are from the same major clade of the Anoplura. The mt genomes of these lice consist of 9–20 minichromosomes; each minichromosome is 1.5–4 kb in size and has 1–8 genes. To understand mt genome fragmentation in the other major clade of the Anoplura, we sequenced the mt genomes of two species of rodent lice in the genus Hoplopleura (family Hoplopleuridae).
Results
We identified 28 mt genes on 10 minichromosomes in the mouse louse, Ho. akanezumi; each minichromosome is 1.7–2.7 kb long and has 1–6 genes. We identified 34 mt genes on 11 minichromosomes in the rat louse, Ho. kitti; each minichromosome is 1.8–2.8 kb long and has 1–5 genes. Ho. akanezumi also has a chimeric minichromosome with parts of two rRNA genes and a full-length tRNA gene for tyrosine. These two rodent lice share the same pattern for the distribution of all of the protein-coding and rRNA genes but differ in tRNA gene content and gene arrangement in four minichromosomes. Like the four genera of blood-sucking lice that have been investigated in previous studies, the Hoplopleura species have four minichromosomes that are only found in this genus.
Conclusions
Our results indicate that fragmented mt genomes were present in the most recent common ancestor of the two major clades of the blood-sucking lice, which lived ~75 million years ago. Intra-genus variation in the pattern of mt genome fragmentation is common in the blood-sucking lice (suborder Anoplura) and genus-specific minichromosomes are potential synapomorphies. Future studies should expand into more species, genera and families of blood-sucking lice to explore further the phylogenetic utility of the novel features associated with fragmented mt genomes.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-751) contains supplementary material, which is available to authorized users.
Keywords: Mitochondrial genome, Genome fragmentation, Minichromosome, Chromosome evolution, Sucking lice
Background
The suborder Anoplura contains 540 species of blood-sucking lice that parasitize over 840 species of eutherian mammals [1–3]. Blood-sucking lice are of medical and veterinary significance as parasites and vectors of disease agents [4–7]. According to Light et al. [8] and Smith et al. [9], the blood-sucking lice evolved from chewing lice ~92 million years ago (Mya) and diversified into two major clades ~75 Mya. One major clade includes the lice of bovids (family Linognathidae), rabbits, rodents and shrews (genera Hoplopleura and Pterophthirus of the family Hoplopleuridae; genera Haemodipsus, Linognathoides, Neohaematopinus and Sathrax of the family Polyplacidae), and sea lions and seals (family Echinophthiridae). The other major clade includes the lice of humans and gorillas (families Pediculidae and Pthiridae), monkeys (family Pedicinidae), pigs and horses (family Haematopinidae), and rodents (genus Ancistroplax of the family Hoplopleuridae; genera Fahrenholzia, Polyplax and Lemurpediculus of the family Polyplacidae). Like Kim [10], Light et al. [8] divided the blood-sucking lice into two major clades, although it differed from Kim [9] in the placement of the family Echinophthiridae, and in recognizing the families Hoplopleuridae and Polyplacidae as polyphyletic.
The mitochondrial (mt) genomes of eight species of blood-sucking lice have been sequenced entirely or near entirely in previous studies: 1) the human body louse, Pediculus humanus [11]); 2) the human head louse, Pe. capitis, and the human pubic louse, Pthirus pubis [12]; 3) the domestic pig louse, Haematopinus suis, and the wild pig louse, Ha. apri [13]; 4) the horse louse, Ha. asini [14]; and 5) the lice of the greater bandicoot rat and the Asian house rat, Polyplax asiatica and Po. spinulosa [15]. All of these lice have fragmented mt genomes that depart radically from the typical single-chromosome mt genomes seen in bilateral animals [16, 17]. The mt genes of these lice are on 9–20 minichromosomes; each minichromosome is 1.5–4 kb in size and has 1–8 genes. There is substantial variation in the extent of mt genome fragmentation and the distribution of mt genes over the minichromosomes among these lice [13]. Indeed, the fragmentation pattern varies even between species of the same genus [14, 15]. It is evident that recombination between minichromosomes played a role in generating the high degree of variation in the extent and the pattern of mt genome fragmentation among the blood-sucking lice [12, 14].
The three human lice (families Pediculidae and Pthiridae), the two pig lice and the horse louse (family Haematopinidae), and the two Polyplax rat lice (Polyplacidae), whose mt genomes have been sequenced, are from the same major clade of the Anoplura [8]. To understand mt genome fragmentation in the other major clade of the Anoplura, we sequenced the mt genomes of a mouse louse and a rat louse in the genus Hoplopleura (family Hoplopleuridae): Ho. akanezumi and Ho. kitti. Hoplopleuridae is the most species-rich family of the Anoplura with 162 described species, and Hoplopleura is the most species-rich genus in the family Hoplopleuridae with 141 described species found on rodents and pikas [2, 3, 8]. Hoplopleura species are in a major clade different from that of the human lice (families Pediculidae and Pthiridae), the pig lice and the horse louse (family Haematopinidae), and the Polyplax rat lice (Polyplacidae) [8]. Ho. akanezumi infests the Cheverier’s field mouse, Apodemus chevrieri, and five other Apodemus species of mice [3, 18] . Ho. kitti infests the Bower’s white-toothed rat, Berylmys bowersi, and two other species of rats, Berylmys berdmorei and Leopoldamys edwardsi [3, 18]. We found that both Ho. akanezumi and Ho. kitti have fragmented mt genomes. These two rodent lice, however, differ in the pattern of mt genome fragmentation. Like other genera of blood-sucking lice, the Hoplopleura species also have minichromosomes that are only found in this genus.
Methods
Collection of rodents and lice
The Chevrier’s field mouse lice, Ho. akanezumi, were collected at Cangshan Mountain, Dali city, Yunnan, China (sample No. 249). The Bower’s white-toothed rat lice, Ho. kitti, were collected in Jinping county, Yunnan, China (sample No. 344). The rodents were caught with trap-cages set outdoors (farmlands, scrublands and woodlands). Alive rodents trapped were placed individually in pre-marked cotton bags and transferred to laboratory for species identification and parasitological check. Blood-sucking lice on the body surface of each rodent host were collected and preserved in 95% ethanol at –20°C prior to DNA extraction. Samples of Ho. akanezumi and Ho. kitti and their rodent hosts were deposited in the Institute of Pathogens and Vectors, Dali University. The capture of rodents was approved by health authorities in Yunnan province, China. Animal protocols and procedures were approved by the animal ethics committees at Guizhou University and Dali University (2004C0049M, 30460125).
DNA extraction, mitochondrial genome amplification and sequencing
Total DNA was extracted from individual louse specimens with DNeasy Tissue kit (QIAGEN). A 452-bp fragment of mt rrnS gene and a 360-bp fragment of mt rrnL gene were initially amplified by polymerase chain reaction (PCR) with primer pairs 12SA–12SB and 16SF–Lx16SR (Additional file 1) for Ho. akanezumi. These two pairs of primers target conserved sequence motifs that are highly conserved among arthropods. The rrnS and rrnL fragments were sequenced directly using Sanger method at the Tiangen Biotech, Beijing (TBB). Two pairs of specific primers for Ho. akanezumi, 12S249F–12S249R and 16S249F–16S249R, were designed from sequences of the rrnS and rrnL fragments. The two specific primers in each pair go outbound and are 1 bp and 89 bp respectively from each other. PCRs with these specific primers amplified two near full-length mt minichromosomes of Ho. akanezumi that contain rrnS and rrnL respectively; these amplicons (1.7 kb and 2.1 kb in size) were sequenced using Sanger method at the TBB. Another pair of primers specific to Ho. akanezumi, 249F–249R, was designed from conserved non-coding sequences that flank the coding regions of the two minichromosomes above. The PCR with 249F–249R primers produced a mixture of amplicons ranging from 0.4 to 2 kb in size, expected from the coding regions of the whole set of mt minichromosomes of Ho. akanezumi (Figure 1A). These amplicons were sequenced with Illumina Hiseq 2000 platform at the BGI Hong Kong. The PCR strategy used in this study was developed from the observations we made in previous studies on the human lice, the pig lice, the Polyplax rat lice and the horse louse that each mt minichromosome has a distinct coding region but a well-conserved non-coding region [11–15].
Figure 1.
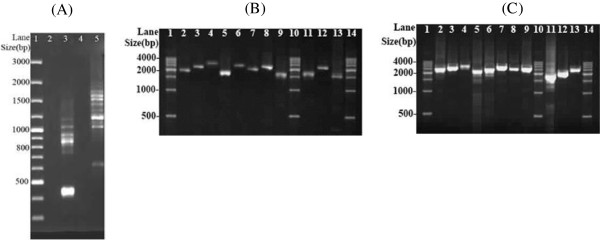
PCR amplification of the mitochondrial (mt) minichromosomes of the Hoplopleura rodent lice. (A) Lane 1: GeneRuler®100 bp DNA Ladder (Thermo Scientific). Lane 3: PCR amplicons generated with primer pair 249F-249R that spans the coding region of each mt minichromosome of Hoplopleura akanezumi. Lane 5: PCR amplicons generated with primer pair 344F-344R that spans the coding region of each mt minichromosome of Hoplopleura kitti. (B) PCR verification of the mt minichromosomes of Ho. akanezumi. Lane 1, 10 and 14: 500 bp DNA Ladder (TIANGEN). Lane 2-13: PCR amplicons from the 10 minichromosomes and a chimeric mt minichromosome of Ho. akanezumi: atp8-atp6- trnN, trnE-cob- trnS 1 -trnS 2, trnI- cox1, trnD-trnY 1 - cox2, trnR-nad4L-trnP- cox3 -trnA-trnT, nad2, trnK- nad4, trnC- nad6 -trnW-trnL 2 (taa), rrnS, trnY 2 - rrnL -trnV, chimeric trnY 2 - prrnL - prrnS. (C) PCR verification of the mt minichromosomes of Ho. kitti. Lane 1, 10 and 13: 500 bp DNA Ladder (TIANGEN). Lane 2-13: PCR amplicons from the 11 minichromosomes of Ho. kitti: atp8-atp6- trnN, trnE -cob-trnS 1 -trnS 2, trnI - cox1, trnD-trnY- cox2 -trnT, trnR-nad4L-trnP- cox3 -trnA, trnQ- nad1 -trnG-nad3, nad2, trnK -nad4, trnC- nad6 -trnW-trnL 2 (taa), rrnS, trnM-trnL 1 (tag)- rrnL -trnV. Genes from which PCR primers were designed are in bold.
For Ho. kitti, a 532-bp fragment of mt cox1 gene, a 452-bp fragment of mt rrnS gene and a 360-bp fragment of mt rrnL gene were amplified initially by PCR with primer pairs mtd6–mtd11, 12SA–12SB and 16SF–Lx16SR (Additional file 1). These three pairs of primers target conserved sequence motifs in cox1, rrnS and rrnL; the PCR amplicons were sequenced using Sanger method at the TBB. Three pairs of specific primers, cox344F–cox344R, 12S344F–12S344R and 16S344F–16S344R were designed from sequences of the cox1, rrnS and rrnL fragments respectively. The specific primers in each pair go outbound and are 2 bp, 32 bp and 49 bp respectively from each other. PCRs with these three pairs of specific primers amplified three near full-length mt minichromosomes of Ho. kitti that contain cox1, rrnS and rrnL respectively; these amplicons (2.7 kb, 2.0 kb and 2.4 kb in size) were sequenced using Sanger method at the TBB. Another pair of primers specific to Ho. kitti, 344F–344R, was designed from conserved non-coding sequences that flank the coding regions of the three minichromosomes above. The PCR with primer pair 344F–344R produced a mixture of amplicons ranging from 0.6 to 2 kb in size, expected from the coding regions of all mt minichromosomes of Ho. kitti (Figure 1A). These amplicons were sequenced with Illumina Hiseq 2000 platform at the BGI-HK.
Taq DNA Polymerase (Tiangen Biotech) was used in the initial short PCRs with the following cycling conditions: 94°C for 1 min; 40 cycles of 98°C for 10 sec, 45°C for 30 sec, 72°C for 1 min; and a final extension of 72°C for 2 min. LA Taq (TakaRa) was used in the long PCRs with the cycling conditions: 94°C for 1 min; 35 cycles of 98°C for 10 sec, 60–65°C (depending on primers) for 30–40 sec, 68°C for 3 min; and a final extension of 72°C for 6 min. Positive and negative controls were run with each PCR experiment. PCR amplicons were checked by agarose gel (1%) eletrophoresis; the sizes of PCR amplicons were estimated by comparing with molecular markers. PCR products were purified with Wizard SV Gel/PCR clean-up system (Promega).
Assembly of Illumina sequence-reads, gene identification and verification of individual mitochondrial minichromosomes
Purified PCR amplicons generated above with primers 249F–249R and 344F–344R from the coding regions of the mt minichromosomes of Ho. akanezumi and Ho. kitti were sequenced with Illumina Hiseq 2000 platform at the BGI-HK. Illumina sequence-reads were assembled into contigs with Geneious 6.1.7 [19]. The assembled parameters were minimum overlap identity 98% and minimum overlap 50 bp. tRNA genes were identified using tRNAscan-SE [20] and ARWEN [21]. Protein-coding genes and rRNA genes were identified with Basic Local Alignment Search Tool (BLAST) searches of GenBank [22, 23]. Identical sequences shared between genes were identified with Wordmatch [24]. Sequence alignments were with Clustal X [25]. The size and circular organization of each mt minichromosome of Ho. akanezumi and Ho. kitti identified by sequence-read assembly were verified by PCR (Figure 1B, C) using outbound primers designed from the coding region of each minichromosome (Additional file 2). The forward primer and reverse primer in each pair were next to each other with a small gap or no gap in between. PCRs with these primers amplified each minichromosome in full or near full length if it had a circular organization. PCR set-up, cycling conditions, agarose gel electrophoresis and size measurement were the same as described above. Positive and negative controls were run for all PCR tests. The nucleotide sequences of the mt genomes of Ho. akanezumi and Ho. kitti have been deposited in GenBank under accession numbers KJ648922-KJ648943.
Results
Mitochondrial genome of Hoplopleura akanezumi, the louse of the Chevrier’s field mouse
We obtained 609,880 sequence-reads from the mt genome of Ho. akanezumi with Illumina Hiseq platform (Table 1). These sequence-reads are 90 bp long each. We assembled the sequence-reads into contigs and identified 28 of the 37 mt genes that are typical of bilateral animals in Ho. akanezumi; these 28 genes are on 10 minichromosomes. Each minichromosome is 1.7–2.6 kb in size, consists of a coding region and a non-coding region, and has a circular organization (Figure 1B). The coding region of each minichromosome contains one to six genes, and varies in size from 678 bp for trnC-nad6-trnW-trnL2 minichromosome to 1,596 bp for trnI-cox1 minichromosome (Table 1; Figure 2A) (Note: minichromosomes are named after their genes hereafter). Eight of the 10 minichromosomes of Ho. akanezumi have one protein-coding or rRNA gene each; the other two minichromosomes have two protein-coding genes each. The 16 tRNA genes we identified are on eight of the 10 minichromosomes; each minichromosome has one to four tRNA genes except nad2 minichromosome and rrnS minichromosome, which have no tRNA genes (Figure 2A; Additional file 3). Each of the 28 mt genes identified in Ho. akanezumi is present on only one minichromosome except trnY2, which is also present in the chimeric minichromosome (see below). All of the 28 mt genes of Ho. akanezumi that we found have the same orientation of transcription relative to the non-coding region (Figure 2A).
Table 1.
Mitochondrial minichromosomes of Hoplopleura akanezumi and Hoplopleura kitti identified by Illumina sequencing
| Minichromosome | Size of coding region (bp) | Number of Illumina sequence-reads |
|---|---|---|
| atp8-atp6-N (atp8-atp6-N) | 898 (924) | 31926 (99377) |
| E-cob-S 1 -S 2 (E-cob-S 1 -S 2 ) | 1309 (1304) | 2539 (52216) |
| I-cox1 (I-cox1) | 1596 (1644) | 6545 (35537) |
| D-Y 1 -cox2 (D-Y-cox2-T) | 810 (904) | 55030 (42531) |
| R-nad4L-P-cox3-A-T (R-nad4L-P-cox3-A) | 1390 (1250) | 1706 (59511) |
| (Q-nad1-G-nad3) | (1447) | (51409) |
| nad2 (nad2) | 984 (990) | 22613 (50604) |
| K-nad4 (K-nad4) | 1322 (1317) | 2059 (41629) |
| C-nad6-W-L 2 (C-nad6-W-L 2 ) | 678 (685) | 33296 (60815) |
| rrnS (rrnS) | 690 (695) | 166399 (88891) |
| Y 2 -rrnL-V (M-L 1 -rrnL-V) | 1263 (1299) | 50213 (60799) |
| chimeric Y 2 -prrnS-prrnL | 433 | 237554 |
| Total | 11373 (12459) | 609880 (643319) |
Note: Gene arrangements and numbers outside brackets are for Hoplopleura akanezumi and those in brackets are for Hoplopleura kitti.
Figure 2.
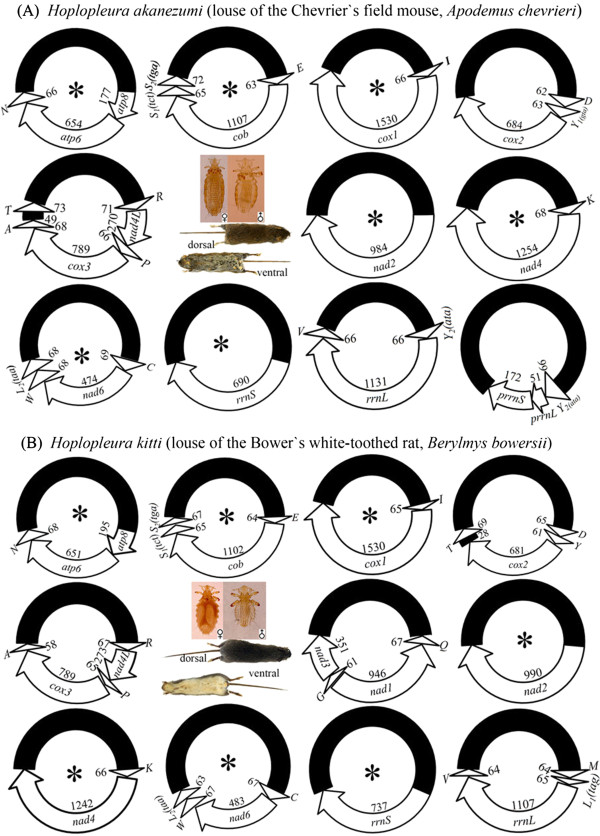
The mitochondrial (mt) genomes of Hoplopleura akanezumi (A) and Hoplopleura kitti (B). Each minichromosome has a coding region (with gene name, transcription orientation and length indicated) and a non-coding region (in black). Minichromosomes are in alphabetical order by the names of their protein coding and rRNA genes. Abbreviations of gene names are: atp6 and atp8 (for ATP synthase subunits 6 and 8), cox1-3 (for cytochrome coxidase subunits 1-3), cob (for cytochrome b), nad1-4 and nad4L (for NADH dehydrogenase subunits 1-6 and 4 L), rrnS and rrnL (for small and large subunits of ribosomal RNA). tRNA genes are shown with the single-letter abbreviations of their corresponding amino acids. Minichromosomes that have identical gene content and gene arrangement between the two Hoplopleura species are indicated with asterisk symbols “*”.
We found a chimeric minichromosome in Ho. akanezumi (Figure 2A). This chimeric minichromosome consists of a coding region and a non-coding region, and has a circular organization (Figure 1B). The coding region of the chimeric minichromosome is 289 bp and contains parts of the two rRNA genes, prrnL and prrnS (note: p for partial), which are only 5% (i.e. 51 bp) and 24% (i.e. 172 bp) of the full-length rrnL and rrnS, respectively. A tRNA gene, trnY2, is upstream prrnL in the chimeric minichromosome and has the same sequence and length as its counterpart in the trnY2-rrnL-trnV minichromosome (Figure 2A, Table 1).
We sequenced the full-length non-coding regions of the rrnS minichromosome and trnY2-rrnL-trnV minichromosome of Ho. akanezumi, 972 bp and 808 bp long, respectively (Figure 3A). The non-coding regions of these two minichromosomes have 78.7% identity to each other. As in other sucking lice [11–15], an AT-rich motif (60 bp, 80% A and T) is present in the non-coding regions of Ho. akanezumi upstream the 5’-end of the coding region, whereas a GC-rich motif (44 bp, 82% G and C) is present downstream the 3’-end of the coding region (Figure 3A). A 72-bp motif repeated four times in tandem in the NCR of rrnS minichromosome of Ho. akanezumi; the four repeat units have 94% identity to each other. No tandem repetitive sequences were found in the NCR of trnY2-rrnL-trnV minichromosome. In addition to the full-length NCR sequences of the two minichromosomes, we also sequenced parts of the NCRs of the other eight minichromosomes and the chimeric minichromosome upstream and downstream of the coding regions, 74–121 bp and 37–128 bp respectively (Additional file 4). Two highly conserved sequence-motifs, 74 bp and 37 bp long respectively, are present in the sections of the NCRs upstream and downstream the coding regions of all of the minichromosomes and the chimeric minichromosome of Ho. akanezumi (Additional file 4).
Figure 3.

Alignment of nucleotide sequences in the non-coding regions of the mitochondrial minichromosomes of Hoplopleura akanezumi (A) and Hoplopleura kitti (B). 249F and 249R are the primers used to amplify the coding regions of all mitochondrial minichromosomes of Ho. akanezumi. 344F and 344R are the primers used to amplify the coding regions of all mitochondrial minichromosomes of Ho. kitti.
Mitochondrial genome of Hoplopleura kitti, the louse of the Bower’s white-toothed rat
We obtained 643,319 sequence-reads from the mt genome of Ho. kitti by Illumina sequencing (Table 1). As above for Ho. akanezumi, these sequence-reads are 90 bp each in length. We assembled these sequence-reads into contigs and identified 34 mt genes typical of bilateral animals in Ho. kitti. These genes are on 11 minichromosomes; each minichromosome is 1.8–2.8 kb in size, has a circular organization and consists of a coding region and a non-coding region (Figure 2B; Figure 1C). As in Ho. akanezumi, eight of the 11 minichromosomes of Ho. kitti have one protein or rRNA-coding gene each; the other three minichromosomes have two protein-coding genes each. The 19 tRNA genes are on nine of the 11 minichromosomes; each minichromosome has one to three tRNA genes except rrnS minichromosome and nad2 minichromosome, which have no tRNA genes (Figure 2B; Additional file 3). Each minichromosome has a coding region and a non-coding region. The coding region of each minichromosome contains one to five genes, and varies in size from 685 bp for trnC-nad6-trnW-trnL2 minichromosome to 1,644 bp for trnI-cox1 minichromosome. With the exception of nad1 and trnQ, all of the mt genes of Ho. kitti have the same orientation of transcription relative to the NCRs (Figure 2B).
We sequenced the full-length NCRs of three mt minichromosomes of Ho. kitti: rrnS, trnM-trnL1-rrnL-trnV and trnI-cox1, 1,249 bp, 1,107 bp and 1,181 bp respectively. The NCRs of these three minichromosomes have 87–93% identity to each other. Tandem repetitive sequences were found in the NCRs of all three minichromosomes. A 60-bp motif repeated three times in tandem; these three repeat units have 87–90% identity to each other. A 72-bp motif repeated twice in tandem; the two repeat units have 93% identity to each other.
As above in Ho. akanezumi and other sucking lice, an AT-rich motif (119 bp, 76% A and T) is present in the NCRs upstream the 5’-end of the coding region, and a GC-rich motif (50 bp, 79% G and C) is present downstream the 3’-end of the coding region in Ho. kitti (Figure 3B). Additional to the full-length NCR sequences, we also sequenced parts of the NCRs upstream and downstream of the coding regions of the other eight minichromosomes of Ho. kitti, 140–326 bp and 48–449 bp respectively. Two highly conserved sequence-motifs, 140 bp and 48 bp long respectively, are present in the sections of the non-coding regions upstream and downstream of the coding regions of all of the 11 minichromosomes in Ho. kitti (Additional file 5).
Discussion
Intra-genus variation in the pattern of mt genome fragmentation in blood-sucking lice
The two species of Hoplopleura rodent lice have the same pattern for the distribution of all of the protein-coding and rRNA genes and most tRNA genes on their minichromosomes (Figure 2). Each minichromosome has a single protein-coding or rRNA gene except for three minichromosomes that have two protein-coding genes each: atp8–atp6, nad4L–cox3, and nad1–nad3 (Figure 2). Seven minichromosomes of Ho. akanezumi and Ho. kitti have identical gene content and gene arrangement; the other four minichromosomes, however, differ between the two species, providing further evidence for intra-genus variation in the pattern of mt genome fragmentation in the blood-sucking lice. Two recent studies also revealed variation in the pattern of mt genome fragmentation within two other genera of the blood-sucking lice. Dong et al. [15] showed that two Polyplax species of rat lice differ in gene content and gene arrangement for nine of the 11 mt minichromosomes; the variation, however, was limited to tRNA genes only. Song et al. [14] showed that the horse louse, Haematopinus asini, differs from the pig lice, Ha. suis and Ha. apri, in gene content and gene arrangement for three of the nine minichromosomes; the variation is not limited to tRNA genes but in protein-coding and rRNA genes as well. Taken together, we conclude that intra-genus variation in the pattern of mt genome fragmentation is common in both major clades of the blood-sucking lice; such variation may involve tRNA genes, protein-coding genes and rRNA genes.
What is the ancestral pattern of mt genome fragmentation of the blood-sucking lice?
According to Light et al. [8] and Smith et al. [9], the blood-sucking lice (suborder Anoplura) evolved from chewing lice ~92 Mya and diversified into two major clades ~75 Mya. The Hoplopleura species, which we investigated in the current study, are in the major clade with the lice of bovids (family Linognathidae), rabbits and shrews (genus Pterophthirus of the family Hoplopleuridae; genera Haemodipsus, Linognathoides, Neohaematopinus and Sathrax of the family Polyplacidae), and sea lions and seals (family Echinophthiridae). The other major clade includes the lice of humans and gorillas (families Pediculidae and Pthiridae), monkeys (family Pedicinidae), pigs and horses (family Haematopinidae), and rodents (genus Ancistroplax of the family Hoplopleuridae; genera Fahrenholzia, Polyplax and Lemurpediculus of the family Polyplacidae). Light et al. [8] and Smith et al. [9] used molecular data (18S, EF-1α, and cox1 gene sequences) of species from eight of the 15 families of the Anoplura and is consistent with an early study by Kim [10], which used morphological data, in dividing the blood-sucking lice into two major clades.
The presence of fragmented mt genomes in both major clades of the blood-sucking lice indicates that the most recent common ancestor (MRCA) of these lice already had a fragmented mt genome. Although the data available now are not sufficient yet for us to establish the exact ancestral fragmentation pattern of that MRCA, we can infer that the gene-arrangement characters that are present in the Hoplopleura species and the species from the other major clade to be ancestral to the blood-sucking lice. These characters include atp8-atp6-N, E-cob, cob-S1, I-cox1, D-Y-cox2, R-nad4L-P-cox3, cox3-A, K-nad4, and M-L1-rrnL-V; each of these characters is shared by at least one species from each of the two major clades of the blood-sucking lice (Table 2).
Table 2.
Mitochondrial gene-arrangement characters inferred to be ancestral for blood- sucking lice (suborder Anoplura)
| Species of insects | Order/suborder | atp8-atp6-N | E-cob | cob-S 1 | I-cox1 | D-Y-cox2 | R-nad4L-P-cox3 | cox3-A | K-nad4 | M-L 1 -rrnL-V |
|---|---|---|---|---|---|---|---|---|---|---|
| Hoplopleura akanezumi (rat louse) | Phthiraptera/Anoplura | + | + | + | + | + | + | + | + | – |
| Hoplopleura kitti (rat louse) | Phthiraptera/Anoplura | + | + | + | + | + | + | + | + | + |
| Pediculus capitis (human head louse) | Phthiraptera/Anoplura | – | – | – | – | – | – | + | + | – |
| Pediculus humanus (human body louse) | Phthiraptera/Anoplura | – | – | – | – | – | – | + | + | – |
| Pthirus pubis (human pubic louse) | Phthiraptera/Anoplura | – | – | + | – | – | – | + | – | – |
| Polyplax asiatica (rat louse) | Phthiraptera/Anoplura | – | + | – | – | + | – | + | + | + |
| Polyplax spinulosa (rat louse) | Phthiraptera/Anoplura | – | + | – | – | + | + | – | + | – |
| Haematopinus suis (domestic pig louse) | Phthiraptera/Anoplura | + | + | – | + | + | – | + | + | – |
| Haematopinus apri (wild pig louse) | Phthiraptera/Anoplura | + | + | – | + | + | – | + | + | – |
| Haematopinus asini (horse louse) | Phthiraptera/Anoplura | + | + | – | + | + | – | + | + | – |
| Bothriometopus macrocnemis (screamer louse) | Phthiraptera/Ischnocera | – | – | – | – | – | – | – | + | – |
| Campanulotes bidentatus (pigeon louse) | Phthiraptera/Ischnocera | – | – | – | – | – | – | – | – | – |
| Ibidoecus bisignatus (ibis head louse) | Phthiraptera/Ischnocera | – | – | – | – | – | – | – | – | – |
| Heterodoxus macropus (wallaby louse) | Phthiraptera/Amblycera | – | – | – | – | – | – | – | – | – |
| Lepidopsocid sp. (barklouse) | Psocoptera/Trogiomorpha | – | – | – | – | – | – | – | – | – |
| Haematomyzus elephantis (elephant louse) | Phthiraptera/ Rhynchophthirina | – | – | – | + | – | – | – | + | – |
| Hypothetical ancestor of insects | – | – | – | – | – | – | – | – | – |
Note: “+” is for “presence”; “-” is for “absence”.
Recombination between mt genes and between mt minichromosomes in Hoplopleuralice
Evidence for recombination between mt genes and between mt minichromosomes has been found in human lice, pig lice, a horse louse, and Polyplax rat lice in previous studies. In the human lice, Pe. humanus, Pe. capitis and Pt. pubis, eight pairs of mt genes share stretches of identical sequences, 16–127 bp long, which are much longer than expected by chance, providing unequivocal evidence for recombination between mt genes [11, 12, 26]. Longer-than-expected identical sequences shared between mt genes were also found in the pig lice, Ha. suis and Ha. apri (three pairs of genes [13]), the horse louse, Ha. asini (nine pairs of genes [14]), and the rat lice, Po. asiatica (one pair of genes) and Po. spinulosa (three pairs of genes [15]). It is noteworthy that trnL1 and trnL2 share identical sequences that are much longer than expected by chance in all of these five blood-sucking lice. In the three species of human lice, trnL1 and trnL2 have identical sequences except for the nucleotides at the third anti-codon positions [12]. In the two species of pig lice and the two species of Polyplax rat lice, these two tRNA genes have near identical sequences at the D-arm and the AC-arm except for the nucleotides at the third anti-codon positions, but differ at the T-arm and the AA-arm [13, 15]. In the horse louse, trnL1 and trnL2 share a 15-bp identical sequence at the D-arm [14]. In addition, seven chimeric mt minichromosomes were found in the human body louse, Pe. humanus, providing evidence for recombination between mt minichromosomes [26, 27].
We did not find identical sequences shared by any mt genes, even between trnL1 and trnL2, longer than expected by chance in Ho. akanezumi and Ho. kitti (Table 3). trnL1 and trnL2, both of which were found in Ho. kitti, share only 7 bp identical sequence, which is expected by chance. Thus, there is no evidence for recombination between mt genes in these two Hoplopleura species. We found, however, a chimeric mt minichromosome in Ho. akanezumi, which has part of rrnS gene (prrnS, 172 bp; “p” for “partial” hereafter) and part of rrnL gene (prrnL, 51 bp) (Figure 2). The chimeric mt minichromosome of Ho. akanezumi has similar structure to the Type 1 and Type 2 chimeric minichromosomes of the human body louse [26]. Type 1 chimeric minichromosome of the human body louse has pcox2 and pcox3, whereas Type 2 has patp6 and pnad1 [26]. The partial genes in each of these chimeric minichromosomes of the human body louse were joined at a short homologous sequence, called “microhomology”, a hallmark of the gene junction formed by non-homologous recombination [26, 28, 29]. Intriguingly, we did not find such microhomology in the junction between prrnS and prrnL in the chimeric minichromosome of Ho. akanezumi. It is not clear yet to us whether, or not, the lack of recombination between mt genes in the two Hoplopleura lice and the lack of microhomology in the chimeric minichromosome of Ho. akanezumi are a general feature for the major clade of blood-sucking lice that they represent. More species from this major clade need to be investigated to understand the recombination between mt genes and between mt minichromosomes.
Table 3.
The longest stretches of identical sequence shared by mitochondrial genes in four rat lice, two pig lice, three human lice that have fragmented mitochondrial genomes, and six other species of bilateral animals that have the typical mitochondrial genomes
| Pairs of gene | The longest stretches of identical sequence | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rodent lice | Pig lice | Human lice | Animals with typical mt genome organization | |||||||||||||
| Hoa | Hok | Pa | Ps | Has | Haa | Pc | Pp | Ph | Bm | Cb | Hm | Dy | Ce | Hos | ||
| trnL 1 | trnL 2 | NA | 7 | 28 | 25, 11 | 16, 10 | 16, 10 | 33, 32 | 35, 32 | 33, 32 | 7 | 6 | 7 | 10 | 6 | 6 |
| cob | nad5 | NA | NA | 12 | 36 | 13 | 13 | 12 | 13 | 12 | 12 | 16 | 14 | 13 | 13 | 12 |
| trnA | trnC | 7 | 6 | 6 | 32 | 6 | 6 | 6 | 6 | 6 | NA | 7 | 17 | 7 | 8 | 6 |
| nad4 | nad5 | NA | NA | 12 | 18 | 12 | 12 | 127, 30 | NA | 127, 30 | 13 | 15 | 15 | 16 | 14 | 11 |
| nad5 | rrnL | NA | NA | 13 | 13 | 11 | 10 | 99 | 10 | 99 | 12 | 14 | 13 | 15 | 16 | 10 |
| trnG | trnR | NA | 6 | 5 | 5 | 5 | 5 | 28, 14 | 32, 26 | 28, 14 | 5 | 6 | 7 | 6 | 8 | 6 |
| cox1 | nad4L | 10 | 10 | 9 | 11 | 11 | 11 | 10 | 29 | 10 | 13 | 11 | 14 | 13 | 12 | 10 |
| nad2 | rrnL | 11 | 11 | 10 | 11 | 10 | 11 | 26 | 10 | 26 | 13 | 11 | 14 | 13 | 12 | 10 |
| trnP | trnT | 7 | 8 | 6 | 7 | 26 | 26 | 7 | NA | 7 | 6 | 8 | 8 | 9 | 10 | 7 |
| atp8 | trnG | NA | 7 | 6 | 6 | 6 | 6 | 26 | 9 | 26 | 10 | 11 | 11 | 12 | NA | 6 |
| atp8 | nad2 | 9 | 9 | 12 | 10 | 25 | 25 | 10 | 8 | 10 | 10 | 14 | 12 | 14 | NA | 11 |
| trnI | trnT | 6 | 6 | 10 | 8 | 6 | 6 | 6 | 16 | 6 | 6 | 5 | 7 | 7 | 9 | 6 |
Note: Abbreviations of species names are: Hoa, Hoplopleura akanezumi (louse of the Chevrier’s field mouse); Hok, Hoplopleura kitti (louse of the Bower’s white-toothed rat); Pa, Polyplax asiatica (loues of the greater bandicoot rat); Ps, Polyplax spinulosa (louse of the Asian house rat); Has, Haematopinus suis (domestic pig louse); Haa, Haematopinus apri (wild pig louse); Pc, Pediculus capititis (human head louse); Pp, Pthirus pubis (human pubic louse); Ph, Pediculus humanus (human body louse); Bm, Bothriometopus macrocnemis (screamer louse); Cb, Campanulotes bidentatus (pigeon louse); Hm, Heterodoxus macropus (wallaby louse); Dy, Drosophila yakuba (fruitfly); Ce, Caenorhabditis elegans (roundworm); Hos, Homo sapiens (human); NA, not applicable. Stretches of shared identical sequences longer than expected by chance are indicated in bold.
Are features of fragmented mt genomes useful for understanding phylogeny?
The suborder Anoplura contains 540 species of blood-sucking lice in 50 genera and 15 families [1–3]. The monophyly of the suborder Anoplura was inferred from morphology and has been generally accepted [30]. The monophyly of most of the genera and families, however, has not been tested. Furthermore, phylogenetic relationships among most of the genera and families are not resolved or are controversial. Indeed, only four studies have addressed the high-level phylogenetic relationships, i.e. among genera and among families of the blood-sucking lice: Kim and Ludwig [1] and Kim [10] used morphological characters, whereas Light et al. [8] and Smith et al. [9] used molecular data (18S, EF-1α, and cox1 gene sequences). As discussed above, the findings from the present study and two recent studies [14, 15] showed that intra-genus variation in the pattern of mt genome fragmentation is common among blood-sucking lice. Such variation may provide a novel source of information, additional to morphology and gene sequences, for inferring genus- and family-level phylogenies of the blood-sucking lice, and for testing the phylogenies inferred from morphology and gene sequences. Along this line, we found that each of the five genera of the blood-sucking lice that have been investigated has one to nine minichromosomes that are only present in that genus (Table 4), which could potentially be synapomorphies for these genera. Future studies should expand into other genera and families of the blood-sucking lice to fully evaluate the phylogenetic utility of the features of fragmented mt genomes, such as genus-specific minichromosomes and derived gene-arrangement characters.
Table 4.
Potential genus-level synapomorphic mitochondrial minichromosomes for the blood-sucking lice investigated to date
| Species | E-cob-S 1 -S 2 | C-nad6-W-L 2 | rrnS | E-cob-I | K-nad4-atp8-atp6-N | nad2-I-cox1-L 2 | D-Y-cox2-S 1 -S 2 -P-cox3-A | E-cob-V | Q-nad1 -T-G-nad3-W | cob | R-nad3 | G-nad4L-V | F-nad6 | L 1 /L 2 -rrnS-C | L 1 -rrnL | S 1 -N-E | T-D-H |
W-
S 2 |
cob
-S 1 |
G-nad3-V-W-S 2 | T-D-H-R-nad4L | F-nad6-E-M | L 1 -rrnS | C |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Hoplopleura akanezumi
(rat louse) |
+ | + | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Hoplopleura kitti (rat louse) | + | + | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Polyplax asiatica (rat louse) | – | – | – | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Polyplax spinulosa (rat louse) | – | – | – | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Haematopinus suis (domestic pig louse) | – | – | – | – | + | + | + | + | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Haemapinus apri (wild pig louse) | – | – | – | – | + | + | + | + | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Haemapinus asini (horse louse) | – | – | – | – | + | + | + | + | + | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – |
| Pediculus capitis (human head louse) | – | – | – | – | – | – | – | – | – | + | + | + | + | + | + | + | + | + | – | – | – | – | – | – |
| Pediculus humanus (human body louse) | – | – | – | – | – | – | – | – | – | + | + | + | + | + | + | + | + | + | – | – | – | – | – | – |
| Pthirus pubis (human pubic louse) | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | – | + | + | + | + | + | + |
Note: neighbor genes are linked by a hyphen “-”; genes in bold, i.e. Q-nad1, have an orientation of transcription opposite to that of other genes; anticodons are: S 1: tct, S 2: tga, L 1: tag, and L 2: taa; “+” is for “presence”; “-” is for “absence”.
Limitations of PCR-based strategies in identifying the complete set of mt chromosomes
The PCR-based strategy we used has its limitations in identifying the complete set of mt chromosomes of the Hoplopleura lice. We did not find nine mt genes that were typical of bilateral animals in Ho. akanezumi: trnF, trnG, trnH, trnL1, trnM, trnQ, nad1, nad3 and nad5. We did not find trnF, trnH and nad5 either in Ho. kitti. These unidentified genes in the Hoplopleura lice are present in the fragmented mt genomes of three human lice, two pig lice, the horse louse, and two Polyplax rat lice [11–15]. The primer pairs, 249F–249R and 344F–344R (Additional file 1), which we used to amplify the coding regions of the mt minichromosomes of Ho. akanezumi and Ho. kitti may not be conserved in the chromosomes that contain these unidentified genes. We cannot exclude either the possibility that macrochromosomes or the typical mt chromosome of animals may exist in the Hoplopleura rodent lice.
A non-PCR-based, shot-gun strategy was used to sequence the mt genome of the human body louse, Pediculus humanus, in conjunction with PCR-based strategy [11, 26]. The shot-gun strategy revealed the mt minichromosomes of the human body louse [11] whereas the PCR-based strategy revealed the less abundant, chimeric mt chromosomes [26]. There is no evidence that the typical mt chromosome of animals is present in the human body louse or other blood-sucking lice. The shot-gun sequencing strategy was possible for the human body louse because of a laboratory strain of this louse that supplied a large number of samples for the sequencing project [11, 27]. For other species of blood-sucking lice, there are no laboratory strains available and it is difficult to collect large number of samples. Thus, PCR-based sequencing strategies become the first option of choice, or the only option for the species of blood-sucking lice collected from wild mammals.
Conclusions
We sequenced the mt genomes of Ho. akanezumi and Ho. kitti, collected from the Chevrier’s field mouse, Apodemus chevrieri, and the Bower’s white-toothed rat, Berylmys bowersi. Both Ho. akanezumi and Ho. kitti have fragmented mt genomes, with the mt genes that we identified distributed on 10 and 11 minichromosomes, respectively. Ho. akanezumi also has a chimeric minichromosome, which has parts of two rRNA genes and a full-length trnY2 gene. These two Hoplopleura rodent lice share the same pattern for the distribution of the protein-coding and rRNA genes but differ in tRNA gene content and gene arrangement in four minichromosomes. Like other genera of blood-sucking lice that have been investigated in previous studies, the Hoplopleura species have four minichromosomes that are only found in this genus. We conclude that fragmented mt genomes were already present in the MRCA of the two major clades of the blood-sucking lice, which lived ~75 Mya. Intra-genus variation in the pattern of mt genome fragmentation is common in the blood-sucking lice. Such variation provides a novel source of information for inferring genus- and family-level phylogenies of the blood-sucking lice, and for testing the phylogenies inferred from morphology and gene sequences. Future studies should expand into other genera and families of the blood-sucking lice to fully explore the phylogenetic utility of the features of fragmented mt genomes.
Availability of supporting data
The nucleotide sequences of the mt genomes of the two Hoplopleura rodent lice supporting the results of this article have been deposited in GenBank (accession numbers KJ648922–KJ648943).
Electronic supplementary material
Additional file 1: PCR primers used to amplify and sequence the mitochondrial genomes of the rodent lice, Hoplopleura akanezumi ( Hoa ) and Hoplopleura kitti ( Hok ). (PDF 92 KB)
Additional file 2: PCR primers used to verify the mitochondrial minichromosomes of the rodent lice, Hoplopleura akanezumi ( Hoa ) and Hoplopleura kitti ( Hok ). (PDF 71 KB)
Additional file 3: Inferred secondary structures of the mitochondrial tRNAs of Hoplopleura akanezumi (Ha) and Hoplopleura kitti (Hk). (PDF 136 KB)
Additional file 4: Alignment of nucleotide sequences of parts of the non-coding regions upstream (A) and downstream (B) of the coding regions of the 10 mitochondrial minichromosomes and a chimeric mitochondrial minichromosomes of Hoplopleura akanezumi. 249F and 249R are the PCR primers used to amplify the coding regions of all mitochondrial minichromosomes of Hoplopleura akanezumi. (PDF 58 KB)
Additional file 5: Alignment of nucleotide sequences of parts of the non-coding regions upstream (A) and downstream (B) of the coding regions of the 11 mitochondrial minichromosomes of Hoplopleura kitti. 344F and 344R are the PCR primers used to amplify the coding regions of all mitochondrial minichromosomes of Hoplopleura kitti. (PDF 255 KB)
Acknowledgments
We acknowledge funding support from The Program for Innovative Research Team in Guizhou ([2009]4003 to DCJ), Natural Science Foundation of China (81160208 to XGG), the Australian Research Council (DP120100240 to RS and SCB), and Australia-China Science & Research Fund (ACSRF00980 to RS).
Abbreviations
- μl
Microliter
- atp6 and atp8
Genes for ATP synthase subunits 6 and 8
- bp
Base pair
- cob
Gene for cytochrome b
- DNA
Deoxyribonucleic acid
- kb
Kilo base pair
- min
Minute
- MRCA
Most recent common ancestor
- mt
Mitochondrial
- Mya
Million years ago
- PCR
Polymerase chain reaction
- RNA
Ribonucleic acid
- rRNA
Ribosomal RNA
- rrnS and rrnL
Genes for small and large subunits of ribosomal RNA
- sec
Second
- T
Thymine
- tRNA
Transfer RNA
- tRNA
Transfer RNA
- trnA or A
tRNA gene for alanine
- trnC or C
tRNA gene for cysteine
- trnD or D
tRNA gene for aspartic acid
- trnE or E
tRNA gene for glutamic acid
- trnF or F
tRNA gene for phenylalanine
- trnG or G
tRNA gene for glycine
- trnH or H
tRNA gene for histidine
- trnI or I
tRNA gene for isoleucine
- trnK or K
tRNA gene for lysine
- trnL1 or L1
tRNA gene for leucine (anticodon NAG)
- trnL2 or L2
tRNA gene for leucine (anticodon YAA)
- trnM or M
tRNA gene for methionine
- trnN or N
tRNA gene for asparagine
- trnP or P
tRNA gene for proline
- trnQ or Q
tRNA gene for glutamine
- trnR or R
tRNA gene for arginine
- trnS1 or S1
tRNA gene for serine (anticodon NCU)
- trnS2 or S2
tRNA gene for serine (anticodon NGA)
- trnT or T
tRNA gene for threonine
- trnV or V
tRNA gene for valine
- trnW or W
tRNA gene for tryptophan
- trnY or Y
tRNA gene for tyrosine
- U
Uracil.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
WGD, SS, XGG, DCJ, SCB and RS designed the research. WGD, SS and RS performed the research. XGG, DCJ and RS contributed reagents and materials. WGD, SS, QY and RS analyzed the data. WGD and RS wrote the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Wen-Ge Dong, Email: dongwenge2740@sina.com.
Simon Song, Email: ssong1@usc.edu.au.
Xian-Guo Guo, Email: xgguo2002@yahoo.com.
Dao-Chao Jin, Email: daochaojin@126.com.
Qianqian Yang, Email: yqian213@gmail.com.
Stephen C Barker, Email: s.barker@uq.edu.au.
Renfu Shao, Email: rshao@usc.edu.au.
References
- 1.Kim KC, Ludwig HW. The family classification of the Anoplura. Syst Entomol. 1978;3:249–284. doi: 10.1111/j.1365-3113.1978.tb00120.x. [DOI] [Google Scholar]
- 2.Durden LA, Musser GG. The sucking lice (Insecta, Anoplura) of the world: a taxonomic checklist with records of mammalian hosts and geographical distributions. Bull Amer Mus Nat Hist. 1994;218:1–90. [Google Scholar]
- 3.Durden LA, Musser GG. The mammalian hosts of the sucking lice (Anoplura) of the world: a host-parasite list. Bull Soc Vector Ecol. 1994;19:130–168. [Google Scholar]
- 4.Nelson WA, Shemanchuk JA, Haufe WO. Haematopinus eurysternus: blood of cattle infested with the short-nosed cattle louse. Exp Parasitol. 1970;28:263–271. doi: 10.1016/0014-4894(70)90096-2. [DOI] [PubMed] [Google Scholar]
- 5.Gibney VJ, Campbell JB, Boxler DJ, Clanton DC, Deutscher GH. Effects of various infestation levels of cattle lice (Mallophaga: Tri-chodectidae and Anoplura: Haematopinidae) on feed efficiency and weight gains of beef heifers. J Econ Entomol. 1985;78:1304–1307. doi: 10.1093/jee/78.6.1304. [DOI] [PubMed] [Google Scholar]
- 6.Otter A, Twomey DF, Crawshaw TR, Bates P. Anaemia and mortality in calves infested with the long-nosed sucking louse (Linognathus vituli) Vet Rec. 2003;153:176–179. doi: 10.1136/vr.153.6.176. [DOI] [PubMed] [Google Scholar]
- 7.Hornok S, Hofmann-Lehmann R, Fernandez de Mera IG, Meli ML, Elek V, Hajtos I, Repasi A, Gonczi E, Tanczos B, Farkas R, Lutz H, de la Fuente J. Survey on blood-sucking lice (Phthiraptera: Anoplura) of ruminants and pigs with molecular detection of Anaplasma and Rickettsia spp. Vet Parasitol. 2010;174:355–358. doi: 10.1016/j.vetpar.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 8.Light JE, Smith VS, Allen JM, Durden LA, Reed RL. Evolutionary history of mammalian sucking lice (Phthiraptera:Anoplura) BMC Evol Biol. 2010;10:292. doi: 10.1186/1471-2148-10-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith VS, Ford T, Johnson KP, Johnson PCD, Yoshizawa K, Light JE. Multiple lineages of lice pass through the K-Pg boundary. Biol Lett. 2011;5:782–785. doi: 10.1098/rsbl.2011.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim KC. Evolutionary parallelism in Anoplura and eutherian mammals. In: Service MW, editor. Biosystematics of Haematophagous Insects. Oxford: Oxford University Press; 1988. pp. 91–114. [Google Scholar]
- 11.Shao R, Kirkness EF, Barker SC. The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus. Genome Res. 2009;19:904–912. doi: 10.1101/gr.083188.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shao R, Zhu XQ, Barker SC, Herd K. Evolution of extensively fragmented mitochondrial genomes in the lice of humans. Genome Biol Evol. 2012;4:1088–1101. doi: 10.1093/gbe/evs088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang HW, Barker SC, Shao R. Substantial variation in the extent of mitochondrial genome fragmentation among blood-sucking lice of mammals. Genome Biol Evol. 2013;5:1298–1308. doi: 10.1093/gbe/evt094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song S, Barker SC, Shao R. Variation in mitochondrial minichromosome composition between blood-sucking lice of the genus Haematopinus that infest horses and pigs. Parasites Vectors. 2014;7:144. doi: 10.1186/1756-3305-7-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong WG, Song S, Jin DC, Guo XG, Shao R. Fragmented mitochondrial genomes of the rat lice, Polyplax asiatica and Polyplax spinulosa: intra-genus variation in fragmentation pattern and a possible link between the extent of fragmentation and the length of life cycle. BMC Genomics. 2014;15:44. doi: 10.1186/1471-2164-15-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boore JL. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lavrov DV. Key transitions in animal evolution : a mitochondrial DNA perspective. Integr Comp Biol. 2007;47:734–743. doi: 10.1093/icb/icm045. [DOI] [PubMed] [Google Scholar]
- 18.Chin TH. Taxonomy and Fauna of Sucking Lice (Anoplura) in China. Beijing: Science Press; 1999. pp. 1–132. [Google Scholar]
- 19.Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laslett D, Canback B. ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics. 2008;24:172–175. doi: 10.1093/bioinformatics/btm573. [DOI] [PubMed] [Google Scholar]
- 22.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gish W, States DJ. Identification of protein coding regions by database similarity search. Nature Genet. 1993;3:266–272. doi: 10.1038/ng0393-266. [DOI] [PubMed] [Google Scholar]
- 24.Rice P, Longden I, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000;16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- 25.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 26.Shao R, Barker SC. Chimeric mitochonrial minichromosomes of the human body louse, Pediculus humans: evidence for homologous and non-homologous recombination. Gene. 2011;473:36–43. doi: 10.1016/j.gene.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 27.Kirkness EF, Haas BJ, Sun W, Braig HR, Perotti MA, Clark JM, Lee SH, Robertson HM, Kennedy RC, Elhaik E, Gerlach D, Kriventseva EV, Elsik CG, Graur D, Hill CA, Veenstra JA, Walenz B, Tubío JM, Ribeiro JM, Rozas J, Johnston JS, Reese JT, Popadic A, Tojo M, Raoult D, Reed DL, Tomoyasu Y, Kraus E, Mittapalli O, Margam VM, Li HM, et al. Genome sequences of the human body louse and its primary endosymbiont provide insights into the permanent parasitic lifestyle. Proc Natl Acad Sci U S A. 2010;27:12168–12173. doi: 10.1073/pnas.1003379107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chu G. Double strand break repair. J Biol Chem. 1997;272:2777–2781. doi: 10.1074/jbc.272.39.24097. [DOI] [PubMed] [Google Scholar]
- 29.Ricchetti M, Fairhead C, Dujon B. Mitochondrial DNA repairs double-strand breaks in yeast chromosomes. Nature. 1999;402:96–100. doi: 10.1038/47076. [DOI] [PubMed] [Google Scholar]
- 30.Lyal CHC. Phylogeny and classification of the Psocodea, with particular reference to the lice (Psocodea: Phthiraptera) Syst Entomol. 1985;10:145–165. doi: 10.1111/j.1365-3113.1985.tb00525.x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: PCR primers used to amplify and sequence the mitochondrial genomes of the rodent lice, Hoplopleura akanezumi ( Hoa ) and Hoplopleura kitti ( Hok ). (PDF 92 KB)
Additional file 2: PCR primers used to verify the mitochondrial minichromosomes of the rodent lice, Hoplopleura akanezumi ( Hoa ) and Hoplopleura kitti ( Hok ). (PDF 71 KB)
Additional file 3: Inferred secondary structures of the mitochondrial tRNAs of Hoplopleura akanezumi (Ha) and Hoplopleura kitti (Hk). (PDF 136 KB)
Additional file 4: Alignment of nucleotide sequences of parts of the non-coding regions upstream (A) and downstream (B) of the coding regions of the 10 mitochondrial minichromosomes and a chimeric mitochondrial minichromosomes of Hoplopleura akanezumi. 249F and 249R are the PCR primers used to amplify the coding regions of all mitochondrial minichromosomes of Hoplopleura akanezumi. (PDF 58 KB)
Additional file 5: Alignment of nucleotide sequences of parts of the non-coding regions upstream (A) and downstream (B) of the coding regions of the 11 mitochondrial minichromosomes of Hoplopleura kitti. 344F and 344R are the PCR primers used to amplify the coding regions of all mitochondrial minichromosomes of Hoplopleura kitti. (PDF 255 KB)