

Abstract
All species continuously evolve to adapt to changing environments. The genetic variation that fosters such adaptation is caused by a plethora of mechanisms, including meiotic recombination that generates novel allelic combinations in the progeny of two parental lineages. However, a considerable number of eukaryotic species, including many fungi, do not have an apparent sexual cycle and are consequently thought to be limited in their evolutionary potential. As such organisms are expected to have reduced capability to eliminate deleterious mutations, they are often considered as evolutionary dead ends. However, inspired by recent reports we argue that such organisms can be as persistent as organisms with conventional sexual cycles through the use of other mechanisms, such as genomic rearrangements, to foster adaptation.
Keywords: adaptation, asexual, genome evolution, meiosis, mitosis, recombination
Introduction
Genomes are highly dynamic and vary considerably between, and often even within, species with respect to structure and content. This dynamics is caused by mutations that are either neutral, and may thus be transient, or mediate increased fitness and became fixed in the population by natural (Darwinian) selection. Variations are caused by a multitude of mechanisms and range from single nucleotide polymorphisms to large-scale genomic rearrangements. The latter may result in translocation, duplication, and deletion of genetic material, thereby affecting chromosomal size and gene content. Even though chromosomal rearrangements have been associated with diseases and reduced fitness, they are also major drivers of evolution. They contribute to adaptation to novel or changing environments and even to speciation by reproductive isolation [1–3].
Although evolution is an ongoing process for any organism, adaptation to dynamic environments is of particular importance for pathogens that need to co-evolve with their hosts in a continuous arms race in which the host tries to detect and intercept the intruder and mount an effective immune response while the pathogen tries to avoid or overcome recognition and evade or suppress such response [4,5]. To this end, pathogens secrete so-called effectors; molecules that interfere with host physiology [6,7]. In turn, hosts evolve immune receptors that can detect effectors or their activities and initiate an immune response. Consequently, pathogens need to continuously modify existing effectors or evolve novel effectors to subvert their host [4,8].
Sexual reproduction has been observed in nearly all branches of the eukaryotic tree of life [9]. It provides an important mechanism to establish genetic variation by combining genetic information of two parental lineages. Before genetic information is transferred to the progeny, meiotic recombination generates novel combinations of existing alleles. It is therefore not surprising that meiotic recombination is considered an important driver for rapid adaptation of pathogens to their hosts [10,11]. Nevertheless, in a considerable proportion of eukaryotic lineages, no sexual cycle has ever been observed [12], e.g. in around 20% of all fungi, a kingdom that contains pathogens of animals and plants. Such lineages that lack a sexual cycle have been considered evolutionary dead-ends [10,13,14]. However, several recent observations challenge this view, since some organisms appear to be successful in the absence of sexual reproduction. For instance, bdelloid rotifers are microscopic animals that have persisted for millions of years with an asexual lifestyle, and recent insight in the genome of a bdelloid rotifer confirmed that its genome structure is incompatible with conventional meiosis due to frequent genomic rearrangements [15]. Similarly, comparative genomics on Verticillium dahliae, a fungal plant pathogen for which a sexual cycle has never been observed, revealed extensive chromosomal rearrangements that establish lineage-specific (LS) genomic regions that are responsible for adaptation to plant hosts and evolution of fungal aggressiveness [11] (Fig. 1). Thus, these organisms evolved means to generate genomic diversity and compensate for the apparent lack of sexual reproduction. A plethora of processes can create genomic variation in eukaryotes, and we hypothesize that commonly observed chromosomal rearrangements provide a pivotal mechanism to promote adaptation.
Figure 1.
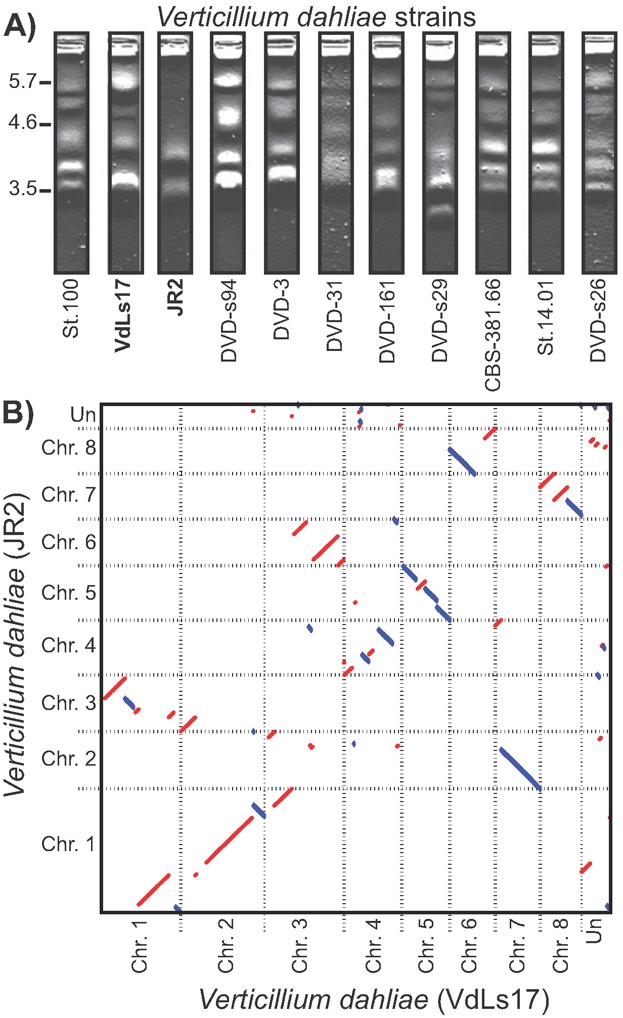
Genomic rearrangements in the asexual fungus Verticillium dahliae. A: Pulsed-field gel electrophoresis shows karyotype variations between 11 V. dahliae strains (bold; strains used in genome alignment). B: Extensive genomic rearrangements between V. dahliae strains JR2 and VdLs17 revealed by whole-genome alignment of the sequenced genomes (forward-forward alignment, red; inversions, blue). Data modified from [11].
A plethora of mechanisms facilitate genome evolution in eukaryotes
Meiotic recombination is a strong driver of genomic diversity by recombining genetic material from two parental lineages. However, various other mechanisms of eukaryotic genome evolution have been described (Fig. 2).
Figure 2.
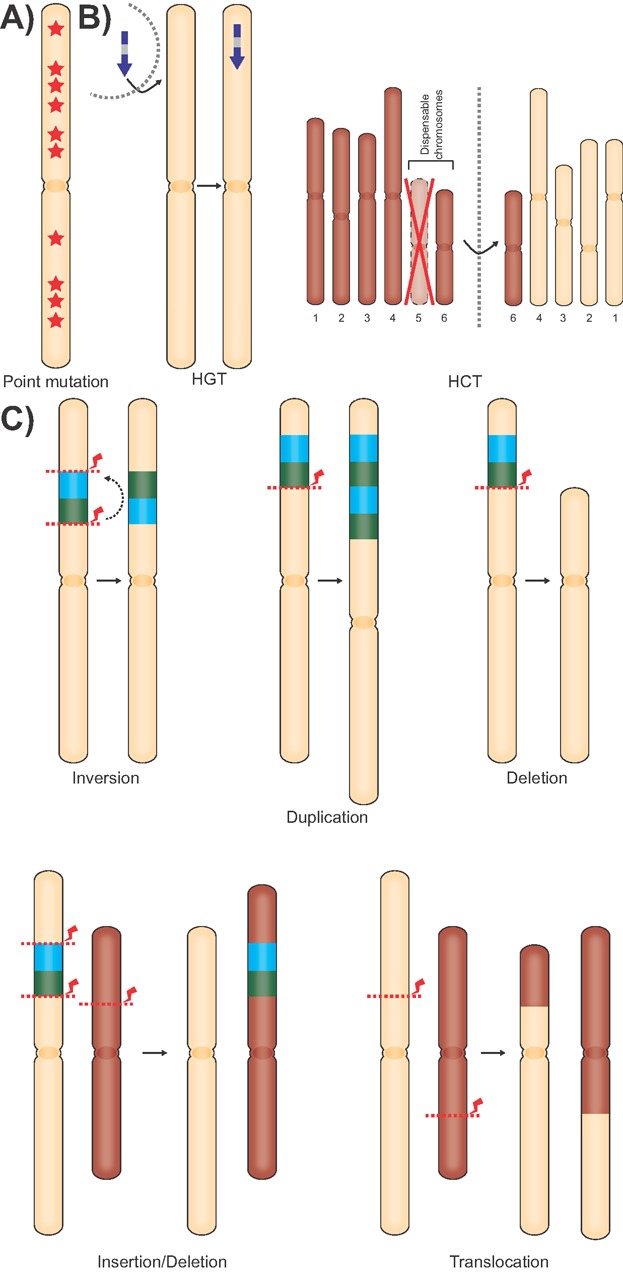
Mechanisms of eukaryotic genome evolution. Several mechanisms can alter the genetic material of eukaryotic species: A: DNA point mutations (stars) can alter single (or few) nucleotides. B: Genetic material can be transferred from a donor lineage over species barriers (dashed line) into the genome of an acceptor lineage. This can either be limited to single genes (horizontal gene transfer; HGT) or comprise entire chromosomes (horizontal chromosome transfer; HCT). C: Inter- or intra-chromosomal rearrangements can lead to a variety of genomic changes. Double-strand breaks (DSBs) are indicated by red arrows and dashed lines.
Replication errors cause single nucleotide polymorphisms
DNA point mutations can be limited to a single or few nucleotides and involve deletions, insertions, and substitutions that are often caused by replication errors (Fig. 2A). The frequency of such mutations is not uniformly dispersed across the genome [16], and accumulation and fixation of (non-synonymous) single nucleotide substitutions has been associated with accelerated evolution. For instance, a single nucleotide polymorphism in Avr4, a gene encoding an effector protein that protects the hyphae of the tomato leaf mould fungus Cladosporium fulvum against the hydrolytic activity of secreted host chitinases, is sufficient to avoid recognition by the immune receptor Cf-4 and reinstall the capability to infect tomato plants [17]. Further evidence that single nucleotide substitutions play an important role in the evolutionary arms race in pathogen-host interaction is provided by the gene encoding the ATR13 effector of the oomycete downy mildew pathogen Hyaloperonospora arabidopsidis and the corresponding immune receptor RPP13 of its host Arabidopsis thaliana, since both are highly polymorphic and under positive diversifying selection [18]. These loci illustrate a co-evolutionary conflict between host and pathogen, where host attempts to intercept the effector are matched by pathogen attempts to evade recognition.
Horizontal DNA transfer can introduce novel traits into genomes
The exchange of genes over species barriers – referred to as horizontal or lateral gene transfer (HGT) – can play an important role in genome evolution (Fig. 2B). A striking example of HGT that affected pathogen evolution is the transfer of the ToxA gene, a host-selective toxin-encoding gene [19], from the wheat pathogen Phaeosphaeria nodorum to Pyrenophora tritici-repentis [20]. This resulted in the ability of P. tritici-repentis to infect wheat and led to the emergence of the wheat spot disease [20]. Using comparative genomics, extensive HGT leading to evolution of the genomes of eukaryotic pathogens has been reported over recent years [21–23]. One example where cross-kingdom transfer from plants to fungi may have occurred is the V. dahliae effector Ave1 that is required for full virulence in tomato. For this effector, only a handful of microbial homologs – in addition to numerous plant homologs – were identified, and their combined phylogeny did not follow species phylogeny [24].
In addition to the transfer of single genes, also the transfer of entire chromosomes has been documented in pathogenic fungi [25,26] (Fig. 2B). This typically involves conditionally dispensable chromosomes that are not required for growth, but confer an advantage when colonizing particular ecological niches. Such dispensable chromosomes have been implicated in pathogenicity of several plant pathogens, such as in Alternaria spp. where they encode biosynthetic genes for host-selective toxins that determine the ability to infect particular plant hosts [27,28]. Horizontal transfer of dispensable chromosomes was experimentally demonstrated in Fusarium oxysporum, a species complex that is composed of non-pathogenic lineages and lineages that cause disease on single to few plant species, collectively affecting a wide range of host plants. Applying comparative genomics to a F. oxysporum strain that causes tomato vascular wilt with Fusarium pathogens of cereals (F. graminearum and F. verticillioides) and pea (F. solani) resulted in the identification of LS genomic regions including four entire chromosomes that are enriched for (candidate) effectors [26]. Through co-incubation of a tomato pathogenic isolate of F. oxysporum and a non-pathogenic isolate, horizontal transfer of LS chromosomes was established, which resulted in pathogenicity on tomato of the strain that was originally non-pathogenic [26]. Thus, horizontal transfer of dispensable chromosomes considerably increases genome plasticity and may contribute to rapid adaptation to novel niches.
Chromosomal rearrangements induce duplications, deletions, inversions, and translocations
The wheat pathogen Mycosphaerella graminicola contains 21 chromosomes of which eight are conditionally dispensable [29,30]. Particularly, the dispensable chromosomes display a high frequency of chromosomal rearrangements and, in the absence of strong positive selection, have been hypothesized to drive divergence [31]. Chromosomal rearrangements induce a multitude of genomic variations such as duplications, deletions, inversions, and translocations, leading to the gain or loss of genomic material [32,33] and are observed in both sexual and asexual fungi [31,34] (Fig. 2C).
Duplication of genetic material immediately leads to genetic redundancy, whereby gene copies can subsequently diverge and evolve toward novel or altered functionality [35] (Fig. 2C). When grown under nutrient limitations, duplications induced by ectopic recombination are commonly observed in yeast [36]. A duplication that occurred independently several times in different populations resulted in additional copies of the high affinity sugar transporters HXT6 and HXT7, which led to increased fitness upon sugar limitation [36,37]. In plant pathogenic fungi and oomycetes, the expansion of a plethora of gene families such as peptidases, glycoside hydrolases, kinases, or transporters has been implicated in the pathogenic lifestyle [38–44].
Loss of genetic material by deletion can reduce the fitness of an organism (Fig. 2C). However, also the opposite situation can occur, for instance when a pathogen loses genes encoding molecules that are recognized by host immune receptors. For example, the deletion of the Avr9 effector gene from the genome of C. fulvum overcomes recognition by tomato plants carrying the Cf-9 immune receptor [45]. Similarly, the Avr-Pita effector of the rice blast pathogen Magnaporthe oryzae is frequently lost to avoid recognition by the Pita immune receptor [46]. Frequent losses, but also duplications, of Avr-Pita genes are mediated by translocations between different chromosomes [47]. These translocations together with transfer of genetic material between strains, provide a mechanism to recover Avr-Pita if the pathogen encounters hosts that lack the Pita immune receptor [47]. Therefore, the dynamic evolution of the Avr-Pita effector provides an interesting example how pathogens can overcome the apparent irreversible loss of genetic material.
Inversions are intra-chromosomal rearrangements that do not necessarily result in loss of genetic material (Fig. 2C). However, inversions can have a considerable impact on meiotic reproduction by reducing recombination frequencies. Firstly, inversions may interfere with the proper pairing of homologous chromosomes during meiosis, and thereby reduce recombination in these regions. Secondly, if meiotic recombination after pairing of homologous chromosomes occurs, chromosomal recombinants may not be stable due to deletions or duplications, which eventually results in suppression of recombination. The suppression of recombination in the region of the inversion fixates gene combinations that otherwise might have been unlinked by meiotic recombination. Together with the reduced homogenizing effect of meiotic recombination between sister chromosomes from different parental lineages, mutations accumulate and lead to divergence in the region of inversion. If genes within the inversion confer adaptation to the local environment, the inversion accumulates and becomes fixed within the population [48,49]. Thus, inversions may increase adaptation, for instance to particular habitats, and support spatial isolation within the population. Moreover, larger inversions can also affect genes involved in reproductive isolation, and, together with enhanced divergence, drive speciation. Inversions, therefore, seem to be strong promoters of reproductive isolation [3,48].
Transposable elements promote genome evolution
The genomes of many eukaryotes contain a high amount of transposable elements (TEs) – DNA sequences that can change their position within the genome. TEs can impact the gene content of a genome by inducing gene knockouts, modulating gene regulation or causing double-strand DNA breaks (DSBs) by TE excision.
In addition to these active roles, the major contribution of TEs toward genome evolution is passive. Non-faithful repair of DSB is facilitated by highly similar sequences such as abundant or propagating TEs. TE-mediated genome rearrangements therefore decrease synteny, as was shown for the rice blast fungus M. oryzae [50,51]. Repair of DSBs that are induced by excision of TEs can cause loss of genetic material [52], which has been hypothesized to cause extensive gene loss, especially of effector genes, in the wheat powdery mildew pathogen Blumeria graminis [52].
Genome rearrangements are not only mediated by TEs, but also by other highly repetitive elements such as micro- and minisatellites; short nucleotide repeats that differ in the length of the repeat unit [53]. Moreover, genome rearrangements have been observed in proximity to highly abundant tRNA genes in yeast [54]. It is yet unclear if the highly repetitive nature of the tRNA genes themselves or the preferred insertion of TE elements in proximity to these tRNA genes is the mediator of ectopic recombination [54,55].
The high abundance and their high levels of similarity make TEs and other repetitive sequences obvious ectopic substrates in naturally occurring DSB repair pathways [56]. Moreover, increasing evidence suggests mobilization of TEs by environmental stresses leading to genome evolution [57].
DNA repair pathways play a central role in the establishment of genomic diversity
Many of the outlined evolutionary mechanisms, and especially the genomic rearrangements, are associated with spontaneous or induced DNA damage that can lead to DSBs. To date, several repair pathways that facilitate repair of DSBs have been characterized (Fig. 3). Repair of DSBs play important roles in fostering genomic diversity during meiotic and mitotic recombination, and errors during these processes can promote extensive genomic rearrangements (Fig. 2).
Figure 3.
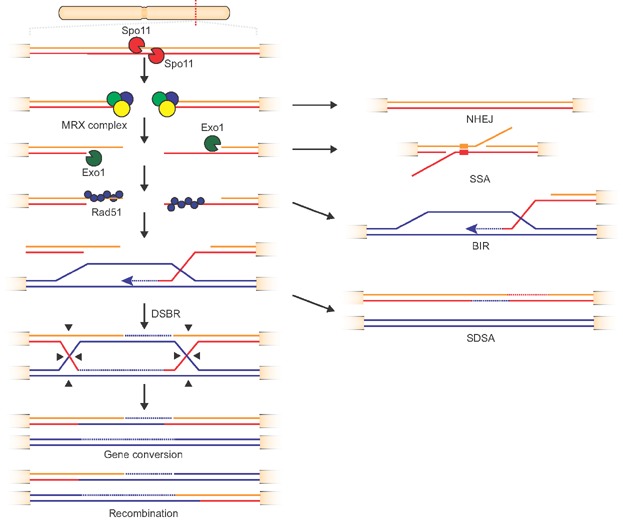
Double-strand break repair pathways. Spontaneous or Spo11-induced double-strand breaks (DSBs) are pre-processed by the MRX complex and repaired by various pathways. Non-homologous end joining (NHEJ) will ligate DSBs. After further processing by the nucleases Exo1 and binding of Rad51, several break repair pathways such as double-strand break repair (DSBR), the synthesis-dependent strand annealing (SDSA), break-induced replication (BIR) and Rad51-independent single-strand annealing (SSA) can operate on DSBs.
DSBs are faithfully repaired by several repair pathways depending on their nature and timing during the cell cycle [58–60]. In vegetative (mitotic) cells, spontaneous DSBs are repaired by homologous recombination (HR) or non-homologous end joining (NHEJ) to maintain genome integrity. In meiosis, DSBs are repaired by HR between sister chromatids, a process that is crucial to generate diversity by recombining genetic information [59]. Moreover, HR plays an additional role by physically linking sister chromatids thereby ensuring correct chromosomal pairing and segregation [59,61]. In contrast to spontaneously induced DSBs, DSBs in meiosis are actively induced by the meiosis-specific topoisomerase-like DNA transesterase protein Spo11 [58,62]. Even though the faithful repair of DSBs and the highly regulated active promotion of DSBs during meiosis is crucial, errors in DSB repair pathways and their regulation can promote ectopic chromosomal rearrangements.
Before the repair of DSBs by HR or NHEJ is initiated, DSBs need to be recognized and pre-processed [63] (Fig. 3). Initially, the 5′ ends of the DNA are trimmed to generate short free 3′ ends on both sides of the breaks by the so-called MRX (Mre11-Rad50-Xrs2) complex, a heterotrimeric protein complex with nuclease activity.
NHEJ is an error-prone mechanism that efficiently ligates ends of DSBs that lack extended sequence similarity [64] (Fig. 3). If more than one DSB occurs, NHEJ can result in deletion, inversion, and translocation within and between chromosomes [65]. NHEJ can occur during the entire cell cycle because it does not require the presence of homologous sequences [64]. However, its activity is suppressed during meiosis to reduce deleterious effects on genome integrity [66]. Interestingly, the yeast Lachancea kluyveri lacks the genes involved in this mechanism and displays a low degree of genomic rearrangements [67].
HR relies on significant regions of homology to faithfully repair DSBs. Therefore, after exposure of short 3′ overhangs of both sides of the break by the MRX complex, the exo-nuclease Exo1 exposes longer stretches of single-stranded DNA by removing nucleotides of one of the strands in 5′–3′ direction (Fig. 3). DSB repair can be directed into several different sub-pathways that differ fundamentally in frequency and outcome between meiotic and mitotic recombination [59]. Whereas cross-overs are the main outcome of meiotic recombination, they are generally rare during mitotic recombination and gene conversion is the predominant outcome [58].
In most of the HR pathways, the highly conserved eukaryotic protein Rad51 binds single-stranded DNA after pre-processing by the MRX complex and the exonuclease Exo1 (Fig. 3). Rad51 promotes pairing of homologous double-strands and the invasion of the template strand (called D-loop) that is used as a primer for DNA synthesis. In the double-strand break repair (DSBR) pathway, Rad51 together with Rad52 promotes capturing of the second DNA strand leading to a mobile cross-over junction between four strands of DNA (so-called double-Holliday junction) [68] that will result either in a cross-over or in gene conversion. During mitosis, the synthesis-dependent strand annealing (SDSA) pathway is evoked more often, since no cross-overs are formed. During SDSA, the initially invading strand can be displaced and subsequently anneal to the sequence on the other side of the DSB. If only a single free DNA end is available, break-induced replication (BIR) is a frequent repair mechanism. In BIR, the D-loop forms a replication fork and uses homologous sequences as template for leading/lagging strand synthesis. Single-strand annealing (SSA), the only pathway that does not utilize Rad51, can operate in the absence of a homologous double strand. SSA aligns homologous stretches within the single-stranded DNA formed by trimming. Subsequently, these can anneal and the breaks can be ligated, frequently causing losses and rearrangements if several independent DSBs occur.
DNA repair is tightly regulated to maintain genome integrity
Due to the deleterious effect of most genomic rearrangements, the detection and repair of DSBs are tightly regulated [59,69]. Not surprisingly, mutations affecting central components of DSB repair pathways are responsible for an increase in genomic rearrangements. Mutations in individual components of the MRX complex increase the rate of chromosomal rearrangements by several hundreds [70]. Similarly, non-deleterious mutations of Rad51 lead to spontaneous chromosomal rearrangements [71].
The chromatin organization and the recruitment of DSB repair proteins are tightly linked since both are determined by histone modification and/or DNA methylation [72,73]. In the yeast Schizosaccharomyces pombe the chromatin structure is altered by the methylation of histones (H3K9), a process that depends on the RNA interference (RNAi) machinery [74]. Mutations in central components of the RNAi machinery result in improper chromosome segregation [74]. In contrast, DNA and histone methylation occurs independent of the RNAi machinery in the filamentous fungus Neurospora crassa [75]. Instead, methylation of AT-rich DNA is promoted by a multi-protein complex known as DCDC (Dim-Cul4-DDB1 Complex) that contains five proteins and is capable of histone methylation (H3K9) [75]. Subsequently, histone methylation is detected and leads to the recruitment of a DNA methyltransferase (Dim-2) that mediates DNA methylation [75]. Mutations in genes encoding DCDC components cause errors in segregation of chromosomes, leading to chromosomal loss, and hypersensitivity to DNA mutagens [75].
TEs are powerful facilitators of evolution but may at the same time be harmful to the individual organism. Therefore, several complementary mechanisms to control TE activity have evolved. In many fungi, duplicated DNA is actively mutated by a process called repeat-induced point mutation (RIP) [76]. RIP components detect duplicated DNA stretches and actively induce C to T point mutations, thereby locally increasing the AT content. The DCDC is recruited to these AT-rich regions leading to the methylation of H3K9 and subsequently to the Dim-2 mediated DNA methylation, and thereby directing heterochromatin formation on these sites [75,77]. In other eukaryotes, TE activity can also be silenced by RNAi at the transcript level [78] as well as by DNA methylation [79]. Mutations modifying the DNA methylation machinery in A. thaliana lead to mobilization of TEs [79].
A structured genome facilitates genome evolution
Eukaryotic genomes are not just a random sequence of genes; they are structured and contain clusters of co-expressed genes or genes that operate in the same process, such as the biosynthesis of particular compounds [80]. Co-expressed neighboring genes are often packed in chromatin structures that are defined by a distinct composition of chromatin binding proteins and likely act as a regulatory unit [81]. Within such chromatin areas chromosomal rearrangements rarely occur, whereas their frequency is higher outside these areas [81]. Moreover, chromosomal rearrangements depend on the spatial organization within chromosomes [82].
Structured genomes are common in eukaryotes and have been described in plants, animals, and fungi [83–85]. Structured genomes are also frequently observed in pathogenic organisms. The oomycete plant pathogen Phytophthora infestans possesses a bipartite genome that contains gene-sparse regions that are transposon-rich and harbor effector genes alongside gene-rich and transposon-sparse regions containing the core genome with its household genes [39,86]. The fungal pathogen Leptosphaeria maculans, the causal agent of stem canker on Brassica, displays a similar genome organization with AT- and transposon-rich regions that contain effector genes [87]. Using a simple evolutionary model with rearrangements mediated by TEs as a mutational operation, it was demonstrated that structured genomes evolve naturally from a random genome over time [88]. These structured genomes contain islands of “core” genes involved in essential functions as well as regions of genes that play a role in adaptation and interaction with the environment. This structure fosters short-term adaptations by increasing the probability of favorable mutations using chromosomal rearrangements [88]. The common occurrence of structured genomes therefore highlights their likely evolutionary advantage to foster adaptations. Dispensable chromosomes that determine pathogenicity in various species can be seen as the most extreme case of genome structuring.
Genome rearrangements foster evolution in the absence of sex
Recent findings suggest that the absence of an obvious sexual lifestyle does not necessarily result in limited genetic diversity, and that in particular genomic rearrangements can foster adaptation (Figs. 1 and 2). For instance, the recently sequenced genome of the persistent (several millions of years) asexual metazoan bdelloid rotifer Adineta vaga revealed a genomic structure that is dominated by genomic rearrangements, mainly through mitotic recombination [15]. Interestingly, this structure is incompatible with meiotic recombination since allelic regions, that normally pair during meiosis, are rearranged; in 20 cases these are found on the same chromosomes [15]. Abundant gene conversion, likely initiated by DSBs, appears to be a mechanism to eliminate deleterious mutations that may otherwise accumulate in the absence of meiotic recombination [15]. Moreover, A. vaga has been subject to HGT as up to 8% of its genes are likely of non-metazoan origin, a frequency as high as reported for some bacteria [15,89]. Frequent genomic rearrangements, gene conversion and HGTs likely contribute to diversification and homogenization in the absence of sex, thereby revealing that sexual reproduction is not required for evolutionary success and persistence [15].
Non-conventional modes of reproduction can drive genetic diversity
Many fungal species have initially been considered to lack a conventional sexual cycle, and were thus seen as strictly asexual (clonal). However, several of these species were later discovered to have cryptic, hence hidden, sexual cycles, or other modes of reproduction [90,91] (Fig. 4). For instance, comparative genomics identified many meiosis-specific genes, as well as the mating type locus that encodes genes that are critically required for sexual reproduction, in the apparently asexual fungus Candida glabrata, suggesting it has a cryptic sexual cycle [92]. Similarly, comparative genomics identified components of the meiotic machinery in apparently asexual mycorrhizal Glomus spp., suggesting that these are cryptic sexual fungal species [93]. In some cases, a sexual cycle has even been observed for apparently asexual species, such as for the saprotrophic and opportunistic mammalian pathogenic fungus Aspergillus fumigatus [94]. In addition to rare, hidden, cycles of sexual reproduction that involve two opposite mating types, non-conventional modes of reproduction are also observed in presumed asexual species. For instance, Candida albicans was considered as an obligate diploid that reproduced strictly asexual, but was later found to undergo parasexual reproduction as well as unisexual reproduction [95,96]. Parasexuality involves the fusion of two hyphae that form a genetically unstable polyploidy nucleus that quickly returns to the initial state by concerted chromosomal loss [97]. Intriguingly, some of the meiosis-specific genes appear to be important for parasexuality, which has been extensively studied under laboratory conditions in C. albicans and can lead to extensive recombination of homologous chromosomes and supernumerary chromosomes [97]. Unisexual reproduction has originally been described for the basidiomycete fungal pathogen Cryptococcus neoformans and involves a regular sexual cycle between partners of the same mating type [98,99] (Fig. 4). Similar to C. albicans, two mating types of C. neoformans are maintained in the population, although the distribution is skewed to one of the two [100]. Unisexual reproduction frequently generates aneuploidy, which can be linked to phenotypic changes and drives adaptation [99]. Interestingly, it has recently been hypothesized that unisexual reproduction may even be the original ancestral form of sexual reproduction to which sexes have evolved later [91]. With these alternative modes of reproduction in mind, one can wonder whether genuinely asexual, and truly clonal, fungal species really exist.
Figure 4.
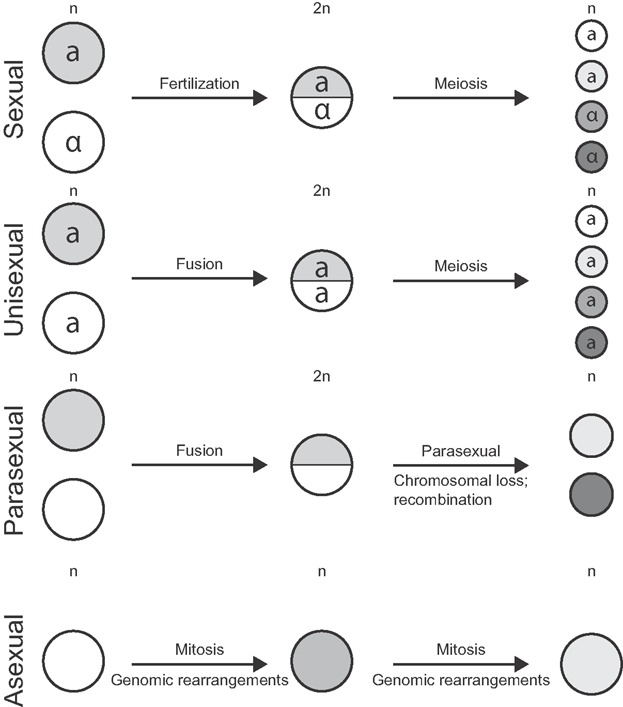
Genomic rearrangements create karyotype diversity. Genomic rearrangements alter the genetic makeup (different shades of cells indicate distinct genetic makeup) and differently foster evolution during sexual, unisexual, parasexual, and asexual reproduction. During sexual reproduction, two cells from opposite mating types (a and α) fuse. Meiotic recombination leads to novel combinations of alleles, and rearrangements can further increase genetic variation. During unisexual reproduction, two cells of the same mating type fuse and meiotic recombination may generate novel allelic combinations. During parasexual reproduction two cells, independent of their mating type, fuse which is occasionally followed by the fusion of nuclei. These fused nuclei are genetically unstable and undergo non-meiotic recombination and chromosomal loss. During asexual reproduction, genomic rearrangements during mitosis, facilitated by repetitive elements and horizontal gene transfer, can generate genetic variation.
Genome rearrangements facilitate adaptation in Verticillium dahliae
V. dahliae is a soil-borne fungal plant pathogen that can infect a broad range of hundreds of host plants through the roots, after which the fungus colonizes the water-transporting xylem vessels to cause wilt disease [101]. Even though two mating types have been described for this fungus, their distribution within the population is heavily skewed toward one of the two, and sexual mating has never been observed in vivo or in vitro [102,103]. Since V. dahliae is a non-motile soil-borne microbe with little to no opportunity to spread, the chance that two opposite mating types encounter each other is extremely limited. Therefore, V. dahliae is considered to be a strictly asexual species that propagates clonally. Nevertheless, sequencing and comparative genomics of 11 V. dahliae strains revealed the occurrence of HGT of the effector gene Ave1 and of extensive inter- and intra-chromosomal rearrangements [11,24] (Fig. 1). These rearrangements have generated highly dynamic LS regions that contain hundreds of genes. Moreover, these LS regions are enriched for in planta-expressed genes. Targeted deletion of several highly in planta-expressed genes compromised fungal aggressiveness, thus providing evidence for a role of LS regions in host colonization and niche adaptation. Therefore, it can be speculated that the genomic plasticity that is established through extensive chromosomal rearrangements drives the evolutionary arms race of this pathogen with one of its hosts, tomato, by facilitating aggressiveness and adaptation.
Similar to C. neoformans, the V. dahliae population shows a heavily skewed mating type distribution, justifying the question whether cryptic sexuality, parasexuality, or even unisexuality may occur. Meiotic recombination during sexual reproduction can generate significant genetic diversity. However, the extent of chromosomal rearrangements as observed between lineages of V. dahliae does not appear to be compatible with a sexual lifestyle [11], unless perhaps with lineages with similar genome structures. Considering the biology of V. dahliae as a soil-borne pathogen that has little to no opportunity to spread, this would entail reproductive isolation, and ultimately perhaps even speciation. Nevertheless, it cannot be excluded that the observed asexual V. dahliae strains continuously emerge from an underlying sexually propagating population. Notably, the diploid crustacean Daphnia pulex occurs as a mixed population containing asexual and sexual lineages. A recent population genomics study revealed that current asexual lineages of D. pulex are young (considerably younger than 1,250 years), exhibit no signs of purging of deleterious mutations, and have high rates of gene conversion and deletion [104]. A rate of mitotic recombination (gene conversion and cross-over) that exceeds the mutation rate is beneficial for asexual taxa due to purging of deleterious and fixation of beneficial mutations [105,106]. However, the high rate of gene conversions and, in particular, gene deletions in the asexual lineages of D. pulex leads to rapid loss of heterozygosity, in turn resulting in the exposure of pre-existing deleterious mutations that lead to genetic deterioration [104,106]. Thus, a high turnover of asexual lineages, which can continuously emerge from sexual relatives, occurs due to decreased longevity of these lineages [104]. A presumably mixed population structure in V. dahliae presents an interesting scenario, since the population would have a high evolutionary potential: sexual reproduction can rapidly create genetic variability and asexual reproduction quickly spreads successfully selected genetic variation, e.g. novel or altered effector genes [10]. Additionally, parasexual as well as unisexual reproduction can create a fair amount of genetic variation, especially in the absence of a compatible mating partner (Fig. 4). However, both types of reproduction involve faithful pairing of homologous chromosomes, which might be compromised by the extensive genomic rearrangements observed between V. dahliae lineages (Fig. 1), and is therefore limited to lineages with similar genomic structures.
So far, cryptic sex or alternative sexual cycles have not yet been observed in V. dahliae. Thus, further understanding of the different molecular and genetic mechanisms that promote genome evolution and genetic diversity, in particular the extensive genomic rearrangements, are required. Furthermore, it still needs to be assessed whether the genome of V. dahliae contains all the genes that are required for meiosis. As Verticillium is a relatively small genus that consists of ten species of soil-born fungi, only a handful of which are successful plant pathogens [107], perhaps the non-pathogenic species display reduced levels of rearrangements and, consequently, do not have the genetic flexibility that allows them to successfully participate in the arms race with plant hosts.
Mechanisms that facilitate genomic rearrangements
The DSB-inducing protein Spo11, which was initially thought to be meiosis-specific, plays a central role in the genetic recombination during the parasexual cycle in C. albicans: deletion of Spo11 compromised recombination, indicating a possible evolutionary connection between these processes [97]. Similarly, a role for Spo11 has been described during the unisexual reproduction of C. neoformans [108]. Interestingly, A. vaga as well as V. dahliae contain a potential ortholog of Spo11 ([15], Seidl MF unpublished). Even though parasexuality as well as unisexuality can generate variability through genetic recombination it has not yet been observed to generate genomic rearrangements as extensive as those observed in V. dahliae. Therefore, reverse genetics using targeted mutagenesis of Spo11, together with other proteins involved in genetic stability and meiotic or mitotic recombination, needs to reveal the role of these components in the establishment of chromosomal rearrangements in V. dahliae.
TEs prompt a danger for asexual organisms since they harbor the potential of unconstrained propagation if not controlled by RNAi, DNA methylation or RIP [15]. Notably, V. dahliae and A. vaga contain only a small repertoire of TEs when compared with other fungi or metazoans [15,41,109]. TEs are not randomly distributed in V. dahliae and occur in clusters, as was similarly found in M. oryzae [41,50,109]. Interestingly, the occurrence of TEs and genes involved in pathogenicity is increased in the proximity of the sites where genomic rearrangements occur [11]. Even though TEs do not occur as frequent as in many sexual propagating pathogens with large genomes [110], they may nevertheless act as the substrate for ectopic rearrangements. Together with a structured genome that includes LS regions that favor chromosomal rearrangements for short-term adaptation, TE-mediated rearrangements are major candidates to drive genome evolution in this pathogen [11,41,88].
Conclusions
We postulate that genomic rearrangements are a driver of adaptation in organisms that are not engaged in conventional sex (Figs. 1 and 4). Although the underlying mechanisms remain unknown, candidate mechanisms can be assigned, as genome integrity depends on the repair of DSBs and mitotic and meiotic recombination, complex processes that are highly linked (Fig. 3). Furthermore, TE activity can mediate genomic rearrangements, which may be regulated by epigenetic modifications such as DNA methylation and histone modification [79]. Central to our hypothesis is the finding that genomic rearrangements in V. dahliae provide an advantage in the evolutionary arms race with its hosts. Verticillium is a relatively small genus that contains only a handful of successful plant pathogenic species [107]. Comparative genomics of multiple strains of pathogenic and non-pathogenic species within the Verticillium genus will reveal the genetic diversity and the occurrence and extent of genomic rearrangements linked to adaptation and virulence. Moreover, systematic population genomic studies complemented with mating experiments should reveal the presence of cryptic sexual, unisexual, or parasexual propagating lineages. Co-evolution experiments involving pathogen lineages with hosts in changing environments might reveal cryptic or alternative sexual cycles and further elucidate their contribution to adaptation. Thus, further research will reveal the molecular mechanisms that allow organisms without a conventional sexual cycle to break evolutionary limitations and foster diversity to adapt to changing environments as well as the time frame wherein they act.
Acknowledgments
The authors acknowledge support by the Research Council for Earth and Life Sciences (ALW) of the Netherlands Organization for Scientific Research (NWO). The authors thank Lidija Berke, David E. Cook, and Ronnie de Jonge for critically reading the manuscript and two anonymous reviewers for their helpful comments and suggestions.
Glossary
- BIR
break-induced replication
- DSB
double-strand DNA break
- DSBR
double-strand break repair
- HGT
horizontal gene transfer
- HR
homologous recombination
- LS
lineage-specific
- NHEJ
non-homologous end joining
- SDSA
synthesis-dependent strand annealing
- SSA
single-strand annealing
- TE
transposable element
References
- 1.Noor MAF, Grams KL, Bertucci LA, Reiland J. Chromosomal inversions and the reproductive isolation of species. Proc Natl Acad Sci USA. 2001;98:12084–8. doi: 10.1073/pnas.221274498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Polakova S, Blume C, Zarate JA, Mentel M, et al. Formation of new chromosomes as a virulence mechanism in yeast Candida glabrata. Proc Natl Acad Sci USA. 2009;106:2688–93. doi: 10.1073/pnas.0809793106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stukenbrock EH. Evolution, selection and isolation: a genomic view of speciation in fungal plant pathogens. New Phytol. 2013;199:895–907. doi: 10.1111/nph.12374. [DOI] [PubMed] [Google Scholar]
- 4.Chisholm ST, Coaker G, Day B, Staskawicz BJ. Host-microbe interactions: shaping the evolution of the plant immune response. Cell. 2006;124:803–14. doi: 10.1016/j.cell.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 5.Thomma BPHJ, Nurnberger T, Joosten MHAJ. Of PAMPs and effectors: the blurred PTI-ETI dichotomy. Plant Cell. 2011;23:4–15. doi: 10.1105/tpc.110.082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Jonge R, Bolton MD, Thomma BP. How filamentous pathogens co-opt plants: the ins and outs of fungal effectors. Curr Opin Plant Biol. 2011;14:400–6. doi: 10.1016/j.pbi.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Bozkurt TO, Schornack S, Banfield MJ, Kamoun S. Oomycetes, effectors, and all that jazz. Curr Opin Plant Biol. 2012;15:483–92. doi: 10.1016/j.pbi.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 8.Jones JDG, Dangl JL. The plant immune system. Nature. 2006;444:323–9. doi: 10.1038/nature05286. [DOI] [PubMed] [Google Scholar]
- 9.de Visser JAGM, Elena SF. The evolution of sex: empirical insights into the roles of epistasis and drift. Nat Rev Genet. 2007;8:139–49. doi: 10.1038/nrg1985. [DOI] [PubMed] [Google Scholar]
- 10.McDonald BA, Linde C. Pathogen population genetics, evolutionary potential, and durable resistance. Annu Rev Phytopathol. 2002;40:349–79. doi: 10.1146/annurev.phyto.40.120501.101443. [DOI] [PubMed] [Google Scholar]
- 11.de Jonge R, Bolton MD, Kombrink A, van den Berg GCM, et al. Extensive chromosomal reshuffling drives evolution of virulence in an asexual pathogen. Genome Res. 2013;23:1271–82. doi: 10.1101/gr.152660.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heitman J, Kronstad JW, Taylor JW, Casselton LA. Sex in fungi: molecular determination and evolutionary implications. Washington, DC: ASM Press; 2007. [Google Scholar]
- 13.Smith JM. Evolution: contemplating life without sex. Nature. 1986;324:300–1. doi: 10.1038/324300a0. [DOI] [PubMed] [Google Scholar]
- 14.Lynch M, Bürger R, Butcher D, Gabriel W. The mutational meltdown in asexual populations. J Hered. 1993;84:339–44. doi: 10.1093/oxfordjournals.jhered.a111354. [DOI] [PubMed] [Google Scholar]
- 15.Flot J-F, Hespeels B, Li X, Noel B, et al. Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga. Nature. 2013;500:453–7. doi: 10.1038/nature12326. [DOI] [PubMed] [Google Scholar]
- 16.Baer CF, Miyamoto MM, Denver DR. Mutation rate variation in multicellular eukaryotes: causes and consequences. Nat Rev Genet. 2007;8:619–31. doi: 10.1038/nrg2158. [DOI] [PubMed] [Google Scholar]
- 17.Joosten MHAJ, Cozijnsen TJ, de Wit PJGM. Host resistance to a fungal tomato pathogen lost by a single base-pair change in an avirulence gene. Nature. 1994;367:384–6. doi: 10.1038/367384a0. [DOI] [PubMed] [Google Scholar]
- 18.Allen RL, Bittner-Eddy PD, Grenville-Briggs LJ, Meitz JC, et al. Host-parasite coevolutionary conflict between Arabidopsis and downy mildew. Science. 2004;306:1957–60. doi: 10.1126/science.1104022. [DOI] [PubMed] [Google Scholar]
- 19.Ciuffetti LM. A single gene encodes a selective toxin causal to the development of tan spot of wheat. Plant Cell. 1997;9:135–44. doi: 10.1105/tpc.9.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesen TL, Stukenbrock EH, Liu Z, Meinhardt S, et al. Emergence of a new disease as a result of interspecific virulence gene transfer. Nat Genet. 2006;38:953–6. doi: 10.1038/ng1839. [DOI] [PubMed] [Google Scholar]
- 21.Richards TA, Talbot NJ. Plant parasitic oomycetes such as Phytophthora species contain genes derived from three eukaryotic lineages. Plant Signal Behav. 2007;2:112–4. doi: 10.4161/psb.2.2.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Richards TA, Soanes DM, Jones MDM, Vasieva O, et al. Horizontal gene transfer facilitated the evolution of plant parasitic mechanisms in the oomycetes. Proc Natl Acad Sci USA. 2011;108:15258–63. doi: 10.1073/pnas.1105100108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gardiner DM, McDonald MC, Covarelli L, Solomon PS, et al. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog. 2012;8:e1002952. doi: 10.1371/journal.ppat.1002952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Jonge R, Peter van Esse H, Maruthachalam K, Bolton MD, et al. Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc Natl Acad Sci USA. 2012;109:5110–5. doi: 10.1073/pnas.1119623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coleman JJ, Rounsley SD, Rodriguez-Carres M, Kuo A, et al. The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 2009;5:e1000618. doi: 10.1371/journal.pgen.1000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma L-J, van der Does HC, Borkovich KA, Coleman JJ, et al. Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature. 2010;464:367–73. doi: 10.1038/nature08850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomma BPHJ. Alternaria spp.: from general saprophyte to specific parasite. Mol Plant Pathol. 2003;4:225–36. doi: 10.1046/j.1364-3703.2003.00173.x. [DOI] [PubMed] [Google Scholar]
- 28.Hu J, Chen C, Peever T, Dang H, et al. Genomic characterization of the conditionally dispensable chromosome in Alternaria arborescens provides evidence for horizontal gene transfer. BMC Genomics. 2012;13:171. doi: 10.1186/1471-2164-13-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wittenberg AHJ, Van der Lee TAJ, Ben M'Barek S, Ware SB, et al. Meiosis drives extraordinary genome plasticity in the haploid fungal plant pathogen Mycosphaerella graminicola. PLoS One. 2009;4:e5863. doi: 10.1371/journal.pone.0005863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodwin SB, Ben M'Barek S, Dhillon B, Wittenberg AHJ, et al. Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 2011;7:e1002070. doi: 10.1371/journal.pgen.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stukenbrock EH, Jørgensen FG, Zala M, Hansen TT, et al. Whole-genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola. PLoS Genet. 2010;6:e1001189. doi: 10.1371/journal.pgen.1001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schubert I. Chromosome evolution. Curr Opin Plant Biol. 2007;10:109–15. doi: 10.1016/j.pbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 33.Schubert I, Lysak MA. Interpretation of karyotype evolution should consider chromosome structural constraints. Trends Genet. 2011;27:207–16. doi: 10.1016/j.tig.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 34.Croll D, McDonald BA. The accessory genome as a cradle for adaptive evolution in pathogens. PLoS Pathog. 2012;8:e1002608. doi: 10.1371/journal.ppat.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Presgraves DC. Evolutionary genomics: new genes for new jobs. Curr Biol. 2005;15:R52–3. doi: 10.1016/j.cub.2004.12.053. [DOI] [PubMed] [Google Scholar]
- 36.Gresham D, Desai MM, Tucker CM, Jenq HT, et al. The repertoire and dynamics of evolutionary adaptations to controlled nutrient-limited environments in yeast. PLoS Genet. 2008;4:e1000303. doi: 10.1371/journal.pgen.1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown CJ, Todd KM, Rosenzweig RF. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol. 1998;15:931–42. doi: 10.1093/oxfordjournals.molbev.a026009. [DOI] [PubMed] [Google Scholar]
- 38.Tyler BM. Phytophthora genome sequences uncover evolutionary origins and mechanisms of pathogenesis. Science. 2006;313:1261–6. doi: 10.1126/science.1128796. [DOI] [PubMed] [Google Scholar]
- 39.Haas BJ, Kamoun S, Zody MC, Jiang RHY, et al. Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature. 2009;461:393–8. doi: 10.1038/nature08358. [DOI] [PubMed] [Google Scholar]
- 40.Spanu PD, Abbott JC, Amselem J, Burgis TA, et al. Genome expansion and gene loss in powdery mildew fungi reveal tradeoffs in extreme parasitism. Science. 2010;330:1543–6. doi: 10.1126/science.1194573. [DOI] [PubMed] [Google Scholar]
- 41.Klosterman SJ, Subbarao KV, Kang S, Veronese P, et al. Comparative genomics yields insights into niche adaptation of plant vascular wilt pathogens. PLoS Pathog. 2011;7:e1002137. doi: 10.1371/journal.ppat.1002137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seidl MF, Van den Ackerveken G, Govers F, Snel B. A domain-centric analysis of oomycete plant pathogen genomes reveals unique protein organization. Plant Physiol. 2011;155:628–44. doi: 10.1104/pp.110.167841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seidl MF, Van den Ackerveken G, Govers F, Snel B. Reconstruction of oomycete genome evolution identifies differences in evolutionary trajectories leading to present-day large gene families. Genome Biol Evol. 2012;4:199–211. doi: 10.1093/gbe/evs003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang RHY, de Bruijn I, Haas BJ, Belmonte R, et al. Distinctive expansion of potential virulence genes in the genome of the oomycete fish pathogen Saprolegnia parasitica. PLoS Genet. 2013;9:e1003272. doi: 10.1371/journal.pgen.1003272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van Kan JA, Van den Ackerveken GF, De Wit PJ. Cloning and characterization of cDNA of avirulence gene avr9 of the fungal pathogen Cladosporium fulvum, causal agent of tomato leaf mold. Mol Plant Microbe Interact. 1991;4:52–9. doi: 10.1094/mpmi-4-052. [DOI] [PubMed] [Google Scholar]
- 46.Orbach MJ, Farrall L, Sweigard JA, Chumley FG, et al. A telomeric avirulence gene determines efficacy for the rice blast resistance gene Pi-ta. Plant Cell. 2000;12:2019–32. doi: 10.1105/tpc.12.11.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chuma I, Isobe C, Hotta Y, Ibaragi K, et al. Multiple translocation of the AVR-Pita effector gene among chromosomes of the rice blast fungus Magnaporthe oryzae and related species. PLoS Pathog. 2011;7:e1002147. doi: 10.1371/journal.ppat.1002147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kirkpatrick M, Barton N. Chromosome inversions, local adaptation and speciation. Genetics. 2006;173:419–34. doi: 10.1534/genetics.105.047985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lowry DB, Willis JH. A widespread chromosomal inversion polymorphism contributes to a major life-history transition, local adaptation, and reproductive isolation. PLoS Biol. 2010;8:e1000500. doi: 10.1371/journal.pbio.1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thon M, Pan H, Diener S, Papalas J, et al. The role of transposable element clusters in genome evolution and loss of synteny in the rice blast fungus Magnaporthe oryzae. Genome Biol. 2006;7:R16. doi: 10.1186/gb-2006-7-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Robberecht C, Voet T, Esteki MZ, Nowakowska BA, et al. Nonallelic homologous recombination between retrotransposable elements is a driver of de novo unbalanced translocations. Genome Res. 2013;23:411–8. doi: 10.1101/gr.145631.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wicker T, Oberhaensli S, Parlange F, Buchmann JP, et al. The wheat powdery mildew genome shows the unique evolution of an obligate biotroph. Nat Genet. 2013;45:1092–6. doi: 10.1038/ng.2704. [DOI] [PubMed] [Google Scholar]
- 53.Piazza A, Serero A, Boulé J-B, Legoix-Né P, et al. Stimulation of gross chromosomal rearrangements by the human CEB1 and CEB25 minisatellites in Saccharomyces cerevisiae depends on G-quadruplexes or Cdc13. PLoS Genet. 2012;8:e1003033. doi: 10.1371/journal.pgen.1003033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gordon JL, Byrne KP, Wolfe KH. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae genome. PLoS Genet. 2009;5:e1000485. doi: 10.1371/journal.pgen.1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chalker DL, Sandmeyer SB. Ty3 integrates within the region of RNA polymerase III transcription initiation. Genes Dev. 1992;6:117–28. doi: 10.1101/gad.6.1.117. [DOI] [PubMed] [Google Scholar]
- 56.Hedges DJ, Deininger PL. Inviting instability: transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat Res. 2007;616:46–59. doi: 10.1016/j.mrfmmm.2006.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Capy P, Gasperi G, Biémont C, Bazin C. Stress and transposable elements: co-evolution or useful parasites. Heredity (Edinb) 2000;85:101–6. doi: 10.1046/j.1365-2540.2000.00751.x. [DOI] [PubMed] [Google Scholar]
- 58.Andersen SL, Sekelsky J. Meiotic versus mitotic recombination: two different routes for double-strand break repair: the different functions of meiotic versus mitotic DSB repair are reflected in different pathway usage and different outcomes. BioEssays. 2010;32:1058–66. doi: 10.1002/bies.201000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012;40:5795–818. doi: 10.1093/nar/gks270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kohl KP, Sekelsky J. Meiotic and mitotic recombination in meiosis. Genetics. 2013;194:327–34. doi: 10.1534/genetics.113.150581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pâques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Keeney S. Spo11 and the formation of DNA double-strand breaks in meiosis. Genome Dyn Stab. 2008;2:81–123. doi: 10.1007/7050_2007_026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mimitou EP, Symington LS. DNA end resection: many nucleases make light work. DNA Repair (Amst) 2009;8:983–95. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Guirouilh-Barbat J, Huck S, Bertrand P, Pirzio L, et al. Impact of the KU80 pathway on NHEJ-induced genome rearrangements in mammalian cells. Mol Cell. 2004;14:611–23. doi: 10.1016/j.molcel.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 66.Joyce EF, Paul A, Chen KE, Tanneti N, et al. Multiple barriers to nonhomologous DNA end joining during meiosis in Drosophila. Genetics. 2012;191:739–46. doi: 10.1534/genetics.112.140996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gordon JL, Byrne KP, Wolfe KH. Mechanisms of chromosome number evolution in yeast. PLoS Genet. 2011;7:e1002190. doi: 10.1371/journal.pgen.1002190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nimonkar AV, Sica RA, Kowalczykowski SC. Rad52 promotes second-end DNA capture in double-stranded break repair to form complement-stabilized joint molecules. Proc Natl Acad Sci USA. 2009;106:3077–82. doi: 10.1073/pnas.0813247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu Rev Biochem. 2008;77:229–57. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 70.Chen C, Kolodner RD. Gross chromosomal rearrangements in Saccharomyces cerevisiae replication and recombination defective mutants. Nat Genet. 1999;23:81–5. doi: 10.1038/12687. [DOI] [PubMed] [Google Scholar]
- 71.Kim TM, Ko JH, Hu L, Kim SA, et al. RAD51 mutants cause replication defects and chromosomal instability. Mol Cell Biol. 2012;32:3663–80. doi: 10.1128/MCB.00406-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.van Attikum H, Gasser SM. Crosstalk between histone modifications during the DNA damage response. Trends Cell Biol. 2009;19:207–17. doi: 10.1016/j.tcb.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 73.Papamichos-Chronakis M, Peterson CL. Chromatin and the genome integrity network. Nat Rev Genet. 2013;14:62–75. doi: 10.1038/nrg3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hall IM, Noma KI, Grewal SIS. RNA interference machinery regulates chromosome dynamics during mitosis and meiosis in fission yeast. Proc Natl Acad Sci USA. 2003;100:193–8. doi: 10.1073/pnas.232688099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lewis ZA, Adhvaryu KK, Honda S, Shiver AL, et al. DNA methylation and normal chromosome behavior in Neurospora depend on five components of a histone methyltransferase complex, DCDC. PLoS Genet. 2010;6:e1001196. doi: 10.1371/journal.pgen.1001196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Selker EU. Repeat-induced gene silencing in fungi. Adv Genet. 2002;46:439–50. doi: 10.1016/s0065-2660(02)46016-6. [DOI] [PubMed] [Google Scholar]
- 77.Lewis ZA, Honda S, Khlafallah TK, Jeffress JK, et al. Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res. 2009;19:427–37. doi: 10.1101/gr.086231.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Buchon N, Vaury C. RNAi: a defensive RNA-silencing against viruses and transposable elements. Heredity (Edinb) 2006;96:195–202. doi: 10.1038/sj.hdy.6800789. [DOI] [PubMed] [Google Scholar]
- 79.Miura A, Yonebayashi S, Watanabe K, Toyama T, et al. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;411:212–4. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- 80.Hurst LD, Pál C, Lercher MJ. The evolutionary dynamics of eukaryotic gene order. Nat Rev Genet. 2004;5:299–310. doi: 10.1038/nrg1319. [DOI] [PubMed] [Google Scholar]
- 81.de Wit E, Braunschweig U, Greil F, Bussemaker HJ, et al. Global chromatin domain organization of the Drosophila genome. PLoS Genet. 2008;4:e1000045. doi: 10.1371/journal.pgen.1000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Filipski J, Mucha M. Structure, function and DNA composition of Saccharomyces cerevisiae chromatin loops. Gene. 2002;300:63–8. doi: 10.1016/s0378-1119(02)00848-x. [DOI] [PubMed] [Google Scholar]
- 83.Cohen BA, Mitra RD, Hughes JD, Church GM. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet. 2000;26:183–6. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- 84.Blumenthal T, Evans D, Link CD, Guffanti A, et al. A global analysis of Caenorhabditis elegans operons. Nature. 2002;417:851–4. doi: 10.1038/nature00831. [DOI] [PubMed] [Google Scholar]
- 85.Williams EJB. Coexpression of neighboring genes in the genome of Arabidopsis thaliana. Genome Res. 2004;14:1060–7. doi: 10.1101/gr.2131104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Raffaele S, Farrer RA, Cano LM, Studholme DJ, et al. Genome evolution following host jumps in the Irish potato famine pathogen lineage. Science. 2010;330:1540–3. doi: 10.1126/science.1193070. [DOI] [PubMed] [Google Scholar]
- 87.Rouxel T, Grandaubert J, Hane JK, Hoede C, et al. Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat-induced point mutations. Nat Commun. 2011;2:202. doi: 10.1038/ncomms1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Crombach A, Hogeweg P. Chromosome rearrangements and the evolution of genome structuring and adaptability. Mol Biol Evol. 2007;24:1130–9. doi: 10.1093/molbev/msm033. [DOI] [PubMed] [Google Scholar]
- 89.Syvanen M. Evolutionary implications of horizontal gene transfer. Annu Rev Genet. 2012;46:341–58. doi: 10.1146/annurev-genet-110711-155529. [DOI] [PubMed] [Google Scholar]
- 90.Heitman J. Evolution of eukaryotic microbial pathogens via covert sexual reproduction. Cell Host Microbe. 2010;8:86–99. doi: 10.1016/j.chom.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feretzaki M, Heitman J. Unisexual reproduction drives evolution of eukaryotic microbial pathogens. PLoS Pathog. 2013;9:e1003674. doi: 10.1371/journal.ppat.1003674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wong S, Fares MA, Zimmermann W, Butler G, et al. Evidence from comparative genomics for a complete sexual cycle in the “asexual” pathogenic yeast Candida glabrata. Genome Biol. 2003;4:R10. doi: 10.1186/gb-2003-4-2-r10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Halary S, Malik SB, Lildhar L, Slamovits CH, et al. Conserved meiotic machinery in Glomus spp., a putatively ancient asexual fungal lineage. Genome Biol Evol. 2011;3:950–8. doi: 10.1093/gbe/evr089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.O'Gorman CM, Fuller HT, Dyer PS. Discovery of a sexual cycle in the opportunistic fungal pathogen Aspergillus fumigatus. Nature. 2009;457:471–4. doi: 10.1038/nature07528. [DOI] [PubMed] [Google Scholar]
- 95.Alby K, Schaefer D, Bennett RJ. Homothallic and heterothallic mating in the opportunistic pathogen Candida albicans. Nature. 2009;460:890–3. doi: 10.1038/nature08252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Miller MG, Johnson AD. White-opaque switching in Candida albicans is controlled by mating-type locus homeodomain proteins and allows efficient mating. Cell. 2002;110:293–302. doi: 10.1016/s0092-8674(02)00837-1. [DOI] [PubMed] [Google Scholar]
- 97.Forche A, Alby K, Schaefer D, Johnson AD, et al. The parasexual cycle in Candida albicans provides an alternative pathway to meiosis for the formation of recombinant strains. PLoS Biol. 2008;6:e110. doi: 10.1371/journal.pbio.0060110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin X, Hull CM, Heitman J. Sexual reproduction between partners of the same mating type in Cryptococcus neoformans. Nature. 2005;434:1017–21. doi: 10.1038/nature03448. [DOI] [PubMed] [Google Scholar]
- 99.Ni M, Feretzaki M, Li W, Floyd-Averette A, et al. Unisexual and heterosexual meiotic reproduction generate aneuploidy and phenotypic diversity de novo in the yeast Cryptococcus neoformans. PLoS Biol. 2013;11:e1001653. doi: 10.1371/journal.pbio.1001653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kwon-Chung KJ, Bennett JE. Distribution of alpha and alpha mating types of Cryptococcus neoformans among natural and clinical isolates. Am J Epidemiol. 1978;108:337–40. doi: 10.1093/oxfordjournals.aje.a112628. [DOI] [PubMed] [Google Scholar]
- 101.Fradin EF, Thomma BPHJ. Physiology and molecular aspects of Verticillium wilt diseases caused by V. dahliae and V. albo-atrum. Mol Plant Pathol. 2006;7:71–86. doi: 10.1111/j.1364-3703.2006.00323.x. [DOI] [PubMed] [Google Scholar]
- 102.Usami T, Itoh M, Amemiya Y. Mating type gene MAT1-2-1 is common among Japanese isolates of Verticillium dahliae. Physiol Mol Plant Pathol. 2008;73:133–7. [Google Scholar]
- 103.Usami T, Itoh M, Amemiya Y. Asexual fungus Verticillium dahliae is potentially heterothallic. J Gen Plant Pathol. 2009;75:422–7. [Google Scholar]
- 104.Tucker AE, Ackerman MS, Eads BD, Xu S, et al. Population-genomic insights into the evolutionary origin and fate of obligately asexual Daphnia pulex. Proc Natl Acad Sci USA. 2013;110:15740–5. doi: 10.1073/pnas.1313388110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mandegar MA, Otto SP. Mitotic recombination counteracts the benefits of genetic segregation. Proc Biol Sci. 2007;274:1301–7. doi: 10.1098/rspb.2007.0056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Xu S, Omilian AR, Cristescu ME. High rate of large-scale hemizygous deletions in asexually propagating Daphnia: implications for the evolution of sex. Mol Biol Evol. 2011;28:335–42. doi: 10.1093/molbev/msq199. [DOI] [PubMed] [Google Scholar]
- 107.Inderbitzin P, Bostock RM, Davis RM, Usami T, et al. Phylogenetics and taxonomy of the fungal vascular wilt pathogen Verticillium, with the descriptions of five new species. PLoS One. 2011;6:e28341. doi: 10.1371/journal.pone.0028341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Feretzaki M, Heitman J. Genetic circuits that govern bisexual and unisexual reproduction in Cryptococcus neoformans. PLoS Genet. 2013;9:e1003688. doi: 10.1371/journal.pgen.1003688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Amyotte SG, Tan X, Pennerman K, del Mar Jimenez-Gasco M, et al. Transposable elements in phytopathogenic Verticillium spp.: insights into genome evolution and inter- and intra-specific diversification. BMC Genomics. 2012;13:314. doi: 10.1186/1471-2164-13-314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raffaele S, Kamoun S. Genome evolution in filamentous plant pathogens: why bigger can be better. Nat Rev Microbiol. 2012;10:417–30. doi: 10.1038/nrmicro2790. [DOI] [PubMed] [Google Scholar]