

Abstract
Mining modern genomics for cancer therapies is predicated on weeding out “bystander” alterations (nonconsequential mutations) and identifying “driver” mutations responsible for tumorigenesis and/or metastasis. We used a direct in vivo RNA interference (RNAi) strategy to screen for genes that upon repression predispose mice to squamous cell carcinomas (SCCs). Seven of our top hits—including Myh9, which encodes nonmuscle myosin IIa—have not been linked to tumor development, yet tissue-specific Myh9 RNAi and Myh9 knockout trigger invasive SCC formation on tumor-susceptible backgrounds. In human and mouse keratinocytes, myosin IIa's function is manifested not only in conventional actin-related processes but also in regulating posttranscriptional p53 stabilization. Myosin IIa is diminished in human SCCs with poor survival, which suggests that in vivo RNAi technology might be useful for identifying potent but low-penetrance tumor suppressors.
Squamous cell carcinomas of the head and neck (HNSCCs) are the sixth most common human cancer worldwide, with frequent, often aggressive recurrence and poor prognosis (1). Established genetic and epigenetic alterations include EGFR amplifications; Trp53, H-RAS, NOTCH, PI3K/Akt, and BRCA1 mutations; and transforming growth factor–β (TGF-β) pathway repression (1). Recent whole-exome sequencing efforts have revealed hundreds of additional “lower-penetrance” HNSCC mutations with unknown functional importance (2, 3).
To functionally test putative “driver mutations” that confer a selective advantage and contribute to tumor progression, researchers have used RNA interference (RNAi) followed by allografting of transduced cultured cancer cells. However, orthotopic transplantations necessitate immunocompromised animals and generate wound responses, which can confound physiological relevance. We have circumvented these difficulties with noninvasive, ultrasound-guided in utero lentiviral-mediated RNAi delivery, selectively transducing single-layered surface ectoderm of living mouse embryos at embryonic day (E) 9.5 (4).
We first showed that mice transduced with Brca1 short hairpin RNAs (shRNAs) that silence BRCA1 expression recapitulate the Brca1-knockout phenotype and develop spontaneous skin and oral SCCs with long latency (5) (fig. S1). We accelerated tumor growth by testing hairpins in epithelial-specific, TGFβ-Receptor-II conditional knockout (cKO) mice (K14-Cre;TβRIIfloxed/floxed), which lose TGF-β signaling in skin, oral, and anogenital epithelia and display enhanced SCC susceptibility (6, 7). On this TβRII-cKO background, Brca1 knockdown generated SCCs with increased frequency and a factor of 4 decrease in latency (fig. S1). Having validated our sensitized approach, we devised a pooled shRNA format to carry out an in vivo screen for additional putative suppressors of SCCs (Fig. 1A).
Fig. 1. Direct in vivo shRNA screen for HNSCC tumor suppressors.

(A) Schematic of pooled shRNA screen. (B) Tumor-free survival of mice of the indicated genotype transduced at E9.5 with the shRNA library targeting putative HNSCC genes (n = number per group; P < 0.0001, log-rank test). (C) Representative pie charts showing percent representation of a particular shRNA within an individual tumor compared to surrounding healthy skin. (D) Top-scoring tumor suppressor candidates and corresponding numbers of tumors showing significant enrichment.
We selected 1762 shRNA lentiviruses targeting 347 mouse genes that either (i) had human orthologs carrying recurring HNSCC somatic mutations (2, 3) or (ii) were deregulated by a factor of ≥2 in tumor-initiating stem cells purified from SCCs initiated by oncogenic HRas (8) (table S1). We also included positive (Brca1 shRNA) and negative (scrambled nontargeting shRNA) controls. After infecting E9.5 epidermis, we isolated E12.5 genomic DNA and verified by Illumina sequencing that shRNA representations correlated nicely with their individual abundance within our initial test pool (fig. S2).
To ensure a coverage of >500 individual clones per shRNA, we infected 74 genotypically matched TβRII-cKO or TβRIIfl/fl control embryos with our lentivirus pool and monitored pups into adulthood. As expected (6), ~5% of the TβRII-cKO mice developed anogenital SCCs. Scrambled shRNA expression did not affect these statistics, nor did transduction with a “control pool” of 1000 random shRNAs. In contrast, all 28 library-transduced TβRII-cKO mice developed lesions within skin, the oral cavity, and/or mucocutaneous junctions at eyelids (Fig. 1B). These findings underscore the efficacy of our approach and document the enrichment of our test shRNA library for SCC tumor suppressors.
Most lesions displayed histopathological features of SCCs with varying degrees of differentiation and local invasion; a few were squamous papillomas or epidermal hyperplastic lesions (figs. S3 to S5). Sequencing revealed that most lesions harbored one or two shRNAs highly enriched relative to initial pool representation and to surrounding healthy skin; gratifyingly, this included the positive control Brca1 shRNA (Fig. 1C). Eight candidate tumor suppressors displayed highly enriched, multiple independent shRNAs in three or more tumors (Fig. 1D). Surprisingly, Trp53 was not among them. Subsequent analyses revealed that the Trp53 hairpins in our pool were inefficient, and when substituted with a more potent Trp53 shRNA, SCCs developed in our assay (fig. S6).
About 40% of tumors were enriched for four out of five shRNAs against Myh9, encoding non-muscle myosin IIa heavy chain (Fig. 1, C and D). Tested individually, Myh9 shRNAs markedly reduced myosin IIa protein, and efficiency correlated strongly with tumor multiplicity (fig. S7). Myh9 shRNA–transduced TβRII-cKO mice displayed an “open eye at birth” phenotype and sparse hair coat, and although initially normal, signs of epidermal hyperproliferation appeared with age (fig. S8).
TβRII-cKO mice transduced with any of three different Myh9 shRNAs developed multiple poorly differentiated myosin IIa–deficient skin SCCs and HNSCCs with median latencies of 3 to 7 months (Fig. 2A). Tumors displayed hallmarks of human SCCs, including invasion into subcutaneous fat, underlying muscle, and salivary glands as well as colonization to the draining lymph nodes (fig. S9). They even formed distant lung metastases (Fig. 2B). Finally, ~30% of TβRIIfl/fl mice (no Cre) transduced with Myh9 shRNAs but not scrambled shRNAs developed skin SCCs after 1 year, indicating that Myh9 loss alone might be sufficient to promote spontaneous tumor development (Fig. 2A).
Fig. 2. Functional validation of Myh9 as a bona fide tumor suppressor.
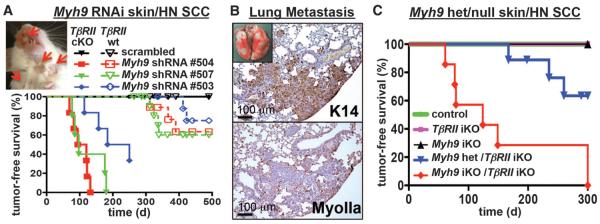
(A) Tumor-free survival of mice of the indicated genotype and shRNA transduction (n > 6 for each genotype, P < 0.0001). Insert shows skin lesions (arrows) on 4-month-old Myh9 shRNA–transduced TβRII-cKO mouse. (B) Myh9 knockdown results in pulmonary metastases in TβRII-cKO mice. Metastatic lesions are immunoreactive for epithelial keratin 14 and negative for myosin IIa. (C) Tumor-free survival of Myh9/TβRII inducible knockout (iKO) as well as Myh9 heterozygous/TβRII iKO and control mice (n = 6, P < 0.001, log-rank test).
To further validate Myh9 as an SCC tumor suppressor, we crossed Myh9fl/fl mice to our epithelial-specific K14-Cre and tamoxifen-regulated K14CreER deleter strains. Embryologically, Myh9-cKO mice recapitulated the “open eye at birth” and hair phenotypes (fig. S10, A to C). In adult mice, inducible deletion of even one Myh9 allele concomitantly with TβRII ablation resulted in multiple invasive SCCs on the back, ears, and anal regions (Fig. 2C and fig. S10, C to E). Control littermates remained tumor-free during this time.
The pronounced invasion and distant metastases were linked to Myh9 knockdown. Indeed, epithelial outgrowth from skin explants was markedly enhanced when TβRIIfl/fl or TβRII-cKO embryos were transduced with Myh9 but not control shRNAs (fig. S11). Similar results were obtained in vitro with scratch-wound and transwell migration as-says independent of TGF-β signaling status. Moreover, reducing Myh9 levels profoundly affected invasion of cells through a Matrigel-coated filter (fig. S12).
These results were consistent with the well-established role for actin-myosin networks in regulating cellular movements. More puzzling was our discovery that, although Myh9 knockdown accelerated tumor development and malignant progression upon TβRII ablation and under conditions favoring HRas mutations (fig. S13, A to E), it had very little effect on tumor latency or multiplicity in mice carrying an epithelial-specific Trp53 gain-of-function mutation (Fig. 3A) (9). This context dependency raised the possibility that myosin IIa deficiency and Trp53 mutations may partially overlap in function—a notion further supported by similar latencies and penetrance of spontaneous SCCs appearing in Myh9 knockdown, Trp53 mutant, and Trp53 mutant–Myh9 knockdown mice (fig. S13F). Such similarities were also observed when Myh9 and Trp53 knockdowns were tested on the SCC-susceptible TβRII-cKO background (fig. S13G).
Fig. 3. A noncanonical role for myosin IIa in nuclear retention of activated p53.

(A) Tumor-free survival of conditional Trp53 mutant mice transduced with the indicated shRNA (n > 6 for each genotype; no significant survival change could be observed). (B) Myh9 knockdown (kd) but not scrambled control shRNA (c) diminishes p53 and targets p21 (CDKN2) levels in response to the DDR inducer doxorubicin. Myosin IIa and GAPDH levels are shown as controls. (C and D) Lack of nuclear p53 in Myh9-cKO versus control (Ctrl) littermate skins 6 hours after γ irradiation (5 Gy). (C) Immunofluorescence (boxed regions show DAPI-stained nuclei in blue); (D) immunoblot analysis. Myosin IIa and GAPDH levels are shown as controls. (E) Quantitative polymerase chain reaction of p53 target genes illustrates the effects of Myh9 knockdown on the p53 pathway. (F) p53 immunoblot of lysates from DDR-induced keratinocytes treated with vehicle (V), blebbistatin (B), Rho kinase inhibitor Y27632 (Y), or latrunculin B (L). GAPDH levels are shown as controls. (G) Nuclear p53 is not retained when DDR-induced Myh9 knockdown primary keratinocytes are exposed to blebbistatin (B). Lamin A/C, IκBα, and γH2AX are controls for nuclear fraction, cytoplasmic fraction, and DDR, respectively. Nuclear export inhibitor leptomycin B rescues the ability of Myh9-deficient cells to retain p53 in the nucleus.
To test for possible epistatic interactions, we treated primary keratinocytes with doxorubicin, which introduces double-strand DNA breaks, thereby triggering the DNA damage response (DDR) pathway and p53 activation (Fig. 3B) (10). Notably, expression of different Myh9 shRNAs in keratinocytes significantly delayed and reduced p53 activation after doxorubicin treatment (Fig. 3B and fig. S14A). This was also true for Myh9fl/fl keratinocytes transduced in vitro with lentiviral Cre and for epidermis from γ-irradiated Myh9-cKO and Myh9 knockdown mice (Fig. 3, C and D, and fig. S14, B to D).
Relative to controls, Myh9-deficient keratinocytes also failed to induce p53-responsive genes such as p21, Fas, Bax, Mdm2, and 14-3-3σ (Fig. 3, B, D, and E). These effects were specific to the p53 pathway, because control and Myh9 knockdown keratinocytes responded equally to other stimuli such as epidermal growth factor (EGF) (fig. S15). Moreover, such effects were not observed with shRNAs against other non-muscle myosin II genes [Myh10 (myosin IIb) and Myh14 (myosin IIc)], nor were they dependent on TGF-β signaling (fig. S16).
Probing deeper, we discovered that the myosin II adenosine triphosphatase (ATPase) inhibitor blebbistatin phenocopied Myh9 loss-of-function effects on DDR-induced p53 activation (Fig. 3F and fig. S17A). Consistent with a role for myosin IIa's ATPase function, inhibition of Rho kinase, an upstream regulator of myosin II's ATPase activity, similarly dampened the DNA damage–induced p53 response. However, latrunculin-mediated inhibition of F-actin polymerization did not elicit these effects, raising the tantalizing possibility that they may be independent of myosin IIa's conventional role (Fig. 3F and fig. S17B).
The initial steps of the DDR were unperturbed, as judged by stress-induced phosphorylation of the histone variant H2AX and activation of DNA checkpoint kinases Chk1 and Chk2 (fig. S17B). Additionally, Myh9 ablation did not affect Trp53 transcription (Fig. 3E). However, in the presence of proteasome inhibitor MG132, p53 protein levels were comparably induced by DDR in both Myh9 knockdown and control keratinocytes (fig. S17C).
Seeking how myosin IIa deficiency might affect p53 stability, we discovered that p53's nuclear accumulation after DNA damage did not occur upon blebbistatin treatment (Fig. 3G). Inhibiting the nuclear export receptor Crm1 restored nuclear p53 accumulation as well as transactivation of p53 target genes such as p21 (Fig. 3G and fig. S17D). Thus, when myosin IIa is defective, even though the p53 pathway can be induced in response to DNA damage, it fails to do so because of an inability of p53 to remain and/or accumulate in the nucleus.
Given the possible clinical relevance of nuclear export inhibitors as a means to overcome p53 effects in myosin IIa–defective tumors, we verified that p53 activation is similarly compromised upon myosin IIa inhibition in primary human keratinocytes (Fig. 4A). We then surveyed myosin IIa's status in >350 human skin, HNSCC, and control tissues. In contrast to normal and hyperplastic skin, which consistently displayed strong immunolabeling, 24% of skin SCCs and 31% of HNSCCs showed weak or no immunolabeling (Fig. 4B and fig. S18A). Myosin IIa was diminished in a number of early-stage (grade I) SCCs, which suggests that its loss may constitute an early event in tumor progression (fig. S18B). Additionally, when skin SCCs were analyzed according to TβRII and P-Smad2 status (6), a substantial fraction (~83%) of myosin IIa–negative tumors showed signs of concomitant loss of TGF-β signaling (fig. S18C). An initial analysis of The Cancer Genome Atlas (TCGA) showed that low MYH9 mRNA expression significantly correlated with reduced survival of HNSCC patients (Fig. 4C), whereas patients with high MYH9 mRNA expression or MYH9 amplifications showed no survival changes (fig. S19, A to C). As larger patient cohorts are analyzed, it will be interesting to see whether this correlation proves diagnostically relevant.
Fig. 4. MYH9 is a bona fide tumor suppressor in human HNSCC.
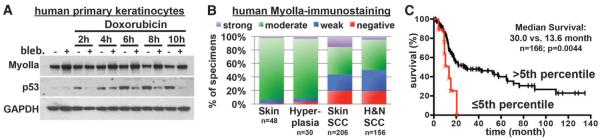
(A) p53 induction in primary human keratinocytes treated with the myosin ATPase inhibitor blebbistatin and with the DDR inducer doxorubicin. GAPDH levels are shown as loading control. (B) Myosin IIa quantifications on 362 samples of human healthy skin, skin SCCs, and HNSCCs. A substantial fraction of cases show absent or reduced myosin IIa expression. (C) Decreased MYH9 mRNA expression correlates with shortened survival. Kaplan-Meier analysis compares overall survival of TCGA HNSCC patients stratified according to the lowest (≤5th percentile) MYH9 expression versus the rest (>5th percentile) (n = 166, P = 0.0044, log-rank test).
Among 302 sequenced HNSCCs, 16 non-synonymous MYH9 mutations surfaced. Computational analyses of evolutionary conservation patterns yielded a functional impact score (FIS), which assigns a probability that a specific amino acid mutation will alter protein function (11). Intriguingly, 15 out of 16 of these MYH9 mutations gave a high or medium FIS score (fig. S20), and when all TCGA mutations were ranked according to FIS, MYH9 ranked 16th among the ~15,000 genes mutated in HNSCCs (P = 0.000026, false discovery rate q = 0.024) and MYH9 was found to be mutated above background also in some other cancers (fig. S21A and tables S2 and S3).
Although this cohort of cancer-associated MYH9 mutations is relatively small, they showed statistically significant clustering to the highly conserved ATPase switch II region (fig. S20A; P = 0.0015). One conserved mutation (Ala454) was already known to compromise ATPase activity in Dictyostelium myosin II (12). Using site-directed point mutagenesis, we further corroborated the relevance of several other human mutants. Notably, although MYH9 Glu475 → Lys and Glu530 → Lys mutants retained their ability to localize to stress fibers, they exerted dominant-negative effects on DDR-induced p53 activation (fig. S22).
Given that Myh9 heterozygosity increased SCC susceptibility in mouse epithelia (Fig. 2C), it was also intriguing that ~15% of all HNSCCs showed hemizygous MYH9 loss. Monoallelic MYH9 loss was also common (~30%) in several other cancers (table S4). That said, no significant correlation existed between MYH9 hemizygosity or MYH9 mutations and poor HNSCC prognosis (fig. S19, D and E). Hence, although provocative, these findings await further experimental and clinical evaluation.
Finally, contrary to our expectations, MYH9 mutations in human HNSCCs were not mutually exclusive with Trp53 mutations (mostly gain-of-function). Although such mutual exclusiveness with oncogenic alteration of established p53 regulators (e.g., MDM2 amplifications) can exist (e.g., among glioblastomas), this paradigm does not apply to HNSCCs (fig. S23). We speculate that in HNSCCs, p53 gain-of-function mutations could confer additional effects beyond inactivation of wild-type p53, such as metastasis promotion.
Like any RNAi screen, our strategy relied on knockdown efficiency and hence invariably missed some relevant genes (e.g., Trp53). It also missed such genes as Notch1, which functions nonautonomously in SCC progression (13). These caveats aside, this methodology evaluated function within the native environment, prior to gross perturbations in tissue architecture that invariably accompany cancer progression. This unearthed some lower-penetrance genes hitherto not associated with cancer. At first glance, the inverse relation between a well-known actin-based mechano-sensitive motor and metastatic SCCs was puzzling, as dominant active Rho kinase and/or extracellular matrix stiffness can promote transformation in some cell lines and animal models (14). Although primary human cancer cells are considerably more pliable (15), and indeed our results document transforming potential, myosin IIa's most striking link to cancer appears to be in a newfound role in regulating p53 stabilization and nuclear retention. These findings highlight the utility of in vivo RNAi (16) to integrate cancer genomics and mouse modeling for rapid discovery, validation, and functional characterization of potent but low-penetrance tumor suppressors.
Supplementary Material
Acknowledgments
We thank members of the Fuchs laboratory for helpful discussions, especially C. Luxenburg for helpful experimental suggestions and S. Dewell for deep sequencing; S. Karlsson for TβRIIfl/fl mice; and T. Golub for sharing HNSCC data with us prior to publication. Special thanks go to the TCGA patients, the TCGA Program, and the cBIO Portal for Cancer Genomics. D.S. is an Emerald Foundation Young Investigator. D.S. was supported by the American Association for Cancer Research and the Austrian Program for Advanced Research and Technology of the Austrian Academy of Sciences. D.S. and A.S. are fellows of the Human Frontier Science Program Organization. E.F. is an Investigator of the Howard Hughes Medical Institute. Supported by NIH grant R37-AR27883 (E.F.) and Emerald Foundation Inc. The Rockefeller University has applied for a patent regarding the use of nuclear export inhibitors for the treatment of human squamous cell carcinomas, with D.S. and E.F. as inventors.
Footnotes
Supplementary Materials www.sciencemag.org/content/343/6168/309/suppl/DC1
References and Notes
- 1.Leemans CR, Braakhuis BJ, Brakenhoff RH. Nat. Rev. Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 2.Stransky N, et al. Science. 2011;333:1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agrawal N, et al. Science. 2011;333:1154–1157. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beronja S, Livshits G, Williams S, Fuchs E. Nat. Med. 2010;16:821–827. doi: 10.1038/nm.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berton TR, et al. Oncogene. 2003;22:5415–5426. doi: 10.1038/sj.onc.1206825. [DOI] [PubMed] [Google Scholar]
- 6.Guasch G, et al. Cancer Cell. 2007;12:313–327. doi: 10.1016/j.ccr.2007.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu SL, et al. Genes Dev. 2006;20:1331–1342. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schober M, Fuchs E. Proc. Natl. Acad. Sci. U.S.A. 2011;108:10544–10549. doi: 10.1073/pnas.1107807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caulin C, et al. J. Clin. Invest. 2007;117:1893–1901. doi: 10.1172/JCI31721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schramek D, et al. Nat. Genet. 2011;43:212–219. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- 11.Reva B, Antipin Y, Sander C. Nucleic Acids Res. 2011;39:e118. doi: 10.1093/nar/gkr407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murphy CT, Rock RS, Spudich JA. Nat. Cell Biol. 2001;3:311–315. doi: 10.1038/35060110. [DOI] [PubMed] [Google Scholar]
- 13.Demehri S, Turkoz A, Kopan R. Cancer Cell. 2009;16:55–66. doi: 10.1016/j.ccr.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DuFort CC, Paszek MJ, Weaver VM. Nat. Rev. Mol. Cell Biol. 2011;12:308–319. doi: 10.1038/nrm3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fritsch A, et al. Nat. Phys. 2010;6:730–732. [Google Scholar]
- 16.Beronja S, et al. Nature. 2013;501:185–190. doi: 10.1038/nature12464. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.