

Abstract
We present a novel fragment-based approach that tackles some of the challenges for chemical biology of predicting protein function. The general approach, which we have termed biofragments, comprises two key stages. First, a biologically relevant fragment library (biofragment library) can be designed and constructed from known sets of substrate-like ligands for a protein class of interest. Second, the library can be screened for binding to a novel putative ligand-binding protein from the same or similar class, and the characterization of hits provides insight into the basis of ligand recognition, selectivity, and function at the substrate level. As a proof-of-concept, we applied the biofragments approach to the functionally uncharacterized Mycobacterium tuberculosis (Mtb) cytochrome P450 isoform, CYP126. This led to the development of a tailored CYP biofragment library with notable 3D characteristics and a significantly higher screening hit rate (14 %) than standard drug-like fragment libraries screened previously against Mtb CYP121 and 125 (4 % and 1 %, respectively). Biofragment hits were identified that make both substrate-like type-I and inhibitor-like type-II interactions with CYP126. A chemical-fingerprint-based substrate model was built from the hits and used to search a virtual TB metabolome, which led to the discovery that CYP126 has a strong preference for the recognition of aromatics and substrate-like type-I binding of chlorophenol moieties within the active site near the heme. Future catalytic analyses will be focused on assessing CYP126 for potential substrate oxidative dehalogenation.
Keywords: biofragments, cytochrome P450, ligand binding, protein models, tuberculosis
Introduction
Predicting the functional roles of proteins remains a major challenge in chemical biology.[1] Advances in high-throughput genome sequencing techniques have seen the rate at which new proteins are identified far exceed that at which they can be biochemically characterized, and it has been reported that less than one percent of proteins have experimentally validated annotations.[1b, 2] The traditional techniques used to assign protein function primarily involve a combination of bioinformatics-based methods, for example, sequence similarity to previously characterized proteins, genomic context, transcriptional patterns, or experimental phenotypes of deletion or knockdown mutants.[1b–d] Newer computational 3D-structure-based methods, involving protein modeling and alignment to find structural similarities (globally or at known or predicted functional sites) and virtual ligand docking, have expanded in recent years but are not yet widely used, and the reliability of docking is also still in question.[1a, 3] For instance, in Mycobacterium tuberculosis (Mtb), the pathogen responsible for tuberculosis (TB) disease, there are 3933 protein-coding genes.[4] Many of these genes are assumed to have essential functions, such as in DNA replication, transcription, translation, and cell-division but this annotation is only on the basis of homologues from other bacteria,[5] and this technique has led to several cases of misassignment.[6] About one third of all Mtb gene products have no functional data at all, assigned as unknown or conserved hypothetical proteins, and without functional classification.[5] For example, in the proline-glutamate (PE)/proline-proline-glutamate (PPE) protein family, which represents about 10 % of the Mtb proteome, enzymatic activity has been demonstrated only for LipY (Rv3097c) as a triacylglycerol hydrolase.[7]
Given these challenges, novel approaches for establishing protein function are very much needed. We attempted to transfer the technique of fragment-based ligand discovery (FBLD) to this field. FBLD is now an established method for developing small-molecule ligands as chemical tools and leads for drug development.[3b, 8] At its heart, this method involves the structure-guided design and synthesis of potent ligands from weak-binding low-molecular-weight fragment molecules (typically <250 Da).[3b, 8] There are two primary advantages to this method: first, because of the low complexity of small fragments, a significantly larger proportion of chemical space can be explored with a relatively small fragment library (usually 102–103 fragments), compared with the approximately 105–106 larger molecules (Mw 300–500 Da) typical in a high-throughput screen (HTS).[3b, 8] Second, fragment hits must make high-quality interactions with the target to bind with sufficient affinity for detection.[3b The quality of these interactions is shown quantitatively by hits having high ligand efficiency (where ligand efficiency (LE) equals the negative ΔG of binding divided by the number of non-hydrogen atoms (NHA) in the fragment).[9]
Herein, we describe a novel fragment-based approach for predicting the function of putative ligand-binding proteins, a method which we have termed “biofragments”. This approach encompasses two main phases: the first phase is the design and construction of more biologically relevant fragment libraries (biofragment libraries) based on known sets of substrate-like ligands for a specific protein class of interest. This step is essential because of the well-recognized disparity between commercial fragment libraries and natural products—commercial fragments have a structural makeup that is largely biased toward readily available small, flat, heterocyclic molecules, whereas natural products have a prevalence of stereogenic centers and even include reactive functional groups.[3b, 10] In the first step of the method, a set of known ligands of both the protein of interest (where available) as well as other related proteins is compiled. Subsequently, a fragment library is assembled that samples the substructural chemical space present in the ligands and is hence expected to increase fragment hit rates. This approach uses the concept of chemogenomics,[10d, e, 11] which maps chemical space to biological space in a systematic manner. Herein, we have applied this method to fragments, both in the library design phase and in the step of selecting putative substrate scaffolds.
The second main phase is the fragment-based screening[3b of the biofragment library for binding to an uncharacterized protein from the same or similar family from which the biofragments were derived. The characterization of hits (at a pharmacophore level or from determination of their exact structural-binding mode) provides insights into the structural determinants for ligand recognition, the probable structural characteristics of endogenous substrate(s), and hence insight into the functional role of the enzyme. In this regard, it has been shown previously that fragments made by breaking down known endogenous ligands display considerable fidelity of their binding mode and interactions, and can provide an understanding of the substructural energetic contributions for binding, to identify hot spots at protein–ligand binding sites.[12]
As a proof-of-concept, our biofragments approach was applied to the functionally uncharacterized Mtb cytochrome P450 enzyme, CYP126. Cytochrome P450 enzymes have a remarkably diverse repertoire of possible catalytic reactions and substrates,[13] making them an ideal model to test our approach. CYP126 (Rv0778) is located near essential Mtb genes encoding enzymes involved in the de novo biosynthesis of purine, and CYP126 is also part of a putative operon with a probable adenylosuccinate lyase, PurB.[5a] However, CYP126 also shares notable homology (35 % identity) with the Mtb cholesterol hydroxylases CYP124 and 125, and it is highly conserved across actinobacteria (including both pathogenic and nonpathogenic strains), which suggests that it may participate in an important general function.[14] Further information about the function of CYP126 is, at this stage, not available, which made it a suitable test system for our approach.
To investigate CYP126, we constructed a CYP biofragments library and, through fragment-based screening, biofragment hits were identified that make both substrate-like type-I and inhibitor-like type-II interactions with the enzyme. A chemical-fingerprint-based substrate model was built from the hits and used to search a virtual TB metabolome, this led to the discovery that CYP126 preferentially binds chlorophenol scaffolds in a substrate-like fashion, close to the heme, and suggests that its endogenous substrate may maintain a similar motif upon further interrogation.
Results and Discussion
Design, construction, and analysis of a CYP biofragment library
A CYP-focused biologically relevant fragment library (“CYP biofragment library”) was designed starting from knowledge of CYP ligands found in the protein data bank (PDB;[15] Figure 1). The PDB was used so that structural binding information would be available for comparison to potential biofragment-bound crystal structures. All PDB CYP ligands were extracted and ions, salts, gases, buffer additives, heme groups or other metal complexes, and azoles were removed, along with any ligands labeled as drugs or inhibitors. The in silico filtering process retained 43 bacterial and 24 eukaryotic CYP substrates and substrate-like/mimetic ligands (see Table S1 in the Supporting Information), and these were grouped into six major structural classes (see Figure S1).
Figure 1.
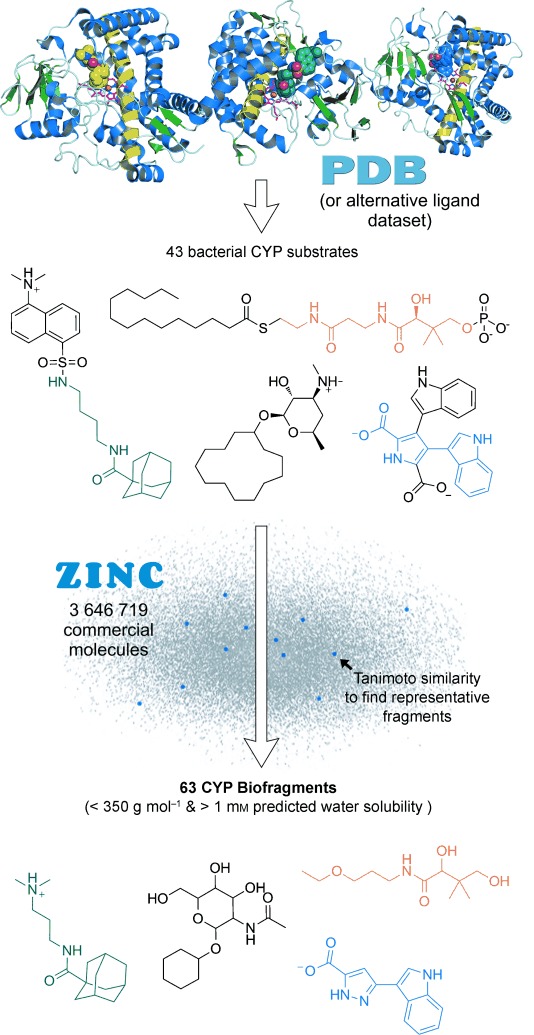
Outline of the biofragment library design process, as used to build a novel CYP biofragment library. Representative CYP biofragments are shown comprising aromatic and aliphatic rings, cages, and chains. The PDB was employed here, but an alternative ligand databank could also be used.
Next, a database of commercially available fragment molecules was created by compiling the ChemBridge, ChemDiv, Enamine, Life Chemicals, Ryan Scientific, and Specs supplier datasets (3 646 719 unique compounds) from the ZINC repository,[16] and filtered for those molecules with a predicted solubility in water greater than 1 mm[17] and a molecular weight Mw<250 Da or Mw<350 Da. The two most similar commercial fragments representing each bacterial CYP ligand (henceforth termed CYP biofragments) were then found by passing the fragment database over the ligand database and calculating the Tanimoto similarity coefficient[18] for every combination. CYP biofragments that would not be amenable to 1D 1H NMR spectroscopy screening (i.e. no downfield proton NMR signal >2 ppm) were manually excluded, and the biofragments were purchased from their respective suppliers. In total, 63 biofragments were compiled in this way for experimental screening.
The composition of the new biofragment library was compared to a conventional commercial drug-like fragment library in regards to physicochemical properties and chemical diversity, using analytical procedures reported previously[10f (Figure 2). Percentage frequency distributions of the libraries by Mw and c log P (calculated logarithm of partition coefficient) show similarities in size and hydrophilicity based on the fragments rule-of-three constraints used in the construction of both libraries (Figure 2 A,B). A bimodal distribution is observed for the Mw of the biofragments (Figure 2 A), and this is probably reflective of the two commercially available fragment databases that were combined during the process of biofragment design (Mw<250 or <350 Da). Significant contrast was observed when plotting the library distributions of the ratio of sp3- to sp2-hybridized carbons in each molecule (Figure 2 C). The traditional commercial fragments contain a high proportion of sp2 carbons, primarily from aromatics, but the CYP biofragments are richer in sp3 centers. A principal moment-of-inertia (PMI) plot[10f, 19] corroborates this finding (Figure 2 D), showing the conventional fragments lying heavily in 2D space, whereas the biofragments have more uniform coverage of molecular shape and structural diversity (by visual inspection). This is a direct consequence of designing the library to resemble nonaromatic natural CYP ligands (see Figure 1 and Figure S1). This theme also appears in the context of drug discovery, where more challenging drug targets (e.g., protein–protein interactions) have pushed efforts towards improving the structural diversity of in-house fragment libraries and also towards increasing the three-dimensional structures available.[3b, 10c, 11] Overall, the above analysis affirms the differing properties of biologically relevant fragment libraries that originate from the first phase in the biofragments approach; the importance of this will be shown later by significantly increased hit rates.
Figure 2.
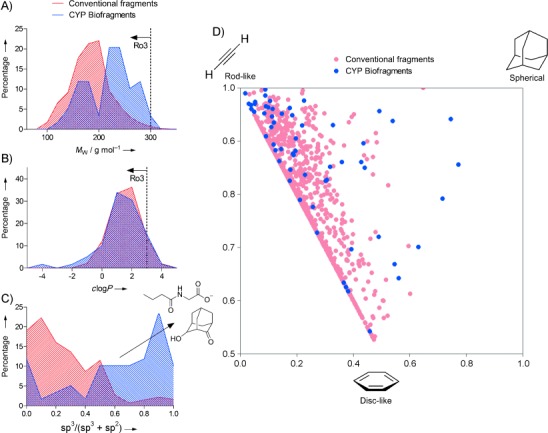
Chemical diversity and physicochemical properties of the CYP biofragments in comparison to a traditional drug-like fragment library. A)–C) Percentage frequency distributions of the two libraries by Mw, c log P, and the ratio of sp3- to sp2-hybridized carbon atoms in each incorporated molecule. Dashed lines show rule-of-three (Ro3) cut-offs. The Mw and c log P distributions are similar, but the biofragments are significantly richer in sp3 centers (arrow in (C)). Exemplar biofragments with several sp3-hybridized carbon atoms are illustrated. D) Rotational inertia similarity (principal moment-of-inertia, PMI) analysis of the libraries. The areas most similar to a true rod, disc, or sphere are indicated. Note the biofragments have significantly more uniform coverage of 3D molecular space than the conventional library fragments.
Biofragment screening by NMR spectroscopy
The 63 CYP biofragments were screened for binding to CYP126 by CPMG,[20] STD,[21] and WaterLOGSY[22] ligand-detected 1D 1H NMR spectroscopy. The antifungal agent, ketoconazole, is known to bind to CYP126 in a typical azole–heme type-II coordination mode by using heme absorbance shift assays (Kd=1.4 μm; K. J. McLean et al., unpublished results), and ketoconazole was used here in displacement experiments to indicate hits that might bind within the CYP126 active site (displacement in CPMG only). Nine hits were identified in total by NMR that were displaced by ketoconazole from the active site (Figure 3 A,B). This represents a hit rate for the screen of 14 %, which is significantly higher than for a standard commercial drug-like fragment library that was screened previously against Mtb CYP121 and 125 (4 and 1 %, respectively[23]). This confirms that biologically related small molecules would be more likely to bind and be recognized by biomacromolecular targets.[10a] The hits identified by ketoconazole displacement were all aromatic compounds, suggesting that CYP126 might preferentially recognize aromatic moieties within its catalytic site.
Figure 3.
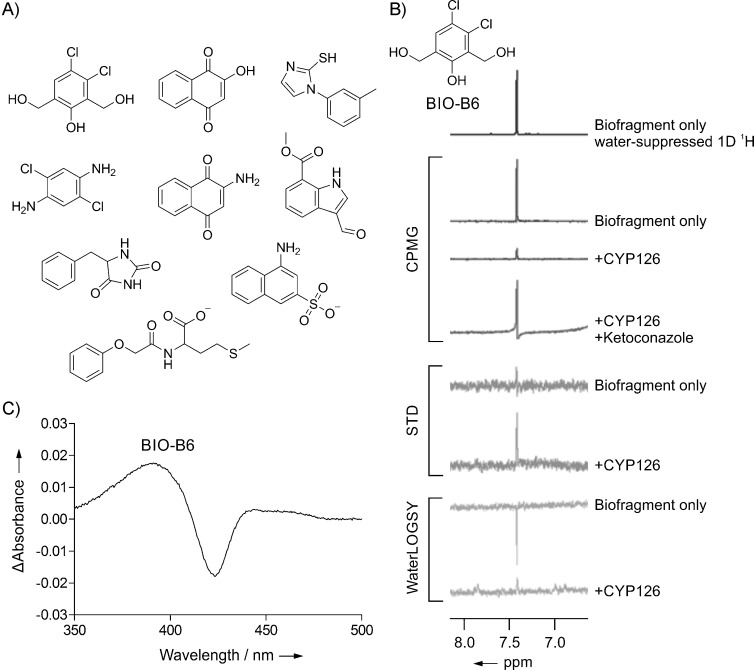
CYP biofragment screening against Mtb CYP126 by NMR spectroscopy and heme-absorbance-shift assay. A) Nine biofragment hits displaceable by ketoconazole were identified by using CPMG, STD, and WaterLOGSY NMR screening experiments. B) NMR spectra for the representative biofragment hit BIO-B6 (1 mm) in the presence and absence of CYP126 (15 μm) or CYP126 (15 μm) plus ketoconazole (250 μm). Only the fragment resonances in the aromatic region are shown. The decreased (CPMG) or increased (STD and WaterLOGSY) biofragment signals in the presence of CYP126 indicates protein binding, and this interaction is reduced by the addition of ketoconazole (CPMG only). C) Absorbance difference spectra for CYP126 (5 μm) with biofragment hit BIO-B6 (1 mm) inducing a type-I blue shift in the Soret absorbance band of the heme.
Biofragment screening by heme absorbance shift
To give further information on the type of interaction the biofragment NMR hits make within the CYP126 active site and whether they bind in close proximity to the heme, a spectrophotometric heme-absorbance-shift assay[13a, 23a] was performed for all nine hits. Two biofragments (BIO-A7 and BIO-B10) were found to induce a type-II red shift in the CYP126 heme Soret absorbance peak (likely from heme-coordination; see Figure S2), and a single hit (BIO-B6) gave a substrate-like type-I blue shift (Figure 3 C). Looking at the structures of these hits, we envisaged that BIO-A7 could coordinate the heme iron atom through its arylamine[23a] and BIO-B10 possibly through the lone pair of the thioether sulfur atom.[24] It is intriguing that CYP126 could support a thioether coordination mode because this has been noted as a rather unusual CYP binding interaction.[24] Of greatest interest, however, is the type-I shift induced by the chlorophenol BIO-B6, which is typically associated with substrate-dependent displacement of the weakly bound resting water molecule from the distal position of the CYP heme iron atom for the first step of the CYP catalytic cycle.[13a] There are only four chlorophenols in our traditional drug-like fragment library (0.30 % of the total library size), which illustrates the difficultly of reaching the same conclusion from a conventional library. Subsequent efforts were focused on exploring the basis of this BIO-B6–CYP126 interaction. In particular, we attempted to experimentally characterize the BIO-B6–CYP126 structural binding interaction by crystallography, but we were only able to obtain native CYP126 crystals with the enzymatic active site occluded by a dimer interface (K. J. McLean et al., unpublished results).
Construction, search, and selection of a virtual TB metabolome and screening by using a heme-absorbance-shift assay
We hypothesized that fingerprint-based characterization of the biofragment hits (particularly BIO-B6) could be used to provide insights into possible classes of substrate for CYP126 based on previously known TB metabolites. A virtual TB metabolome was constructed from the KEGG PATHWAY database[25] and searched by Tanimoto similarity to BIO-B6. An additional Naïve Bayes substrate model based on Molprint2D fingerprints of all nine NMR hits was constructed[26] and used as a secondary search. Twenty-three TB metabolite matches (0.60 % of the total metabolome) were then screened against CYP126 by heme-absorbance-shift assay as described above for the original biofragment hits (Figure 4). This screen identified a substrate-like type-I hit, pentachlorophenol TB23, and a chloroaniline type-II hit, TB8 (Figure 4 B and Figure S3). A complete titration heme-absorbance-shift assay was performed for TB23, successfully confirming it has high binding affinity and ligand efficiency (Figure 4 B; Kd=150 μm, LE=0.43 kcal mol−1 NHA−1). Both of these metabolites are aromatic chlorobenzenes, like biofragments BIO-B6 and BIO-A7, which further supports an apparent preference by CYP126 for binding this type of motif close to the heme and implies that this scaffold could be consistent in the endogenous substrate. BIO-B6 and TB23, which induce a substrate-like type-I shift, are both chlorophenols. The selection of the negatively charged pentachlorophenol TB23 over the other similarly assayed non-heme-coordinating chlorophenols and benzyl alcohols is also intriguing (Figure 4 A), which indicates that the additional electron-withdrawing chlorine atoms promote CYP126 active-site recognition. While an exact pentachlorophenol metabolite pathway has not been explicitly reported for Mtb, the pathway appears as part of the Mtb H37Rv KEGG dataset, based on extension of the well-known chlorocyclohexane and chlorobenzene biodegradation processes found in microorganisms from diverse environments.[27] In this pathway, pentachlorophenol is dehalogenated oxidatively to tetrachlorohydroquinone.[27b, d, 28] This metabolic process has recently been shown to be CYP-mediated in the white rot fungus, Phanerochaete chrysosporium,[28a] by human cytochrome P450 3A4,[28b and CYP involvement is also suspected in Mucor ramosissimus[29] and Mycobacterium chlorophenolicum.[30]
Figure 4.
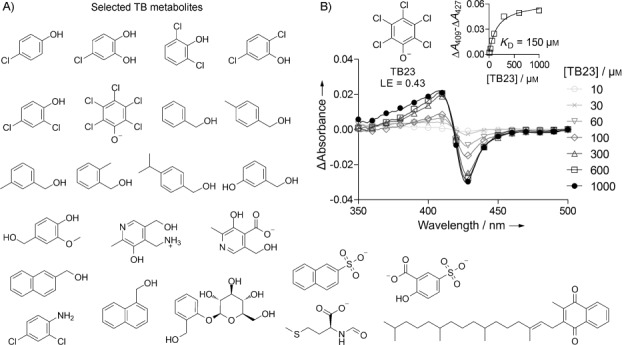
TB metabolite screening against Mtb CYP126 by heme-absorbance-shift assay. A) Commercially available TB metabolites matched from the fingerprints represented by the CYP biofragment NMR hits. Only those matches that were viable for screening by heme absorbance shift (see the Experimental Section) are shown. Chemical formulas are drawn in their predominant protonation state at physiological pH. B) Absorbance difference spectra for CYP126 (5 μm) titrated with various concentrations of TB23. The substrate-like type-I blue shift in the Soret absorbance band of the heme induced by TB23 is shown as a ΔAmax at 409 nm and ΔAmin at 427 nm in the difference spectra. Inset: the shift in the absorbance band of the heme (quantified as ΔAmax−ΔAmin) as a function of TB23 concentration (squares) with the one-site binding equilibrium model fitted for calculating the Kd (line).
Conclusions
Herein, we presented a novel fragment-based approach, biofragments, to help address the fundamental problem in biology of assigning function to proteins. The approach has two main stages: first, a biologically relevant fragment library (biofragment library) is constructed based on known sets of substrate-like ligands for the protein class of interest. Second, the biofragment library is screened (fragment-based screening cascade) for binding to a novel putative ligand-binding protein from that class. The characterization of hits provides insight into the basis of ligand recognition, selectivity, and function. We applied this approach to the functionally uncharacterized Mtb CYP isoform, CYP126. The designed CYP biofragment library had notable 3D characteristics and its screening hit rate against CYP126 (of 14 %) was significantly higher than for the conventional drug-like fragment libraries versus Mtb CYP121 and 125 (which were 4 and 1 %, respectively[23]). Overall, the biofragment hits and their follow-up TB metabolites (found by a chemical fingerprint-based substrate model search of a virtual TB metabolome), indicate a strong preference for the recognition of aromatics by CYP126 and the substrate-like type-I binding of chlorophenol moieties within the active site near the heme. Future studies will assess CYP126 for potential substrate oxidative dehalogenation or similar CYP-mediated reactions.[13a, 27b, 28a, 31] These findings confirm our hypothesis that biologically related fragments are more likely to bind to bio-macromolecules, and suggest that biofragments could also be used to improve the hit rates for screens against traditionally difficult target classes, such as protein–protein interactions or structured nucleic acids.[3b In summary, we conclude that the biofragments approach is a novel method to deconvolute protein function, adding to the previously established value of fragment-based approaches in drug discovery.
Experimental Section
All biological and computational experimental methods are given in the Supporting Information.
Acknowledgments
We acknowledge funding from the EC (as part of the NM4TB project) and the BBSRC (grants BB/I019227/1 to C.A. and BB/I019669/1 to A.W.M.). S.A.H. was supported by a Sir Mark Oliphant Cambridge Australia Scholarship awarded by the Cambridge Trusts. E.H.M. was supported by the NIH-Oxford-Cambridge Scholars Program. This research was supported in part by the Intramural Research Program of the NIH, NIAID. A.B. thanks Unilever for funding.
Supporting Information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re-organized for online delivery, but are not copy-edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
miscellaneous_information
References
- 1a.Lee D, Redfern O, Orengo C. Nat. Rev. Mol. Cell Biol. 2007;8:995. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 1b.Gabaldón T, Huynen MA. Cell. Mol. Life Sci. 2004;61:930. doi: 10.1007/s00018-003-3387-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c.Eisenberg D, Marcotte EM, Xenarios I, Yeates TO. Nature. 2000;405:823. doi: 10.1038/35015694. [DOI] [PubMed] [Google Scholar]
- 1d.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Science. 1999;285:751. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 2.Barrell D, Dimmer E, Huntley RP, Binns D, O'Donovan C, Apweiler R. Nucleic Acids Res. 2009;37:D396. doi: 10.1093/nar/gkn803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3a.Pierri CL, Parisi G, Porcelli V. Biochim. Biophys. Acta Proteins Proteomics. 2010;1804:1695. doi: 10.1016/j.bbapap.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 3b.Scott DE, Coyne AG, Hudson SA, Abell C. Biochemistry. 2012;51:4990. doi: 10.1021/bi3005126. [DOI] [PubMed] [Google Scholar]
- 3c.Anand P, Sankaran S, Mukherjee S, Yeturu K, Laskowski R, Bhardwaj A, Bhagavat R, Consortium O, Brahmachari SK, Chandra N. PLoS One. 2011;6:e27044. doi: 10.1371/journal.pone.0027044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4a.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, et al. Nature. 1998;393:537. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 4b.Camus JC, Pryor MJ, Medigue C, Cole ST. Microbiology. 2002;148:2967. doi: 10.1099/00221287-148-10-2967. [DOI] [PubMed] [Google Scholar]
- 5a.Lew JM, Kapopoulou A, Jones LM, Cole ST. Tuberculosis. 2011;91:1. doi: 10.1016/j.tube.2010.09.008. [DOI] [PubMed] [Google Scholar]
- 5b.Mehaffy MC, Kruh-Garcia NA, Dobos KM. J. Proteome Res. 2012;11:17. doi: 10.1021/pr2008658. [DOI] [PubMed] [Google Scholar]
- 6a.Larrouy-Maumus G, Biswas T, Hunt DM, Kelly G, Tsodikov OV, de Carvalho LP. Proc. Natl. Acad. Sci. USA. 2013;110:11320. doi: 10.1073/pnas.1221597110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6b.Zuo Y, Deutscher MP. Nucleic Acids Res. 2001;29:1017. doi: 10.1093/nar/29.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7a.Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. J. Biol. Chem. 2006;281:3866. doi: 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b.Akhter Y, Ehebauer MT, Mukhopadhyay S, Hasnain SE. Biochimie. 2012;94:110. doi: 10.1016/j.biochi.2011.09.026. [DOI] [PubMed] [Google Scholar]
- 8.Davies TG, Hyvönen M, editors. Fragment-Based Drug Discovery and X-Ray Crystallography. Heidelberg: Springer; 2012. [Google Scholar]
- 9a.Kuntz ID, Chen K, Sharp KA, Kollman PA. Proc. Natl. Acad. Sci. USA. 1999;96:9997. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b.Hopkins AL, Groom CR, Alex A. Drug Discovery Today. 2004;9:430. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 10a.Larsson J, Gottfries J, Muresan S, Backlund A. J. Nat. Prod. 2007;70:789. doi: 10.1021/np070002y. [DOI] [PubMed] [Google Scholar]
- 10b.Lovering F, Bikker J, Humblet C. J. Med. Chem. 2009;52:6752. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 10c.Over B, Wetzel S, Grütter C, Nakai Y, Renner S, Rauh D, Waldmann H. Nat. Chem. 2013;5:21. doi: 10.1038/nchem.1506. [DOI] [PubMed] [Google Scholar]
- 10d.van der Horst E, Peironcely JE, IJzerman AP, Beukers MW, Lane JR, van Vlijmen HW, Emmerich MT, Okuno Y, Bender A. BMC Bioinf. 2010;11:316. doi: 10.1186/1471-2105-11-316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10e.Bender A, Young DW, Jenkins JL, Serrano M, Mikhailov D, Clemons PA, Davies JW. Comb. Chem. High Throughput Screening. 2007;10:719. doi: 10.2174/138620707782507313. [DOI] [PubMed] [Google Scholar]
- 10f.Hung AW, Ramek A, Wang Y, Kaya T, Wilson JA, Clemons PA, Young DW. Proc. Natl. Acad. Sci. USA. 2011;108:6799. doi: 10.1073/pnas.1015271108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Graaf C, Vischer HF, de Kloe GE, Kooistra AJ, Nijmeijer S, Kuijer M, Verheij MH, England PJ, van Muijlwijk-Koezen JE, Leurs R, de Esch IJP. Drug Discovery Today. 2013;18:323. doi: 10.1016/j.drudis.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 12a.Ciulli A, Williams G, Smith AG, Blundell TL, Abell C. J. Med. Chem. 2006;49:4992. doi: 10.1021/jm060490r. [DOI] [PubMed] [Google Scholar]
- 12b.Stout TJ, Sage CR, Stroud RM. Structure. 1998;6:839. doi: 10.1016/S0969-2126(98)00086-0. [DOI] [PubMed] [Google Scholar]
- 13a.Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. 3rd. New York: Kluwer Academic/Plenum; 2005. [Google Scholar]
- 13b.Hudson SA, McLean KJ, Munro AW, Abell C. Biochem. Soc. Trans. 2012;40:573. doi: 10.1042/BST20120062. [DOI] [PubMed] [Google Scholar]
- 14.Ouellet H, Johnston JB, Ortiz de Montellano PR. Arch. Biochem. Biophys. 2010;493:82. doi: 10.1016/j.abb.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bernstein FC, Koetzle TF, Williams GJ, Meyer EF, Jr, Brice MD, Rodgers JR, Kennard O, Shimanouchi T, Tasumi M. J. Mol. Biol. 1977;112:535. doi: 10.1016/s0022-2836(77)80200-3. [DOI] [PubMed] [Google Scholar]
- 16.Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. J. Chem. Inf. Model. 2012;52:1757. doi: 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warr WA. J. Comput.-Aided Mol. Des. 2012;26:801. doi: 10.1007/s10822-012-9577-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crisman TJ, Bender A, Milik M, Jenkins JL, Scheiber J, Sukuru SC, Fejzo J, Hommel U, Davies JW, Glick M. J. Med. Chem. 2008;51:2481. doi: 10.1021/jm701314u. [DOI] [PubMed] [Google Scholar]
- 19.Sauer WH, Schwarz MK. J. Chem. Inf. Comput. Sci. 2003;43:987. doi: 10.1021/ci025599w. [DOI] [PubMed] [Google Scholar]
- 20.Hajduk PJ, Olejniczak ET, Fesik SW. J. Am. Chem. Soc. 1997;119:12257. [Google Scholar]
- 21.Mayer M, Meyer B. Angew. Chem. 1999;111:1902. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 1999;38:1784. [Google Scholar]
- 22.Dalvit C, Pevarello P, Tato M, Veronesi M, Vulpetti A, Sundstrom M. J. Biomol. NMR. 2000;18:65. doi: 10.1023/a:1008354229396. [DOI] [PubMed] [Google Scholar]
- 23a.Hudson SA, McLean KJ, Surade S, Yang YQ, Leys D, Ciulli A, Munro AW, Abell C. Angew. Chem. 2012;124:9445. doi: 10.1002/anie.201202544. [DOI] [PubMed] [Google Scholar]
- Angew. Chem. Int. Ed. 2012;51:9311. [Google Scholar]
- 23b.Hudson SA, Surade S, Coyne AG, McLean KJ, Leys D, Munro AW, Abell C. ChemMedChem. 2013;8:1451. doi: 10.1002/cmdc.201300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martell JD, Li H, Doukov T, Martasek P, Roman LJ, Soltis M, Poulos TL, Silverman RB. J. Am. Chem. Soc. 2010;132:798. doi: 10.1021/ja908544f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25a.Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. Nucleic Acids Res. 2012;40:D109. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b.Kanehisa M, Goto S. Nucleic Acids Res. 2000;28:27. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26a.Bender A, Mussa HY, Glen RC, Reiling S. J. Chem. Inf. Comput. Sci. 2004;44:170. doi: 10.1021/ci034207y. [DOI] [PubMed] [Google Scholar]
- 26b.Bender A, Mussa HY, Glen RC, Reiling S. J. Chem. Inf. Comput. Sci. 2004;44:1708. doi: 10.1021/ci0498719. [DOI] [PubMed] [Google Scholar]
- 27a.Camacho-Pérez B, Ríos-Leal E, Rinderknecht-Seijas N, Poggi-Varaldo HM. J. Environ. Manage. 2012;95:S306. doi: 10.1016/j.jenvman.2011.06.047. [DOI] [PubMed] [Google Scholar]
- 27b.Hackett JC, Sanan TT, Hadad CM. Biochemistry. 2007;46:5924. doi: 10.1021/bi700365x. [DOI] [PubMed] [Google Scholar]
- 27c.Kao CM, Prosser J. J. Hazard. Mater. 1999;69:67. doi: 10.1016/s0304-3894(99)00060-6. [DOI] [PubMed] [Google Scholar]
- 27d.Fetzner S, Lingens F. Microbiol. Rev. 1994;58:641. doi: 10.1128/mr.58.4.641-685.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28a.Ning D, Wang H. PLoS One. 2012;7:e45887. doi: 10.1371/journal.pone.0045887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b.Mehmood Z, Williamson MP, Kelly DE, Kelly SL. Chemosphere. 1996;33:759. doi: 10.1016/0045-6535(96)00212-3. [DOI] [PubMed] [Google Scholar]
- 29.Szewczyk R, Dlugonski J. Int. Biodeterior. Biodegrad. 2009;63:123. [Google Scholar]
- 30.Uotila JS, Salkinoja-Salonen MS, Apajalahti JH. Biodegradation. 1991;2:25. doi: 10.1007/BF00122422. [DOI] [PubMed] [Google Scholar]
- 31.Isin EM, Guengerich FP. Biochim. Biophys. Acta Gen. Subj. 2007;1770:314. doi: 10.1016/j.bbagen.2006.07.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
miscellaneous_information