

Abstract
Protein quantification without isotopic labels has been a long-standing interest in the proteomics field. However, accurate and robust proteome-wide quantification with label-free approaches remains a challenge. We developed a new intensity determination and normalization procedure called MaxLFQ that is fully compatible with any peptide or protein separation prior to LC-MS analysis. Protein abundance profiles are assembled using the maximum possible information from MS signals, given that the presence of quantifiable peptides varies from sample to sample. For a benchmark dataset with two proteomes mixed at known ratios, we accurately detected the mixing ratio over the entire protein expression range, with greater precision for abundant proteins. The significance of individual label-free quantifications was obtained via a t test approach. For a second benchmark dataset, we accurately quantify fold changes over several orders of magnitude, a task that is challenging with label-based methods. MaxLFQ is a generic label-free quantification technology that is readily applicable to many biological questions; it is compatible with standard statistical analysis workflows, and it has been validated in many and diverse biological projects. Our algorithms can handle very large experiments of 500+ samples in a manageable computing time. It is implemented in the freely available MaxQuant computational proteomics platform and works completely seamlessly at the click of a button.
Mass-spectrometry-based proteomics has become an increasingly powerful technology not only for the identification of large numbers of proteins, but also for their quantification (1–3). Modern mass spectrometer hardware, in combination with increasingly sophisticated bioinformatics software for data analysis, is now ready to tackle the proteome on a global, comprehensive scale and in a quantitative fashion (4–6).
Stable isotope-based labeling methods are the gold standard for quantification. However, despite their success, they inherently entail extra preparation steps, whereas label-free quantification is by its nature the simplest and most economical approach. Label-free quantification is in principle applicable to any kind of sample, including materials that cannot be directly metabolically labeled (for instance, many clinical samples). In addition, there is no limit on the number of samples that can be compared, in contrast to the finite number of “plexes” available for label-based methods (7).
A vast literature on label-free quantification methods, reviewed in Ref. 3 and Refs. 8–13, and associated software projects (14–31) already exist. These computational methods include simple additive prescriptions to combine peptide intensities (32, 33), reference-peptide-based estimates (34), and statistical frameworks utilizing additive linear models (35, 36). However, major bottlenecks remain: Most methods require measurement of samples under uniform conditions with strict adherence to standard sample-handling procedures, with minimal fractionation and in tight temporal sequence. Also, many methods are tailored toward a specific biological question, such as the detection of protein interactions (37), and are therefore not suitable as generic tools for quantification at a proteome scale. Finally, the modest accuracy of their quantitative readouts relative to those obtained with stable-isotope-based methods often prohibits their use for biological questions that require the detection of small changes, such as proteome changes upon stimulus.
Metabolic labeling methods such as SILAC1 (38) excel because of their unparalleled accuracy and robustness, which are mainly due to stability with regard to variability in sample processing and analysis steps. When isotope labels are introduced early in the workflow, samples can be mixed, and any sample-handling issues equally affect all proteins or peptides. This allows complex biochemical workflows without loss of quantitative accuracy. Conversely, any up-front separation of proteins or peptides potentially poses serious problems in a label-free approach, because the partitioning into fractions is prone to change slightly in the analysis of different samples. Chemical labeling (39–41) is in principle universally applicable, but because the labels are introduced later in the sample processing, some of the advantages in robustness are lost. Depending on the label used, it can also be uneconomical for large studies.
High mass resolution and accuracy and high peptide identification rates have been key ingredients in the success of isotope-label-based methods. These factors contribute similarly to the quality of label-free quantification. An increased identification rate directly improves label-free quantification because it increases the number of data points and allows “pairing” of corresponding peptides across runs. Although high mass accuracy aids in the identification of peptides (42), it is the high mass resolution that is crucial to accurate quantification. This is because the accurate determination of extracted ion currents (XICs) of peptides is critical for comparison between samples (43). At low mass resolution, XICs of peptides are often contaminated by nearby peptide signals, preventing accurate intensity readouts. In the past, this has led many researchers to use counts of identified MS/MS spectra as a proxy for the ion intensity or protein abundance (44). Although the abundance of proteins and the probability of their peptides being selected for MS/MS sequencing are correlated to some extent, XIC-based methods should clearly be superior to spectral counting given sufficient resolution and optimal algorithms. These advantages are most prominent for low-intensity protein/peptide species, for which a continuous intensity readout is more information-rich than discrete counts of spectra. Therefore, we here apply the term “label-free quantification” strictly to XIC-based approaches and not to spectral counting.
In this manuscript, we describe the MaxLFQ algorithms, part of the MaxQuant software suite, that solve two of the main problems of label-free protein quantification. We introduce “delayed normalization,” which makes label-free quantification fully compatible with any up-front separation. Furthermore, we implemented a novel approach to protein quantification that extracts the maximum ratio information from peptide signals in arbitrary numbers of samples to achieve the highest possible accuracy of quantification.
MaxLFQ is a generic method for label-free quantification that can be combined with standard statistical tests of quantification accuracy for each of thousands of quantified proteins. MaxLFQ has been available as part of the MaxQuant software suite for some time and has already been successfully applied to a variety of biological questions by us and other groups. It has delivered excellent performance in benchmark comparisons with other software solutions (31).
EXPERIMENTAL PROCEDURES
Proteome Benchmark Dataset
An Escherichia coli K12 strain was grown in standard LB medium, harvested, washed in PBS, and lysed in BugBuster (Novagen Merck Chemicals, Schwalbach, Germany) according to the manufacturer's protocol. HeLa S3 cells were grown in standard RPMI 1640 medium supplemented with glutamine, antibiotics, and 10% FBS. After being washed with PBS, cells were lysed in cold modified RIPA buffer (50 mm Tris-HCl, pH 7.5, 1 mm EDTA, 150 mm NaCl, 1% N-octylglycoside, 0.1% sodium deoxycholate, complete protease inhibitor mixture (Roche)) and incubated for 15 min on ice. Lysates were cleared by centrifugation, and after precipitation with chloroform/methanol, proteins were resuspended in 6 m urea, 2 m thiourea, 10 mm HEPES, pH 8.0. Prior to in-solution digestion, 60-μg protein samples from HeLa S3 lysates were spiked with either 10 μg or 30 μg of E. coli K12 lysates based on protein amount (Bradford assay). Both batches were reduced with dithiothreitol and alkylated with iodoacetamide. Proteins were digested with LysC (Wako Chemicals, GmbH, Neuss, Germany) for 4 h and then trypsin digested overnight (Promega, GmbH, Mannheim, Germany). Digestion was stopped by the addition of 2% trifluroacetic acid. Peptides were separated by isoelectric focusing into 24 fractions on a 3100 OFFGEL Fractionator (Agilent, GmbH, Böblingen, Germany) as described in Ref. 45. Each fraction was purified with C18 StageTips (46) and analyzed via liquid chromatography combined with electrospray tandem mass spectrometry on an LTQ Orbitrap (Thermo Fisher) with lock mass calibration (47). All raw files were searched against the human and E. coli complete proteome sequences obtained from UniProt (version from January 2013) and a set of commonly observed contaminants. MS/MS spectra were filtered to contain at most eight peaks per 100 mass unit intervals. The initial MS mass tolerance was 20 ppm, and MS/MS fragment ions could deviate by up to 0.5 Da (48). For quantification, intensities can be determined alternatively as the full peak volume or as the intensity maximum over the retention time profile, and the latter method was used here as the default. Intensities of different isotopic peaks in an isotope pattern are always summed up for further analysis. MaxQuant offers a choice of the degree of uniqueness required in order for peptides to be included for quantification: “all peptides,” “only unique peptides,” and “unique plus razor peptides” (42). Here we chose the latter, because it is a good compromise between the two competing interests of using only peptides that undoubtedly belong to a protein and using as many peptide signals as possible. The distribution of peptide ions over fractions and samples is shown in supplemental Fig. S1.
Dynamic Range Benchmark Dataset
The E. coli K12 strain was grown in standard LB medium, harvested, washed in PBS, and lysed in 4% SDS, 100 mm Tris, pH 8.5. Lysates were briefly boiled and DNA sheared using a Sonifier (Branson Model 250). Lysates were cleared by centrifugation at 15,000 × g for 15 min and precipitated with acetone. Proteins were resuspended in 8 m urea, 25 mm Tris, pH 8.5, 10 mm DTT. After 30 min of incubation, 20 mm iodoacetamide was added for alkylation. The sample was then diluted 1:3 with 50 mm ammonium bicarbonate buffer, and the protein concentration was estimated via tryptophan fluorescence emission assay. After 5 h of digestion with LysC (Wako Chemicals) at room temperature, the sample was further diluted 1:3 with ammonium bicarbonate buffer, and trypsin (Promega) digestion was performed overnight (protein-to-enzyme ratio of 60:1 in each case). E. coli peptides were then purified by using a C18 Sep Pak cartridge (Waters, Milford, MA) according to the manufacturer's instructions. UPS1 and UPS2 standards (Sigma-Aldrich) were resuspended in 30 μl of 8 m urea, 25 mm Tris, pH 8.5, 10 mm DTT and reduced, alkylated, and digested in an analogous manner, but with a lower protein-to-enzyme ratio (12:1 for UPS1 and 10:1 for UPS2, both LysC and trypsin). UPS peptides were then purified using C18 StageTips. E. coli and UPS peptides were quantified based on absorbance at 280 nm using a NanoDrop spectrophotometer (Fisher Scientific). For each run, 2 μg of E. coli peptides were then spiked with 0.15 μg of either UPS1 or UPS2 peptides, and about 1.6 μg of the mix was then analyzed via liquid chromatography combined with mass spectrometry on a Q Exactive (Thermo Fisher). Data were analyzed with MaxQuant as described above for the proteome dataset. All files were searched against the E. coli complete proteome sequences plus those of the UPS proteins and common contaminants.
Retention Time Alignment and Identification Transfer
To increase the number of peptides that can be used for quantification beyond those that have been sequenced and identified by an MS/MS database search engine, one can transfer peptide identifications to unsequenced or unidentified peptides by matching their mass and retention times (“match-between-runs” feature in MaxQuant). A prerequisite for this is that retention times between different LC-MS runs be made comparable via alignment. The order in which LC-MS runs are aligned is determined by hierarchical clustering, which allows one to avoid reliance on a single master run. The terminal branches of the tree from the hierarchical clustering typically connect LC-MS runs of the same or neighboring fractions or replicate runs, as they are the most similar. These cases are aligned first. Moving along the tree structure, increasingly dissimilar runs are integrated. The calibration functions that are needed to completely align LC-MS runs are usually time-dependent in a nonlinear way. Every pair-wise alignment step is performed via two-dimensional Gaussian kernel smoothing of the mass matches between the two runs. Following the ridge of the highest density region determines the recalibration function. At each tree node the resulting recalibration function is applied to one of the two subtrees, and the other is left unaltered.
Unidentified LC-MS features are then assigned to peptide identifications in other runs that match based on their accurate masses and aligned retention times. In complex proteomes, the high mass accuracy on current Orbitrap instruments is still insufficient for an unequivocal peptide identification based on the peptide mass alone. However, when comparing peptides in similar LC-MS runs, the information contained in peptide mass and recalibrated retention time is enough to transfer identifications with a sufficiently low FDR (in the range of 1%), which one can estimate by comparing the density of matches inside the match time window to the density outside this window (49).
The matching procedure takes into account the up-front separation, in this case isoelectric focusing of peptides into 24 fractions. Identifications are only transferred into adjacent fractions. If, for instance, for a given peptide sequenced in fraction 7, isotope patterns are found to match by mass and retention time in fractions 6, 8, and 17, the matches in fraction 17 are discarded because they have a much greater probability of being false. The same strategy can be applied to any other up-front peptide or protein separation (e.g. one-dimensional gel electrophoresis). All matches with retention time differences of less than 0.5 min after recalibration are accepted. Further details on the alignment and matching algorithms, including how to control the FDR of matching, will be described in a future manuscript.
Software and Data Availability
The label-free software MaxLFQ is completely integrated into the MaxQuant software (42) and can be activated by one additional click. It is freely available to academic and commercial users as part of MaxQuant and can be downloaded via the Internet. MaxQuant runs on Windows desktop computers with Vista or newer operating systems, preferably the 64-bit versions. There is a large user community at the MaxQuant Google group.
All downstream analysis was done using our in-house developed Perseus software, which is also freely available from the MaxQuant website.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the dataset identifier PXD000279.
RESULTS
Proteome-wide Benchmark Dataset
Evaluation of the accuracy of a label-free workflow at a proteome scale requires a dataset with known ratios. To this end we produced a benchmark dataset by mixing whole, distinguishable proteomes in defined ratios. Combined trypsin-digested lysates of HeLa cells and E. coli cells were extensively separated via isoelectric focusing into 24 fractions as described (45) and analyzed via LC-MS/MS in three replicates (“Experimental Procedures”). This was repeated with the same quantity of HeLa, but admixed with a 3-fold increased amount of E. coli lysate. In the resulting six samples all human proteins therefore should have had one-to-one ratios and all E. coli proteins should have had a ratio of three to one between replicate groups.
Raw data were processed with MaxQuant (42) and its built-in Andromeda search engine (50) for feature extraction, peptide identification, and protein inference. Peptide and protein FDRs were both set at 1%. MaxQuant identified a total of 789,978 isotope clusters through MS/MS sequencing. Transferring identifications to other LC-MS runs by matching them to unidentified features based on their masses and recalibrated retention times increased the number of quantifiable isotope patterns more than 2-fold (“match-between-runs,” “Experimental Procedures”).
A Novel Solution to the Normalization Problem
A major challenge of label-free quantification with prefractionation is that separate sample processing inevitably introduces differences in the fractions to be compared. In principle, correct normalization of each fraction can eliminate this error. However, the total peptide ion signals, necessary in order to perform normalization of the LC MS/MS runs of each fraction, are spread over several adjacent runs. Therefore one cannot sum up the peptide ion signals before one knows the normalization coefficients for each fraction.
We solve this dilemma by delaying normalization. After summing up intensities with normalization factors as free variables, we determine their quantities via a global optimization procedure based on achieving the least overall proteome variation.
Formally, we want to determine normalization coefficients Nj, which multiply all intensities in the jth LC-MS run (j runs from 1 to 144 in our example). The normalization is done purely from the data obtained and without the addition of external quantification standards or reliance on a fixed set of “housekeeping” proteins. Directly adjusting the normalization coefficients Nj for each of the fractions so that the total signal is equalized leads to errors if the fractionation is slightly irreproducible or if the mass spectrometric responses in the jth run are different from average. Therefore, we wish to summarize the peptide ion signals over the fractions in each sample. This, however, already requires the determination of the run-specific normalization factors Nj. We exploit the fact that the majority of the proteome typically does not change between any two conditions so that the average behavior can be used as a relative standard. This concept is also applied in label-based methods (e.g. for the normalization of SILAC ratios in MaxQuant). After summing the peptide ion signals across fractions with as-yet unknown Nj factors, we determined these factors in a nonlinear optimization model that minimized overall changes for all peptides across all samples (Fig. 1). For this we defined the total intensity of a peptide ion P in sample A as
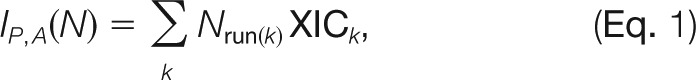 |
where the index k runs over all isotope patterns for peptide ion P in sample A. Here, different charge modification states are treated separately. The sum is understood as a generalized summation that can be the regular sum or the maximum over fractions. Also, for the XIC several choices exist, including total three-dimensional peak volume or area of the cross-section at the retention time when the maximum intensity is reached, which was used for this study. The quantity
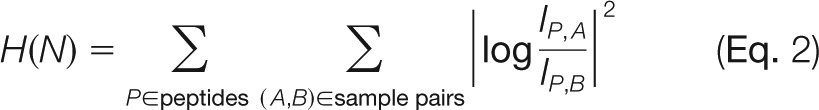 |
is the sum of all squared logarithmic fold changes between all samples and summed over all peptide ions (see Fig. 1). We minimized H(N) numerically with respect to the normalization coefficients Nj via Levenberg–Marquardt optimization (51) in order to achieve the least possible amount of differential regulation for the bulk of the proteins. This procedure is compatible with any kind of prefractionation and also is insensitive toward irreproducibility in processing. The computational effort for this procedure grows quadratically with the number of samples to be compared, which may hamper the analysis of very large datasets containing hundreds of samples. In these cases, however, a heuristic may be employed to estimate normalization coefficients by considering only a subset of possible pair-wise combinations of samples (see subsection “Fast Label-free Normalization of Large Datasets”). In principle, weighting factors can be included in the sum for H(N) in order to penalize low-intensity ions. Here we refrained from this in order to keep the parameterization of the model simple.
Fig. 1.

Schematic construction of the function H(N) to be minimized in order to determine the normalization coefficients for each LC-MS/MS run. Intensity distributions of three peptides (orange, green, and red) over samples and fractions are indicated by the sizes of the circles. H(N) is the sum of the squared logarithmic changes in all samples (A, B, C, …) for all peptides (P, Q, R, …). When using the fast normalization option, only a subset of all possible pairs of samples will be considered.
Extraction of Maximum Peptide Ratio Information
Another principal problem in label-free quantification is the selection of the peptide signals that should contribute to the optimal determination of the protein signal across the samples. A simple solution to this problem is to add up all peptide signals for each protein and then compare protein ratios. Alternatively, peptide intensities may be averaged, or only the top n intense species may be taken (31). However, these solutions discard the individual peptide ratios and thus do not extract the maximum possible quantification information. Instead, ratios derived from individual peptide signals should be taken into account, rather than a sum of intensities, because the XIC ratios for each peptide are already a measurement of the protein ratio. The very same concept is applied in label-based methods such as SILAC and contributes to their accuracy.
Due to stochastic MS/MS sequencing and differences in protein abundances across samples, peptide identifications are often missing in specific samples. One way to nevertheless obtain a signal for each peptide in every sample is to integrate the missing peptide intensities over the mass retention time plane using the integration boundaries from the samples in which the peptide has been identified. In this case, noise level effectively substitutes for the signal. Care has to be taken not to under- or overestimate the true ratios in either of these approaches. Yet another possibility is to restrict quantification to peptides that have a signal in all samples. Although this works well when comparing two samples, it becomes impractical when the number of samples is large—for example, requiring a peptide signal to be present in all of 100 clinical samples would likely eliminate nearly all peptides from quantification.
We propose a novel method for protein quantification that does not suffer from the problems described above (Fig. 2). We want to use only common peptides for pair-wise ratio determination without losing scalability for large numbers of samples. We achieve this for each protein by first calculating its ratio between any two samples using only peptide species that are present in both (Figs. 2A and 2B). Then the pair-wise protein ratio is calculated as follows, taking the pair-wise ratio of the protein in samples B and C in Fig. 2 as an example: First the intensities of peptides occurring in both samples are employed to calculate peptide ratios. In this case, peptide species P2, P3, and P6 are shared (Fig. 2C). The pair-wise protein ratio rCB (Fig. 2D) is then defined as the median of the peptide ratios, to protect against outliers. We then proceed to determine all pair-wise protein ratios. In the example in Fig. 2, we require a minimal number of two peptide ratios in order for a given protein ratio to be considered valid. This parameter is configurable in the MaxQuant software. Setting a higher threshold will lead to more accurate quantitative values, at the expense of more missing values.
Fig. 2.

Algorithm constructing protein intensity profiles for one protein from its peptide signals. A, an exemplary protein sequence. Peptides with an XIC-based quantification are indicated in magenta. B, the five peptide sequences give rise to seven peptide species. For this purpose, a peptide species is a distinct combination of peptide sequence, modification state, and charge, each of which has its own occurrence pattern over the different samples. C, occurrence matrix of peptide species in the six samples. D, matrix of pair-wise sample protein ratios calculated from the peptide XIC ratios. Valid/invalid ratios are colored in green/red based on a configurable minimum ratio count cut-off. If a sample has no valid ratio with any other sample, like sample F, the intensity will be set to zero. E, system of equations that needs to be solved for the protein abundance profile. F, the resulting protein abundance profile for one protein. The absolute scale is adapted to match the summed-up raw peptide intensities.
At this point we have constructed a triangular matrix containing all pair-wise protein ratios between any two samples, which is the maximal possible quantification information. This matrix corresponds to an overdetermined system of equations for the underlying protein abundance profile (IA, IB, IC, …) across the samples (Fig. 2E). We perform a least-squares analysis to reconstruct the abundance profile optimally satisfying the individual protein ratios in the matrix based on the sum of squared differences
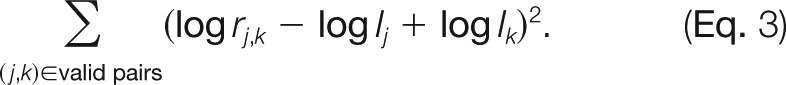 |
Then we rescale the whole profile to the cumulative intensity across samples, thereby preserving the total summed intensity for a protein over all samples (Figs. 2E and 2F). This procedure is repeated for all proteins, resulting in an accurate abundance profile for each protein across the samples. The computational effort grows quadratically with the number of samples in which a protein is present; however, it is readily parallelizable at the protein level.
All resulting profiles are written into the MaxQuant output tables in columns starting with “LFQ intensity.”
Quantification Results for the Proteome Benchmark Set
To apply the algorithms to the E. coli and HeLa cell mixture, we required a protein to have non-zero intensity in two out of the three replicates for each condition. In addition, protein groups had to be unambiguously assignable to one species; this was the case for 3453 human and 1556 E. coli proteins (supplemental Table S1). In Fig. 3, we compare the performance of MaxLFQ against that of two other frequently used quantitative metrics: spectral counting and summed peptide intensities. Both were also extracted by MaxQuant, so we do not introduce biases due to the search engine and the set of identified peptides, and only benchmark conceptually different metrics of quantification. For each case, we averaged the three replicates of each experimental condition and plotted the log ratios against the log of the summed peptide intensity, which can be used as a proxy for absolute protein abundance (52–54). In all cases, human and E. coli proteins formed distinct clouds, but with different degrees of overlap. Spectral count ratio clouds were clearly separated only for the most abundant proteins (Figs. 3A and 3D). In the low-intensity region, spectral counts became discrete values, and their log ratios adopted a very wide distribution with pronounced overlap of human and E. coli proteins. Furthermore, a systematic distortion was observable that resulted in a general overestimation of the ratios of low-intensity proteins. Ratios of summed peptide intensities already allowed almost complete separation of human and E. coli proteins across the entire abundance range, with some overlap occurring only in the lower half (Figs. 3B and 3E). This demonstrates a clear advantage of intensity-based approaches. When we used our MaxLFQ algorithm, the overlap of the populations was further reduced relative to the summed intensity approach, and the number of extreme outliers was markedly reduced (Figs. 3C and 3F). We quantified the widths of the distributions and the degree of overlap (Figs. 3G–3I), which demonstrated that MaxLFQ performed best not only by generating the narrowest distributions, but also by most accurately recapitulating the expected fold change of three between the population averages.
Fig. 3.

Quantification results for the proteome benchmark dataset. Replicate groups were filtered for two out of three valid values and averaged, and the log ratios of the E. coli (orange)/human (blue) 3:1 versus 1:1 samples were plotted against the logarithm of summed peptide intensities from the 1:1 sample as a proxy for absolute protein abundance. A, quantification using spectral counts. B, quantification using summed peptide intensities. C, quantification using MaxLFQ. D–F, same as A–C, but colored using density estimation. G, H, histograms of the ratio distributions of human and E. coli proteins obtained using the different quantification methods.
MaxLFQ has the prerequisite that a majority population of proteins exists that is not changing between the samples. How big this population needs to be and what the consequences are if the changing population becomes comparable in size to the non-changing one can be seen in the benchmark dataset itself, in which the changing (E. coli) population comprised 31% of the proteins measured in total. MaxLFQ still operated well under these circumstances. The average factor of three between the changing and non-changing population was recovered well. The only effect of the large size of the changing population was a total shift of all log-ratios such that the non-changing population was centered not exactly at zero but at slightly negative values. However, this had no effect on subsequent tests for finding differentially expressed proteins, as they are all insensitive to global shifts of all values. Regarding samples involving enrichment steps, we refer to our examples of interaction proteomics studies, in which MaxLFQ performed very well. In such datasets, enriched proteins may constitute a large part of the total protein mass (or peak intensity). Still, we routinely observed a dominant population of background binding proteins contributing a large number of peptide features that changed minimally between experimental conditions (even if their intensities were lower). In large pulldown datasets, the background population does not have to be the same over all samples and can be a different one in each pair-wise sample comparison in MaxLFQ.
Analysis at a population level does not in itself provide statistically sound information on the regulation state of individual proteins. In fact, Fig. 3B shows several human proteins that appear to be changing by several-fold. In a clinical context these might have been mistaken for biomarkers without further analysis. We therefore explored different strategies to retrieve significantly changing proteins based either on simple fold change or on the variance of their quantitative signals, ranking the proteins by their highest apparent fold change (highest ratio of average intensities), by their standard t test p value, by their Welch modified t test p value, and finally by their Wilcoxon–Mann–Whitney p value. Because we had full prior knowledge about which proteins were changing (only the E. coli ones), we independently knew the FDR and could construct precision-recall curves for each case to assess performance (Fig. 4A). This revealed that retrieving proteins by ratio (corresponding to a fixed fold change cut-off) was the worst strategy. It had low precision even at small recall values because of its sensitivity to outlier ratios in individual replicates. When sorting proteins by ratios, we found that the fourth protein was a false positive (Fig. 4B, arrow). The Wilcoxon–Mann–Whitney test performed better but also had problems at low recall. Both versions of the t test performed significantly better, and the Welch modified t test was slightly better than the standard t test. At a precision of 0.98, 72% of the E. coli proteins were recalled. With a precision of 95%, which is often used in similar circumstances, the vast majority (88%) of E. coli proteins were retrieved when we used the Welch modified t test.
Fig. 4.
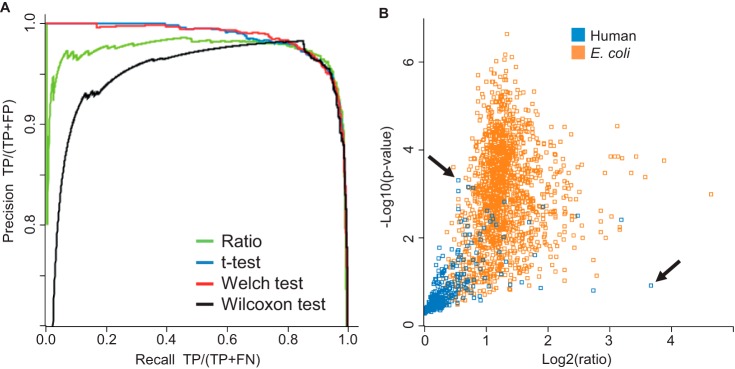
Statistical significance of protein regulation. A, precision-recall curves based on four different strategies. TP, true positives; FP, false positives; FN, false negatives. B, the Welch modified t test p value is plotted logarithmically against the ratio. The vast majority of E. coli proteins (orange) have p values better than 0.05, indicating significant regulation. An extremely small number of human proteins (blue) appear to have a large ratio and significant p value (false positives for quantification). The arrows indicate that the best strategy is to select significantly regulated proteins by t test p value (first false positive after hundreds of correct hits with better p values) rather than fold change (first false positive after three correct hits with higher fold change).
In datasets of practical interest, the true proportion of false positives is not known a priori. As a means to control the FDR and solve potential multiple hypothesis testing problems in real biological datasets, we usually apply permutation-based methods for calculating q-values and global FDRs. These robust strategies have been successfully applied to high-throughput biological data for a long time (55). The advantage of permutation-based methods is that no assumptions need to be made regarding the parametric distributions of intensities or ratios. The significance analysis of microarrays (SAM) method that we apply to most of the biological datasets also utilizes moderation to ensure the stability of the results. Whereas in most real applications the stabilization parameter s0 introduced in Ref. 55 is beneficial, in this particular benchmark dataset it did not improve the performance relative to the original t test statistic. This is presumably because in the benchmark dataset all true ratios were either 1:1 or 1:3, whereas in real applications the true ratio distribution has a dense spectrum of small changes.
Interestingly, about one-third of the proteome was changing in the benchmark dataset, which is a large amount, considering that the normalization was based on the assumption of a dominating population of non-changing proteins. The effect of this can be observed in Fig. 3I. The center of the 1:1 population is shifted to slightly negative values. However, the distance between the means of the 3:1 and 1:1 populations is near the correct value of log2(3). Such a global shift of all ratios will not affect statistical testing, as a test such as the t test is insensitive to such a global shift of all values. If one insists on having a 1:1 distribution centered exactly at 0, one can apply another normalization step in which one subtracts the most frequent value (i.e. the position of the global maximum).
So far we have assessed the measurability of 3:1 changes over the whole accessible dynamic range of protein abundances. Another question of interest is how measurable smaller ratios are. For this purpose we conducted an in silico experiment in which the results of the actual 3:1 experiment were rescaled in order to mimic results obtained with lower mixing ratios. We rescaled the log ratios of all E. coli proteins in the three samples with the 3-fold increased E. coli abundance by adding the constant
to all of these values. Here, mean(E. coli) is the average difference in log intensities between the two replicate groups for the E. coli proteins, mean(human) is the same quantity calculated for all human proteins, and s is a scaling factor between 0 and 1. For s = 1, the original data are recovered, whereas for s = 0 the mean ratio is 1:1 for all proteins, in particular for the E. coli proteins. For a given value of s, the corresponding simulated ratio is r = 3s.
Fig. 5A shows precision-recall curves similar to those in Fig. 4A. This time, only the t test was used for determining significant changes, and we scanned through several values for the simulated ratio r. As an example, we tolerated a proportion of false discoveries (Q, the value estimated by the FDR) of 10% for calling changes significant. Although in that case almost all truly changing proteins are recovered with a ratio of 3, about half of them are still obtained at a ratio of 1.6. Going below a mean ratio change of 1.6 will lead to strong drop in coverage. The FDR threshold that one wishes to apply depends on the experimental situation and on the biological or technological question. There is no a priori given FDR that is applicable to every case. For instance, if pre-screening is done (e.g. to explore regulated pathways or biological processes), a 25% FDR might still be tolerable, whereas in other cases a 5% FDR might not be stringent enough. To get an idea about the relationship between protein ratio and coverage achieved for proteins having this ratio, we plotted this dependence in Fig. 5B for several values of Q. In particular, for low stringency there is a very rapid drop of coverage around a well-defined ratio. For instance, the Q = 0.25 curve has a steep slope around a ratio of 1.4 where it achieves half of the coverage. One could define this “half-coverage point” as the situation for which it still makes sense to look for ratio changes. In Fig. 5C we show the ratio at the point of half-coverage as a function of Q. These ratios can achieve values of far less than 2 for larger values of Q.
Fig. 5.

Statistical significance of small protein ratios. A, precision-recall curves based on a t test on a set of ratios that were simulated in silico by shrinking the experimental ratio of three. B, ratio-coverage plots for these simulated ratios at a set of fixed proportions of false discoveries among the discoveries (Q). One can see a drop in coverage around a given ratio, which is particularly steep for large values of Q. C, simulated ratio at which one achieves half-coverage plotted against the value of Q.
Dynamic Range Benchmark Set
So far, we have demonstrated that MaxLFQ is able to accurately and robustly quantify small fold changes on a proteome scale. This is relevant, for instance, for the analysis of cellular proteome remodeling upon stimulation. Next, we wanted to test the performance of the algorithm in the quantification of high ratios in the range of several orders of magnitude. Such ratios typically occur in the context of interaction proteomics experiments (56), where early mixing of isotope-labeled samples is usually not possible and some of the principal advantages of metabolic labeling are therefore lost. We have recently shown that both SILAC and MaxLFQ generate similar ratio distributions (57), indicating that in such cases MaxLFQ is capable of achieving quantification accuracies comparable to those obtained with SILAC.
As a benchmark dataset for high protein ratios, we made use of the universal protein standard (UPS) (Sigma-Aldrich), a mixture of 48 recombinant human proteins that is available as an equimolar mixture (UPS1) or mixed at defined ratios spanning 5 orders of magnitude (UPS2). This dataset does not contain fractionation and is used for showing that MaxLFQ performs well at high dynamic range quantification in general. We separately digested UPS1 and UPS2 with trypsin and spiked the peptides into a trypsin-digested E. coli lysate. We analyzed each condition in four replicates via single-shot LC-MS/MS. Raw data were processed as described for the proteome benchmark dataset, with some exceptions as outlined below. MaxQuant identified 232,835 isotope clusters by MS/MS, and matching between runs increased the number of quantifiable features by 38%. After protein inference, this resulted in 2200 non-redundant E. coli protein groups. We identified all of the 48 human UPS proteins in all samples containing E. coli with the equimolar UPS1 standard (supplemental Table S2). In the sample of E. coli plus UPS2, 15 of the lower abundant human UPS proteins were never sequenced by MS/MS, but 10 of them could be identified and quantified in at least some of the replicates through matching to the UPS1-containing samples. Applying the same requirement of two shared peptides for each pair-wise comparison (as used in the proteome benchmark dataset) expectedly resulted in missing values for samples in which only individual peptides were found; therefore we lowered this threshold to one. Extreme ratios typically coincide with very different peptide populations identified in the samples to be compared: many in the sample with high protein abundance, of which only a small subset is found in the low-abundance sample. This can make the protein ratio determination rely on very few quantification events, which increases the sensitivity to outliers. To address this issue, we implemented an optional feature called “large ratio stabilization,” which modifies the ratio determination for pair-wise comparisons where the number of peptides quantified in the two samples differs substantially. In a case when fewer than one out of five peptides is shared between samples, the ratio of the summed-up peptide intensities is taken for quantification. If more than two out of five peptides are shared, the median of pair-wise ratios is used. For intermediate cases, we interpolate linearly between these two kinds of ratio determinations. In summary, the protein ratio r is determined by the median of peptide ratios rm and the ratio of summed-up peptide intensities rs by the formula
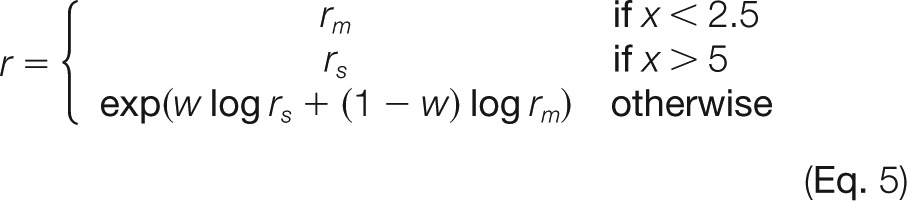 |
where w = (x − 2.5)/2.5 and x is the ratio of the number of peptide features in the sample with the most peptide features to the number of common peptide features. We found that this stabilized the general ratio trend and reduced the outlier sensitivity.
Fig. 6A shows the quantification results for samples containing UPS2 versus UPS1, plotted in the same way as in Fig. 3. UPS proteins are clearly separated from the narrow cloud of E. coli proteins and cluster in groups according to their relative abundances. For further analysis, we subtracted the median of the group of UPS proteins present in equal amounts in both UPS1 and UPS2. In a direct comparison of true ratio versus the MaxLFQ readout (Fig. 6B), we show that within 2 orders of magnitude, we obtained quantification results that were extremely close to the expected values. For ratios of more than 100-fold, we detected increased scatter, but no systematic error that would lead to an over- or underestimation of the ratio (Fig. 6F). Summed intensities yielded very similar results within 2 orders of magnitude (Fig. 6C) but a small systematic underestimation of very large ratios (Fig. 6G). Spectral counts covered 2 orders of magnitude less than intensity-based methods, because there were no MS/MS events for all proteins of the lowest two abundance groups in all UPS2-plus-E. coli samples (Fig. 6D). For proteins covered by MS/MS spectra in both UPS1 and UPS2 samples, there was a pronounced systematic underestimation of the ratio when calculating the ratio of spectral counts (Fig. 6H). This clearly shows that spectral counting suffers from a very narrow dynamic range that is limited by the total number of identified MS/MS spectra. Of note, all methods unanimously detected ratios of less than 10 for the comparison of the group of most abundant proteins in the UPS2 samples. This leads us to speculate that this was not due to a quantification error, but rather due to the composition of the UPS2 peptide mixture. It is possible that the eight most abundant proteins could be slightly underrepresented because of LC-MS saturation effects or incomplete digestion.
Fig. 6.

Quantification results for the dynamic range benchmark dataset. Replicate groups were filtered for three out of four valid values and averaged. A, log ratios of the UPS2 versus UPS1 samples plotted against the logarithm of summed peptide intensities from the UPS1 sample as a proxy for absolute protein abundance. E. coli proteins are plotted in gray and form a narrow population centered on zero. UPS proteins are color-coded by their abundance groups in the UPS2 sample. B–D, to compare the ratio readout against the true ratio, we shifted the population of UPS proteins that were present in UPS1 and UPS2 in equimolar amounts to 1:1 and plotted the log ratio obtained from (B) MaxLFQ, (C) summed intensities, and (D) spectral counts against the log of the true ratio. E, log intensity ratio plotted against log MaxLFQ ratios. F–H, data from B–D plotted as the deviation from the true ratio. Spectral counts show a clear underestimation of ratios across the entire dynamic range and lose 2 orders of magnitude. Summed intensities and MaxLFQ show increased scatter toward ratios of several orders of magnitude. Summed intensities show some degree of systematic underestimation of large ratios, which was not observed for MaxLFQ ratios.
Fast Label-free Normalization of Large Datasets
In the analysis of very large datasets, one of the computationally most expensive steps is the determination of the normalization factors for each LC-MS run by minimizing the quantity H(N) described earlier and depicted in Fig. 1. This quantity contains a sum running over all pairs of samples that grows quadratically with the number of samples. (Note that in the case of pre-fractionation, multiple LC-MS runs contribute to one sample and do not contribute to a further quadratic increase of the computational effort.) One approach would be to do normalization in a more simplistic way and only use the reconstruction of protein abundances based on paired peptide ratios from MaxLFQ. However, because the normalization is crucial with fractionated samples, we wanted to find an algorithm that delivered results very similar to those of the full MaxLFQ computation, but within a much smaller computation time.
Because the resulting minimization problem becomes increasingly overdetermined for larger numbers of samples, we reasoned that a meaningful subset of comparisons would significantly reduce the computing time while still delivering correct normalization factors. Even a linear chain of comparisons in which every sample occurs exactly once would in principle be sufficient to determine all normalization factors. However, this minimal strategy may lead to unstable and error-prone calculations, as the failure or imprecision of a single comparison may propagate into the calculation of all normalization factors. As a compromise considering stability, correctness, and computational efficiency, a reasonable and robust subset of pair-wise comparisons needs to be found. We started by creating a graph with all samples as nodes. A large overlap of peptides between each pair of nodes was interpreted as a small distance between them. A subgraph was then determined in which each node had a minimum number of three nearest neighbors and the average number of neighbors over all nodes was six. All edges that were not needed to fulfill these criteria were removed while making sure that all nodes remained connected. For the sum in H(N) in Fig. 1, only those sample pairs were taken into account that had an edge in this graph, resulting in linear scaling of the computational effort with the number of samples. This “fast” normalization option can be optionally activated in MaxQuant, and the parameters for subgraph determination are adjustable by the user.
DISCUSSION
We have introduced MaxLFQ as a suite of novel algorithms for relative protein quantification without stable isotopes. “Delayed normalization” efficiently solves the problem of how to compare sample fractions that have been handled in slightly different ways and analyzed with different MS performance. Importantly, delayed normalization does not require “household” proteins, which are assumed to be unchanging in the experiment. The only prerequisite is a dominant population of proteins that change minimally between experimental conditions. The second algorithm allows the retrieval of the maximum possible information from peptide ratios across samples, without resorting to arbitrary assignment of the signal when a peptide signal cannot be detected. Finally, a profile of “LFQ” intensities is calculated for each protein as the best estimate satisfying all the pair-wise peptide comparisons. Importantly, this intensity profile retains the absolute scale from the original summed-up peptide intensities. This should readily qualify it as a proxy for absolute protein abundance. MaxLFQ is a generic approach that works independently of the experimental question under investigation, and we have demonstrated equally good performance for the determination of small and very large ratios. For assessing the statistical significance of individual protein ratios, we found that t testing on a dataset with three or more replicates delivered the best results and was superior to a simple fold-change cut-off.
Our laboratory has successfully used MaxLFQ in a number of studies with very diverse biological questions. For instance, in measurements that spanned more than a year, we studied the proteomic differences of rare immunological cell types and found mutually exclusive expressions of pattern recognition receptors (58). We have also followed the proteome rearrangements during colon cancer development and metastasis in the colon mucosa (54). Furthermore, we have used label-free quantification to study protein–protein interactions expressed as GFP-tagged constructs from bacterial artificial chromosomes under endogenous control (56) and screened for interactors of post-translationally modified histone tails in mouse tissues (57). In that case we showed that MaxLFQ achieved similar quantification accuracies as SILAC. Interaction proteomics experiments typically detect specific interactors with enrichment factors on the order of several magnitudes. Here, the general ratio trend is sometimes more important than a very accurate readout of the actual ratio. Such cases offer a straightforward remedy for dealing with missing values: they can simply be imputed as simulated values forming a distribution around the detection limit of measured intensities and serve as the basis for judging enrichment factors. This is a principal advantage over label-based ratio determination, where dealing with infinite ratios is conceptually more difficult.
In a very recent study, we used MaxLFQ to study the secretome of activated immune cells and detected proteins whose abundance was increased by several orders of magnitude in the culture medium upon stimulation (59).
We have already been making MaxLFQ available as part of the MaxQuant software for some time, and other groups have made frequent use of it (60–75). It has also been benchmarked against other software solutions for label-free quantification (31), independently confirming the excellent performance of our software.
Recent advances in mass spectrometer hardware (76, 77) have provided a boost in the depth of standard analyses and enabled near-complete model proteome quantification in minimal measuring time (6). Label-free quantification benefits dramatically from this depth, as it increases the number of quantifiable features present in a given LC-MS run and allows averaging over more peptides for protein quantification. Illustrating this, in our dynamic-range benchmark dataset we recorded one of the largest published E. coli proteomes so far, resulting in a high sequence coverage and hence a very narrow cloud of E. coli protein quantifications.
Some challenges for label-free quantification remain: Sample handling variability needs to be minimized when samples are to be recorded over the course of many months, on different machines, or by different laboratories. Standardization of instrumentation, simplification of sample preparation procedures, and automation using multiwell systems or robotics will help to mitigate this issue. Biological studies that depend on the ultimate accuracy of the ratio readout or on quantitative information about individual peptides, such as post-translationally modified ones, will still rely on isotope labels. In addition, applications that require extreme robustness, such as sample handling in a clinical setting, will likely benefit from spike-in references that serve as internal standards. That said, we expect label-free quantification methods in general and MaxLFQ in particular to gain further momentum in the proteomics community and become the method of choice for many applications. The ease of use of MaxLFQ as part of the MaxQuant software suite should enable our technology to be widely adopted by nonspecialized labs as well.
Supplementary Material
Acknowledgments
We thank all members of the Proteomics and Signal Transduction Group for help and discussions and Francesca Forner, Charo Robles, and Gabriele Stoehr for critical reading of the manuscript.
Footnotes
Author contributions: J.C., M.Y.H., and M.M. designed research; J.C., C.A.L., I.P., N.N., and M.M. performed research; M.Y.H. contributed new reagents or analytic tools; J.C. and M.Y.H. analyzed data; J.C. and M.M. wrote the paper.
* This project was supported by the European Commission's 7th Framework Program PROteomics SPECificat Ion in Time and Space (PROSPECTS, HEALTH-F4-2008-021,648) and by the German Federal Ministry of Education and Research (DiGtoP Consortium, FKZ01GS0861).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- SILAC
- stable isotope labeling by amino acids in cell culture
- MS
- mass spectrometry
- LC-MS
- liquid chromatography–mass spectrometry
- MS/MS
- tandem mass spectrometry
- XIC
- extracted ion current
- LFQ
- label-free quantification
- UPS
- universal protein standard
- FDR
- false discovery rate.
REFERENCES
- 1. Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 [DOI] [PubMed] [Google Scholar]
- 2. Ong S. E., Mann M. (2005) Mass spectrometry-based proteomics turns quantitative. Nat. Chem. Biol. 1, 252–262 [DOI] [PubMed] [Google Scholar]
- 3. Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 [DOI] [PubMed] [Google Scholar]
- 4. Cox J., Mann M. (2007) Is proteomics the new genomics? Cell 130, 395–398 [DOI] [PubMed] [Google Scholar]
- 5. Altelaar A. F., Munoz J., Heck A. J. (2013) Next-generation proteomics: towards an integrative view of proteome dynamics. Nat. Rev. Genet. 14, 35–48 [DOI] [PubMed] [Google Scholar]
- 6. Mann M., Kulak N. A., Nagaraj N., Cox J. (2013) The coming age of complete, accurate, and ubiquitous proteomes. Mol. Cell 49, 583–590 [DOI] [PubMed] [Google Scholar]
- 7. Dephoure N., Gygi S. P. (2012) Hyperplexing: a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci. Signal. 5, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Listgarten J., Emili A. (2005) Statistical and computational methods for comparative proteomic profiling using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 4, 419–434 [DOI] [PubMed] [Google Scholar]
- 9. Domon B., Aebersold R. (2006) Mass spectrometry and protein analysis. Science 312, 212–217 [DOI] [PubMed] [Google Scholar]
- 10. Mueller L. N., Brusniak M. Y., Mani D. R., Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 7, 51–61 [DOI] [PubMed] [Google Scholar]
- 11. Nahnsen S., Bielow C., Reinert K., Kohlbacher O. (2012) Tools for label-free peptide quantification. Mol. Cell. Proteomics 12, 549–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bantscheff M., Lemeer S., Savitski M. M., Kuster B. (2012) Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal. Bioanal. Chem. 404, 939–965 [DOI] [PubMed] [Google Scholar]
- 13. Matzke M. M., Brown J. N., Gritsenko M. A., Metz T. O., Pounds J. G., Rodland K. D., Shukla A. K., Smith R. D., Waters K. M., McDermott J. E., Webb-Robertson B. J. (2013) A comparative analysis of computational approaches to relative protein quantification using peptide peak intensities in label-free LC-MS proteomics experiments. Proteomics 13, 493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mueller L. N., Rinner O., Schmidt A., Letarte S., Bodenmiller B., Brusniak M. Y., Vitek O., Aebersold R., Muller M. (2007) SuperHirn—a novel tool for high resolution LC-MS-based peptide/protein profiling. Proteomics 7, 3470–3480 [DOI] [PubMed] [Google Scholar]
- 15. Bellew M., Coram M., Fitzgibbon M., Igra M., Randolph T., Wang P., May D., Eng J., Fang R., Lin C., Chen J., Goodlett D., Whiteaker J., Paulovich A., McIntosh M. (2006) A suite of algorithms for the comprehensive analysis of complex protein mixtures using high-resolution LC-MS. Bioinformatics 22, 1902–1909 [DOI] [PubMed] [Google Scholar]
- 16. Rauch A., Bellew M., Eng J., Fitzgibbon M., Holzman T., Hussey P., Igra M., Maclean B., Lin C. W., Detter A., Fang R., Faca V., Gafken P., Zhang H., Whiteaker J., States D., Hanash S., Paulovich A., McIntosh M. W. (2006) Computational Proteomics Analysis System (CPAS): an extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. J. Proteome Res. 5, 112–121 [DOI] [PubMed] [Google Scholar]
- 17. May D., Fitzgibbon M., Liu Y., Holzman T., Eng J., Kemp C. J., Whiteaker J., Paulovich A., McIntosh M. (2007) A platform for accurate mass and time analyses of mass spectrometry data. J. Proteome Res. 6, 2685–2694 [DOI] [PubMed] [Google Scholar]
- 18. Jaffe J. D., Mani D. R., Leptos K. C., Church G. M., Gillette M. A., Carr S. A. (2006) PEPPeR, a platform for experimental proteomic pattern recognition. Mol. Cell. Proteomics 5, 1927–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kohlbacher O., Reinert K., Gropl C., Lange E., Pfeifer N., Schulz-Trieglaff O., Sturm M. (2007) TOPP—the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197 [DOI] [PubMed] [Google Scholar]
- 20. Palagi P. M., Walther D., Quadroni M., Catherinet S., Burgess J., Zimmermann-Ivol C. G., Sanchez J. C., Binz P. A., Hochstrasser D. F., Appel R. D. (2005) MSight: an image analysis software for liquid chromatography-mass spectrometry. Proteomics 5, 2381–2384 [DOI] [PubMed] [Google Scholar]
- 21. Johansson C., Samskog J., Sundstrom L., Wadensten H., Bjorkesten L., Flensburg J. (2006) Differential expression analysis of Escherichia coli proteins using a novel software for relative quantitation of LC-MS/MS data. Proteomics 6, 4475–4485 [DOI] [PubMed] [Google Scholar]
- 22. Roy S. M., Becker C. H. (2007) Quantification of proteins and metabolites by mass spectrometry without isotopic labeling. Methods Mol. Biol. 359, 87–105 [DOI] [PubMed] [Google Scholar]
- 23. Katajamaa M., Miettinen J., Oresic M. (2006) MZmine: toolbox for processing and visualization of mass spectrometry based molecular profile data. Bioinformatics 22, 634–636 [DOI] [PubMed] [Google Scholar]
- 24. Leptos K. C., Sarracino D. A., Jaffe J. D., Krastins B., Church G. M. (2006) MapQuant: open-source software for large-scale protein quantification. Proteomics 6, 1770–1782 [DOI] [PubMed] [Google Scholar]
- 25. Smith R. D., Anderson G. A., Lipton M. S., Pasa-Tolic L., Shen Y., Conrads T. P., Veenstra T. D., Udseth H. R. (2002) An accurate mass tag strategy for quantitative and high-throughput proteome measurements. Proteomics 2, 513–523 [DOI] [PubMed] [Google Scholar]
- 26. Old W. M., Meyer-Arendt K., Aveline-Wolf L., Pierce K. G., Mendoza A., Sevinsky J. R., Resing K. A., Ahn N. G. (2005) Comparison of label-free methods for quantifying human proteins by shotgun proteomics. Mol. Cell. Proteomics 4, 1487–1502 [DOI] [PubMed] [Google Scholar]
- 27. Sturm M., Bertsch A., Gropl C., Hildebrandt A., Hussong R., Lange E., Pfeifer N., Schulz-Trieglaff O., Zerck A., Reinert K., Kohlbacher O. (2008) OpenMS—an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Listgarten J., Neal R. M., Roweis S. T., Wong P., Emili A. (2007) Difference detection in LC-MS data for protein biomarker discovery. Bioinformatics 23, e198–e204 [DOI] [PubMed] [Google Scholar]
- 29. Park S. K., Venable J. D., Xu T., Yates J. R., 3rd (2008) A quantitative analysis software tool for mass spectrometry-based proteomics. Nat. Methods 5, 319–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bridges S. M., Magee G. B., Wang N., Williams W. P., Burgess S. C., Nanduri B. (2007) ProtQuant: a tool for the label-free quantification of MudPIT proteomics data. BMC Bioinformatics 8 Suppl 7, S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weisser H., Nahnsen S., Grossmann J., Nilse L., Quandt A., Brauer H., Sturm M., Kenar E., Kohlbacher O., Aebersold R., Malmstrom L. (2013) An automated pipeline for high-throughput label-free quantitative proteomics. J. Proteome Res. [DOI] [PubMed] [Google Scholar]
- 32. Ning K., Fermin D., Nesvizhskii A. I. (2012) Comparative analysis of different label-free mass spectrometry based protein abundance estimates and their correlation with RNA-Seq gene expression data. J. Proteome Res. 11, 2261–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cheng F. Y., Blackburn K., Lin Y. M., Goshe M. B., Williamson J. D. (2009) Absolute protein quantification by LC/MS(E) for global analysis of salicylic acid-induced plant protein secretion responses. J. Proteome Res. 8, 82–93 [DOI] [PubMed] [Google Scholar]
- 34. Polpitiya A. D., Qian W. J., Jaitly N., Petyuk V. A., Adkins J. N., Camp D. G., 2nd, Anderson G. A., Smith R. D. (2008) DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24, 1556–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Karpievitch Y., Stanley J., Taverner T., Huang J., Adkins J. N., Ansong C., Heffron F., Metz T. O., Qian W. J., Yoon H., Smith R. D., Dabney A. R. (2009) A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics 25, 2028–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clough T., Key M., Ott I., Ragg S., Schadow G., Vitek O. (2009) Protein quantification in label-free LC-MS experiments. J. Proteome Res. 8, 5275–5284 [DOI] [PubMed] [Google Scholar]
- 37. Choi H., Glatter T., Gstaiger M., Nesvizhskii A. I. (2012) SAINT-MS1: protein-protein interaction scoring using label-free intensity data in affinity purification-mass spectrometry experiments. J. Proteome Res. 11, 2619–2624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 39. Gygi S. P., Rist B., Gerber S. A., Turecek F., Gelb M. H., Aebersold R. (1999) Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 17, 994–999 [DOI] [PubMed] [Google Scholar]
- 40. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 41. Boersema P. J., Aye T. T., van Veen T. A., Heck A. J., Mohammed S. (2008) Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8, 4624–4632 [DOI] [PubMed] [Google Scholar]
- 42. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 43. Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. (2003) Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 [DOI] [PubMed] [Google Scholar]
- 44. Ishihama Y., Oda Y., Tabata T., Sato T., Nagasu T., Rappsilber J., Mann M. (2005) Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics 4, 1265–1272 [DOI] [PubMed] [Google Scholar]
- 45. Hubner N. C., Ren S., Mann M. (2008) Peptide separation with immobilized pI strips is an attractive alternative to in-gel protein digestion for proteome analysis. Proteomics, Dec. 8 (23–24), 4862–4872 [DOI] [PubMed] [Google Scholar]
- 46. Rappsilber J., Ishihama Y., Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 47. Olsen J. V., de Godoy L. M., Li G., Macek B., Mortensen P., Pesch R., Makarov A., Lange O., Horning S., Mann M. (2005) Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 4, 2010–2021 [DOI] [PubMed] [Google Scholar]
- 48. Cox J., Hubner N. C., Mann M. (2008) How much peptide sequence information is contained in ion trap tandem mass spectra? J. Am. Soc. Mass Spectrom. 19, 1813–1820 [DOI] [PubMed] [Google Scholar]
- 49. Geiger T., Wehner A., Schaab C., Cox J., Mann M. (2012) Comparative proteomic analysis of eleven common cell lines reveals ubiquitous but varying expression of most proteins. Mol. Cell. Proteomics 11, M111.014050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 51. Press W. H., Teukolsky S. H., Vetterling W. T., Flannery B. P. (2007) Numerical Recipes: The Art of Scientific Computing, 3rd ed., Cambridge University Press, Cambridge, UK [Google Scholar]
- 52. de Godoy L. M., Olsen J. V., Cox J., Nielsen M. L., Hubner N. C., Frohlich F., Walther T. C., Mann M. (2008) Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature 455, 1251–1254 [DOI] [PubMed] [Google Scholar]
- 53. Schwanhausser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 54. Wisniewski J. R., Ostasiewicz P., Dus K., Zielinska D. F., Gnad F., Mann M. (2012) Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol. Syst. Biol. 8, 611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tusher V. G., Tibshirani R., Chu G. (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U.S.A. 98, 5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hubner N. C., Bird A. W., Cox J., Splettstoesser B., Bandilla P., Poser I., Hyman A., Mann M. (2010) Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. J. Cell Biol. 189, 739–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Eberl H. C., Spruijt C. G., Kelstrup C. D., Vermeulen M., Mann M. (2013) A map of general and specialized chromatin readers in mouse tissues generated by label-free interaction proteomics. Mol. Cell 49, 368–378 [DOI] [PubMed] [Google Scholar]
- 58. Luber C. A., Cox J., Lauterbach H., Fancke B., Selbach M., Tschopp J., Akira S., Wiegand M., Hochrein H., O'Keeffe M., Mann M. (2010) Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity 32, 279–289 [DOI] [PubMed] [Google Scholar]
- 59. Meissner F., Scheltema R. A., Mollenkopf H. J., Mann M. (2013) Direct proteomic quantification of the secretome of activated immune cells. Science 340, 475–478 [DOI] [PubMed] [Google Scholar]
- 60. Batruch I., Smith C. R., Mullen B. J., Grober E., Lo K. C., Diamandis E. P., Jarvi K. A. (2012) Analysis of seminal plasma from patients with non-obstructive azoospermia and identification of candidate biomarkers of male infertility. J. Proteome Res. 11, 1503–1511 [DOI] [PubMed] [Google Scholar]
- 61. Boerries M., Grahammer F., Eiselein S., Buck M., Meyer C., Goedel M., Bechtel W., Zschiedrich S., Pfeifer D., Laloe D., Arrondel C., Goncalves S., Kruger M., Harvey S. J., Busch H., Dengjel J., Huber T. B. (2013) Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int. 83, 1052–1064 [DOI] [PubMed] [Google Scholar]
- 62. de Godoy L. M., Marchini F. K., Pavoni D. P., Rampazzo R. de C., Probst C. M., Goldenberg S., Krieger M. A. (2012) Quantitative proteomics of Trypanosoma cruzi during metacyclogenesis. Proteomics 12, 2694–2703 [DOI] [PubMed] [Google Scholar]
- 63. Lopez-Contreras A. J., Ruppen I., Nieto-Soler M., Murga M., Rodriguez-Acebes S., Remeseiro S., Rodrigo-Perez S., Rojas A. M., Mendez J., Munoz J., Fernandez-Capetillo O. (2013) A proteomic characterization of factors enriched at nascent DNA molecules. Cell Rep. 3, 1105–1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Smaczniak C., Li N., Boeren S., America T., van Dongen W., Goerdayal S. S., de Vries S., Angenent G. C., Kaufmann K. (2012) Proteomics-based identification of low-abundance signaling and regulatory protein complexes in native plant tissues. Nat. Protoc. 7, 2144–2158 [DOI] [PubMed] [Google Scholar]
- 65. Gamez-Pozo A., Ferrer N. I., Ciruelos E., Lopez-Vacas R., Martinez F. G., Espinosa E., Vara J. A. (2013) Shotgun proteomics of archival triple-negative breast cancer samples. Proteomics Clin. Appl. 7, 283–291 [DOI] [PubMed] [Google Scholar]
- 66. Sakurai H., Kubota K., Inaba S. I., Takanaka K., Shinagawa A. (2013) Identification of a metabolizing enzyme in human kidney by proteomic correlation profiling. Mol. Cell. Proteomics 12, 2313–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Liu N. Q., Braakman R. B., Stingl C., Luider T. M., Martens J. W., Foekens J. A., Umar A. (2012) Proteomics pipeline for biomarker discovery of laser capture microdissected breast cancer tissue. J. Mammary Gland Biol. Neoplasia 17, 155–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tao Y., Fang L., Yang Y., Jiang H., Yang H., Zhang H., Zhou H. (2013) Quantitative proteomic analysis reveals the neuroprotective effects of huperzine A for amyloid beta treated neuroblastoma N2a cells. Proteomics 13, 1314–1324 [DOI] [PubMed] [Google Scholar]
- 69. Craven R. A., Cairns D. A., Zougman A., Harnden P., Selby P. J., Banks R. E. (2013) Proteomic analysis of formalin-fixed paraffin-embedded renal tissue samples by label-free MS: assessment of overall technical variability and the impact of block age. Proteomics Clin. Appl. 7, 273–282 [DOI] [PubMed] [Google Scholar]
- 70. Hogl S., van Bebber F., Dislich B., Kuhn P. H., Haass C., Schmid B., Lichtenthaler S. F. (2013) Label-free quantitative analysis of the membrane proteome of Bace1 protease knock-out zebrafish brains. Proteomics 13, 1519–1527 [DOI] [PubMed] [Google Scholar]
- 71. Tsai S. T., Tsou C. C., Mao W. Y., Chang W. C., Han H. Y., Hsu W. L., Li C. L., Shen C. N., Chen C. H. (2012) Label-free quantitative proteomics of CD133-positive liver cancer stem cells. Proteome Sci. 10, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aye T. T., Soni S., van Veen T. A., van der Heyden M. A., Cappadona S., Varro A., de Weger R. A., de Jonge N., Vos M. A., Heck A. J., Scholten A. (2012) Reorganized PKA-AKAP associations in the failing human heart. J. Mol. Cell. Cardiol. 52, 511–518 [DOI] [PubMed] [Google Scholar]
- 73. Merl J., Ueffing M., Hauck S. M., von Toerne C. (2012) Direct comparison of MS-based label-free and SILAC quantitative proteome profiling strategies in primary retinal Muller cells. Proteomics 12, 1902–1911 [DOI] [PubMed] [Google Scholar]
- 74. Sessler N., Krug K., Nordheim A., Mordmuller B., Macek B. (2012) Analysis of the Plasmodium falciparum proteasome using Blue Native PAGE and label-free quantitative mass spectrometry. Amino Acids 43, 1119–1129 [DOI] [PubMed] [Google Scholar]
- 75. Zelenak C., Foller M., Velic A., Krug K., Qadri S. M., Viollet B., Lang F., Macek B. (2011) Proteome analysis of erythrocytes lacking AMP-activated protein kinase reveals a role of PAK2 kinase in eryptosis. J. Proteome Res. 10, 1690–1697 [DOI] [PubMed] [Google Scholar]
- 76. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Muller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J. J., Cox J., Horning S., Mann M., Makarov A. (2012) Ultra high resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 11, O111.013698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Michalski A., Damoc E., Hauschild J. P., Lange O., Wieghaus A., Makarov A., Nagaraj N., Cox J., Mann M., Horning S. (2011) Mass spectrometry-based proteomics using Q Exactive, a high-performance benchtop quadrupole Orbitrap mass spectrometer. Mol. Cell. Proteomics 10, M111.011015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.